
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mastering palladium-catalyzed cross-coupling reactions: the critical role of in situ pre-catalyst reduction design†
Tommaso
Fantoni‡
 a,
Chiara
Palladino‡
a,
Riccardo
Grigolato
a,
Beatrice
Muzzi
b,
Lucia
Ferrazzano
a,
Alessandra
Tolomelli
a and
Walter
Cabri
*a
a,
Chiara
Palladino‡
a,
Riccardo
Grigolato
a,
Beatrice
Muzzi
b,
Lucia
Ferrazzano
a,
Alessandra
Tolomelli
a and
Walter
Cabri
*a
aTolomelli-Cabri Lab, Center for Chemical Catalysis, Department of Chemistry “Giacomo Ciamician”, University of Bologna, via Gobetti, 85-40129 Bologna, Italy. E-mail: walter.cabri@unibo.it
bICCOM-CNR, Sesto Fiorentino FI, I-50019, Italy
First published on 22nd January 2025
Abstract
Palladium-catalyzed cross-coupling reactions are among the most used methods for carbon–carbon bond formation in the agrochemical and pharmaceutical segments. The key step common to all methodologies based on Pd(0) catalysis is the in situ generation of the active catalyst. This paper describes how to control pre-catalyst reduction in order to generate the target complex species while avoiding phosphine oxidation or, as in the case of the Heck–Cassar–Sonogashira and the Suzuki–Miyaura reactions, reactant consumption via dimerization. For PPh3, DPPF, DPPP, Xantphos, SPhos, RuPhos, XPhos and sSPhos, we identified protocols that are able to maximize reduction via alcohols while preserving ligands and reagents. The correct combination of counterion, ligand, and base allowed the perfect control of the Pd(II) reduction to Pd(0) in the presence of primary alcohols.
Introduction
Palladium-catalyzed cross-coupling reactions are a cornerstone of modern synthetic chemistry, enabling the formation of carbon–carbon and carbon–heteroatom bonds with high efficiency and selectivity.1 These reactions are particularly valuable in the pharmaceutical and agrochemical industries2 due to their versatility and ability to create complex molecules with precise control over functional groups. Among them, key reactions include the Suzuki–Miyaura (SM),3 Heck–Cassar–Sonogashira (HCS),4 Mizoroki–Heck (MH),5 Stille,6 and Buchwald–Hartwig (BH)7 couplings. The preferred approach for managing Pd(0)-catalyzed reactions involves the use of Pd(II) salts like palladium(II) acetate (Pd(OAc)2) or palladium(II) chloride (PdCl2/PdCl2(ACN)2) to be combined with the correct ligand to generate pre-catalysts.Simple Pd(II) complexes are available, stable at room temperature, and cost-effective compared to preformed pre-catalysts or the direct use of Pd(0) complexes, making them a practical choice for both academic and industrial applications. However, a complete guide to perform efficiently in situ pre-catalyst reduction to generate the active Pd(0) is not available. To address this, several strategies have emerged.8 The use of Pd2(dba)3 allows the generation of the Pd(0)phosphine complex by a simple ligand exchange. Unfortunately, the palladium manipulation increases the costs and the presence of nanoparticles in the Pd2(dba)3 complex is a serious issue.9 On the other hand, well-defined Pd(II) pre-catalysts have been designed to undergo rapid reductive elimination,10 facilitating the formation of the target Pd(0) complex. Although these approaches minimize side reactions and ensure a smoother transition into the catalytic cycle, they have drawbacks limiting their industrial application. In fact, Pd(II) catalysts may have intellectual property protection11 and/or synthesis requires further manipulation of the ligand and the expensive metal, thus decreasing the overall efficiency and increasing costs.
The simple mixing of Pd(II) salts, ligands, auxiliaries, and substrates under standard reaction conditions does not guarantee the efficient formation of the active Pd(0)Ln species necessary to initiate and sustain catalytic cycles in cross-coupling reactions. Fig. 1 describes the first step of the Pd(II) reduction process where different reducing agents enter the palladium coordination sphere.
 | ||
| Fig. 1 Pd(II) pre-catalyst reduction. | ||
Phosphine ligands, which play a key role in many reactions, are sometimes expensive, and not recyclable.
Efficient in situ reduction of Pd(II) to Pd(0) is essential for optimizing reaction performance, reducing palladium usage, decreasing the costs and enhancing sustainability. This is not applicable to very basic phosphines, such as tri-tert-butylphosphine (tBu3P) and tricyclohexylphosphine (Cy3P),12 which are rapidly oxidized. Inefficient reduction can lower catalytic activity, requiring higher palladium loadings to achieve the desired results. The Pd(II)/Pd(0) conversion process has been extensively studied by various scientists, with significant contributions from Amatore/Jutand.13 In these studies, the palladium source was Pd(OAc)2, which is a trimer in the solid state.14 However, for clarity, in the present study, we will describe palladium acetate in its monomeric form since in solution we have never detected the trimeric one.15 Typically, the reduction of the metal occurs at the expense of the phosphine ligand or some reagents. While Amatore/Jutand and many other scientists have studied metal reduction at the expense of phosphine ligands, it has not been clearly described how to avoid scenarios where the phosphine ligand undergoes oxidation to form phosphine oxide altering the ligand-to-metal ratio. Indeed, this oxidative pathway can impact the structure and stability of the desired catalysts. For example, when BINAP or any chiral bidentate phosphine is used as a ligand, the transfer of chiral information is ensured only if the ligand remains unoxidized.16 In addition, the primary risk is the formation of mixed catalysts or nanoparticles,17 which exhibit significantly different reactivity compared to the intended catalysts. Employing a large excess of ligand can overcome the oxidation issue, but this approach can influence the reaction outcome. It can stabilize unreactive complexes or inhibit specific steps of the catalytic cycle that require ligand dissociation.18 Therefore, careful consideration and balance are necessary to optimize the ligand usage without compromising the desired catalytic activity.
Pre-catalyst reduction can also be performed at the expense of reagents with the concomitant formation of impurities. At the industrial level, especially in the pharmaceutical and agrochemical segments, this can be an issue in terms of efficiency because most of the time expensive fragments are consumed. In addition, for example, using a 0.1–1 mol% catalyst loading to produce 1000 tons of the product, as in the case of the fungicide Boscalid,19 generates 1–10 tons of waste in the boronate palladium reduction step as side products.
The combination of the factors discussed above based on a uncontrolled balance between palladium and the ligand can lead to a complete misinterpretation of the reaction data that are not based on the formation of the targeted Pd(0) catalyst. Typical examples are ligand screenings that are generally performed under standard reaction conditions.20 The pre-catalyst reduction efficiency is determined by several components: ligand, base, temperature, and solvent. Moreover, the sequence of addition of pre-catalysts, ligands and auxiliaries affects the efficiency of catalyst formation.
