
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of porous self-assembled homoleptic and heteroleptic Pd2+ cages incorporating silicon-based fluoride acceptors: the way towards nuclear imaging applications†
Marike
Drexler
a,
Ioannis
Kanavos
b,
Daniela
Krauss
a,
Guillermo
Moreno-Alcántar
 a,
Lydia M.
Smith
c,
Madeleine E.
George
c,
Timothy H.
Witney
c,
Ryszard
Lobinski
bd,
Luisa
Ronga
*b and
Angela
Casini
*a
a,
Lydia M.
Smith
c,
Madeleine E.
George
c,
Timothy H.
Witney
c,
Ryszard
Lobinski
bd,
Luisa
Ronga
*b and
Angela
Casini
*a
aChair of Medicinal and Bioinorganic Chemistry, School of Natural Sciences, Technical University of Munich, Garching b., München, Germany. E-mail: angela.casini@tum.de
bUniversité de Pau et des Pays de l'Adour, E2S UPPA, CNRS, IPREM, 2 Av. du Président Pierre Angot, Pau, France. E-mail: luisa.ronga@univ-pau.fr
cSchool of Biomedical Engineering and Imaging Sciences, King's College London, St Thomas’ Hospital, London, SE1 7EH, UK
dChair of Analytical Chemistry, Warsaw University of Technology, ul. Noakowskiego 3, 00-664 Warszawa, Poland
First published on 2nd June 2025
Abstract
Supramolecular self-assembly is a promising tool to develop multifunctional theranostic agents and has recently entered the field of radiochemistry. In this work, lantern-shaped [Pd2L4]4+ metallacages featuring a blood–brain barrier-penetrating peptide were designed for dual-modality imaging. Two silicon-based fluoride acceptors were incorporated in the cage ligand for radiolabeling with fluorine-18, an isotope used in positron emission tomography (PET). Ligands (L1, L2) and the respective homoleptic cages (C1hom [Pd2(L1)4]4+, C2hom [Pd2(L2)4]4+) were fully characterized by NMR spectroscopy, liquid chromatography and high-resolution electrospray mass spectrometry (HR-ESI-MS). Cage stability was assessed in different solvents and concentrations by HPLC. 18F-labelled cages were obtained by radiolabeling the ligands pre-assembly under mild conditions within four hours via [19F]-to-[18F] isotopic exchange. The high lipophilicity of the ligands was also assessed in vitro (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) DpH7.4) and in ovo, using the chick chorioallantoic membrane (CAM) model. Furthermore, formation of host–guest complexes between the metallacages and perrhenate (ReO4−), the ‘cold’ surrogate of radioactive 99mTcO4− (used for single photon computed tomography, SPECT), could be detected by mass spectrometry, although the adduct did not sustain chromatographic separation. To improve stability of the supramolecular system, heteroleptic cages of general formula [Pd2(L)m(L0)n]4+ (m = 1, 2, n = 4 − m) were synthesized by statistical self-assembly and separated by liquid chromatography in good yield. Overall, this study demonstrates the feasible adaptation of supramolecular principles to achieve innovative theranostic agents.
DpH7.4) and in ovo, using the chick chorioallantoic membrane (CAM) model. Furthermore, formation of host–guest complexes between the metallacages and perrhenate (ReO4−), the ‘cold’ surrogate of radioactive 99mTcO4− (used for single photon computed tomography, SPECT), could be detected by mass spectrometry, although the adduct did not sustain chromatographic separation. To improve stability of the supramolecular system, heteroleptic cages of general formula [Pd2(L)m(L0)n]4+ (m = 1, 2, n = 4 − m) were synthesized by statistical self-assembly and separated by liquid chromatography in good yield. Overall, this study demonstrates the feasible adaptation of supramolecular principles to achieve innovative theranostic agents.
Introduction
In the last few decades, supramolecular coordination complexes (SCCs) have been explored in different fields of chemistry due to the broad span of possible applications, including in medicine.1–3 SCCs are discrete molecular arrangements composed by metal nodes bound to organic ligands that come together via coordination-driven self-assembly (CDSA).4,5 A plethora of two-dimensional and three-dimensional shapes can be created upon thoughtful combination of the aforementioned building blocks, which morphologically are classified broadly into three main types, namely metallacycles, metallacages and helicates.6,7 The coordination geometry of the metal ion, ligand curvature, size and number of coordination sites encode the self-assembly of the proposed architecture.3,8 In this context, the case of the metallacages (MCgs) is particularly appealing since their porous nature favors their use to encapsulate and transport small molecules.9 Moreover, exo- and endo-functionalization of the ligands pre- and post-CDSA allows for further tuning of the supramolecular system. The host–guest chemistry and versatility of MCgs towards functionalization led to their investigation as catalysts,10,11 sensors12,13 and gas storage systems14 amongst other possible uses.15 Concerning biomedical applications, MCgs have been initially explored as potential carriers of cargos of pharmaceutical interest16–20 either for therapy or imaging (Fig. 1A), or for their intrinsic cytotoxic properties.21–23 Some MCgs structures have also been incorporated into nanoparticles for multimodal imaging and to improve their stability and targeting capability.24,25 Besides the cytotoxic potential of SCCs, their design can be directed to incorporate other interesting therapeutic modalities, such as photodynamic therapy via generation of reactive oxygen species at the target site.26–28 Recently, an Au(III)-porphyrin-based Ru8-cage was reported for G-quadruplex-DNA-targeted photodynamic therapy, which displays higher cytotoxicity upon irradiation compared to the free porphyrin.29Exo-functionalization of the cage ligand is a suitable strategy for the conjugation of targeting vectors towards cancer cells. As an example, [Pd2L4]4+ metallacages with ligands bioconjugated to integrin-binding RGD-peptides were developed.30 Similarly, MCgs can also be designed to display luminescent properties useful to study their fate in biological systems by optical imaging methods.31–34 | ||
| Fig. 1 Literature examples of MCgs designed for biomedical applications. (A) Self-assembled lantern shaped-[Pd2L2]4+ MCgs encapsulating the anticancer drug cisplatin; (B) tetrahedral Gd(III) MCgs used for MRI; (C) porphyrin-based MCgs designed for trimodal imaging (MRI, PET or aggregation-induced emission) in vivo. | ||
Concerning clinical imaging applications, self-assembled Gd(III)/Fe(III) tetrahedral cages (Fig. 1B), originally designed by Raymond et al.,35 were investigated by Morrow and coworkers as T1 MRI probes in vivo,36–39 showing resistance to dissociation in biological media. In 2018, the group of Lusby and Archibald made use of the encapsulation properties of SCCs and designed a Co(III)4L6 rhomboidal MCg for the incorporation of 99mTcO4−, a γ-emitter that is commonly used for single photon emission computed tomography (SPECT) imaging in the clinic.40 While free 99m-pertechnetate exhibited high thyroid uptake in healthy mice, encapsulation of the oxo-anion resulted in a distinct biodistribution profile, showcasing the utility of drug delivery properties also for radionuclides. Representative examples of MCgs for medicinal applications are included in Fig. 1 and the reader is directed to more comprehensive reviews for further insights.2,41,42
Amongst the most intriguing aspects of MCgs is the possibility to design them as multifunctional systems for targeted concomitant diagnostic and therapeutic (theranostic) applications.43 For example, initial work by Stang, Chen, Cook and coworkers reported on different metallacage systems for therapy and imaging via different approaches, including nuclear imaging modalities or aggregation-induced emission (Fig. 1C).27,44 In this context, the case of “lantern-shaped” cationic [Pd2L4]4+ cages (L = bitopic monodentate N-donor ligand) is noticeable since already a few reports exist on their application in fluorescence microscopy and nuclear imaging (Table S1†).32,33,45–50
In the case of radiolabeled MCgs, three strategies are applicable to integrate the radionuclide in the system: ligand functionalization, encapsulation via host–guest chemistry, and complexation of the radiometal as part of the supramolecular assembly. Thus, our group reported on [Pd2L4]4+ cages whereby the ligand L was bioconjugated to a peptide (AGILKRW, PepH3) capable to cross the blood–brain barrier (BBB), and assembled into a [Pd2L4]4+ cage.48 Upon encapsulation of 99mTcO4−, the MCg was injected in mice in vivo, and brain accumulation of the host–guest complex was detected at 0.42% ID per g 5 min p.i., supporting cage application as brain delivery vehicle.
Previously, a [Pd2L4]4+ cage had already been modified via click chemistry with 18F-trifluoroborate moieties for positron emission tomography (PET) imaging.49 Here, the intactness of the MCg in vivo was corroborated by its different biodistribution with respect to the free ligand by PET imaging. Interestingly, the zwitterionic nature of the trifluoroborate moiety rendered the cage sufficiently soluble in aqueous environment. More recently, (Tyr3)-octreotate, a somatostatin-receptor 2 addressing peptide for targeting neuroendocrine tumors was tethered via amide bond formation to the bispyridyl L ligand.50 A DOTA chelator (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) was also inserted into the ligand sequence to bind Lu-177, a β− and γ-emitting radionuclide used for cancer treatment and therapy control via SPECT. While the sterically demanding ligand only allowed the formation of a [Pd2L2]4+ homoleptic metallacycle, formation of a heteroleptic MCg was achieved by adding a non-functionalized less sterically hindering ligand.50
Thriving to implement self-assembled MCgs as new generation radiopharmaceuticals, we herein report on the synthesis by CDSA of homoleptic and heteroleptic Pd2+-cages featuring a 3,5-bis(3-ethynylpyridine)phenyl backbone and exo-functionalized with the BBB-targeting peptide PepH3 tethered to two different silicon-based fluoride acceptors (SiFAs) moieties for radiolabeling with fluorine-18 for PET imaging (Fig. 2A). PepH3 has already been studied for its BBB translocation capability in vitro and in vivo,51 including bioconjugated to MCgs structures.48 Interestingly, the peptide has been reported to cross the BBB via an active transport mechanism, namely adsorptive-mediated transcytosis (AMT), due to the presence of positively charged amino acid residues in its sequence.52 Here, the physiologically more stable D-enantiomer of PepH3 (DPepH3) was applied.53 The suitability of this design strategy was challenged by exploring the fundamentals of homoleptic and heteroleptic CDSA (Fig. 2B). The lipophilic character of the ligands (logDpH7.4) was determined, and in one case, also investigated in ovo in glioblastoma U87 tumors engrafted on the chicken chorioallantoic membrane (CAM) model by PET. Further, the obtained homoleptic MCgs were characterized by different methods, including 1H NMR spectroscopy and high-resolution electrospray ionization mass spectrometry (HR-ESI-MS). In addition, the encapsulation properties of the homoleptic systems were investigated by using the guest molecule perrhenate as a cold surrogate for 99mTcO4−. Further, a protocol for the synthesis of 18F-labeled MCgs via pre-assembly labeling was optimized, which held radiochemically pure MCgs. In parallel, the synthesis of heteroleptic MCgs, of general formula [Pd2(L)m(L0)n]4+ (m = 1, 2, n = 4 − m) was achieved by statistical self-assembly of two different types of ligands,54 followed by chromatographic separation.