Our previous research on the HCS reaction using triphenylphosphine (PPh3) allowed carrying out straightforward mechanistic studies through DFT calculations, kinetic studies, NMR experiments, and the isolation of Pd(II) complexes.21 However, transitioning to bidentate phosphines or Buchwald's first-generation ligands introduces significant complexity as proved by the inconclusive 31P NMR spectra, due to the poor control over the formation of the Pd(0) catalyst which leads to unexpected Pd(0) complexes. The variations in ligand properties, such as steric and electronic effects, further complicate the formation and stability of the active Pd(0) species, affecting the catalytic cycle and reaction efficiency. To address these challenges, systematic studies and a combination of experimental and computational evaluations have been performed to understand and control the behavior of these ligands in the catalytic system.
This paper aims to shed light on the Pd(II) reduction process by studying the effects of ligands, salts, bases, and reagents in order to perfectly control the process, maximizing the rapid formation of the active Pd(0) catalyst, avoiding substrate consumption and preventing the formation of nanoparticles by maintaining the correct metal/ligand ratio. In particular, we have focused the study on the HCS, SM, MH and Stille reactions.
Results and discussion
We have considered as the palladium source the stable and largely available Pd(OAc)2, the readily available PdCl2(ACN)2 (instead of the commonly used PdCl2) and, in the case of DPPF as the ligand, PdCl2(DPPF). The two acetate and chloride counterions exhibit completely different behaviors, which are directly linked to the strength of the Pd–X bond. Thus, the effects of ligands, bases and the reaction conditions had to be studied with both salts. Phosphine ligands are widely used in coordination chemistry.22 Among them, we decided to investigate the following: (i) the monodentate triphenylphosphine (PPh3) which is popular thanks to its low cost and availability; (ii) bidentate phosphine ligands, namely 1,1′-bis(diphenylphosphino)ferrocene (DPPF), 1,3-bis(diphenylphosphino)propane (DPPP), and van Leuween's large bite-angle phosphine 4,5-bis(diphenylphosphino)-9,9-dimethyl-xanthene (Xantphos);23 and (iii) basic monodentate Buchwald's phosphines such as 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos).24 The reduction process was evaluated using 31P NMR and DFT calculations25 and for selected reduction processes the detection of nanoparticles was also investigated. The Pd(II) reduction was studied in dimethylformamide (DMF), a polar aprotic solvent able to solubilize all the pre-catalysts with the only exception of Pd(OAc)2/Xantphos that required the use of tetrahydrofuran (THF). In DMF or THF, we also carried out the reaction with 30% of N-hydroxyethyl pyrrolidone (HEP) as a cosolvent to reduce Pd(II) via oxidation of the primary alcohol moiety (Fig. 2, mechanism A).26 The effect of HEP is similar to that of primary alcohols with the difference that product extraction does not require extensive quantities of organic solvents.27 The reduction was carried out in the presence of several bases, such as N,N,N′,N′-tetramethyl guanidine (TMG), triethylamine (TEA), Cs2CO3, K2CO3, and pyrrolidine (for details, see Tables S2 and S3†). | ||
| Fig. 2 Reduction mechanism A via oxidation of a primary alcohol. | ||
Cross-coupling partners as reductive agents
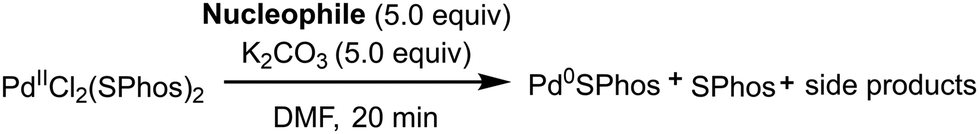
In order to understand which partner in the different cross-coupling reactions could be consumed in the presence of a pre-catalyst, we have used as a model the pre-catalyst generated in situ from PdCl2(ACN)2 and two equivalents of SPhos. In fact, under these conditions, the stable complex PdCl2(SPhos)2 is rapidly formed (Table 1, entry 1). The effect of the excess reactant on the reduction of the metal, generating monoligated Pd(0)SPhos + free SPhos, can be evaluated by 31P NMR. We decided to limit the monitoring to 20 minutes after the addition of 5 equiv. of the reactant at 25 °C or 60 °C, since the catalyst reduction is rapidly completed. The stoichiometry of the investigation was in some way conservative, since in standard cross-coupling reactions, the ratio between palladium and the aryl halide is usually >100. The reagents investigated were styrene for the Heck reaction, tributylphenylstannane for the Stille reaction, phenyl boronic acid for the Suzuki–Miyaura reaction, and phenylacetylene for the Heck–Cassar–Sonogashira reaction. All the experiments were carried out in DMF using K2CO3 as the base. The pre-catalyst proved to be stable under HCS and Stille conditions (entries 2–5) and no traces of Pd(0)SPhos or free SPhos were observed. On the other hand, while the pre-catalyst was stable at room temperature in the presence of phenyl boronate and phenylacetylene (entries 6 and 8), at 60 °C, it was quantitatively reduced to Pd(0)SPhos by both reagents (entries 7 and 9). In other words, the risk of reactant consumption in reducing the pre-catalyst has to be considered only during Suzuki or HCS reactions. In addition to carrying out cross-coupling reactions with low palladium loading according to the green chemistry principles, it is necessary to fulfil other sustainability requirements like avoiding reagent excess or the formation of byproducts that affects both the yield and the product purification processes. In particular, as for HCS and Suzuki reactions, the definition of a fast alternative method to control the pre-catalyst reduction is a must, since the formation of bis-alkyne and biphenyl, respectively, should be avoided (Fig. 3, mechanisms B and C). | ||
| Fig. 3 Reduction mechanisms B and C, via alkyne and boronate, respectively. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Reactant | T (°C) | Reaction | Pd(0)/Pd(II)b |
| a Reactions were carried out with 0.013 mmol in 600 μL of DMF for 20 minutes. b Conversion of Pd(II) into Pd(0) was calculated by 31P NMR with an internal standard 20 minutes after the addition of the reactant. | ||||
| 1 | — | 25 | — | 0/100 |
| 2 | Styrene | 25 | MH | 0/100 |
| 3 | Styrene | 60 | MH | 0/100 |
| 4 | PhSnBu3 | 25 | Stille | 0/100 |
| 5 | PhSnBu3 | 60 | Stille | 0/100 |
| 6 | PhB(OH)2 | 25 | SM | 0/100 |
| 7 | PhB(OH)2 | 60 | SM | 100/0 |
| 8 | PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH CH |
25 | HCS | 0/100 |
| 9 | PhCCH |
60 | HCS | 100/0 |
The appropriate PPh3/Pd(II) ratio was established to be 3/1, to avoid the formation of palladium nanoparticles and also to compensate for the reduced amount of available phosphine due to oxidation. The PdCl2(PPh3)2 precomplex was stable in DMF (Table 2, entry 1) and it was reduced to Pd(0) only after the addition of TMG with the concomitant formation of OPPh3 (entry 2). TMG and secondary amines can coordinate with palladium, displacing 1 equiv. of PPh3. This mechanism facilitates the oxidation of PPh3 (Fig. 4, mechanism D). In contrast, no reaction occurred in the presence of TEA even at 80 °C (entry 3). With inorganic bases, the pre-catalyst reduction was slower than the one promoted by TMG. Partial conversion was observed in 20 minutes only at 60 °C (entries 4–6), following mechanism E (Fig. 4). By adding HEP in a 1/2 ratio with DMF, the supplementary reduction pathway via mechanism A allowed for complete pre-catalyst reduction at 25 °C with TMG and Cs2CO3 (entries 7 and 8).
 | ||
| Fig. 4 Reduction mechanisms D and E, via displacement of the ligand and counterion, respectively. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X | Sol. | Base | T (°C) | Mech. | Pd(0)/Pd(II)b | P/OHc |
| a Reactions were carried out with 0.013 mmol in 600 μL of solvent for 20 minutes. b Conversion of Pd(II) into Pd(0) was calculated by 31P NMR with an internal standard 20 minutes after the addition of the base. c P/OH is the ratio between the reduction via phosphine (P) and alcohol (OH) and “n.d.” means not determined. d DMF/HEP were used in a 2/1 ratio. | |||||||
| 1 | Cl | DMF | — | 60 | — | 0/100 | — |
| 2 | Cl | DMF | TMG | 25 | D | 100/0 | 100/0 |
| 3 | Cl | DMF | TEA | 80 | — | 0/100 | — |
| 4 | Cl | DMF | Cs2CO3 | 25 | — | 0/100 | — |
| 5 | Cl | DMF | Cs2CO3 | 60 | E | 34/66 | 100/0 |
| 6 | Cl | DMF | K2CO3 | 60 | E | 12/88 | 100/0 |
| 7 | Cl | DMF/HEPd | TMG | 25 | A/D | 100/0 | n.d. |
| 8 | Cl | DMF/HEPd | Cs2CO3 | 25 | A | 100/0 | 0/100 |
| 9 | Cl | DMF/HEPd | K2CO3 | 25 | A | 28/72 | 0/100 |
| 10 | Cl | DMF/HEPd | K2CO3 | 60 | A/D | 100/0 | n.d. |
| 11 | AcO | DMF | — | 25 | E | 42/58 | 100/0 |
| 12 | AcO | DMF | — | 60 | E | 100/0 | 100/0 |
| 13 | AcO | DMF | TMG | 25 | D/E | 100/0 | 100/0 |
| 14 | AcO | DMF | Cs2CO3 | 25 | D | 43/57 | 100/0 |
| 15 | AcO | DMF/HEPd | Cs2CO3 | 25 | A/E | 100/0 | n.d. |
| 16 | AcO | DMF/HEPd | K2CO3 | 25 | A/E | 100/0 | n.d. |

Complete reduction was observed with K2CO3 only when the temperature was increased to 60 °C (entries 9 and 10).
Pre-catalyst reduction by phosphine oxidation
PPh3 is the reference standard as it is the cheapest and most popular phosphine used as a palladium ligand (Table 2).As expected, the reduction of Pd(OAc)2 was much faster than the one of PdCl2. In DMF, indeed, even in the absence of a base, Pd(0) was partially formed at 25 °C and it was then completely obtained at 60 °C (entries 11 and 12). With TMG, the reduction was completed at 25 °C (entry 13). These data indicate that the base is playing a key role in accelerating the reduction process with Pd(OAc)2.
With the inorganic bases and Pd(OAc)2, 100% reduction was achieved at 25 °C only in the presence of HEP (entries 14–16). While for the above-described experiments, the preferred pathways could be envisaged, in a few cases (entries 7, 10, 15, and 16), it was not possible to determine the predominant reduction mechanism clearly. In general, the acetate can easily dissociate from the metal generating a cationic palladium species that is stabilized by excess inorganic salts, following mechanism E. These results suggested that with PPh3 it is difficult to avoid phosphine oxidation, and the only exceptions were the reaction with chloride as the counterion and Cs2CO3 or K2CO3 as the base at 25 °C (entries 8 and 9).
Bidentate ligands, DPPP, DPPF, and Xantphos
Bidentate ligands are widely used in palladium cross-coupling reactions and for this reason DPPF, DPPP and Xantphos were also investigated. These ligands are characterized by completely different bite angles, respectively, of 91° for DPPP, 96° for DPPF and 112° for Xantphos, with the one of DPPP being close to the perfect angle for a square planar complex.28 The three phosphines are also different in terms of basicity, DPPP being more basic than the others because of the presence of an alkyl moiety. The use of only 1 equiv. of bidentate ligand can generate nanoparticles and less stable catalysts, and for this reason, all the experiments have been carried out using 2 equiv. There are several papers describing the beneficial effects of cross-coupling reactions when the bidentate phosphine is oxidized to the corresponding monophosphine oxide.29,30 Thus, we evaluated if the combination of base and solvent can generate one of the target catalysts reported in Fig. 5. | ||
| Fig. 5 Complexes 1, 2, and 3 are potential Pd(0) species, generated during pre-catalyst reduction in the presence of DPPF or DPPP. | ||
Using 31P NMR with Pd(DPPF)X2 it was possible to discriminate between reduction mechanisms A and D/E, comparing Pd0(DPPF) 1a that is in equilibrium with Pd0(DPPF)24a,31 Pd0(DPPF)DPPF(O) 2a, DPPF and DPPF(0) (for details, see Fig. S73–S96†). Pd(DPPF)Cl2 is stable in DMF at 60 °C (Table 3, entry 1) and can be efficiently and rapidly reduced in the presence of a base (entries 2–4) such as TMG via mechanism D or the inorganic ones via mechanism E. The presence of HEP favors reduction (entries 5–8). However, only with K2CO3 at 25 °C was the reduction selectively achieved via mechanism A (entry 7). With Pd(OAc)2 the pre-catalyst reduction was efficient even in the absence of a base (entries 9 and 10) while in the presence of any base, the reduction was completed at 25 °C within 20 minutes (entries 11–13). Summing up, independent of the Pd(II) source in the presence of HEP, K2CO3 at 25 °C was able to selectively generate Pd0(DPPF) (entries 7 and 12).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X | Sol. | Base | T (°C) | Mech. | Pd(0)/Pd(II)b | P/OHc |
| a Reactions were carried out with 0.013 mmol in 600 μL of solvent for 20 minutes. b Conversion of Pd(II) into Pd(0) was calculated by 31P NMR with an internal standard 20 minutes after the addition of the base. c P/OH is the ratio between the reduction via phosphine (P) and alcohol (OH). d DMF/HEP were used in a 2/1 ratio. | |||||||
| 1 | Cl | DMF | — | 60 | — | 0/100 | — |
| 2 | Cl | DMF | TMG | 25 | D | 100/0 | 100/0 |
| 3 | Cl | DMF | Cs2CO3 | 60 | E | 100/0 | 100/0 |
| 4 | Cl | DMF | K2CO3 | 60 | E | 100/0 | 100/0 |