 | ||
| Fig. 2 (A) Design and synthesis of BBB-targeted ligands L1 and L2 by Fmoc-SPPS. The exo-functionalization consists of a BBB shuttle peptide DPepH3, a silicon-based fluoride acceptor (SiFA) for radiolabeling with the PET nuclide fluoride-18 and a PEG linker (4-((2-(2-(2-aminoethoxy)ethoxy)ethyl)amino)-4-oxobutanoic acid) that acts as a spacer. (B) Coordination-driven self-assembly of homoleptic and heteroleptic MCgs, Chom and Chet. Reaction conditions: 4 eq. ligand (in total), 2 eq. [Pd(MeCN)4](BF4)2, DMSO, RT, 1 h. (C) 1H NMR (top) and 1H-DOSY NMR (bottom) spectra in DMSO-d6 (400 MHz) of C2hom (purple) compared to L2 (blue). The α-pyridinyl protons a and b shift upon metal coordination as explained by the scheme. (D) Mass spectra of C2hom (top) and the host–guest complex ReO4−⊂C2hom (bottom) obtained by DI-HR-ESI-MS. Corresponding theoretical and experimental isotopic patterns are shown in the graph insets. | ||
Experimental
General information
All reagents and solvents were purchased commercially and were used without further purification. The synthesis of SiFAs building block was achieved as previously reported,55,56 and their purity >95% were determined by reverse-phase high performance liquid chromatography (RP-HPLC). Ligand L0 (2-(3,5-bis(pyridin-3-ylethynyl)phenyl)acetic acid) was synthesized according to literature-known procedure.57 The product was obtained as a white solid in a total yield of 66% and 97% purity by RP-HPLC.Fluoride-18 in target water ([18O]H2O) was obtained from Klinikum rechts der Isar (Munich, Germany) and Guys and St Thomas’ Hospital (London, UK).
1H NMR and 1H-DOSY NMR spectra were recorded on a Bruker Avance III HD 400 NMR instrument by Bruker Cooperations (Billerica, USA). Chemical shifts are given in parts per million (ppm) and are referenced to the residual proton signal of the solvent. The coupling constants J are reported in hertz (Hz) and the signal splitting patterns are expressed as singlet (s), doublet (d), doublet of doublets (dd), triplet (t), doublet of triplets (dt), doublet of quartets (dq) and multiplet (m).
Analytical and semi-preparative RP-HPLC measurements were performed using gradient systems by Shimadzu Corp. (Kyoto, Japan), consisting of a SPD-20A dual wavelength UV/Vis detector (l = 220 nm, l = 254 nm), two LC-20AD gradient pumps, a CBM-20A communication unit and a CTO-20A column oven. For analytical RP-HPLC measurements a flow rate of 1 mL min−1 and for semi-preparative RP-HPLC measurements a flow rate of 10 mL min−1 was used. Quality controls of the ligands and analysis of cage formation reactions were performed using a MultoKrom® 100-5 C-18 column (150 × 4.6 mm, 5 μm particle size) from CS-Chromatographie Service GmbH (Langerwehe, Germany). For analytical separation of ligands and cages, different gradients of H2O (0.1% trifluoroacetic acid (TFA)) (A) and MeCN (2% H2O, 0.1% TFA) (B) were used during RP-HPLC measurements. For semi-preparative purification of ligands, different gradients of H2O (0.1% TFA) (A) and MeCN (5% H2O, 0.1% TFA) (B) were used.
Quality controls of radiolabeled products was conducted using a gradient system by Shimadzu Corp. (Kyoto, Japan), consisting of an SPD-20A dual wavelength UV/Vis detector (l = 220 nm, l = 254 nm), two LC-20AD gradient pumps, a HERM LB500 (NaI scintillation crystal) as well as a FlowStar2 LB514 radio detector from Berthold Technologies GmbH (Bad Wilbad, Germany) and a CBM-20A communication unit. Radiolabeled ligands were analysed using a MultoKrom® 100-5 C-18 column (125 × 4 mm, 5 μm particle size) with a constant flow rate of 1 mL min−1. Analysis of the radiolabeled cages was performed using a MultoHigh® Bio 300-5 C4 column (150 × 4.6 mm, 5 μm particle size) at a flow rate of 1 mL min−1. Radio thin-layer chromatography (TLC) was performed on a ScanRAM radio TLC detector by LabLogic (Sheffield, UK) and analysed with the Laura software.
Direct-infusion (DI) and liquid chromatography (LC) high-resolution electrospray-ionization mass spectrometry (HR-ESI-MS) was applied for the analysis of ligands and MCgs using an Orbitrap Q-Exactive Plus Mass Spectrometer from Thermo Fisher Scientific. For LC-MS, the MS system was coupled to Dionex ultimate 3000 series UHPLC from Thermo Fisher Scientific for LC-MS. The column used for the C2hom analysis was a SUPELCO BIOshell A400 Protein C4 (3.4 μm, 400 Å, 15 cm × 1 mm) from Sigma-Aldrich. The mobile phases were A: H2O and B: MeCN, both with 0.1% formic acid (FA). The flow rate was 0.05 mL min−1 and sample elution was performed by using a gradient from 20% to 40% of B over 3 min, followed by a gradient from 40% to 80% of B over 15 min. Sample injection volume was 10 μL. For the C1het analysis the column used was an AcclaimTM 120: C18 (5 μm, 120 Å, 4.6 mm × 100 mm), Dionex Bonded Silica Products from Thermo Fisher Scientific. The mobile phases were A: H2O and B: ACN, both with 0.1% formic acid (FA). The flow rate was 0.5 mL min−1 and sample elution was performed by using a gradient from 20% to 40% of B over 3 min, followed by a gradient from 40% to 80% of B over 15 min. Ionization was performed using an ESI source operating in positive ion mode with a capillary voltage of 3.50 kV and capillary temperature 320 °C. Sheath gas, auxiliary gas and sweep gas flow rate were set at 20, 12 and 1 (arbitrary units), respectively. Auxiliary gas temperature was set at 100 °C.
For DI, the sample was infused at 0.01 mL min−1 and ionization was performed using an ESI source operating in positive ion mode with a capillary voltage of 3.50 kV and capillary temperature of 300 °C. Sheath gas, auxiliary gas and sweep gas flow rate were set at 20, 12 and 0 (arbitrary units), respectively. Auxiliary gas temperature was set at 50 °C.
Ligand synthesis
Fmoc-deprotection. The N-terminal Fmoc-protected DPepH3, immobilized on resin, (60.0 μmol, 1.0 eq.) was deprotected by addition of 20% piperidine in dimethyl formamide (DMF, 5 mL) at RT. The deprotection step was carried out twice (1 × 15 min, 1 × 5 min) and afterwards the resin was washed with DMF (6 × 5 mL).
Fmoc-DDap(Dde)-OH (N-alpha-(9-fluorenylmethyloxycarbonyl)-N-beta-[(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl]-D-2,3-diaminopropionic acid) (90.0 μmol, 1.5 eq.), TBTU (N,N,N′,N′-tetramethyluronium-tetrafluorborate) (90.0 μmol, 1.5 eq.), HOAt (1-hydroxy-7-azabenzotriazole) (90.0 μmol, 1.5 eq.) and 2,4,6-trimethylpyridine (300 μmol, 5.0 eq.) were dissolved in 5 mL DMF. After 2 min preincubation, the solution was added to the deprotected resin-bound peptide and shaken for 2 h at RT. Afterwards the resin was washed with DMF (6 × 5 mL) and N-methyl-2-pyrrolidone (NMP, 6 × 5 mL). For a coupling in branched position the Dde-protection group was cleaved by adding a solution of NH2OH·HCl (6.0 mmol, 100 eq.) and imidazole (4.5 mmol, 75 eq.) in 7 mL NMP/DCM (5/2). After shaking for 2 h at RT, the resin was washed with NMP (6 × 5 mL) and DMF (6 × 5 mL).
Amide coupling. Fmoc-(SiFA)BA (72.0 μmol, 1.2 eq.), TBTU (90.0 μmol, 1.5 eq.) and HOAt (90.0 μmol, 1.5 eq.) were dissolved in 5 mL DMF. After the addition of DIPEA (N,N-diisopropylethylamine) (210 μmol, 3.5 eq.) the solution was preincubated for 30 min. The coupling solution was added to the deprotected resin-bound compound and the reaction mixture was shaken at RT for 2.5 h. Afterwards the resin was washed with DMF (6 × 5 mL).
Fmoc-Ebes (N-[8-(9-fluorenylmethyloxycarbonyl)amino-3,6-dioxaoctyl]succinamic acid) (90.0 μmol, 1.5 eq.) and L0 (90.0 μmol, 1.5 eq.) were coupled consecutively to the resin-bound compound by first performing Fmoc-deprotection and then amide coupling.
Resin cleavage. The ligand was cleaved off the resin by shaking with 2 × 5 mL of a mixture of TFA/triisopropyl silane (TIPS)/H2O (95/2.5/2.5) for 2 × 1 h at RT. The ligand solution was collected in a round-bottom flask and stirred for 1 h before being concentrated in an N2 stream. Then, 5 mL of TFA was added and the mixture was stirred at RT for another 2 h. Afterwards, the TFA was removed again in an N2 stream and the ligand was purified via semi-preparative RP-HPLC and lyophilized.
D PepH3-DDap((SiFA)BA)-Ebes-L0 was obtained as a white solid (11.5 mg, 11% yield, 98% purity).
RP-HPLC (10–90% B): tR = 9.7 min.
1H NMR (400 MHz, DMSO-d6) δ (ppm) = 10.78 (s), 8.78 (d, J = 3.1 Hz, 2H), 8.61 (d, J = 6.5 Hz, 2H), 8.49 (d, J = 4.4 Hz), 8.26 (d, J = 6.6 Hz), 8.21 (d, J = 5.9 Hz), 8.14–8.08 (m), 8.04–7.97 (m), 7.98–7.92 (m), 7.89–7.79 (m), 7.68 (s), 7.67–7.61 (m), 7.61–7.53 (m), 7.52–7.43 (m), 7.39 (s), 7.32 (d, J = 7.1 Hz), 7.11 (d, J = 2.6 Hz), 7.08–7.01 (m), 7.00–6.92 (m), 4.43 (dq, J = 13.6, 6.7 Hz), 4.33–3.92 (m), 3.81–3.58 (m), 3.53–3.46 (m), 3.43 (t, J = 5.8 Hz), 3.36 (t, J = 5.9 Hz), 3.20 (dt, J = 30.4, 5.7 Hz), 3.12–2.92 (m), 2.72 (d, J = 7.5 Hz), 2.36 (s), 1.85–1.36 (m), 1.25 (d, J = 7.4 Hz), 1.11 (s), 1.01 (d, J = 1.1 Hz), 0.85 (d, J = 6.4 Hz), 0.83–0.73 (m).
MS (ESI positive): calculated monoisotopic mass for C90H124FN19O14Si: 1741.93; found by ESI-MS: m/z = 582 [M + 3H]3+, 872 [M + 2H]2+.