| 5 | Cl | DMF/HEPd | TMG | 25 | A/D | 100/0 | 91/9 |
| 6 | Cl | DMF/HEPd | Cs2CO3 | 25 | A/E | 100/0 | 30/70 |
| 7 | Cl | DMF/HEPd | K2CO3 | 25 | A | 100/0 | 0/100 |
| 8 | Cl | DMF/HEPd | K2CO3 | 60 | A/E | 100/0 | 78/22 |
| 9 | AcO | DMF | — | 25 | E | 20/80 | 100/0 |
| 10 | AcO | DMF | — | 60 | E | 100/0 | 100/0 |
| 11 | AcO | DMF | TMG | 25 | D | 100/0 | 100/0 |
| 12 | AcO | DMF/HEPd | K2CO3 | 25 | A | 100/0 | 0/100 |
| 13 | AcO | DMF/HEPd | Cs2CO3 | 25 | A/E | 100/0 | 45/55 |

The DPPP pre-catalyst generated with PdCl2(ACN)2 was perfectly stable at 60 °C in DMF (Table 4, entry 1). Interestingly, the addition of TMG did not promote metal reduction (entry 2). While TMG is able to compete with DPPF in coordinating PdCl2, promoting mechanism D (Table 3, entry 2), in the case of the more basic DPPP, TMG was not able to compete with the phosphine in coordinating Pd(II) and mechanism D was completely inhibited even at 60 °C (Table 4, entry 2). Only by moving to inorganic bases like Cs2CO3 and K2CO3 at 60 °C was the reduction completed via mechanism E (entries 3 and 4). The addition of HEP allowed switching to alcohol-based mechanism A at 25 °C (Fig. 2), generating selectively 2b (entries 5 and 6). On moving to acetate as the counterion, the trend was identical to that of DPPF, with the reduction taking place even in the absence of a base via mechanism E (entries 7 and 8), and it was accelerated at 25 °C by the addition of inorganic bases and HEP as a cosolvent (entries 9 and 10). Only by using PdCl2 in the presence of HEP and inorganic bases (K2CO3 or Cs2CO3) at 25 °C could ligand oxidation be avoided (entries 5 and 6).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X | Sol. | Base | T (°C) | Mech.b | Pd(0)/Pd(II)b | P/OHc |
| a Reactions were carried out with 0.013 mmol in 600 μL of solvent for 20 minutes. b Conversion of Pd(II) into Pd(0) was calculated by 31P NMR with an internal standard 20 minutes after the addition of the base. c P/OH is the ratio between the reduction via phosphine (P) and alcohol (OH) and “n.d.” means not determined. d DMF/HEP were used in a 2/1 ratio. | |||||||
| 1 | Cl | DMF | — | 60 | — | 0/100 | — |
| 2 | Cl | DMF | TMG | 60 | — | 0/100 | — |
| 3 | Cl | DMF | Cs2CO3 | 60 | E | 100/0 | 100/0 |
| 4 | Cl | DMF | K2CO3 | 60 | E | 100/0 | 100/0 |
| 5 | Cl | DMF/HEPd | Cs2CO3 | 25 | A | 100/0 | 0/100 |
| 6 | Cl | DMF/HEPd | K2CO3 | 25 | A | 55/45 | 0/100 |
| 7 | AcO | DMF | — | 25 | E | 11/89 | 100/0 |
| 8 | AcO | DMF | — | 60 | E | 100/0 | 100/0 |
| 9 | AcO | DMF/HEPd | Cs2CO3 | 25 | A/E | 100/0 | n.d. |
| 10 | AcO | DMF/HEPd | K2CO3 | 25 | A/E | 100/0 | n.d. |

In Table 5 the experiments with Xantphos are reported. The pre-catalyst with chloride as the counterion was stable even in the presence of bases (entries 1–4). Upon addition of NaOAc, a rapid exchange with chloride promoted palladium reduction via mechanism E (see entries 5 and 9). Eastgate and Blackmond, in an interesting paper resulting from the collaboration between academia and industry, pointed out the role of Xantphos monophosphine oxide in a CH activation reaction as an “hemilabile” efficient ligand 3c.29
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X | Sol. | Base | T (°C) | Mech.b | Pd(0)/Pd(II)b | P/OHc |
| a Reactions were carried out with 0.013 mmol in 600 μL of solvent for 20 minutes. b Conversion of Pd(II) into Pd(0) was calculated by 31P NMR with internal standard 20 minutes after the addition of the base. c P/OH is the ratio between the reduction via phosphine (P) and alcohol (OH). d DMF/HEP were used in a 2/1 ratio. | |||||||
| 1 | Cl | DMF | — | 60 | — | 0/100 | — |
| 2 | Cl | DMF | TMG | 60 | — | 0/100 | — |
| 3 | Cl | DMF | Cs2CO3 | 60 | — | 0/100 | — |
| 4 | Cl | DMF | K2CO3 | 60 | — | 0/100 | — |
| 5 | Cl | DMF | NaOAc | 60 | E | 100/0 | 100/0 |
| 6 | Cl | DMF/HEPd | Cs2CO3 | 60 | A | 100/0 | 0/100 |
| 7 | Cl | DMF/HEPd | K2CO3 | 60 | A | 100/0 | 0/100 |
| 8 | AcO | THF | — | 25 | — | 0/100 | — |
| 9 | AcO | THF | — | 60 | E | 100/0 | 100/0 |
| 10 | AcO | THF | TMG | 25 | — | 0/100 | — |
| 11 | AcO | THF | K2CO3 | 25 | — | 0/100 | — |
| 12 | AcO | THF | Cs2CO3 | 25 | E | 40/60 | 100/0 |
| 13 | AcO | THF/HEPd | Cs2CO3 | 25 | A/E | 100/0 | 47/53 |
| 14 | AcO | THF/HEPd | K2CO3 | 25 | A | 100/0 | 0/100 |

The catalyst (4 mol%) was generated using dimethyl acetamide and the conditions very similar to entry 5 in Table 5. The role of the acetate was not only critical for the cross-coupling step but also for palladium reduction via mechanism E and the selective formation of the monophosphine oxide. In the presence of HEP, the inorganic salts allowed the achievement of the reduction of the pre-catalyst via mechanism A (entries 6 and 7). Since the combination Pd(OAc)2/Xantphos is not soluble in DMF, THF was used. Under these conditions, complete catalyst reduction occurred at 60 °C (Table 5, entries 8 and 9), and the process was accelerated in the presence of Cs2CO3 (entry 12) at 25 °C but not with TMG and K2CO3 (entries 10 and 11). Again, the addition of HEP was able to favor Pd(II) reduction via mechanism A at 25 °C (entries 13 and 14). Interestingly, when the Xantphos ligand was combined with K2CO3 as a base, with both PdCl2 and Pd(OAc)2, it was possible to reduce the metal without phosphine oxidation at 60 °C and 25 °C, respectively, in the presence of HEP (entries 6 and 13). With Cs2CO3, only with chloride as the counterion was it possible to achieve complete reduction via mechanism A at 60 °C (entry 6).