RP-HPLC (10–90% B): tR = 9.5 min.
1H NMR (400 MHz, DMSO-d6) δ (ppm) = 10.79 (s), 8.79 (s, 2H), 8.73 (d, J = 6.5 Hz), 8.61 (dd, J = 4.9, 1.7 Hz, 2H), 8.52–8.43 (m), 8.39–8.30 (m), 8.28–8.17 (m), 8.00 (dt, J = 7.9, 1.8 Hz), 7.98–7.78 (m), 7.75–7.62 (m), 7.61–7.53 (m), 7.53–7.45 (m), 7.39 (s), 7.32 (d, J = 7.9 Hz), 7.12 (d, J = 2.4 Hz), 7.09–7.01 (m), 7.00–6.92 (m), 4.44 (dt, J = 19.4, 6.9 Hz), 4.33 (d, J = 5.9 Hz), 4.30–3.93 (m), 3.74 (d, J = 5.8 Hz), 3.50 (dd, J = 4.3, 2.6 Hz), 3.43 (t, J = 5.7 Hz), 3.37 (t, J = 5.9 Hz), 3.29–3.14 (m), 3.14–2.94 (m), 2.73 (d, J = 7.5 Hz), 2.46–2.29 (m), 2.24 (d, J = 0.6 Hz), 2.08 (s), 1.97–1.90 (m), 1.71–1.58 (m), 1.56–1.42 (m), 1.37 (d, J = 7.1 Hz), 1.23 (s), 1.11 (s), 1.03–0.97 (m), 0.88–0.70 (m).
MS (ESI positive): calculated monoisotopic mass for C88H121FN18O13Si: 1684.91; found by ESI-MS: m/z = 562 [M + 3H]3+, 844 [M + 2H]2+, 1686 [M + H]+.
Synthesis of metallacages by CDSA and RP-HPLC analysis
To achieve homoleptic MCgs assembly, ligand L1 or L2 (2.0 eq., 5 mM in DMSO) and [Pd(MeCN)4](BF4)2 (1.0 eq., 20 mM in DMSO) were mixed in DMSO to reach a theoretical cage concentration of 1 mM. Instead, CDSA of the heteroleptic cages was achieved by either mixing 1.0 eq. of L1 (5 mM in DMSO) and 3.0 eq. of L0 (5 mM in DMSO) for C1het, or 2.7 eq. of L2 (5 mM in DMSO) and 1.3 eq. of L0 (5 mM in DMSO) for C2het, with 2.0 eq. of [Pd(MeCN)4](BF4)2 (20 mM in DMSO) and adding DMSO to reach a theoretical cage concentration of 1 mM.In all cases, the reaction was complete after 1 h at RT and analysed by analytical RP-HPLC. For the homoleptic cages a gradient of 40–70% B in 15 min was applied. In the case of the heteroleptic systems, the reaction resulted in a mixture of heteroleptic coordination species with different ligand ratios. Therefore, two different gradients were tested to achieve the best separation: (i) 30–60% B over 15 min with a flow rate of 1 mL min−1 and 0.1% TFA as additive, and (ii) 20%–40% B over 3 min, followed by a gradient 40%–80% over 15 min with a flow rate of 0.5 mL min−1 and 0.1% FA as additive.
[Pd 2 ( L1 ) 4 ](BF 4 ) 4 ( C1 hom )
RP-HPLC (40–70% B in 15 min): tR = 8.9 min. Yield 88.7%.
[Pd 2 ( L2 ) 4 ](BF 4 ) 4 ( C2 hom )
RP-HPLC (40–70% B in 15 min): tR = 7.3 min. Yield 82.1%.
Stability studies of homoleptic cages
For stability studies, C1hom and C2hom (20 μL, 1 mM) were added to 180 μL of different solvents (DMSO, H2O, MeCN, MeCN/H2O (1/1, +0.1% TFA)), phosphate buffered saline (PBS, pH 7.4) and saline solution (0.9 wt% NaCl), respectively, to achieve a cage concentration of 0.1 mM. The stability was determined by RP-HPLC after incubation at different times (0 min, 30 min, 60 min, 4 h, 24 h).CDSA and encapsulation studies by 1H and 1H-DOSY NMR spectroscopy
The NMR samples were prepared using ∼7 mg of ligand L1/L2 (2.0 eq.) dissolved in 400 μL of DMSO-d6, and 1H NMR as well as 1H-DOSY NMR spectra were recorded. To the ligand solution, [Pd(MeCN)4](BF4)2 (1.0 eq.) was added in 25 μL DMSO-d6. 1H NMR and 1H-DOSY NMR spectra were recorded after 1 h at RT to observe homoleptic cage formation.To monitor CDSA of the heteroleptic cages, ∼2 mg of L1 (1.0 eq.) and 3.0 eq. of L0 for C1het, or ∼6 mg of L2 (2.7 eq.) and 1.3 eq. of L0 for C2het, were dissolved in 400 μL of DMSO-d6 in an NMR tube. Thus, 1H NMR as well as 1H-DOSY NMR spectra were recorded. To the ligand solution was added 2.0 eq. of [Pd(MeCN)4](BF4)2 in 25 μL DMSO-d6 and after 1 h at RT, 1H NMR and 1H-DOSY NMR spectra of the formed homoleptic cages C1het and C2het were measured. Note that a mixture of different heteroleptic coordination species results from the reaction.
To study the encapsulation properties of the homoleptic MCgs, the cages were prepared in DMSO-d6/D2O (1/1). 2.0 eq. of NaReO4 in 25 μL DMSO-d6/D2O (1/1) was added to the cage solution, and after 30 min at RT, 1H NMR spectra were recorded.
[Pd 2 ( L1 ) 4 ](BF 4 ) 4 ( C1 hom )
1H NMR (400 MHz, DMSO-d6) δ (ppm) = 10.78 (s), 9.77 (s, 2H), 9.41 (s, 2H), 8.52–8.44 (m), 8.27–8.17 (m), 8.13–8.04 (m), 8.04–7.91 (m), 7.90–7.75 (m), 7.73–7.60 (m), 7.58 (d, J = 4.8 Hz), 7.47 (t, J = 5.4 Hz), 7.40 (s), 7.32 (d, J = 8.1 Hz), 7.11 (s), 7.09–7.01 (m), 7.00–6.92 (m), 4.50–4.37 (m), 4.33–3.84 (m), 3.46 (s), 3.42–3.28 (m), 3.19–2.92 (m), 2.75–2.69 (m), 2.34 (s), 2.07 (s), 1.75 (s), 1.46 (dt, J = 20.2, 7.5 Hz), 1.25 (t, J = 7.1 Hz), 1.11 (s), 1.06–0.93 (m), 0.88–0.72 (m).
[Pd 2 ( L2 ) 4 ](BF 4 ) 4 ( C2 hom )
1H NMR (400 MHz, DMSO-d6) δ (ppm) = 10.80 (s), 9.78 (s, 2H), 9.43 (s, 2H), 8.72 (d, J = 6.2 Hz), 8.48 (t, J = 5.8 Hz), 8.33 (d, J = 5.6 Hz), 8.24 (d, J = 7.8 Hz), 8.06–7.77 (m), 7.68 (d, J = 5.6 Hz), 7.58 (d, J = 7.6 Hz), 7.51 (d, J = 5.4 Hz), 7.42 (s), 7.32 (d, J = 8.1 Hz), 7.11 (d, J = 2.3 Hz), 7.10–7.01 (m), 7.01–6.92 (m), 4.51–4.38 (m), 4.32 (d, J = 5.7 Hz), 4.29–4.11 (m), 3.76 (s), 3.46 (s), 3.41–3.29 (m), 3.24–2.92 (m), 2.72 (d, J = 7.2 Hz), 2.42–2.27 (m), 2.24 (s), 2.08 (s), 1.92 (s), 1.75 (s), 1.71–1.56 (m), 1.57–1.41 (m), 1.36 (d, J = 7.0 Hz), 1.21 (d, J = 3.0 Hz), 1.11 (s), 0.99 (s), 0.88–0.70 (m).
Synthesis of metallacages by CDSA and analysis by mass spectrometry
Stock solutions of L0, L1, L2 and [Pd(MeCN)4](BF4)2 (20 mM) were prepared in DMSO. The reaction of ligand L1 or L2 with the Pd(II) precursor [Pd(MeCN)4](BF4)2 mixed in a 2:1 ratio in DMSO resulted in the homoleptic coordination cage C1hom or C2hom within 1 h at RT. To achieve heteroleptic C1het system, the stock solutions (20 mM) of L1 and L0 (20 mM) were mixed in a 1:3 ratio in DMSO (final concentrations 3 and 9 mM, respectively). Similarly, for C2het stock solutions (20 mM) of L2 and L0 were mixed in a 1:2 ratio in DMSO (final concentrations 4 and 8 mM, respectively). The ligand mixtures were stirred for 1 h at RT with 0.5 eq. (6 mM) of the Pd(II) precursor [Pd(MeCN)4](BF4)2 to achieve a theoretical cage concentration of 3 mM. The cage mixtures were then diluted to a final cage concentration of 2 μM, using H2O with 5% (v/v) MeCN and 0.1% (v/v) FA, and used for RP-HPLC, DI- and LC-HR-ESI-MS analysis.
Encapsulation studies by mass spectrometry
For the encapsulation of ReO4−, C2hom (3 mM in DMSO) was diluted with MilliQ water in 67% of H2O. Stock solution of NaReO4 (10 mM) in MilliQ water was freshly prepared, and an aliquot added to each cage solution to achieve cage: NaReO4 ratio of 1:2 (0.9 mM:1.8 mM) in 72% of H2O. After 30 min incubation at RT, the mixtures were diluted to a cage concentration of 4 μM, using H2O with 5% (v/v) of MeCN and 0.1% (v/v) FA, and used for DI-HR-ESI-MS or LC-HR-ESI-MS analysis.
Radiolabeling of ligands and cages
18 F- D PepH3- D Dap((SiFA)BA)-Ebes- L0 ( 18 F-L1 )
RCY: 44%, RCP: 98%.
Radio RP-HPLC (10–80% B in 15 min): tR = 12.4 min.
18 F- D PepH3-(SiFA)SeFe-Ebes- L0 ( 18 F-L2 )
RCY: 47%, RCP: 99%.
Radio RP-HPLC (30–60% B in 15 min): tR = 10.4 min.
18 F-[Pd 2 ( L1 ) 4 ](BF 4 ) 4 ( 18 F-C1 hom )
Radio RP-HPLC (40–70% B in 15 min): tR = 9.6 min.
18 F-[Pd 2 ( L2 ) 4 ](BF 4 ) 4 ( 18 F-C2 hom )
Radio RP-HPLC (40–70% B in 15 min): tR = 7.7 min.