Pre-catalyst reduction by SPhos
Since we have carried out extensive catalytic studies with the corresponding water-soluble sulfonate ligand sSPhos,26 we decided to study the pre-catalyst generated in situ by this phosphine. In addition, SPhos is frequently used in Suzuki–Miyaura reactions.24 The main characteristic of this ligand is that the Pd(II) complex coordinates with two SPhos ligands while Pd(0) coordinates with only one in the twelve-electron complex.18,26b,32 For this reason we decided to use 2 equiv. to stabilize the complexes in the case of phosphine oxidation.The reduction via phosphorus oxidation using PdCl2 did not take place even in the presence of bases (Table 6, entries 1–4). However, adding HEP enabled reduction via primary alcohol oxidation of the pre-catalyst in the presence of bases, with inorganic ones proving to be more efficient (entries 5–8). Also with Pd(OAc)2, the reduction took place in DMF with or without the bases (entries 9–12). In the presence of HEP, Pd(0) was selectively generated via mechanism A with only K2CO3 at 25 °C (entry 14).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X | Sol. | Base | T (°C) | Mech.b | Pd(0)/Pd(II)b | P/OHc |
| a Reactions were carried out with 0.013 mmol in 600 μL of solvent for 20 minutes. b Conversion of Pd(II) into Pd(0) was calculated by 31P NMR with an internal standard 20 minutes after the addition of the base. c P/OH is the ratio between the reduction via phosphine (P) and alcohol (OH) and “n.d.” means not determined. d DMF/HEP were used in a 2/1 ratio. | |||||||
| 1 | Cl | DMF | — | — | — | 0/100 | — |
| 2 | Cl | DMF | TMG | 60 | — | 0/100 | — |
| 3 | Cl | DMF | Cs2CO3 | 60 | — | 0/100 | — |
| 4 | Cl | DMF | K2CO3 | 60 | — | 0/100 | — |
| 5 | Cl | DMF/HEPd | Cs2CO3 | 25 | A | 56/44 | 0/100 |
| 6 | Cl | DMF/HEPd | Cs2CO3 | 60 | A | 100/0 | 0/100 |
| 7 | Cl | DMF/HEPd | K2CO3 | 60 | A | 100/0 | 0/100 |
| 8 | Cl | DMF/HEPd | TMG | 60 | A | 15/85 | 0/100 |
| 9 | AcO | DMF | — | 60 | E | 29/71 | 100/0 |
| 10 | AcO | DMF | TMG | 60 | E | 15/85 | 100/0 |
| 11 | AcO | DMF | Cs2CO3 | 25 | E | 71/29 | 100/0 |
| 12 | AcO | DMF | K2CO3 | 25 | E | 54/46 | 100/0 |
| 13 | AcO | DMF/HEPd | Cs2CO3 | 25 | A/E | 100/0 | 42/58 |
| 14 | AcO | DMF/HEPd | K2CO3 | 25 | A | 100/0 | 0/100 |
| 15 | AcO | DMF/HEPd | TMG | 60 | A/E | 31/69 | n.d. |

Heck–Cassar–Sonogashira and Suzuki–Miyaura: mechanism A versus mechanisms B and C
The definition of the ideal conditions for mechanism A allowed the avoidance of, in HCS and SM cross-couplings, pre-catalyst reduction at the expense of alkynes or boronates. Pd(II)(SPhos)2Cl2 generated using PdCl2(ACN)2 and SPhos was rapidly reduced to the corresponding Pd(0) catalyst in HEP with K2CO3 as the base at room temperature in the presence of phenyl acetylene or phenylboronic acid (Fig. 6). Under these stressed conditions, with 20 mol% of palladium and a 1/1 ratio of PhI and the reactants, the homocoupling products resulting from pre-catalyst reduction through mechanism B (1,4-diphenylbuta-1,3-diyne) or C (4,4′-dimethyl-1,1′-biphenyl) were not detected. After pre-catalyst reduction in preparative HCS and SM reactions, the temperature can be increased to the desired level to ensure optimal efficiency and progress of the couplings. Using this protocol, homocoupling side products have never been observed, and the palladium metal loading was minimized.21,27 | ||
| Fig. 6 Heck–Cassar–Sonogashira and Suzuki–Miyaura cross-coupling effects of the presence of the reactants in HEP. | ||
Protocol variants
Monodentate Buchwald-type ligands are among the best for cross-coupling reactions and, for this reason, the reaction scope was expanded, targeting reduction mechanism A, for the generation of twelve-electron catalysts.18The PdCl2(ACN)2/ligand/alcohol/base protocol worked perfectly in several green solvent combinations33 with SPhos via mechanism A (Table 7, entries 1–5). The protocol was successfully applied to 2-dicyclohexylphosphino-2′,6′-diisopropoxy-1,1′-biphenyl (RuPhos)34 and 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (XPhos). With XPhos it was necessary to use toluene as a cosolvent because of the solubility of the ligand. As expected, the complex reduction was less efficient with organic bases like TMG and PYR. The presence of these bases that enter the coordination sphere of Pd(II) destabilized Pd(SPhos)2Cl2 and the formation of Pd(0)SPhos was less efficient (entries 8 and 9). The amount of Pd(0)SPhos did not increase over time and macroscopically, we observed the formation of palladium black after 1 h. The formation of palladium black suggests the rapid formation of soluble nanoparticles likely due to ligand loss. This negative outcome was observed with a conservative base excess at only 5 equivalents, whereas a typical reaction features a base/ligand ratio greater than 100.
| Entrya | Ligand | Base | Solvent | Pd(0)/Pd(II)b |
|---|---|---|---|---|
| a Reactions were carried out with 0.013 mmol in 600 μL of solvent for 20 minutes. b Conversion of Pd(II) into Pd(0) was calculated by 31P NMR with an internal standard 20 minutes after the addition of the base. | ||||
| 1 | SPhos | K2CO3 | Anisole/EtOH 2/1 | 100/0 |
| 2 | SPhos | K2CO3 | CPME/EtOH 2/1 | 100/0 |
| 3 | SPhos | K2CO3 | MeTHF/EtOH 2/1 | 100/0 |
| 4 | SPhos | K2CO3 | Anisole/MeOH 2/1 | 100/0 |
| 5 | SPhos | K2CO3 | Anisole/HEP 2/1 | 100/0 |
| 6 | RuPhos | K2CO3 | Anisole/EtOH 2/1 | 100/0 |
| 7 | XPhos | K2CO3 | Toluene/EtOH 2/1 | 100/0 |
| 8 | SPhos | PYR | Anisole/EtOH 2/1 | 25/0 |
| 9 | SPhos | TMG | Anisole/EtOH 2/1 | 28/0 |
| 10 | sSPhos | K2CO3 | HEP/H2O 4/1 | 100/0 |
| 11 | sSPhos | K2CO3 | EtOH/H2O 4/1 | 100/0 |
| 12 | sSPhos | PYR | EtOH/H2O 4/1 | 52/0 |
| 13 | sSPhos | K2CO3 | IPA/H2O 4/1 | 0/100 |
The use of sodium 2′-dicyclohexylphosphino-2,6-dimethoxy-1,1′-biphenyl-3-sulfonatehydrate (sSPhos) allowed the introduction of water as a cosolvent in the green protocol (see entries 10 and 11).26b,35,36 Also in this case the use of PYR generated only 52% of the expected Pd(0)sSPhos complex within 20 min (entry 12). Secondary alcohols like isopropanol (IPA), used to replace HEP or EtOH, were not able to reduce the pre-catalyst (entry 13). The solvent mixtures described in Table 6 were not optimized but simply demonstrated the general applicability of using alcohols for Pd(II) pre-catalyst reduction, provided the pre-catalyst was soluble at those concentrations and inorganic bases were preferred.
Mechanism A, DFT studies
Density functional theory (DFT) calculations were performed for mechanism A using the B3LYP/DEF2-TZVP level of theory,37 focusing on the cationic Pd(II) complex. We have excluded the counterion and the second SPhos in the Pd(II) precatalyst to prevent calculation inaccuracies arising from their presence. Therefore, a simplified Pd(II)–SPhos complex with DMF as the ligand was chosen as the reference model to evaluate the reduction process (Fig. 7). This approach generalizes the model for a variety of palladium systems. The initial stage in mechanism A involves the coordination of the carbonate and the alcohol, forming complex A with an energy of 6.9 kcal mol−1. This coordination makes the alcohol's proton more acidic, allowing the deprotonation by a base to yield B via the transition state TS0, which has an energy barrier of 5.2 kcal mol−1. The overall activation energy required for alcohol deprotonation is calculated to be 12.1 kcal mol−1, with the transition state influenced by the specific coordination of the carbonate to the palladium centre. Our calculations revealed that the most stable configuration involves both oxygen atoms of the carbonate coordinating to the palladium complex. The reaction pathway proceeds through beta-hydride elimination at TS1, with a very low energy barrier of 0.6 kcal mol−1, resulting in the formation of intermediate C that is by far the more stable complex. The DFT studies using PYR as the base showed a similar trend with higher transition energies (see Fig. S159†). In fact, the overall energy to reach the corresponding TS0 is 15.2 kcal, confirming that the reduction with PYR was less efficient. | ||
| Fig. 7 DFT-calculated reaction profile and solution-state Gibbs free energies (ΔGDMF, kcal mol−1) at the B3LYP/DEF2-TZVP level of theory at 298 K for stationary points of mechanism A. | ||
DFT studies identified the deprotonation process as the rate-determining step of mechanism A. This observation was confirmed by a kinetic isotope effect (KIE) study using 31P NMR (Fig. 8). In fact, the reaction with CH3OH was consistently faster than the one in CD3OD, with a KIE of 1.6.
 | ||
| Fig. 8 Kinetic isotope effect: CH3OH versus CD3OD. | ||
Conclusion
The results of this study led to the development of efficient protocols for synthesizing Pd(0)Ln complexes by simply mixing Pd(II) salts with phosphine ligands. The research explored various classes of phosphine ligands, alongside investigating the effects of palladium counterions, bases, and temperature on the reduction process. These factors were carefully chosen to prevent the oxidation of phosphines and the dimerization of reactants during Heck–Cassar–Sonogashira and Suzuki–Miyaura cross-couplings. Through Density Functional Theory (DFT) studies, the mechanism of the process was examined, revealing that the deprotonation that generates the oxygen–palladium sigma bond in the Pd(II) complex was the rate-determining step. Different classes of ligands were found to require specific reaction conditions to ensure high catalyst efficiency and optimal reaction performance. PdCl2 (or PdCl2(ACN)2) provides better control over metal reduction and catalyst stability than Pd(OAc)2 when primary alcohols and inorganic bases like K2CO3 are used. This approach allowed for robust and controlled catalyst formation. Furthermore, the best protocol developed for the ligand SPhos was successfully extended to other Buchwald ligands, demonstrating its versatility across various organic solvent mixtures, as long as a primary alcohol was present and the catalyst was soluble.Author contributions
TF and CP contributed equally to the investigation. TF, CP and RG performed the reactions and analysis. TF performed the DFT studies. AT and LF performed data reviewing. WC was responsible for conceptualization. The manuscript was written through contributions of all authors. All authors have given approval for the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI† or at data repository https://doi.org/10.6092/unibo/amsacta/8177.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DFT calculations for this work were performed at CINECA, through the Italian Super Computing Resource Allocation – ISCRA. The CINMPIS consortium (Interuniversity Research National Consortium for the Development of Innovative Methodologies for Synthetic Processes) and the C3-Center for Chemical Catalysis of the University of Bologna are also gratefully acknowledged.References
- (a) M. O. Kitching, T. J. Colacot and V. Snieckus, Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed; (b) T. Fantoni, A. Tolomelli and W. Cabri, A translation of the twelve principles of green chemistry to guide the development of cross-coupling reactions, Catal. Today, 2022, 397–399, 265–271 CrossRef CAS.
- Organometallic Chemistry in Industry: A Practical Approach, ed. T. J. Colacot and C. C. C. J. Seechurn, Wiley-VCH, Weinheim, 2020 Search PubMed.