Lipophilicity determination (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) DpH7.4)
DpH7.4)
The octanol-PBS partition coefficient (logDpH7.4 values) were determined by addition of 0.5 MBq of the labeled ligand to a 1.5 mL LoBind Eppendorf tube, filled with 500 μL octanol and 500 μL PBS (n = 8). The tubes were vortexed for 3 min at RT and centrifuged (9000 rpm, 5 min, RT) using a Heraeus Pico 17 centrifuge by Thermo Scientific (Waltham, USA) (24 × 1.5/2.0 mL rotor with ClickSeal-Lid). Out of each tube 200 μL of each solvent layer were taken out and the activity was quantified separately using a γ-counter by PerkinElmer Inc. (Langerwehe, Germany). Lipophilicity of the cages could not be determined due to stability issues upon vortexing of their solution.
logDpH7.4 (18F-L1) = 2.53 ± 0.05.
logDpH7.4 (18F-L2) = 2.07 ± 0.07.
Cell culture
U87 cells were cultivated at 37 °C (+5% CO2) in MEME (Sigma Life Science) containing 10% fetal bovine serum (Thermo Fisher), 1% L-glutamine (Sigma Life Science) and 1% penicillin–streptomycin (Sigma Aldrich). The cells were split 1/6 twice a week using trypsin–EDTA solution (Thermo Fisher) for detachment. The cells were tested Mycoplasma-free every month.In ovo studies
All in ovo experiments were carried out on fertilized Dekalb white or brown eggs (Henry Stewart & co. Ltd, UK). Experiments took place before embryonic day 14 (E14). The eggs were incubated for up to 14 days at 12–14 °C in a wine cooler (Haller) with humidified atmosphere until use. To grow tumors on the chick chorioallantoic membrane (CAM), eggs were cleaned with Brinsea disinfectant (100×) and moved to an incubator (Brinsea) with a temperature of 38.7 °C and 48% humidity. The first day of incubation at this temperature was embryonic day 0 (E0). To loosen the CAM from the eggshell the incubator trays were slowly tilted from one side to the other until E3. On E3, eggs were taken from the incubator for window cutting. The eggs were rolled to prevent the CAM sticking to the shell and placed on a cushioned holder. They were pierced at the wide base where the air cell is located and approximately 5 mL of albumin was removed through the hole using a syringe with a 19G needle. The hole was then resealed with Scotch Magic tape. Next, a square of tape was placed onto one side of the egg and four rectangularly arranged holes were punched into the shell through the tape. A rectangular window (1 × 2 cm) was made with sharp dissection scissors by carefully cutting three sides of a rectangle into the shell using the holes for orientation and without damaging the inner shell membrane. The window was closed with tape and the eggs placed in the incubator again until E7. U87 cells were maintained as described previously and cell culture media was renewed 24 h prior to harvesting. On the day of inoculations (E7), cells were detached from the flask, resuspended in media and aliquots with 3 × 106 cells were prepared. The aliquots were centrifuged for 3 min at 500g, 4 °C in a Centrifuge 5424 R (Eppendorf) and stored on ice. Meanwhile, Matrigel® Matrix Basement Membrane (Corning) was defrosted on ice. Eggs were taken from the incubator, placed on an egg holder and the windows were opened to locate the CAM. After dabbing the CAM dry with a sterile lens tissue, a suspension of the U87 cell pellet in 20 μL of Matrigel was pipetted onto the CAM. The eggs were closed with tape, labelled accordingly and placed in the incubator for a further 7 days. On E14 the eggs were removed from the incubator and placed on a cushioned holder. For tumor imaging the shell window was enlarged to allow for direct injection of the radiotracer into a CAM blood vessel.Results and discussion
Ligands synthesis and characterization
To achieve the assembly of the desired radiolabelled metallacages, the bispyridinyl ligand L0 (3,5-bis(3-ethynylpyridine)phenyl)acetic acid (Fig. S1–S4†) was functionalized via amide bond formation with a PEG linker (Ebes) acting as spacer and further attached to a SiFA synthon for 18F-radiolabeling, as well as tethered to the BBB-shuttle peptide DPepH3 (Fig. 2A). Two different SiFAs were selected: a monofunctional SiFA-benzoic acid ((SiFA)BA) prosthetic group55 which can be attached in a branched fashion between the peptide and the PEG linker as55 in L1, and a new bifunctional silicon-based fluoride acceptor ((SiFA)SeFe) recently developed in our group.56 SiFAs are important synthons in radiochemistry since they can undergo 18F-fluorination under mild conditions compatible, for example, with the presence of bioactive molecules in the radiotracer. Unfortunately, a major challenge is the high lipophilic character of the SiFA moiety, which can lead to an unfavourable slow hepatic excretion.59,60 The latter decreases the image quality and increases off-target radiation dose to the abdomen. To partly overcome this problem, ((SiFA)BA) was developed featuring a carboxylic functionality.55 An important progress was achieved with the ((SiFA)SeFe) synthon, whereby the amino acid-like bifunctionality increases its hydrophilic character, and also enables its direct incorporation in a ligand sequence in a bridged fashion as in L2. This feature reduces the steric demand and further counterbalances the SiFA lipophilicity.56Ligands L1 and L2 were synthesized via semi-automated fluorenylmethoxycarbonyl solid-phase peptide synthesis (Fmoc-SPPS) by consecutive amide coupling on a Rink amide resin starting from DPepH3 (Fig. 2A). After resin cleavage and semi-preparative RP-HPLC purification, the ligands were obtained in moderate yield of 11% and 17% for L1 and L2, respectively and excellent RP-HPLC purity (98%). Full characterization of L1 and L2 was carried out by HR-ESI-MS as well as by 1H NMR and 1H-DOSY NMR spectroscopy (Fig. S5–S8 and S9–S12,† for L1 and L2, respectively).
Metallacage synthesis and characterization
NMR spectroscopy is the most applied method to assess the formation of MCgs by CDSA. The 1H NMR spectra of C1hom and C2hom are shown in Fig. S15 and S18,† respectively. In both cases, the ligand's α-pyridyl protons Ha and Hb are exposed to a new electronic environment upon metal coordination, which results in their deshielding shift in the 1H NMR spectrum.61 Exemplary, the pyridyl protons of L2 shift from 8.79 ppm and 8.61 ppm to 9.78 ppm and 9.43 ppm, respectively, indicating the formation of a supramolecular species (Fig. 2C). Additional 1H-DOSY NMR spectra of the cages revealed, as expected, a decrease in the diffusion coefficient upon CDSA (Fig. S16, S19† and Fig. 2C). The SEGWE calculator62 was used to determine the cages’ solvodynamic radii from the corresponding diffusion coefficients, which amount to 2.29 and 1.80 nm for C1hom and C2hom, respectively. The Pd2L4 assemblies were further investigated by DI-HR-ESI-MS or LC-HR-ESI-MS. Successful CDSA of C1hom was confirmed by a peak at 661.8748 m/z, attributed to the 11+ charge state of C1hom adducts with formic acid (FA) and acetonitrile (MeCN) molecules from sample preparation (Fig. S14† and Table 1). Concerning cage C2hom, the mass spectrum obtained by DI shows a number of peaks corresponding to C1hom adducts with FA and solvent molecules, or to Pd2(L2)3 species (Fig. 2D top). Formation of the coordination cage C2hom was also revealed by LC-HR-ESI-MS analysis, whereby a peak at tR = 13.94 min in the total ion chromatogram (TIC) corresponds to species with m/z value of 637.3228, attributed to the 11+ charge state of the C2hom adduct with formic scid related mass (Fig. 3 and Table 1). The LC-HR-ESI-MS also shows the presence of a Pd2(L2)3 coordination complex at tR = 14.37 min (m/z 670.8255, z = +8). Finally, the TIC spectrum demonstrates presence of free ligand L2 at tR = 14.60 min (data not shown). These results suggest the possible detachment of the fourth ligand in the MCg structure induced by the chromatographic elution. Nevertheless, it is evident that all the possible supramolecular species can be fully separated and properly characterized.
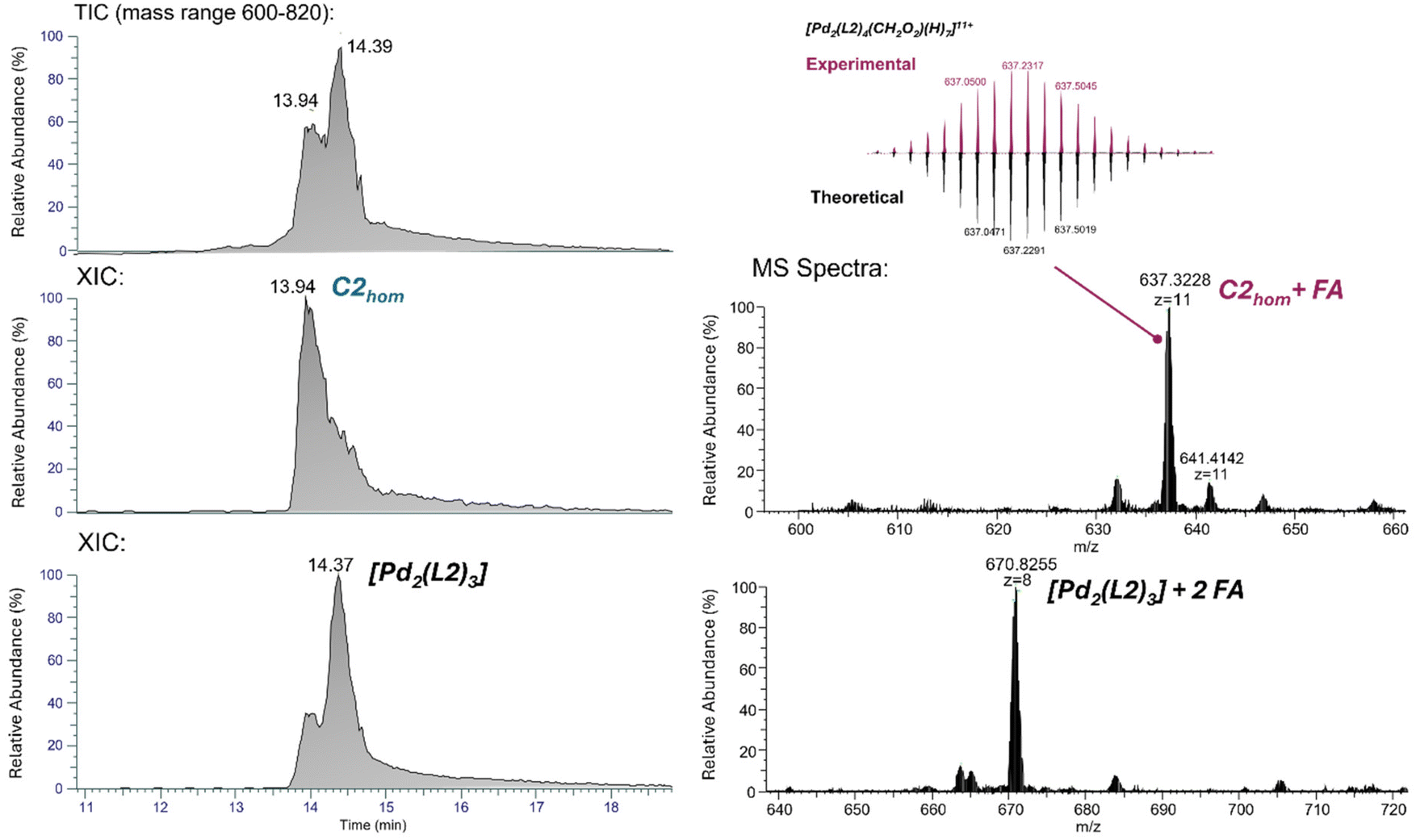 | ||
| Fig. 3 Characterization of the homoleptic cage C2hom by LC-HR-ESI-MS. Left: TIC (mass range between 600–820 m/z) and extracted ion chromatograms (XIC) of ions m/z 637.32 (z = 11, C2hom at tR = 13.94 min) and 670.83 (z = 8, [Pd2(L2)3] + 2 FA at tR = 14.37 min). Right: mass spectrum of C2hom + FA at tR = 13.94 min (comparison of theoretical and isotopic pattern is provided as insert) and [Pd2(L2)3] + 2 FA at tR = 14.37 min. The mobile phases were A: H2O and B: MeCN, both with 0.1% formic acid (FA). The flow rate was 0.05 mL min−1 and sample elution was performed by using a gradient from 20% to 40% of B over 3 min, followed by a gradient from 40% to 80% of B over 15 min. | ||
| Compound | Formula | m/z | Observed adducts | |
|---|---|---|---|---|
| Theoretical | Measured | |||
| C1hom | C360H496F4N76O56Pd2Si4 | 661.8758 (z = 11) | 661.8747 | [M4+ + FA + ACN + 7H+]11+ |
| 664.2367 | [M4+ + 2FA + Na + 7H+]11+ | |||
| [Pd2(L1)3] | C270H372F3N57O42Pd2Si3 | 686.4560 (z = 8) | 686.4570 | [M4+ + FA + 4H+]8+ |
| 692.3329 | [M4+ + 2FA + 4H+]8+ | |||
| 695.2059 | [M4+ + 2FA + Na+ + 4H+]8+ | |||
| 700.5811 | [M4+ + 2DMSO + 4H+]8+ | |||
| 704.9545 | [M4+ + Cl− + 2DMSO + 5H+]8+ | |||
| 709.2046 | [M4+ + 2Cl− + 2DMSO + 6H+]8+ | |||
| C2hom | C352H484F4N72O52Si4Pd2 | 637.3200 (z = 11) | 637.3203 | [M4+ + FA + 7H+]11+ |
| 641.0483 | [M4+ + FA + ACN + 7H+]11+ | |||
| 643.5020 | [M4+ + 2FA + Na + 7H+]11+ | |||
| 705.5519 (z = 10) | 705.5536 | [M4+ + 2FA + 6H+]10+ | ||
| ReO4⊂ C2hom | C352H484F4N72O56Si4Pd2Re | 655.9506 (z = 11) | 655.9501 | [M3+ + 8H+]11+ |
| [Pd2(L2)3] | C264H363F3N54O39Si3Pd2 | 670.8237 (z = 8) | 670.8237 | [M4+ + 2FA + 4H+]8+ |
| 683.4458 | [M4+ + 2DMSO + Cl− + 5H+]8+ | |||
| 687.8202 | [M4+ + 2DMSO + 2Cl− + 6H+]8+ | |||
| ReO4⊂ [Pd2(L2)3] | C264H363F3N54O43Si3Pd2Re | 696.4408 (z = 8) | 696.4399 | [M3+ + FA + 5H+]8+ |
| C1het | C156H166FN25O20Pd2Si | 594.2134 (z = 5) | 594.2142 | [M4+ + H+]5+ |
| 742.7652 (z = 4) | 742.7651 | [M4+]4+ | ||
| C2het | C154H163FN24O19SiPd2 | 582.8092 (z = 5) | 582.8102 | [M4+ + H+]5+ |
| 605.8088 | [M4+ + 2FA + Na]5+ | |||
| [Pd2(L2)2(L0)2] | C220H270F2N40O30Si2Pd2 | 609.2671 (z = 7) | 609.2677 | [M4+ + 3H+]7+ |
| 625.4096 | [M4+ + 2FA + Na+ + 2H+]7+ | |||
Next, stability studies were conducted in solvents relevant for different assays, radiolabeling and analytic procedures. Therefore, cage solutions were prepared as described in the Experimental section (1 mM theoretical concentration) and diluted 10-fold with DMSO, H2O, MeCN, HPLC solvent (MeCN/H2O (1/1) +0.1% TFA), 0.9 wt% NaCl, and PBS (pH 7.4), respectively, and analysed via RP-HPLC (Fig. 4 and Fig. S21, S22†). At this concentration, precipitation and partial disassembly of the cage were observed in PBS and isotonic NaCl solution, although less pronounced in the latter case over 1 h. This poses a problem for preclinical studies as salts are naturally present in physiological conditions, as well as for encapsulation of 99mTcO4−, which is eluted from the 99Mo/99Tc-generator with saline. Instead, both MCgs exhibited sufficient stability in water with 97% of C1hom and 75% of C2hom being intact after 4 h. In general, C2hom appears to be less stable than C1hom despite the predicted reduced steric hindrance of the exo-functionalities. The presence of coordinating solvents like DMSO or MeCN induced formation of new supramolecular assemblies over 24 h, leaving only ca. 50% (or lower) remaining intact cage (Fig. 4). Of note, addition of water to MeCN as used in the HPLC conditions, increased the stability of the cages over time. Interestingly, an effect of concentration on cage stability was also recorded. For example, a 10-fold more concentrated solution of C2hom (1 mM) remained intact in DMSO solution for at least 24 h (Fig. S20†). Overall, these studies show that many factors can affect the stability of Pd2L4 MCgs, including steric effects of the exo-functionalities, cage surface charges which can be sensitive to the ionic strength of the medium, as well as interactions of the Pd(II) centres with nucleophilic molecules (e.g. DMSO). In the specific case of C1hom and C2hom, the obtained results suggest that both electrostatic and nucleophilic interactions may play a major role in destabilizing the cages in solution.
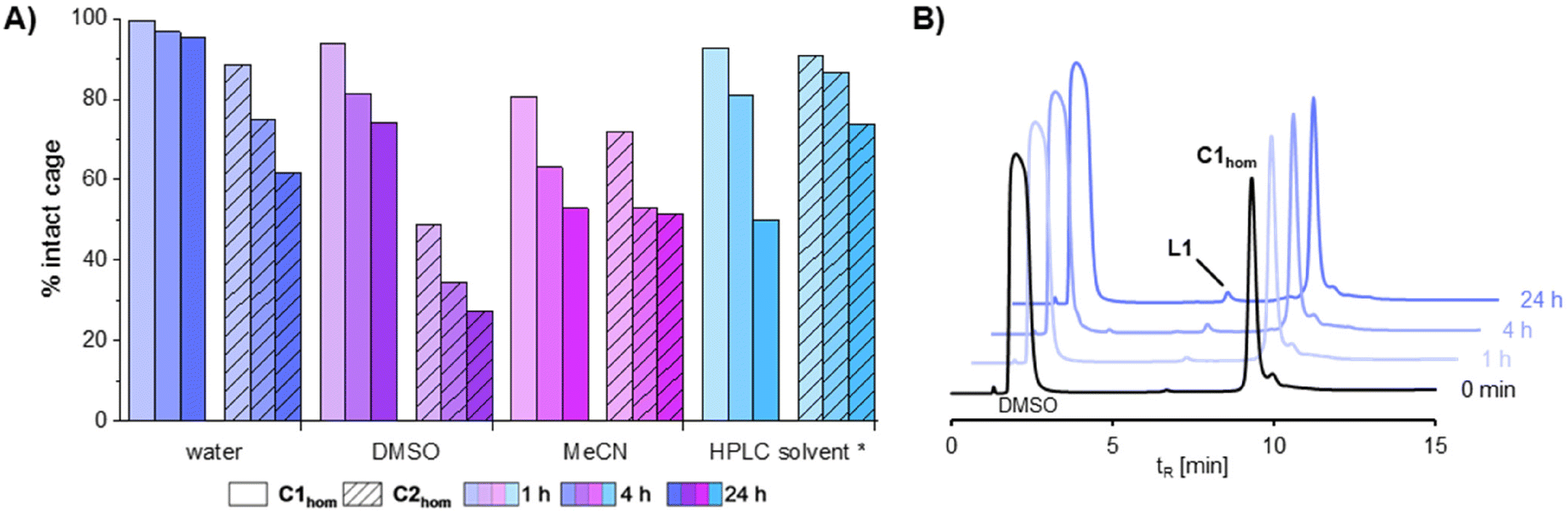 | ||
| Fig. 4 (A) Stability study of C1hom and C2hom in different solvents analysed by RP-HPLC. The theoretical cage concentration after dilution with the respective solvent was 0.1 mM. *MeCN/H2O (1/1) +0.1% TFA. (B) Stability of C1hom in Milli-Q H2O monitored over 24 h by RP-HPLC (40–70% B in 15 min). | ||
To get further insights into the effect of ligand concentration on the CDSA, C1hom and C2hom were self-assembled in DMSO directly to reach a theoretical concentration of 0.1 mM, which resulted in only partial cage formation (65% and 57% conversion to C1hom and C2hom, respectively) (Fig. S23 and S24†), hence indicating that CDSA is concentration dependent and can result in different coordination species. This finding needs to be kept in mind especially when entering new fields such as radiochemistry, where reactions are performed in μmol or even nmol scale.
Overall, both homoleptic metallacages C1hom and C2hom were successfully assembled and characterized using three different analytical techniques. However, further improvement in cage stability, which might be affected by the complex and sterically demanding peptidic ligands, is needed.
:3 – [Pd2(L1)(L0)3]4+ – 1 eq. of L1 and 3 eq. of L0 were reacted with [Pd(MeCN)4](BF4)4 in DMSO for 1 hour at RT. 1H NMR spectroscopy revealed a deshielding of the α-pyridinyl protons to 9.76 and 9.40 ppm, respectively, compared to the reference spectra of L1 and L0 (Fig. S25†). Moreover, the 1H-DOSY NMR spectrum showed more than one newly formed species, upon addition of the Pd2+ precursor, suggesting a mixture of supramolecular assemblies (Fig. S26†). The self-assembly process was further investigated by RP-HPLC, where the species could be separated and analysed by MS (Fig. S27A and B†). By comparing the chromatogram with those of the corresponding ligands and homoleptic cages – including C0hom [Pd2(L0)4]4+ and C1hom – several peaks of similar intensity could already be attributed (tR(L0) = 3.2 min, tR(L1) = 11.5 min, tR(C0hom) = 8.3 min). MS analysis of the peak at 11.1 min (25%) associated it to the target C1het species. The remaining peaks at 9.4 and 12.7 min could not be clearly identified by MS, but were likely to originate from heteroleptic CDSA leading to a statistical distribution of metallacages with different L1/L0 ratios (Fig. S27†). Interestingly, when changing the HPLC gradient from 30–60% B in 15 min (1 mL min−1) to 20–40% B in 3 min and 40–80% B in 15 min (0.5 mL min−1), a different chromatogram was obtained (Fig. S28†), whereby only a mean peak at tR = 6.7 min (62.4%) was observed, corresponding to C1het. In order to identify the different assemblies, the mixture was further analysed by LC-HR-ESI-MS, using the optimized HPLC gradient (Fig. 5 and Table 1). In the obtained TIC graph, three main peaks could be resolved. At tR = 7.5 min, the desired [Pd2(L1)(L0)3]4+C1het was the dominant species with m/z of 594.2142 and 742.7651 corresponding to the 5+ and 4+ charge state, respectively. Additional species eluting at tR = 8.4 and 9.3 min corresponded to free L0 and L1 ligands, respectively.
 | ||
| Fig. 5 Characterization of the heteroleptic cage [Pd2(L1)(L0)3]4+ (C1het) by LC-HR-ESI-MS. TIC (mass range between 200–2000 m/z) and XIC of ions m/z 594.21 (z = 5, C1het at tR = 7.37 min), m/z 399.1131 (z = 1, L0 at tR = 8.38 min) and m/z 872.48 (z = 2, L1, tR = 9.31 min). Right: mass spectrum of C1het at tR = 7.37 min (comparison of theoretical and isotopic pattern is provided as insert), L0 at tR = 8.38 min and L1 at tR = 9.31 min. The mobile phases were A: H2O and B: ACN, both with 0.1% formic acid (FA). The flow rate used was 0.5 mL min−1 and sample elution was performed by using a gradient from 20% to 40% of B over 3 min, followed by a gradient from 40% to 80% of B over 15 min. | ||
For the assembly of the heteroleptic [Pd2(L2)(L0)3]4+ (C2het) cage, tuning of the ratio L2/L0 was required. Eventually, a mixture of heteroleptic species was obtained by combining 2.7 eq. of L2 and 1.3 eq. of L0 and reacting with [Pd(MeCN)4](BF4)4 in DMSO for 1 hour at RT, as evidenced by 1H and 1H-DOSY NMR spectroscopy (Fig. S29 and S30†). The mixture of heteroleptic assemblies could be unpicked again by RP-HPLC using different gradients (Fig. S31 and S32†). Even in this case, the optimized gradient (20–40% B in 3 min and 40–80% B in 15 min (0.5 mL min−1)) enabled the separation of a main peak at tR = 6.8 min (58.7%) corresponding to C2het (Fig. S32†). Analysing directly the mixture by DI-HR-ESI-MS, the main desired species (C2het) [Pd2(L2)(L0)3]4+ (m/z = 582.8102, z = +5) and [Pd2(L1)2(L0)2]4+ (m/z = 609.2677, z = +7) were identified, including comparison of the experimental and theoretical isotopic patterns (Fig. S33† and Table 1).
Encapsulation studies
One of the strategies to radiolabel metallacages is via encapsulation of the radioactive guest molecule. The encapsulation of 99mTcO4− and stability of different host–guest systems in vivo have already been demonstrated successfully by the groups of Lusby and Archibald as well as by Correia and Casini.48 In both studies perrhenate was used as a ‘cold’ surrogate for pertechnetate and the host–guest interaction was investigated either by NMR spectroscopy or via in silico methods. Following these promising results, we investigated the encapsulation of perrhenate in our MCgs. Thus, 2 eq. of NaReO4 were added to solutions of C1hom and C2hom in D2O/DMSO-d6 (1/1) and 1H NMR spectra were recorded after 30 min. Deuterated water was added to support the encapsulation of the oxo-anion, which interacts with the host cavity via electrostatic forces and hydrogen bonding.63 For C1hom a deshielding of 0.01 ppm of the Ha proton was observed upon perrhenate addition and in the case of C2hom the Ha proton was shifted by 0.03 ppm (Fig. S34 and S35†). No significant Hb shift was detected for both cage-perrhenate mixtures. Compared to the proton upfield shifts of a similar host–guest system (Ha: ∼0.08 ppm, Hb: ∼0.04 ppm),48 this result indicates poor encapsulation of perrhenate under these conditions, due to the high concentration of DMSO in the media, which does not reflect the clinical setting. Therefore, the ability of the cages to encapsulate perrhenate in predominantly aqueous media was additionally evaluated by DI-HR-ESI-MS in the case of C2hom. The comparison of the mass spectra of C2hom and the host–guest system (ReO4−)⊂C2hom in Fig. 2D, unambiguously revealed the formation of the host–guest adduct through the appearance of signals at m/z 655.9501 and 721.3443, which can be attributed to the 11+ and 10+ charge state of (ReO4−)⊂C2hom, respectively (Table 1). While the encapsulation is not quantitative, this result confirms some affinity of ReO4− for the cage cavity in aqueous solution, at concentrations more relevant to the clinical setting, in the μM range. However, it should also be noted that at variance with previously reported studies on similar MCgs,48 we were not able to preserve any host–guest complex following separation by LC.Fluoride-18 radiolabeling and logDpH7.4 determination
Next, the ligands were radiolabeled with fluoride-18 by [19F]-to-[18F] isotopic exchange. A Kryptofix-free synthesis route using cartridges as depicted in Fig. 6A was chosen.58 This method achieved 60–70% radiochemical conversion (RCC) and resulted in the radiolabeled compounds 18F-L1 and 18F-L2 obtained in 44–47% radiochemical yield (RCY) and 98–99% radiochemical purity (RCP) as evidenced by radio TLC and radio RP-HPLC (Fig. S36–S39†). Since lipophilicity is an important parameter in radiotracers development, the distribution coefficient (logDpH7.4) was determined for both ligands. High lipophilicity could cause low tumor accumulation in vivo due to an increased nonspecific liver uptake that elevates the off-target radiation level to the abdomen.60 On the other hand, a study, in which the physicochemical properties of around 300 central nervous system (CNS) drugs were analysed and compared to non-CNS drugs, revealed that positive logDpH7.4 values (logDpH7.4 in the range 2–3) can be beneficial for BBB-crossing.64
 | ||
| Fig. 6 (A) Schematic representation of the 18F-radiolabeling procedure of the ligands and CDSA of radiolabeled homoleptic MCgs. For fixation either a silica-based ion exchange cartridge (QMA) or a copolymer-based reverse phase cartridge (HLB) was used. (B) Radio chromatograms of 18F-C1hom and 18F-C2hom after 4 h. Chromatograms of the ligands (blue) and the co-injected non-radiolabelled cages (purple) are inserted as comparison. (C) CDSA of 18F-C1hom and 18F-C2hom monitored over 4 h by radio RP-HPLC. | ||
In the case of L1, a logDpH7.4 of 2.53 ± 0.05 of the radiolabeled ligand was determined. To assess the effects of such high lipophilicity in vivo, the organ uptake of 18F-L1 was evaluated in the CAM model (Fig. 7A). This preclinical tool can be used for high-throughput tumor imaging in a time- and cost-effective way and is in line with the ethical principles of reduction, refinement and replacement (3Rs).65 Glioblastoma tumors from U87 cells were grown on the CAM of fertilized chicken eggs. After injection of approximately 3 MBq of radiolabeled ligand, a 60 min dynamic PET scan was performed, and representative time points are depicted in Fig. 7B. While, this model was not intended for BBB-translocation experiments, evaluation of the maximal brain uptake was attempted over time up to 60 min p.i. (Fig. 7C and D). Overall, the obtained values are in accordance with published data for the BBB shuttle peptide PepH3 (0.31 ± 0.07% ID per g),51 as well as a PepH3-conjugated MCgs (0.42 ± 0.06% ID per g)48 evaluated in mice models in vivo. However, further studies are necessary to validate these data, possibly using bimodal PET/MRI imaging to better delineate the brain region. Moreover, long blood circulation and very high liver uptake demand further optimization of the ligand. In addition, bone uptake of 3.55 ± 0.91% ID per g might indicate defluorination of the ligand.66 Despite these hurdles, good tumor uptake was measured ex vivo, reaching 3.94 ± 1.31% ID per g at 60 min p.i. In the future, tumor accumulation could even be enhanced by combining the BBB-penetrating peptide with a glioblastoma-targeting ligand in a heteroleptic approach.
 | ||
| Fig. 7 In ovo evaluation of 18F-L1. (A) Schematic representation of the chick CAM model. (B) Representative in ovo PET images from a 60 min dynamic scan (n = 4). The liver (L) is indicated. (C) Time-activity curve of the chick embryo brain. (D) Ex ovo biodistribution one hour p.i. | ||
In the case of L2, as expected, the logDpH7.4 was 2.07 ± 0.07, which is a substantial reduction compared to the (SiFA)BA ligand. Nevertheless, its evaluation in the CAM model was not performed, due to the predicted high liver uptake; moreover, our focus was to investigate the formation of radiolabeled cages.
Synthesis of 18F-labeled homoleptic metallacages
Since the DPepH3-conjugated ligands L1 and L2 are modified with a SiFA moiety, labeling of the metallacage structure with F-18 is possible via exo-functionalization. Here, the radiolabeling can either proceed before CDSA (pre-assembly), during CDSA (one-pot) or after CDSA (post-assembly).43 In our case, to avoid cage disassembly in the relatively ‘harsh’ radiofluorination conditions, the ligands were radiolabeled pre-assembly and then CDSA was performed to form the homoleptic cages. Thus, after labeling 30 nmol of ligand (1 eq.) with ∼200 MBq of fluoride-18 and elution in EtOH, the solvent was fully evaporated and CDSA was conducted by adding 2 eq. of [Pd(MeCN)4](BF4)2 in 6 μL of DMSO (Fig. 6A). These conditions enabled the optimal concentration of each component (mM range) for successful CDSA. The self-assembly was monitored by radio RP-HPLC at 15 min, 60 min and 4 h (Fig. 6C). As depicted in Fig. 6B, 73% conversion to 18F-C1hom (tR = 9.6 min) was reached after 4 h with 18% remaining 18F-L1 (tR = 6.7 min), while full conversion to 18F-C2hom (tR = 7.7 min) was achieved. In both cases slight defluorination (18F-C1hom: 9%, 18F-C2hom: 3%) occurred as evidenced by the F-18 peak at tR = 2.4 min. Further purification of the reaction mixtures by analytical radio RP-HPLC or via cartridges was not carried out due to the short half-life of F-18 of 109.7 min, as well as possible instability of the radiolabeled cages.Conclusions
In this work, we aimed at further substantiating the applicability of supramolecular coordination complexes, namely porous MCgs, for radiopharmaceutical applications. These nano-structures can benefit from the self-assembly multicomponent design for functionalization and pharmacokinetics modulation, but at the same time their size range (1-few nm) enables their precise characterization by classical separation and analytical methods; thus, offering an ideal versatile chemical space to achieve next generation nuclear imaging agents.Here, two bispyridinyl ligands featuring a BBB shuttle peptide- and different silicon-based fluoride acceptor synthons (L1 and L2) were successfully synthesized by solid-phase peptide synthesis and radiolabeled with fluorine-18. The lipophilicity of the ligands was determined in vitro, via standard logDpH7.4 determination, and for L1 also ex vivo using the CAM model. Afterwards, formation of the respective homoleptic metallacages C1hom and C2hom was achieved by CDSA and confirmed by NMR spectroscopy, RP-HPLC and HR-ESI-MS. Importantly, 18F-radiolabeled homoleptic cages were achieved by labeling the ligands pre-assembly and via formation of the cages in situ. The optimization of the isotopic exchange protocol was an important step towards the future applicability of MCgs for PET imaging in a clinical setting.
Additionally, partial encapsulation of ReO4− into the cages following mixing of the components was evidenced by HR-ESI-MS; however, the extent and stability of the (ReO4−)⊂MCg host–guest adducts appeared less pronounced with respect to previously reported Pd2L4 cage structures.48 Thus, further ligand optimization is necessary to validate the suitability of these porous systems for encapsulation of anionic radioactive counterparts, such as 99mTcO4− for SPECT imaging or the β−-emitting 188ReO4−.
In order to improve the stability of the homoleptic Pd-based cages in solution, heteroleptic MCgs were also synthesized by statistical self-assembly and characterized by ESI-MS. The obtained results showed that [Pd2L(L0)3]4+ (C1/2het) and [Pd2L2(L0)2]4+ were the main assembled species.
In the future, an option to increase MCg's stability and to enable optimized radiofluorination of the cages post-assembly, could be replacing Pd2+ with the more kinetically inert Pt2+ to achieve [Pt2L4]4+ cages. The latter have already shown to be more stable with respect to ligand exchange than their Pd-based counterparts,67,68 which would also improve their stability in physiological conditions. Alternatively, organometallic building blocks endowed with higher metal–carbon bond stability could also be integrated in the cage scaffold.1
Overall, our results highlight the current challenges in the construction of targeted theranostics based on SCCs for future clinical applications in nuclear medicine. While much effort still needs to be spent on the development of metallacages for biomedical applications, their design flexibility and easiness of functionalization offer a unique toolbox for the obtainment of multimodal theranostics.
Author contributions
A. C. and L. R. were responsible for the project conceptualization and supervision. M. D. and A. C. wrote the first draft of the manuscript and all co-authors contributed to the Results and discussion sections. M. D., I. K. and A. C. designed the graphics. M. D. and D. K. performed the synthesis and radiolabeling of ligands and cages. I. K., L. R. and R. L. carried out high-resolution mass spectrometry studies and performed the data analysis. G. M.-A. and A. C. provided expertise in MS and NMR data analysis. M. D., L. M. S., M. E. G. and T. H. W. contributed to, and supervised, the in ovo studies.Data availability
The data supporting this article have been included as part of the ESI.† Raw data for this article, including chromatograms, NMR spectra, MS data, gamma counter data for logD determination, in ovo imaging data are available at Zenodo at [https://doi.org/10.5281/zenodo.15358484].
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge funding from EU Horizon Europe Research and Innovation program, EIC Pathfinder Open network “SMARTdrugs” (grant agreement ID: 101129886). Funding for T. H. W. was provided by UK Research and Innovation (UKRI) under the UK Government's Horizon Europe funding guarantee (grant no. 10091247) to T. H. W., arising from the above mentioned EIC Pathfinder grant. The TUM International Graduate School of Science and Engineering (IGSSE) is kindly acknowledged for financial support (International Project Team: Supramolecular Organometallic and Coordination Complexes for Medical Applications, SOCoMED). This research has also received funding from the European Union's Horizon H2020 Research and Innovation under the Marie Skłodowska-Curie grant agreement no. 945416 through the PhD fellowship of IK. The authors thank Dr Simon Godin for training and technical assistance with LC-MS/MS instrument.References
- A. Pöthig and A. Casini, Recent Developments of Supramolecular Metal-based Structures for Applications in Cancer Therapy and Imaging, Theranostics, 2019, 9, 3150 CrossRef PubMed.
- A. Casini, B. Woods and M. Wenzel, The Promise of Self-Assembled 3D Supramolecular Coordination Complexes for Biomedical Applications, Inorg. Chem., 2017, 56, 14715 CrossRef CAS PubMed.
- H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, Biomedically Relevant Self-Assembled Metallacycles and Metallacages, J. Am. Chem. Soc., 2019, 141, 14005 CrossRef CAS PubMed.
- D. L. Caulder and K. N. Raymond, Supermolecules by Design, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Controllable Coordination-Driven Self-Assembly: From Discrete Metallocages to Infinite Cage-Based Frameworks, Acc. Chem. Res., 2015, 48, 201–210 CrossRef CAS PubMed.
- J. Malina, P. Scott and V. Brabec, Recognition of DNA/RNA bulges by antimicrobial and antitumor metallohelices, Dalton Trans., 2015, 44, 14656–14665 RSC.
- J. Malina, H. Kostrhunova, P. Scott and V. Brabec, FeII Metallohelices Stabilize DNA G-Quadruplexes and Downregulate the Expression of G-Quadruplex-Regulated Oncogenes, Chem. – Eur. J., 2021, 27, 11682–11692 CrossRef CAS PubMed.
- S. Pullen, J. Tessarolo and G. H. Clever, Increasing structural and functional complexity in self-assembled coordination cages, Chem. Sci., 2021, 12, 7269–7293 RSC.
- G. Moreno-Alcántar and A. Casini, Bioinorganic supramolecular coordination complexes and their biomedical applications, FEBS Lett., 2023, 597, 191 CrossRef PubMed.
- W.-X. Gao, H.-N. Zhang and G.-X. Jin, Supramolecular catalysis based on discrete heterometallic coordination-driven metallacycles and metallacages, Coord. Chem. Rev., 2019, 386, 69–84 CrossRef CAS.
- R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Supramolecular Coordination Cages for Artificial Photosynthesis and Synthetic Photocatalysis, Chem. Rev., 2023, 123, 5225–5261 CrossRef CAS PubMed.
- N. Dey and C. J. E. Haynes, Supramolecular Coordination Complexes as Optical Biosensors, ChemPlusChem, 2021, 86, 418–433 CrossRef CAS PubMed.
- S. Ganta and D. K. Chand, Multi-Stimuli-Responsive Metallogel Molded from a Pd2L4-Type Coordination Cage: Selective Removal of Anionic Dyes, Inorg. Chem., 2018, 57, 3634–3645 CrossRef CAS PubMed.
- M. M. Deegan, M. R. Dworzak, A. J. Gosselin, K. J. Korman and E. D. Bloch, Gas Storage in Porous Molecular Materials, Chem. – Eur. J., 2021, 27, 4531–4547 CrossRef CAS PubMed.
- E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Design and Applications of Water-Soluble Coordination Cages, Chem. Rev., 2020, 120, 13480–13544 CrossRef PubMed.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Stimuli-responsive Pd2L4 metallosupramolecular cages: towards targeted cisplatin drug delivery, Chem. Sci., 2012, 3, 778 RSC.
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, The “Complex-in-a-Complex” Cations [(acac)2M⊂Ru6(p-iPrC6H4Me)6(tpt)2(dhbq)3]6+: A Trojan Horse for Cancer Cells, Angew. Chem., Int. Ed., 2008, 47, 3773 CrossRef CAS PubMed.
- J. W. Yi, N. P. E. Barry, M. A. Furrer, O. Zava, P. J. Dyson and B. Therrien, et al., Delivery of Floxuridine Derivatives to Cancer Cells by Water-Soluble Organometallic Cages, Bioconjugate Chem., 2012, 23, 461–471 CrossRef CAS PubMed.
- A. Schmidt, V. Molano, M. Hollering, A. Pöthig, A. Casini and F. E. Kühn, Evaluation of New Palladium Cages as Potential Delivery Systems for the Anticancer Drug Cisplatin, Chem. – Eur. J., 2016, 22, 2253 CrossRef CAS PubMed.
- J. Han, A. Schmidt, T. Zhang, H. Permentier, G. M. M. Groothuis and R. Bischoff, et al., Bioconjugation strategies to couple supramolecular exo-functionalized palladium cages to peptides for biomedical applications, Chem. Commun., 2017, 53, 1405 RSC.
- X. Wang, Q. Su, Z. Zhang, J. Yang, Y. Zhang and M. Zhang, Biotinylated platinum(II) metallacage towards targeted cancer theranostics, Chem. Commun., 2020, 56, 8460–8463 RSC.
- G. Yu, B. Zhu, L. Shao, J. Zhou, M. L. Saha and B. Shi, et al., Host−guest complexation-mediated codelivery of anticancer drug and photosensitizer for cancer photochemotherapy, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6618–6623 CrossRef CAS PubMed.
- Y. Li, F. Huang, P. J. Stang and S. Yin, Supramolecular Coordination Complexes for Synergistic Cancer Therapy, Acc. Chem. Res., 2024, 57, 1174–1187 CrossRef CAS PubMed.
- J. Zhang, W. Ma, B. Yang, T. Shi, S. Liao and Y. Li, et al., Biomimetic Metallacage Nanoparticles with Aggregation-Induced Emission for NIR-II Fluorescence Imaging-Guided Synergistic Immuno-Phototherapy of Tumors, ACS Appl. Mater. Interfaces, 2024, 16, 69028–69044 CrossRef CAS PubMed.
- D. Xu, Y. Li, S. Yin and F. Huang, Strategies to address key challenges of metallacycle/metallacage-based supramolecular coordination complexes in biomedical applications, Chem. Soc. Rev., 2024, 53, 3167–3204 RSC.
- R. D. Mukhopadhyay, Y. Kim, J. Koo and K. Kim, Porphyrin Boxes, Acc. Chem. Res., 2018, 51, 2730–2738 CrossRef CAS PubMed.
- G. Yu, S. Yu, M. L. Saha, J. Zhou, T. R. Cook and B. C. Yung, et al., A discrete organoplatinum(II) metallacage as a multimodality theranostic platform for cancer photochemotherapy, Nat. Commun., 2018, 9, 4335 CrossRef PubMed.
- X. Lin, F. Chen, X. Yu, H. Wang, H. Qiu and Y. Li, et al., Phenylthiol-BODIPY-based supramolecular metallacycles for synergistic tumor chemo-photodynamic therapy, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203994119 CrossRef CAS PubMed.
- T. Rodríguez-Prieto, D. Wragg, N. Heiduk, M. Park, N. Strittmatter and R. A. Fischer, et al., A Golden Touch in the Design of Multifunctional Porphyrin Metallacages: Host–Guest Chemistry for Drug-Target Interactions, CCS Chem., 2024, 6, 1662–1671 CrossRef.
- J. Han, A. F. B. Räder, F. Reichart, B. Aikman, M. N. Wenzel and B. Woods, et al., Bioconjugation of Supramolecular Metallacages to Integrin Ligands for Targeted Delivery of Cisplatin, Bioconjugate Chem., 2018, 29, 3856 CrossRef CAS PubMed.
- M. L. Saha, X. Yan and P. J. Stang, Photophysical Properties of Organoplatinum(II) Compounds and Derived Self-Assembled Metallacycles and Metallacages: Fluorescence and its Applications, Acc. Chem. Res., 2016, 49, 2527–2539 CrossRef CAS PubMed.
- B. Woods, D. Döllerer, B. Aikman, M. N. Wenzel, E. J. Sayers and F. E. Kühn, et al., Highly luminescent metallacages featuring bispyridyl ligands functionalised with BODIPY for imaging in cancer cells, J. Inorg. Biochem., 2019, 199, 110781 CrossRef CAS PubMed.
- B. Aikman, R. Bonsignore, B. Woods, D. Doellerer, R. Scotti and C. Schmidt, et al., Highly-fluorescent BODIPY-functionalised metallacages as drug delivery systems: synthesis, characterisation and cellular accumulation studies, Dalton Trans., 2022, 51, 7476–7490 RSC.
- E. O. Bobylev, R. A. Knol, S. Mathew, D. A. Poole, I. Kotsogianni and N. I. Martin, et al., In vivo biodistribution of kinetically stable Pt2L4 nanospheres that show anti-cancer activity, Chem. Sci., 2023, 14, 6943–6952 RSC.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Supramolecular Catalysis in Metal–Ligand Cluster Hosts, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- A. Dissanayake, J. A. Spernyak and J. R. Morrow, An octahedral coordination cage with six Fe(III) centers as a T1 MRI probe, Chem. Commun., 2024, 60, 12249–12252 RSC.
- P. R. Sahoo, J. A. Spernyak, S. G. Turowski and J. R. Morrow, Self-Assembled Iron(III) Coordination Cage as an MRI-Active Carrier for a Gold(I) Drug, Bioconjugate Chem., 2024, 35, 1618–1626 CAS.
- R. Wang, L. An, J. He, M. Li, J. Jiao and S. Yang, A class of water-soluble Fe(III) coordination complexes as T1 -weighted MRI contrast agents, J. Mater. Chem. B, 2021, 9, 1787–1791 RSC.
- G. E. Sokolow, M. R. Crawley, D. R. Morphet, D. Asik, J. A. Spernyak and A. J. R. McGray, et al., Metal−Organic Polyhedron with Four Fe(III) Centers Producing Enhanced T1 Magnetic Resonance Imaging Contrast in Tumors, Inorg. Chem., 2022, 61, 2603–2611 CrossRef CAS PubMed.
- B. P. Burke, W. Grantham, M. J. Burke, G. S. Nichol, D. Roberts and I. Renard, et al., Visualizing Kinetically Robust CoIII4L6 Assemblies in Vivo: SPECT Imaging of the Encapsulated [99mTc]TcO4, Anion, J. Am. Chem. Soc., 2018, 140, 16877 CrossRef CAS PubMed.
- A. Ahmedova, Biomedical Applications of Metallosupramolecular Assemblies—Structural Aspects of the Anticancer Activity, Front. Chem., 2018, 6, 620 CrossRef CAS PubMed.
- J. Xu, J. Wang, J. Ye, J. Jiao, Z. Liu and C. Zhao, et al., Metal–Coordinated Supramolecular Self–Assemblies for Cancer Theranostics, Adv. Sci., 2021, 8, 2101101 CrossRef CAS PubMed.
- G. Moreno-Alcántar, M. Drexler and A. Casini, Assembling a new generation of radiopharmaceuticals with supramolecular theranostics, Nat. Rev. Chem., 2024, 8, 893–914 CrossRef PubMed.
- G. Yu, T. R. Cook, Y. Li, X. Yan, D. Wu and L. Shao, et al., Tetraphenylethene-based highly emissive metallacage as a component of theranostic supramolecular nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13720–13725 CrossRef CAS PubMed.
- A. B. S. Elliott, J. E. M. Lewis, H. van der Salm, C. J. McAdam, J. D. Crowley and K. C. Gordon, Luminescent Cages: Pendant Emissive Units on [Pd2L4]4+ “Click” Cages, Inorg. Chem., 2016, 55, 3440 CrossRef CAS PubMed.
- A. Schmidt, M. Hollering, M. Drees, A. Casini and F. E. Kühn, Supramolecular exo-functionalized palladium cages: fluorescent properties and biological activity, Dalton Trans., 2016, 45, 8556 RSC.
- A. Schmidt, M. Hollering, J. Han, A. Casini and F. E. Kühn, Self-assembly of highly luminescent heteronuclear coordination cages, Dalton Trans., 2016, 45, 12297 RSC.
- B. Woods, R. D. M. Silva, C. Schmidt, D. Wragg, M. Cavaco and V. Neves, et al., Bioconjugate Supramolecular Pd2+ Metallacages Penetrate the Blood Brain Barrier In Vitro and In Vivo, Bioconjugate Chem., 2021, 32, 1399 CrossRef CAS PubMed.
- R. Cosialls, C. Simó, S. Borrós, V. Gómez-Vallejo, C. Schmidt and J. Llop, et al., PET Imaging of Self–Assembled 18F–Labelled Pd2L4 Metallacages for Anticancer Drug Delivery, Chem. – Eur. J., 2023, 29, e202202604 CrossRef CAS PubMed.
- S. Deiser, M. Drexler, G. Moreno-Alcántar, M. Irl, C. Schmidt and T. Günther, et al., Synthesis of 177Lu-Labeled, Somatostatin-2 Receptor-Targeted Metalla-Assemblies: Challenges in the Design of Supramolecular Radiotherapeutics, Inorg. Chem., 2023, 62, 20710–20720 CrossRef CAS PubMed.
- V. Neves, F. Aires-da-Silva, M. Morais, L. Gano, E. Ribeiro and A. Pinto, et al., Novel Peptides Derived from Dengue Virus Capsid Protein Translocate Reversibly the Blood–Brain Barrier through a Receptor-Free Mechanism, ACS Chem. Biol., 2017, 12, 1257 CrossRef CAS PubMed.
- D. Wu, Q. Chen, X. Chen, F. Han, Z. Chen and Y. Wang, The blood–brain barrier: Structure, regulation and drug delivery, Signal Transduction Targeted Ther., 2023, 8, 217 CrossRef PubMed.
- M. Cavaco, J. Valle, R. da Silva, J. D. G. Correia, M. A. R. B. Castanho and D. Andreu, et al., DPepH3, an Improved Peptide Shuttle for Receptor-independent Transport Across the Blood-Brain Barrier, Curr. Pharm. Des., 2020, 26, 1495 CrossRef CAS PubMed.
- J. E. M. Lewis, Molecular engineering of confined space in metal–organic cages, Chem. Commun., 2022, 58, 13873–13886 RSC.
- L. Iovkova, B. Wängler, E. Schirrmacher, R. Schirrmacher, G. Quandt and G. Boening, et al., para–Functionalized Aryl–di–tert–butylfluorosilanes as Potential Labeling Synthons for 18F Radiopharmaceuticals, Chem. – Eur. J., 2009, 15, 2140–2147 CrossRef CAS PubMed.
- S. Deiser, S. Fenzl, V. König, M. Drexler, L. M. Smith and M. E. George, et al., (SiFA)SeFe: A Hydrophilic Silicon-Based Fluoride Acceptor Enabling Versatile Peptidic Radiohybrid Tracers, J. Med. Chem., 2024, 67, 14077–14094 CrossRef CAS PubMed.
- B. Woods, M. N. Wenzel, T. Williams, S. R. Thomas, R. L. Jenkins and A. Casini, Exo-Functionalized Metallacages as Host-Guest Systems for the Anticancer Drug Cisplatin, Front. Chem., 2019, 7, 68 CrossRef CAS PubMed.
- T. Günther, N. Holzleitner, D. Di Carlo, N. Urtz-Urban, C. Lapa and H.-J. Wester, Development of the First 18F-Labeled Radiohybrid-Based Minigastrin Derivative with High Target Affinity and Tumor Accumulation by Substitution of the Chelating Moiety, Pharmaceutics, 2023, 15, 826 CrossRef PubMed.
- A. Höhne, L. Mu, M. Honer, P. A. Schubiger, S. M. Ametamey and K. Graham, et al., Synthesis, 18F-Labeling, and in Vitro and in Vivo Studies of Bombesin Peptides Modified with Silicon-Based Building Blocks, Bioconjugate Chem., 2008, 19, 1871–1879 CrossRef PubMed.
- C. Wängler, B. Waser, A. Alke, L. Iovkova, H.-G. Buchholz and S. Niedermoser, et al., One-Step 18F-Labeling of Carbohydrate-Conjugated Octreotate-Derivatives Containing a Silicon-Fluoride-Acceptor (SiFA): In Vitro and in Vivo Evaluation as Tumor Imaging Agents for Positron Emission Tomography (PET), Bioconjugate Chem., 2010, 21, 2289–2296 CrossRef PubMed.
- F. Kaiser, A. Schmidt, W. Heydenreuter, P. J. Altmann, A. Casini and S. A. Sieber, et al., Self-Assembled Palladium and Platinum Coordination Cages: Photophysical Studies and Anticancer Activity, Eur. J. Inorg. Chem., 2016, 2016, 5189 CrossRef CAS.
- R. Evans, G. Dal Poggetto, M. Nilsson and G. A. Morris, Improving the Interpretation of Small Molecule Diffusion Coefficients, Anal. Chem., 2018, 90, 3987–3994 CrossRef CAS PubMed.
- E. Puig, C. Desmarets, G. Gontard, M. N. Rager, A. L. Cooksy and H. Amouri, Capturing a Square Planar Gold(III) Complex Inside a Platinum Nanocage: A Combined Experimental and Theoretical Study, Inorg. Chem., 2019, 58, 3189 CrossRef CAS PubMed.
- A. K. Ghose, T. Herbertz, R. L. Hudkins, B. D. Dorsey and J. P. Mallamo, Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery, ACS Chem. Neurosci., 2012, 3, 50–68 CrossRef CAS PubMed.
- L. M. Smith, H. E. Greenwood, W. E. Tyrrell, R. S. Edwards, V. de Santis and F. Baark, et al., The chicken chorioallantoic membrane as a low-cost, high-throughput model for cancer imaging, npj Imaging, 2023, 1, 1 CrossRef PubMed.
- Y. Li, C. Schiepers, R. Lake, S. Dadparvar and G. R. Berenji, Clinical utility of 18F-fluoride PET/CT in benign and malignant bone diseases, Bone, 2012, 50, 128–139 CrossRef PubMed.
- Z. T. Avery, M. G. Gardiner and D. Preston, Using a New Pt(II) Source to Make Pt(II) Lantern-Shaped Cages, Including Low-Symmetry, Heteroleptic, and Multicavity Examples, Angew. Chem., Int. Ed., 2024, e202418079 Search PubMed.
- Ž. D. Bugarčić, J. Bogojeski and R. van Eldik, Kinetics, mechanism and equilibrium studies on the substitution reactions of Pd(II) in reference to Pt(II) complexes with bio-molecules, Coord. Chem. Rev., 2015, 292, 91–106 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, chromatograms, MS data, gamma counter data, in ovo imaging data. See DOI: https://doi.org/10.1039/d5qi01092f |
| This journal is © the Partner Organisations 2025 |