- N. Miyaura and A. Suzuki, Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC.
- (a) H. A. Dieck and F. R. Heck, Palladium catalyzed synthesis of aryl, heterocyclic and vinylic acetylene derivatives, J. Organomet. Chem., 1975, 53, 259–263 CrossRef; (b) L. Cassar, Synthesis of aryl- and vinyl-substituted acetylene derivatives by the use of nickel and palladium complexes, J. Organomet. Chem., 1975, 93, 253–257 CrossRef CAS; (c) K. Sonogashira, Y. Tohda and N. Hagihara, A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- R. F. Heck, Palladium-catalyzed reactions of organic halides with olefins, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS.
- D. Milstein and J. K. Stille, A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium, J. Am. Chem. Soc., 1978, 100, 3636–3638 CrossRef CAS.
- (a) A. S. Guram, R. A. Rennels and S. L. Buchwald, A Simple Catalytic Method for the Conversion of Aryl Bromides to Arylamines, Angew. Chem., Int. Ed. Engl., 1995, 34, 1348–1350 CrossRef CAS; (b) J. Louie and J. F. Hartwig, Palladium-catalyzed synthesis of arylamines from aryl halides. Mechanistic studies lead to coupling in the absence of tin reagents, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef CAS.
- K. H. Shaughnessy, Development of Palladium Precatalysts that Efficiently Generate LPd(0) Active Species, Isr. J. Chem., 2019, 59, 1–16 CrossRef.
- S. S. Zalesskiy and V. P. Ananikov, Pd2(dba)3 as a Precursor of Soluble Metal Complexes and Nanoparticles: Determination of Palladium Active Species for Catalysis and Synthesis, Organometallics, 2012, 31(6), 2302–2309 CrossRef CAS.
- (a) B. T. Ingoglia, C. C. Wagen and S. L. Buchwald, Biaryl monophosphine ligands in palladium-catalyzed C–N coupling: An updated User's guide, Tetrahedron, 2019, 75, 4199–4211 CrossRef CAS PubMed; (b) K. S. Iyer, K. B. Dismuke Rodriguez, R. M. Lammert, J. R. Yirak, J. M. Saunders, R. D. Kavthe, D. H. Aue and B. H. Lipshutz, Rapid Aminations of Functionalized Aryl Fluorosulfates in Water, Angew. Chem., Int. Ed., 2024, e202411295 CAS.
- N. C. Bruno and S. L. BuchwaldPhosphine-Ligated Palladium Sulfonate Palladacycles, US Pat., US8981086, 2014 Search PubMed.
- T. Allman, R. G. Goel, T. Allman and R. G. Goel, Can. J. Chem., 1982, 60, 716–722 CrossRef CAS.
- Among many other papers we have selected: (a) C. Amatore, A. Jutand and A. Thuilliez, Formation of Palladium(0) Complexes from Pd(OAc)2 and a Bidentate Phosphine Ligand (dppp) and Their Reactivity in Oxidative Addition, Organometallics, 2001, 20, 3241–3249 CrossRef CAS; (b) S. Wagschal, L. A. Perego, A. Simon, A. Franco-Espejo, C. Tocqueville, J. Albaneze-Walker, A. Jutand and L. Grimaud, Formation of XPhos-Ligated Palladium(0) Complexes and Reactivity in Oxidative Additions, Chem. – Eur. J., 2019, 25, 6980–6987 CrossRef CAS PubMed.
- (a) W. A. Carole and T. J. Colacot, Understanding Palladium Acetate from a User Perspective, Chem. – Eur. J., 2016, 22, 1–11 CrossRef; (b) N. W. J. Scott, M. J. Ford, C. Schotes, R. R. Parker, A. C. Whitwood and I. J. S. Fairlamb, The ubiquitous cross-coupling catalyst system ‘Pd(OAc)2′/2PPh3 forms a unique dinuclear PdI complex: an important entry point into catalytically competent cyclic Pd3 clusters, Chem. Sci., 2019, 10, 7898 RSC; (c) M. Montgomery, H. M. O'Brien, C. Mendez-Galvez, C. R. Bromfield, J. P. M. Roberts, A. M. Winnicka, A. Horner, D. Elorriaga, H. A. Sparkes and R. B. Bedford, The surprisingly facile formation of Pd(I)-phosphido complexes from ortho-biphenylphosphines and palladium acetate, Dalton Trans., 2019, 48, 3539–3542 RSC.
- W. Carole, J. Bradley, M. Sarwar and T. J. Colacot, Can Palladium Acetate Lose Its “Saltiness”? Catalytic Activities of the Impurities in Palladium Acetate, Org. Lett., 2015, 17, 5472–5475 CrossRef CAS PubMed.
- (a) F. Ozawa, A. Kubo and T. Hayashi, Catalytic asymmetric arylation of 2,3-dihydrofuran with aryl triflates, J. Am. Chem. Soc., 1991, 113, 1417–1419 CrossRef CAS; (b) T. P. Imamoto, P-Stereogenic Phosphorus Ligands in Asymmetric Catalysis, Chem. Rev., 2024, 124, 8657–8739 CrossRef CAS PubMed; (c) R. Rio, H. Liang, M.-E. L. Perrin, L. A. Perego, L. Grimaud and P.-A. Payard, We Already Know Everything about Oxidative Addition to Pd(0): Do We?, ACS Catal., 2023, 13, 11399–11421 CrossRef.
- A. M. Trzeciak and A. W. Augustyniak, The role of palladium nanoparticles in catalytic C–C cross-coupling reactions Coord, Chem. Rev., 2019, 384, 1–20 CAS.
- S. J. Firsan, V. Sivakumar and T. J. Colacot, Emerging Trends in Cross-Coupling: Twelve-Electron-Based L1Pd(0) Catalysts, Their Mechanism of Action, and Selected Applications, Chem. Rev., 2022, 122, 16983–17027 CrossRef CAS PubMed.
- C. Torborg and M. Beller, Recent Applications of Palladium-Catalyzed Coupling Reactions in the Pharmaceutical, Agrochemical, and Fine Chemical Industries, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- C. M. So, O. Y. Yuen, S. S. Ng and Z. Chen, General Chemoselective Suzuki-Miyaura Coupling of Polyhalogenated Aryl Triflates Enabled by an Alkyl-Heteroaryl-Based Phosphine Ligand, ACS Catal., 2021, 11, 7820–7827 CrossRef CAS.
- C. Palladino, T. Fantoni, L. Ferrazzano, B. Muzzi, A. Ricci, A. Tolomelli and W. Cabri, New Mechanistic Insights into the Copper-Free Heck–Cassar–Sonogashira Cross-Coupling Reaction, ACS Catal., 2023, 13, 12048 CrossRef CAS.
- E. I. Musina, A. S. Balueva and A. A. Karasik, Tertiary phosphines: preparation and reactivity, Organophosphorus Chem., 2022, 51, 1–61 Search PubMed.
- P. C. J. Kamer, P. W. N. M. van Leuween and J. N. H. Reek, Wide Bite Angle Diphosphines: Xantphos Ligands in Transition Metal Complexes and Catalysis, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, Catalysts for Suzuki–Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed.
- T. E. Barder, M. R. Biscoe and S. L. Buchwald, Structural Insights into Active Catalyst Structures and Oxidative Addition to (Biaryl)phosphine–Palladium Complexes via Density Functional Theory and Experimental Studies, Organometallics, 2007, 26, 2183–2192 CrossRef CAS.
- (a) P. R. Melvin, D. Balcells, N. Hazari and A. Nova, Understanding Precatalyst Activation in Cross-Coupling Reactions: Alcohol Facilitated Reduction from Pd(II) to Pd(0) in Precatalysts of the Type (η3-allyl)Pd(L)(Cl) and (η3-indenyl)Pd(L)(Cl), ACS Catal., 2015, 5, 5596–5606 CrossRef CAS; (b) F. Sirindil, R. Pertschi, E. Naulin, D. Hatey, J.-M. Weibel, P. Pale and A. Aurélien Blanc, trans-Dichlorobis(XPhos)palladium(II) Precatalyst for Suzuki–Miyaura Cross-Coupling Reactions of Aryl/Vinyl Sulfonates/Halides: Scope, Mechanistic Study, and Synthetic Applications, ACS Omega, 2022, 7, 1186–1196 CrossRef CAS PubMed.
- (a) L. Ferrazzano, G. Martelli, T. Fantoni, A. Daka, D. Corbisiero, A. Viola, A. Ricci, W. Cabri and A. Tolomelli, Fast Heck-Cassar-Sonogashira (HCS) Reactions in Green Solvents, Org. Lett., 2020, 22, 3969–3973 CrossRef CAS PubMed; (b) T. Fantoni, S. Bernardoni, A. Mattellone, G. Martelli, L. Ferrazzano, P. Cantelmi, D. Corbisiero, A. Tolomelli, W. Cabri, F. Vacondio, F. Ferlenghi, M. Mor and A. Ricci, Palladium Catalyst Recycling for Heck-Cassar-Sonogashira Cross-Coupling Reactions in Green Solvent/Base Blend, ChemSusChem, 2021, 14, 2591–2600 CrossRef CAS PubMed.
- S. Subongkoj, S. Lange, W. Chen and J. Xiao, Effect of diphosphine ligands on ruthenium catalysed asymmetric hydrogenation of ketones, J. Mol. Catal. A: Chem., 2003, 196, 125–129 CrossRef CAS.
- P.-A. Payard, A. Bohn, D. Tocqueville, K. Jaouadi, E. Escoude, S. Ajig, A. Dethoor, G. Gontard, L. A. Perego, M. Vitale, I. Ciofini, S. Wagschal and L. Grimaud, Role of dppf Monoxide in the Transmetalation Step of the Suzuki–Miyaura Coupling Reactions, Organometallics, 2021, 40, 1120–1128 CrossRef CAS.
- Y. Ji, R. E. Plata, C. S. Regens, M. Hay, M. Schmidt, T. Razler, Y. Qiu, P. Geng, Y. Hsiao, T. Rosner, M. D. Eastgate and D. G. Blackmond, Mono-Oxidation of Bidentate Bis-Phosphines in Catalyst Activation: Kinetic and Mechanistic Studies of a Pd/Xantphos-Catalyzed C-H Functionalization, J. Am. Chem. Soc., 2015, 137, 13272–13281 CrossRef CAS PubMed.
- L. M. Alcazar-Roman, J. F. Hartwig, A. L. Rheingold, L. M. Liable-Sands and I. A. Guzei, Mechanistic Studies of the Palladium-Catalyzed Amination of Aryl Halides and the Oxidative Addition of Aryl Bromides to Pd(BINAP)2 and Pd(DPPF)2: An Unusual Case of Zero-Order Kinetic Behavior and Product Inhibition, J. Am. Chem. Soc., 2000, 122, 4618–4630 CrossRef CAS.
- U. Christmann and R. Vilar, Monoligated Palladium Species as Catalysts in Cross-Coupling Reactions, Angew. Chem., Int. Ed, 2005, 44, 366–374 CrossRef CAS PubMed.
- (a) F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Tools and techniques for solvent selection: green solvent selection guides, Chem. Process., 2016, 4, 7 Search PubMed; (b) J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Solvent effects in palladium catalysed cross-coupling reactions, Green Chem., 2019, 21, 2164–2213 RSC.
- (a) M. D. Charles, P. Schultz and S. L. Buchwald, Efficient Pd-Catalyzed Amination of Heteroaryl Halides, Org. Lett., 2005, 7, 3965–3968 CrossRef CAS PubMed; (b) R. Martin and S. L. Buchwald, Palladium-catalyzed Suzuki-Miyaura cross-coupling reactions employing dialkylbiaryl phosphine ligands, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- J. Struwe, L. Ackermann and F. Gallou, Recent progress in copper-free Sonogashira-Hagihara cross-couplings in water, Chem. Catal., 2022, 3, 100485 CrossRef.
- (a) E. B. Landstrom, S. Handa, D. H. Aue, F. Gallou and B. H. Lipshutz, EvanPhos: a ligand for ppm level Pd-catalyzed Suzuki–Miyaura couplings in either organic solvent or water, Green Chem., 2018, 20, 3436–3443 RSC; (b) B. S. Takale, R. R. Thakore, S. Handa, F. Gallou, J. Reillyd and B. H. Lipshutz, A new, substituted palladacycle for ppm level Pd-catalyzed Suzuki–Miyaura cross couplings in water, Chem. Sci., 2019, 10, 8825–8831 RSC; (c) N. Akporji, R. R. Thakore, M. Cortes-Clerget, J. Andersen, E. Landstrom, D. H. Aue, F. Gallou and B. H. Lipshutz, N2Phos – an easily made, highly effective ligand designed for ppm level Pd-catalyzed Suzuki–Miyaura cross couplings in water, Chem. Sci., 2020, 11, 5205–5212 RSC.
- S. A. Kondrashova, F. M. Polyancev and S. K. Latypov, DFT Calculations of 31P NMR Chemical Shifts in Palladium Complexes, Molecules, 2022, 27, 2668 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 31P NMR experiments, table with complete data and DFT calculations. See DOI: https://doi.org/10.1039/d4qo02335h |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |