
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron-rich phenanthroline bearing N-heterocyclic imine substituents: synthesis, optical properties, metal coordination†
Jonas H.
Franzen
a,
Xuequan
Zhou
b,
Kelly
Biv
c,
Alessandro
Ajò
b,
Austin
Mencke
c,
Lukas F. B.
Wilm
d,
Michael
Seidl
a,
Thomas S.
Hofer
 a,
Luisa
De Cola
be,
Peter
Brüggeller
a,
Mark E.
Thompson
*c and
Fabian
Dielmann
*a
a,
Luisa
De Cola
be,
Peter
Brüggeller
a,
Mark E.
Thompson
*c and
Fabian
Dielmann
*a
aInstitut für Allgemeine, Anorganische unt Theoretische Chemie, Leopod-Franzens-universität Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: fabian.dielmann@uibk.ac.at
bDepartment of Biochemistry and Molecular Pharmacology, Istituto di Ricerche Farmacologiche Mario Negri, IRCCS, 20156 Milano, Italy. E-mail: luisa.decola@unimi.it
cDepartment of Chemistry, University of Southern California, Los Angeles, CA 90089, USA. E-mail: met@usc.edu
dInsitut für Anorganische und Analytische Chemie, Universität Münster, Corrensstraße 30, 48179 Münster, Germany
eDepartment of Pharmaceutical Science, DISFARM, Università degli Studi di Milano, Via C. Golgi 19, 20133 Milan, Italy
First published on 12th June 2025
Abstract
Polypyridines functionalized with π-donating groups constitute a class of electron-rich ligands with significant relevance in coordination chemistry and catalysis. The incorporation of strongly basic guanidinyl substituents, however, often introduces multiple binding sites, with coordination typically favoring the guanidyl nitrogen atoms. Herein, we report the synthesis and characterization of a new electron-rich 1,10-phenanthroline ligand featuring bulky NHI groups that define a well-structured coordination cavity. Protonation studies and the preparation of a zinc(II) complex reveal that Lewis acids preferentially coordinate at the phenanthroline nitrogen atoms rather than the NHI moiety. The electronic and photophysical properties of the new ligand and its complexes are explored through a combination of computational and experimental methods, demonstrating that its emission and absorption characteristics are highly sensitive to protonation, concentration, and metal coordination.
Introduction
N-heterocyclic imines (NHIs) are a class of cyclic guanidines characterized by an exocyclic imine function attached to a nitrogen-containing heterocycle. Their manifold accessibility takes root in the seminal synthetic protocols reported by Kuhn in 1995 and thereafter.1–6 The use of NHIs as imine substituents or as anionic iminato ligands takes advantage of their capability to act as 4π or 2σ/4π donors, respectively, due to the allocation of electron density from the heterocycle to the exocyclic nitrogen atom (Fig. 1A).7 The effective stabilization of a positive charge within the N-heterocycle renders NHIs stronger π donors compared to aliphatic guanidines. In addition, modifying the N,N′-substituents enables predictable steric control without compromising the potent donor properties. As a result, the coordination chemistry of NHIs towards main group and transition metal elements has been extensively investigated and reviewed in the past.4–10 | ||
| Fig. 1 (A) Mesomeric Lewis structure of the NHI scaffold. (B) and (C) Selected examples of electron-rich pyridine-based ligands with (cyclic) guanidyl substituents. | ||
While these characteristics have been pivotal for the stabilization and isolation of otherwise elusive low-valent main group species,11–22 the exceptional electron-donating capacity of NHIs render them also particularly effective in generating electron-rich compounds.23 For instance, our group has shown that guanidyl and NHI substituents produce superbasic phosphines when attached up to three times to a phosphorus centre.24–27 We also demonstrated that decoration of porphyrins with NHIs can lead to improved selectivity in the electroreduction of CO2.28 Generally, the decoration of arenes with multiple π-donor substituents results in highly electron-rich π systems, which was utilized for the development of organic reducing agents,29–37 dyes,38,39 and redoxactive multidentate ligands.40–43
The influence of para-NHI substitution on the properties of pyridines has been studied using several descriptors, including the methyl cation affinity (MCA),44–47 the molecular electrostatic potential (MESP) topology analysis,48–51 and the Huynh electronic parameter (HEP).52–54 Consistently, these parameters show that the electron-density at the pyridine N atom correlates with the π-donating ability of the NHI group (Fig. 1B). This was experimentally demonstrated through the reversible formation of SO2 and CO2 adducts by the most basic pyridines which are not isolable using unsubstituted pyridine.55–58 The Himmel group demonstrated that the incorporation of multiple guanidinyl groups into pyridine frameworks significantly increases the electron density of the pyridine ring and simultaneously generates ligands with multiple donor sites.38,39,59–61 This is due to the fact that guanidines themselves are highly effective ligands in coordination chemistry.62 As exemplified by compound I, guanidinyl groups frequently serve as the preferred coordination sites for transition metals M (Fig. 1C). Similarly, in the case of bipyridine derivative II, the guanidyl moieties compete with the pyridine nitrogen atoms for coordination to the metal center.41 Moreover, Henkel and colleagues reported the synthesis of the 1,10-phenanthroline-based Janus head pro-ligand III, which can accommodate different transition metal centers at opposite ends of the molecule.63,64
In contrast, the parent 1,10-phenanthroline (phen) ligand is described as electron-deficient species with predominant π acceptor properties due to the large conjugated aromatic system and low-lying π* orbital.65,66 Its rigidity causes the two N atoms to always be oriented cis to each other rendering phen an excellent chelating ligand67 and the synthesis of coordination compounds incorporating almost every element of the periodic table has been realized.65,68 Thus, the application of phen and its derivatives has been broadly explored over many areas of research such as materials chemistry,69–73 medicinal chemistry,74–79 catalysis,80–87 and photochemistry.88–95
As part of our ongoing efforts to develop highly electron-rich ligands,25,28,55 we report the synthesis and characterization of a 1,10-phenanthroline derivative bearing bulky N,N′-di-tert-butylimidazolin-2-ylidenamino substituents adjacent to the metal coordination site.
We hypothesize that, in contrast to the NHI carrying ligands II and III, this ligand will accommodate only a single metal center due to the rigid framework of the phenanthroline core and the steric hindrance provided by the tert-butyl groups. This design will allow for a detailed investigation of the effect of NHI substitution on the optical properties of 1,10-phenanthroline.
Results and discussion
Ligand and complex synthesis
The synthesis of the NHI-substituted 1,10-phenanthroline 1 was attempted in analogy to the reported procedure for the synthesis of N-heterocyclic imines.96,97 Our group recently demonstrated the feasibility of this approach for pyridine based systems using 4-aminopyridine.55 In fact, heating a suspension of 2,9-diamino-1,10-phenanthroline, N,N′-di-tert-butylimidazolin-2-chloroimidazolinium tetrafluoroborate97,98 and KF at 90 °C for three days led to the selective formation of 1·HBF4 which was isolated after filtration and washing with water to remove excess KF in good yields (Scheme 1). 1·HBF4 is a bright yellow, bench-stable solid best soluble in polar organic solvents like DCM and MeCN. In the 1H NMR spectrum, the acidic proton resonance of the 1H+ cation appears at high frequencies (CD2Cl2: 10.61 ppm, MeCN-d3: 10.04 ppm) as a broad singlet indicating a dynamic process involving shuttling of the proton. The sharp aromatic 1H NMR resonances exhibit the typical splitting pattern for (protonated) 1,10-phenanthrolines of two doublets and one singlet.99–101 Likewise, the NHI groups are chemically and magnetically equivalent and the methylene- and tert-butyl groups each appear as a singlet. Attempts to localize the acidic N–H proton using 2D and variable temperature NMR experiments remained unsuccessful (see ESI† for details). In symmetrically substituted phenanthroline ligands, the proton is bound between both N atoms.69,102 A single-crystal X-ray diffraction (SCXRD) analysis of 1·HBF4 revealed that the proton is positioned on either of the two phenanthroline N atoms in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (see ESI† for details) (Fig. 2). The preferential protonation at the pyridine nitrogen rather than the NHI group is consistent with calculated proton affinities of various amino- and imino-substituted pyridines.44,103
:1 ratio (see ESI† for details) (Fig. 2). The preferential protonation at the pyridine nitrogen rather than the NHI group is consistent with calculated proton affinities of various amino- and imino-substituted pyridines.44,103
 | ||
| Scheme 1 Synthesis of 1·HBF4, 1·2HBF4 and the free ligand 1. | ||
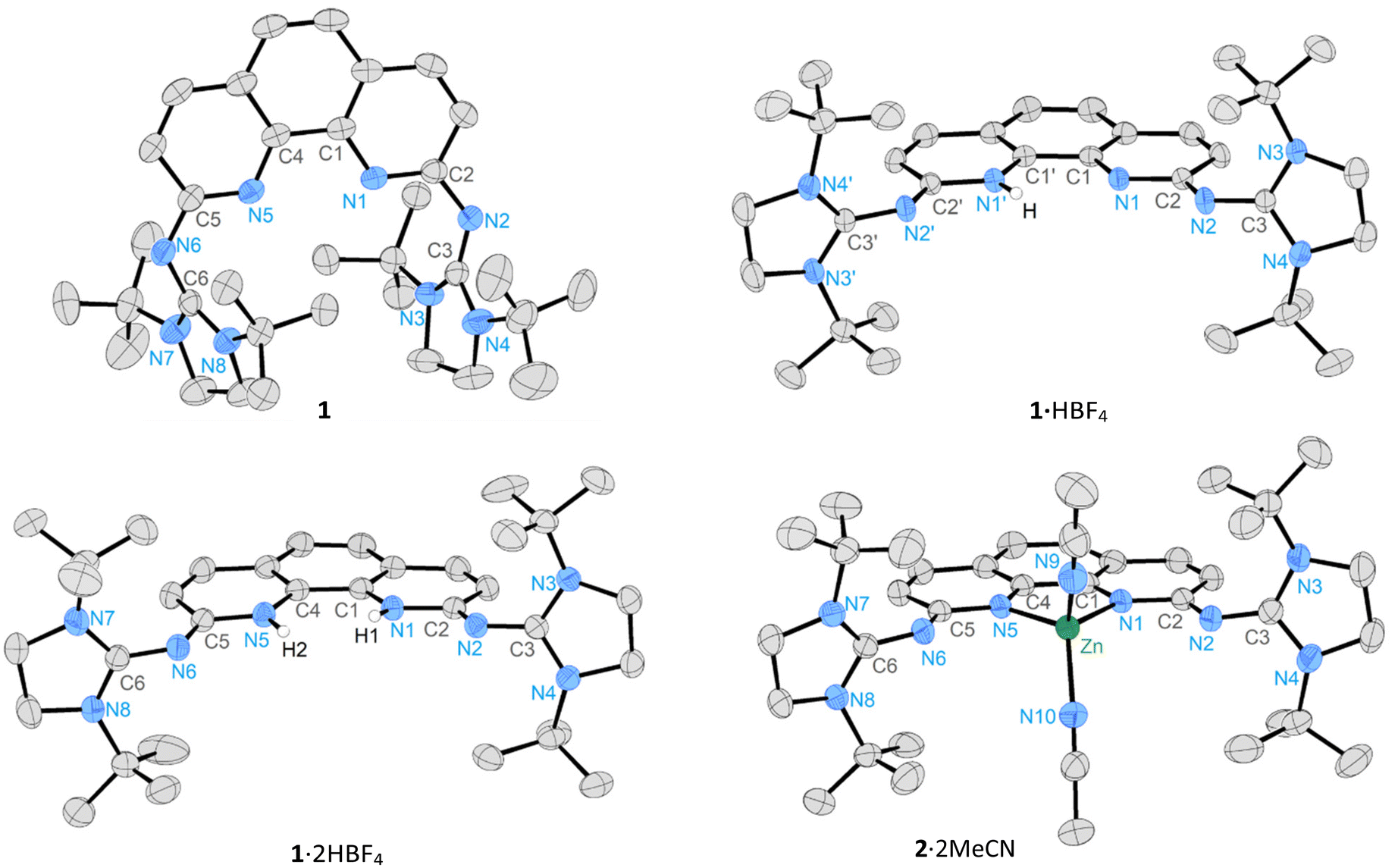 | ||
| Fig. 2 Molecular structures of 1, 1·HBF4 and 1·2HBF4 and 2·2MeCN. Ellipsoids are set at 50% probability. Hydrogen atoms except for the N–H hydrogen atoms and counterions are omitted for clarity. Selected bond lengths (Å) and angles (°): 1: C1–C4 1.451(2), C1–N1 1.363(2), N1–C2 1.339(1), C2–N2 1.348(2), N2–C3 1.307(2), C3–N3 1.367(2), C3–N4 1.367(2), N1–C2–N2 122.2(1), C2–N2–C3 130.3(1); 1·HBF4: C1–C1′ 1.423(3), C1–N1 1.366(3), N1–C2 1.352(3), C2–N2 1.326(3), N2–C3 1.332(3), C3–N3 1.350(3), C3–N4 1.354(3), N1–C2–N2 116.3(2), C2–N2–C3 126.2(2); 1·2HBF4: C1–C4 1.418(2), C1–N1 1.381(2), N1–C2 1.367(2), C2–N2 1.301(2), N2–C3 1.354(2), C3–N3 1.342(2), C3–N4 1.336(2), N1–C2–N2 116.1(1), C2–N2–C3 122.7(1); 2·2MeCN: N1–Zn 1.984(2), N5–Zn 1.984(2), N9–Zn 1.972(3), N10–Zn 1.979(3), C1–C4 1.430(3), N1–C2 1.356(3), C2–N2 1.318(3), N2–C3 1.340(3), C3–N3 1.351(4), C3–N4 1.345(4), N1–C2–N2 116.5(2), C2–N2–C3 123.7(2), N1–Zn–N5 84.23(9), N9–Zn–N10 109.23(11). | ||
DFT calculations at the B3LYP/6-31G(d,p)/SMD(DCM) level of theory confirm that the first protonation of 1 is energetically favored by 52.0 kJ mol−1 at a phenanthroline nitrogen atom compared to protonation at an imine group. The second protonation preferentially occurs at the imine nitrogen rather than the second phenanthroline nitrogen, with an energy advantage of 12.6 kJ mol−1 (see ESI† for details). DFT calculations reveal that the basicity of 1, with a pKBH+ value of 25.0 in acetonitrile (ACN), is significantly enhanced compared to pristine phenanthroline (pKBH+ (ACN) = 13.7).104 This places its basicity within the range of superbases such as DBU (pKBH+ (ACN) = 24.1) and TBD (pKBH+ (ACN) = 26.0).105 This value was further validated by proton transfer experiments using a variety of bases, including DBU, TBD and the phosphazene base P1-tBu(pyrr)3 (pKBH+ (ACN) = 28.4)106 (see ESI† for details). Experimentally, the second protonation was achieved using either HBF4·Et2O or [H(EtO)2][B(C6F5)4].107 The 1H NMR resonances of the resulting dication are significantly broadened, indicating the presence of several tautomers in solution. However, an SCXRD study of 1·2HBF4 revealed that, in the solid state, both protons are bound to the phenanthroline N atoms, forming hydrogen-bonding interactions with acetonitrile solvate molecules in the crystal lattice. Attempts to achieve further protonation and form [1·3H]3+ using [H(OEt2)2][B(C6F5)4] remained unsuccessful. In the 1H NMR spectrum of the mixture, the resonances of [1·2H]2+ and the acid were observed indicating that [1·2H]2+ is less basic than Et2O (see ESI† for more details). The free ligand 1 was isolated as an orange-to-red solid in good yields after deprotonation of 1·HBF4 with KOtBu. 1 is soluble in polar organic solvents like MeCN and DCM, moderately soluble in THF and toluene, and virtually insoluble in n-hexane or water. Nonspecific decomposition of 1 in CDCl3 within minutes and in DCM-d2 over the course of hours was observed by 1H NMR spectroscopy reflecting the high basicity of 1. Measured in C6D6, the phenanthroline 1H NMR resonances of 1 appear at slightly higher frequencies (7.65 ppm, 7.21 ppm, 6.93 ppm) compared to those of the parent 1,10-phenanthroline (7.47 ppm, 7.07 ppm, 6.85 ppm).
The ZnII complex 2·2MeCN was prepared by treatment of 1 with Zn(OTf)2 in MeCN at ambient temperature (Scheme 2). In contrast to phenanthroline complexes III (Fig. 1C), the use of an excess ZnII did not lead to bimetallic complexes due to the close proximity of the bulky NHI groups to the coordination site in 1. Protonation of 2·2MeCN with [H(OEt2)2][B(C6F5)4] was accomplished, but it resulted in partial dissociation of the complex and the release of the cationic ligand 1·H(BC6F5)4. The solid-state structures of 1 and 2·2MeCN were established by SCXRD studies (Fig. 2). For the free ligand 1, the imidazole groups are staggered and rotated (Ø 65.3°) relative to the phenanthroline plane. In contrast, within 1·HBF4 (81.6°), 1·2HBF4 (Ø 84.8°), and 2·2MeCN (Ø 79.0°), the imidazole groups adopt a nearly orthogonal orientation. The N1–C2–N2 bond angles (1: 122.2°, 1·HBF4: 116.3°, 1·2HBF4: 116.1°, 2·2MeCN: 116.5°) and the C2–N2–C3 angles (1: 130.3°, 1·HBF4: 126.2°, 1·2HBF4: 122.7°, 2·2MeCN: 123.7°) are similar. The C–N bond distances (1.301 Å–1.340 Å) are within the range of typical C–N single and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds.108,109 In the solid state structure of III, the NHI groups are staggered in a similar fashion to 1 and rotated (Ø 53.8°) relative to the phenanthroline plane. A comparison of geometrical parameters is shown in Table 1.63,110 Notably, the Cphen–Nimine and the endocyclic CNHI–NNHI bond lengths are shortened in 1 compared to III, while the NimineCNHI bonds are elongated. This indicates a more pronounced π donation of the NHI groups towards the pyridine units in the case of 1.
N double bonds.108,109 In the solid state structure of III, the NHI groups are staggered in a similar fashion to 1 and rotated (Ø 53.8°) relative to the phenanthroline plane. A comparison of geometrical parameters is shown in Table 1.63,110 Notably, the Cphen–Nimine and the endocyclic CNHI–NNHI bond lengths are shortened in 1 compared to III, while the NimineCNHI bonds are elongated. This indicates a more pronounced π donation of the NHI groups towards the pyridine units in the case of 1.
 | ||
| Scheme 2 Synthesis of the ZnII complex 2·2MeCN. | ||
| 1 | III | |
|---|---|---|
| Cphen–Nimine | 1.347(2) Å | 1.398(2) Å |
| NimineCNHI |
1.314(2) Å | 1.284(2) Å |
| CNHI–NNHI | 1.364(2) Å | 1.383(3) Å |
| Cphen–NimineCNHI |
130.3(1)° | 126.3(2)° |
| NimineCNHI–NNHI |
125.2(1)° | 126.3(2)° |
| NNHI–CNHI–NNHI | 109.1(1)° | 107.5(2)° |
Electrochemical investigations
Considering the strong π-donor capability of the NHI groups,7,55,63 we were curious to investigate their effect on the redox potentials and thus studied the electrochemical properties of 1, 1·HB(C6F5)4, 2·2MeCN and phen using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) (see ESI† for details). For 1, two partially reversible one-electron oxidation processes (−0.03 V and 0.38 V vs. Fc/Fc+), one irreversible oxidation process (0.57 V vs. Fc/Fc+) and two irreversible one-electron reductions at low potentials (−3.03 V and −3.18 V vs. Fc/Fc+) are observed (Fig. 3a). The latter are significantly shifted cathodically when compared to phen (−2.63 V, −2.82 V vs. Fc/Fc+) evidencing the increase of electron density within the aromatic system.111 According to the DFT calculations, the HOMO and HOMO−1 of 1 are distributed over the phenanthroline moiety and imine N atoms and are mostly of π character (see Fig. S68 in the ESI†). Thus, the first two oxidation processes likely occur at the aromatic phenanthroline scaffold and the oxidation potential follows the same trend of the phenanthroline ligand in III (Eox1st = −0.26 V vs. Fc/Fc+).63 Similarly, the positive charges are stabilized through resonance structures which involve the NHI groups. However, due to the different substitution pattern, a third irreversible oxidation process is observed in the case of 1 (see Fig. S68 and S69 in the ESI† for details). Within the potential window of the first oxidation event, the process appears to be reversible even at low scan rates (Fig. 3b). Within the potential window including all oxidation events on the other hand, these processes only possess reversible character at higher scan rates. This indicates the formation of a short-lived species upon further oxidation.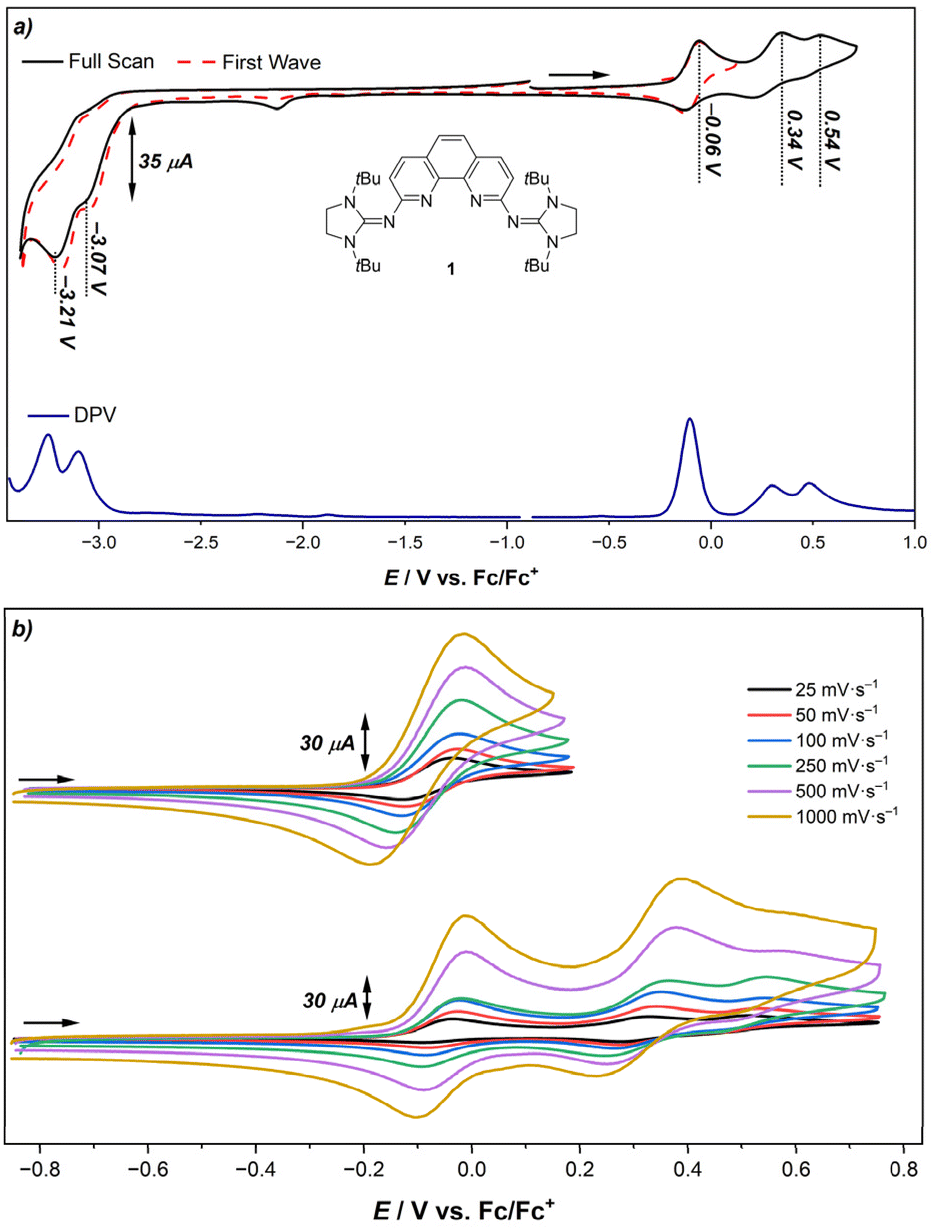 | ||
| Fig. 3 (a) Cyclic voltammogram and differential pulse voltammogram of 1 under inert conditions. (CV: scan rate: 100 mV s−1, in MeCN, electrolyte: [n-Bu4N][PF6] (0.1 M), working electrode: glassy carbon, counter electrode: Pt wire; DPV: step size: 5 mV, pulse time: 0.1 s, pulse size: 25 mV). Internally referenced against Fc*/Fc*+ and reported against Fc/Fc+. (b) Cyclic voltammogram of 1 under inert conditions at different potential windows and different scan rates (in MeCN, electrolyte: [n-Bu4N][PF6] (0.1 M), working electrode: glassy carbon, counter electrode: Pt wire). Internally referenced against Fc*/Fc*+ and reported against Fc/Fc+. | ||
In comparison to 1, the protonated salt 1·HB(C6F5)4 is less electron-rich. Consistently, the first reduction potential is shifted anodically (Ered1st = −2.19 V) and the first oxidation potential is shifted cathodically (Eox1st = 0.53 V). This trend was also observed in the CV studies of the zinc complex 2·2MeCN (Table 2).
| Compound | E red1st | E ox1st |
|---|---|---|
| Phen | −2.67 V | — |
| 1 | −3.07 V | −0.06 V |
| 1·HB(C6F5)4 | −2.22 V | 0.50 V |
| 2·2MeCN | −2.64 V | 0.71 V |
Photophysical properties
The photophysical properties of 1, 1·HBF4 and 2·2MeCN were studied and compared with those of 1,10-phenanthroline (phen) for reference. Phen absorbs light in the UV region (200–320 nm) and displays a weak and short lived fluorescence emission (τ ≤ 1 ns, λem ≈ 360 nm) at 298 K and a long lived phosphorescence (τ = 1.1 s, λem ≈ 460 nm) at 77 K.112–114 Protonation of phen induces a red-shift of the absorption and emission maxima.69,102 This change of photophysical properties of phen and its derivatives in dependence of the pH or upon complexation to a metal center has led to their application as chemosensors.66,115–1181 shows a complex photophysical behavior. As in phen, the luminescence of 1 at 77 K consists of both fluorescence and phosphorescence with dramatically different excited state lifetimes of nanoseconds and seconds, respectively. The short lifetime is consistent with rapid deactivation of the singlet excited state (S1) via intersystem crossing (ISC) to the triplet excited sate (T1). The T1 in contrast is expected to have a very long-lived phosphorescence. Fig. 4 (A, top) shows the absorption spectra of 1 in THF at ambient temperature and at concentrations of 0.5, 1, 2, 3, 5, 10, 20, 30, 50, 100, and 200 μM. For 1, the absorption at 290 nm is shifted to 280 nm in CH2Cl2. This band can be assigned to a spin-allowed π → π* transition of the phenanthroline moiety of 1.113 Compared with unsubstituted 1,10-phenanthroline, where this band occurs at 264 nm in CH2Cl2, it is red shifted in 1. This red shift indicates lowering of the π–π* level due to the charge-transfer character resulting from the electron donation of the NHI groups into the phenanthroline π system. Therefore, the electron-donating properties of the substituents in 1 promote this red shift. This band at 290 nm shows no concentration dependence within the range 0.5 to 200 μM (Fig. 4A, above). By contrast, the second spin-allowed π → π* transition in 1 exhibits a small red shift from 350 nm (0.5 μM) to 360 nm (200 μM). Since this transition is completely lacking in the UV/Vis spectrum of 1,10-phenanthroline,113 it can be attributed to a shifted π → π* band due to the imidazolidine substituents of 1. In order to further characterise the individual UV/Vis excitations, TD-DFT calculations have been carried out. The resulting theoretical spectrum determined at B3LYP/6-31G(d,p) level in implicit dichloromethane was found in good agreement with the experimental reference (see Fig. S106, see in the ESI†). According to the calculations, three excitations giving rise to four dominant absorption bands at 361, 340, 301 and 299 nm are observed. Based on a visual inspection of the associated canonical molecular orbitals, all excitations can be classified to be predominantly π → π* transitions. A linear dependence absorbance versus concentration is observed for 1 (Fig. S80†), following the Beer–Lambert law, indicating that aggregation is not occurring, even at the highest concentrations of 0.2 M. Fig. 4 (A, bottom) shows two main emission peaks at 464 and 550 nm when excited at 360 nm at ambient temperature. At the lowest concentrations, only the peak at 550 nm is present. At higher concentrations (5–200 μM), the emission is blue shifted to a maximum band at 464 nm. At low concentrations the lifetimes measured for the blue component (λmax = 465 nm) has a short component of 0.9 ns (64%) and a longer-lived component of 8.0 ns (36%), while the longer wavelengths (λem = 550 nm) has a lifetime of 1.2 (28%) and 10.2 ns (72%) respectively. At higher concentrations, where the blue emission line dominates the spectrum, only a very long-lived emission decay is observed, on the order of 0.1 to 1.0 s. The concentration dependent photophysics is most likely the result of a triplet–triplet annihilation (TTA) process (Fig. 5). In the TTA process, two molecules in their triplet excited states diffuse together and form a molecule in the ground state (S0) and one in the S1 state (T1 + T1 → S0 + S1 → 2S0 + hν). The lifetimes of the green and blue bands track the TTA rate, not the S1 rate. The S1 states formed by TTA decay by a combination of emission and ISC to the T1, both of which are orders of magnitude faster than the TTA process that generated the S1. The TTA process here is enabled by the long-lived T1 state (τ = 0.1–1 s). Thus, at low concentration the optical density and thus the concentration of T1 states formed on excitation is low and phosphorescence from T1 outcompetes TTA. As the concentration is increased, the optical density increases leading to a higher T1 concentration on excitation and TTA takes place leading to S1 emission being the dominant process. Assuming the emission of 1 changes with concentration due to TTA, it is important to check that all emissions stem from the same excited states at all concentrations. The complete emission range from 420 to 600 nm shows the same excitation peak at 360 nm (Fig. S77†), confirming that the same state formed on light absorption leads to both emission bands. These excitation peaks at 360 nm correspond to the S0→S1 absorption peak (Fig. 4A, top). To make the assignments of the excited states complete, the emissions of 1 in a polystyrene matrix at ambient temperature and in a 2-methyl-THF glass at 77 K were measured (Fig. 6 and Fig. S81†). At ambient temperature fluorescence appears (λmax = 465 nm τave = 3.7 ns), but the major contributor to the emission is the phosphorescence band at λmax = 520 nm (τave = 0.5 s). The emission band at 465 nm also shows a long time decay trace of 0.5 s, matching the value observed at 520 nm. At 77 K phosphorescence also plays a dominant role in the emission spectrum, differing from fluid solution at this concentration. This is typical for 1,10-phenanthroline and its derivatives.113 The fluorescence at 77 K is centered at 450 nm, thus showing a typical rigidochromic effect on cooling,119 when compared with the peak at 464 nm at ambient temperature in polystyrene. | ||
| Fig. 4 The absorption and emission spectra of ligand 1 (A), 1·HBF4 (B), and 2·2MeCN (C) were measured in THF (A) and DMF (B and C), setting concentrations as 0.5, 1, 2, 3, 5, 10, 20, 30, 50, 100, and 200 μM. Emission spectra for an excitation at 360 nm (A) and 350 nm (B and C). | ||
 | ||
| Fig. 5 Jablonski diagram of compound 1 including the intermolecular TTA process with a second molecule of 1 acting as annihilator (ISC = intersystem crossing, TTA = triplet–triplet annihilation, PS = polystyrene). | ||
 | ||
| Fig. 6 Emission spectra of a polystyrene film of 1 (1% w/w) at ambient temperature (left) and 10 μM solution of 1, 1·HBF4 and 2·2MeCN recorded in glassy 2-methyl-THF at 77 K. The excitation wavelength was 360 nm. The dashed line contains both fluorescence and phosphorescence, whereas in the gated spectrum (red line) the fluorescence is suppressed. These spectra are independent on concentration. | ||
The phosphorescence shows a similar rigidochromic effect, with λmax at 513 nm with a shoulder at 540 nm at 77 K. This long-lived phosphorescence in the rigid matrix at 77 K is assigned to the lowest 3π–π* level.112,113,120 Its lifetime is independent of wavelength and for 450, 500, and 550 nm the same lifetimes of 1.2 s have been measured (see Fig. S79 in the ESI†) for an excitation wavelength of 360 nm. Note that in both polystyrene at ambient temperature and 2-methyl-THF at 77 K, diffusion is precluded, preventing TTA-based emission.
To further confirm these assignments, excited lifetime measurements were carried out. We measured the lifetimes of ligand 1 in air or degassed conditions at 0.5 and 100 μM for the peaks at 440, 464 and 550 nm (see Fig. S75 in the ESI†). The wavelength of excitation was 375 nm for all these measurements. At a given concentration (0.5 or 100 μM), the luminescence decay lifetimes at 440 and 464 nm are similar and are not influenced markedly by O2. In contrast, the lifetimes at 550 nm are longer for degassed samples, which is to be expected for the long-lived triplet state. Ligand 1 in low concentration (0.5 μM) showed a longer lifetime than the sample with the high concentration (100 μM), consistent with a faster TTA process at the high concentration (see the ESI† for details on the evaluation of the lifetimes).121 The quantum yield of 1 in THF at ambient temperatures is 2%.
Substituted phenanthrolines show interesting photophysical properties, when coordinated to different metals.122,123 Therefore, the photophysical properties of the protonated ligand 1·HBF4 and the ZnII complex 2·2MeCN were investigated. Fig. 4B shows that neither the absorption (top) nor the emission spectra (bottom) of 1·HBF4 show any indication of concentration dependence comparable to 1. Plots of absorbance versus concentration for 1·HBF4 deviate from linearity at high concentrations, due to limited solubility in DMF, while the plot for the ZnII complex 2·2MeCN remains linear to the highest concentration (see Fig. S85 and S88†). Emission appears to come from phosphorescence only. Electrostatic repulsion of the cationic 1·HBF4 precludes TTA for this emitter. In contrast 2·2MeCN (Fig. 4C) shows TTA-based emission, similar to that observed for 1 in THF, however, this complex does not show phosphorescence even at the lowest concentrations examined.
Both 1·HBF4 and the ZnII complex 2·2MeCN were further studied in glassy 2-methyl-THF at 77 K. The emission of 1·HBF4 and 2·2MeCN at this temperature is dominated by phosphorescence (Fig. 6, two plots on the right). However, the lifetimes of 1·HBF4 are now dependent on wavelengths with biexponential decays with average lifetimes of 0.7 s at 500 nm and 0.5 s at 530 nm were determined for an excitation wavelength of 350 nm (see Fig. S84 in the ESI†). The lifetimes of 2·2MeCN are longer, τave = 1.3 to 1.9 s for 518 and 500 nm, respectively, for an excitation wavelength of 360 nm (see Fig. S87 in the ESI†). These long-lived 3ππ* level112,113,120 are typical for the phenanthroline moiety of 1·HBF4 and 2·2MeCN. The quantum yield is higher for 1·HBF4 (10%) and 2·2MeCN (4.0%) than for 1 (2.0%) in THF. The lifetimes of 1·HBF4 gives a biexponential decay at 450 nm of τ = 3.5 ns (68%) and 15.8 ns (32%), with a longer decay time of 31 ns observed at 500 and 550 nm (see Table S3 in the ESI†). The decay lifetime at 450 nm is largely due to ISC to T1 and is slower than observed for 1. The lifetimes of a 10 μM solution of 2·2MeCN in degassed THF at 298 K are given in Table S4,† again showing very little dependence on the wavelengths of emission (τave = 3.4 ns for the shorter components, see Table S4†).
Titration experiments were also carried out using HOTf as acid for the successive protonation or Zn(OTf)2 for the complexation of 1. Upon addition of 0.25–4.0 equivalents of acid, the spin-allowed π → π* transition of the phenanthroline moiety at 250 nm in DMF increases, whereas the spin-allowed π → π* transition at 350 nm decreases for the same amounts of HOTf (Fig. 7A, above). A comparable increased luminescence during the addition of acid was observed for 2,3,7,8-tetrakis(tetramethylguanidino)-phenazine.38
 | ||
| Fig. 7 (A) UV/Vis (above) and emission (below, excitation at 350 nm) spectra of 1 in DMF, depending on the addition of HOTf (0.5–4.0 equivalents); (B) UV/Vis (above) and emission (below, excitation at 350 nm) spectra of 1 in DMF, depending on the addition of Zn(OTf)2 (1.0–3.0 equivalents). | ||
Upon excitation at 350 nm, the emission at 550 nm increases for added HOTf in the range of 0.25–1.5 equivalents (Fig. 7A, below). In contrast, addition of more than 2.0 equivalents of HOTf hampers the luminescence intensity (Fig. 7A below). Fig. 7B shows that a luminescence enhancement could also be achieved by complexation of 1 with ZnII.
This is a typical example of structure-driven photoluminescence enhancement by ZnII.66,124,125 In the case of 1 after addition of one equivalent ZnII no further increase of luminescence could be observed (Fig. 7B, below). Thus, this effect corresponds to the formation of 2·2MeCN (see Fig. 2), where coordination experiments have shown that no further uptake of ZnII by 2·2MeCN is possible. In a similar vein to the titration experiments, the color change of a DCM solution of 1 upon successive protonation was investigated under inert conditions (Fig. 8). The initially orange solution of 1 gradually turns yellow (1.0 equivalents of HOTf) matching the color of the isolated 1·HBF4 and then fades upon addition of more acid (≤2.0 equivalents), also matching the off-white to pale-yellow color of the isolated 1·2HBF4. Further addition of HOTf under inert conditions (>2.0 equivalents) causes the solution to become slightly yellow and ultimately turbid (see Fig. S92 in the ESI†). This indicates that protonation beyond [1·2H]2+ is potentially feasible in a non-coordinating solvent using excess HOTf.
 | ||
| Fig. 8 Images of 0.15 M solution of 1 in DCM after successive addition of HOTf under inert conditions. | ||
Conclusions
In conclusion, we present the synthesis and characterization of a new electron-rich 1,10-phenanthroline ligand, 1, bearing bulky NHI groups adjacent to the coordination center. Electrochemical studies of 1 are in line with previously reported guanidyl-substituted polypyridine ligands and confirm the increased electron density of the ligand due to the π-donation of the NHI substituents. Coordination experiments with ZnII showed that 1 binds a single metal center, coordinating selectively at the phenanthroline N atoms, which is in contrast to the Janus head pro-type ligand III and the preference of I to coordinate with the guanidyl N atoms. Likewise, the protonation of 1 was theoretically and experimentally confirmed to take place at one of the phenanthroline N atoms. The second protonation, however, is less regioselective, leading to tautomeric forms in solution, with DFT calculations indicating a 12.6 kJ mol−1 energetic preference for protonation at the imine nitrogen atom. Photophysical experiments show that the optical properties of 1 are highly sensitive to protonation, concentration, and metal coordination. Upon protonation at the phenanthroline nitrogen atoms, the emission intensity increases, but it decreases upon binding of a second proton, presumably due to protonation of an imine group.38 The UV/Vis absorption at 350 nm, responsible for the red-to-orange color of 1, fades upon protonation, corresponding to a visible color change. In addition, the absorption and emission of 1 are dependent on concentration due to TTA in solution. This effect is absent in the cases of the ionic compounds 1·HBF4 and 2·2MeCN. Furthermore, coordination with ZnII significantly enhances emission, as steric constraints lead to rigidification of the ligand scaffold.In summary, 1 is an electron-rich ligand with four donor sites positioned within a coordination cavity shaped by bulky tert-butyl groups. Due to the strong π-donating effect of the NHI groups, the phenanthroline nitrogen atoms exhibit significantly enhanced basicity (pKBH+ (ACN) = 25.0) driving preferential coordination of Lewis acids via the phenanthroline core. This electronic effect also significantly impacts the ligand's redox and optical properties. Ongoing studies in our group are focused on further exploring its coordination chemistry and multiple-site binding capabilities.
Author contributions
J. H. F. carried out the experiments and synthesized and characterized the new compounds. L. F. B. W. and J. H. F. developed the synthesis of 1. F. D. directed the investigation. Investigations on the optical properties were performed by X. Z., A. A., K. B., A. M. and P. B. Electrochemical measurements were made by K. B. and A. M. DFT calculations were performed by T. S. H. SCXRD studies were carried out by M. S. The manuscript was written by J. H. F., P. B. and F. D., and edited by F. D., K. B., A. M., M. T. and L. D. C. All authors have given approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
We sincerely thank the Tiroler Wissenschaftsförderung (TWF, F.45075) for funding to realize this project and the German Academic Scholarship Foundation for a PhD fellowship (L. F. B. W.).References
- N. Kuhn, R. Fawzi, M. Steimann, J. Wiethoff, D. Bläser and R. Boese, Synthese und Struktur von 2-Imino-1,3-dimethylimidazolin/Synthesis and Structure of 2-Imino-1,3-dimethylimidazoline, Z. Naturforsch., B, 1995, 50, 1779–1784 CrossRef CAS.
- N. Kuhn, R. Fawzi, M. Steimann, J. Wiethoff and G. Henkel, Synthese und Struktur von [ImNCS 2 K] (ImN = 1,3-Dimethylimidazolin-2-imino): ein neuartiges Kaliumdithiocarbiminat, Z. Anorg. Allg. Chem., 1997, 623, 1577–1582 CrossRef CAS.
- N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Frenking and Y. Chen, 2-Iminoimidazoline—starke Stickstoffbasen als Koordinationspartner in der Anorganischen Chemie, Z. Anorg. Allg. Chem., 2003, 629, 793–802 CrossRef CAS.
- N. Kuhn, R. Fawzi, M. Steimann and J. Wiethoff, Derivate des Imidazols. XXIII. 2-Iminoimidazolin-Derivate des Magnesiums und Aluminiums, Z. Anorg. Allg. Chem., 1997, 623, 554–560 CrossRef CAS.
- N. Kuhn, U. Abram, C. Maichle-Mößmer and J. Wiethoff, Derivate des Imidazols. XXIV [1]. [Li12O2Cl2(ImN)8(THF)4]·8THF: Ein Peroxo–Komplex des Lithiums mit neuartiger Käfigstruktur, Z. Anorg. Allg. Chem., 1997, 623, 1121–1124 CrossRef CAS.
- A. Doddi, M. Peters and M. Tamm, N-Heterocyclic, Carbene Adducts of Main Group Elements and Their Use as Ligands in Transition Metal Chemistry, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
- X. Wu and M. Tamm, Transition metal complexes supported by highly basic imidazolin-2-iminato and imidazolin-2-imine N-donor ligands, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef CAS.
- T. Ochiai, D. Franz and S. Inoue, Applications of N-heterocyclic imines in main group chemistry, Chem. Soc. Rev., 2016, 45, 6327–6344 RSC.
- A. G. Trambitas, T. K. Panda and M. Tamm, Rare Earth Metal Complexes Supported by Ancillary Imidazolin-2-iminato Ligands, Z. Anorg. Allg. Chem., 2010, 636, 2156–2171 CrossRef CAS.
- M. Tamm, S. Randoll, T. Bannenberg and E. Herdtweck, Titanium complexes with imidazolin-2-iminato ligands, Chem, Commun., 2004, 876–877 RSC.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, A crystalline singlet phosphinonitrene: a nitrogen atom-transfer agent, Science, 2012, 337, 1526–1528 CrossRef CAS.
- F. Dielmann, C. E. Moore, A. L. Rheingold and G. Bertrand, Crystalline, Lewis base-free, cationic phosphoranimines (iminophosphonium salts), J. Am. Chem. Soc., 2013, 135, 14071–14073 CrossRef CAS PubMed.
- P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Isolation, characterization and reactivity of three-coordinate phosphorus dications isoelectronic to alanes and silylium cations, Nat. Chem., 2019, 11, 1139–1143 CrossRef CAS PubMed.
- M. A. Wünsche, T. Witteler and F. Dielmann, Lewis Base Free Oxophosphonium Ions: Tunable, Trigonal-Planar Lewis Acids, Angew. Chem., Int. Ed., 2018, 57, 7234–7239 CrossRef PubMed.
- P. Löwe, T. Witteler and F. Dielmann, Lewis base-free thiophosphonium ion: a cationic sulfur atom transfer reagent, Chem. Commun., 2021, 57, 5043–5046 RSC.
- P. Löwe, M. A. Wünsche, F. R. S. Purtscher, J. Gamper, T. S. Hofer, L. F. B. Wilm, M. B. Röthel and F. Dielmann, Terminal methylene phosphonium ions: precursors for transient monosubstituted phosphinocarbenes, Chem. Sci., 2023, 14, 7928–7935 RSC.
- D. Wendel, T. Szilvási, C. Jandl, S. Inoue and B. Rieger, Twist of a Silicon-Silicon Double Bond: Selective Anti-Addition of Hydrogen to an Iminodisilene, J. Am. Chem. Soc., 2017, 139, 9156–9159 CrossRef CAS PubMed.
- D. Wendel, D. Reiter, A. Porzelt, P. J. Altmann, S. Inoue and B. Rieger, Silicon and Oxygen's Bond of Affection: An Acyclic Three-Coordinate Silanone and Its Transformation to an Iminosiloxysilylene, J. Am. Chem. Soc., 2017, 139, 17193–17198 CrossRef CAS PubMed.
- F. S. Tschernuth, F. Hanusch, T. Szilvási and S. Inoue, Isolation and Reactivity of Chlorotetryliumylidenes Using a Bidentate Bis(N-heterocyclic imine) Ligand, Organometallics, 2020, 39, 4265–4272 CrossRef CAS.
- S. V. Hirmer, F. S. Tschernuth, F. Hanusch, R. Baierl, M. Muhr and S. Inoue, Modification of bidentate bis(N-heterocyclic imine) ligands for low-valent main group complexes, Mendeleev Commun., 2022, 32, 16–18 CrossRef CAS.
- D. Franz, E. Irran and S. Inoue, Isolation of a three-coordinate boron cation with a boron-sulfur double bond, Angew. Chem., Int. Ed., 2014, 53, 14264–14268 CrossRef CAS.
- M. W. Lui, C. Merten, M. J. Ferguson, R. McDonald, Y. Xu and E. Rivard, Contrasting reactivities of silicon and germanium complexes supported by an N-heterocyclic guanidine ligand, Inorg. Chem., 2015, 54, 2040–2049 CrossRef CAS PubMed.
- R. F. Weitkamp, B. Neumann, H.-G. Stammler and B. Hoge, Phosphorus-Containing Superbases: Recent Progress in the Chemistry of Electron-Abundant Phosphines and Phosphazenes, Chem. – Eur. J., 2021, 27, 10807–10825 CrossRef CAS.
- F. Buß, M. B. Röthel, J. A. Werra, P. Rotering, L. F. B. Wilm, C. G. Daniliuc, P. Löwe and F. Dielmann, Tris(tetramethylguanidinyl)phosphine: The Simplest Non-ionic Phosphorus Superbase and Strongly Donating Phosphine Ligand, Chem. – Eur. J., 2022, 28, e202104021 CrossRef.
- M. A. Wünsche, P. Mehlmann, T. Witteler, F. Buß, P. Rathmann and F. Dielmann, Imidazolin-2-ylidenaminophosphines as Highly Electron-Rich Ligands for Transition-Metal Catalysts, Angew. Chem., Int. Ed., 2015, 54, 11857–11860 CrossRef PubMed.
- P. Mehlmann, C. Mück-Lichtenfeld, T. T. Y. Tan and F. Dielmann, Tris(imidazolin-2-ylidenamino)phosphine: A Crystalline Phosphorus(III) Superbase That Splits Carbon Dioxide, Chem. – Eur. J., 2017, 23, 5929–5933 CrossRef CAS PubMed.
- P. Rotering, L. F. B. Wilm, J. A. Werra and F. Dielmann, Pyridinylidenaminophosphines: Facile Access to Highly Electron-Rich Phosphines, Chem. – Eur. J., 2020, 26, 406–411 CrossRef CAS PubMed.
- M. Abdinejad, L. F. B. Wilm, F. Dielmann and H. B. Kraatz, Electroreduction of CO2 Catalyzed by Nickel Imidazolin-2-ylidenamino-Porphyrins in Both Heterogeneous and Homogeneous Molecular Systems, ACS Sustainable Chem. Eng., 2021, 9, 521–530 CrossRef CAS.
- J. A. Murphy, Discovery and development of organic super-electron-donors, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS PubMed.
- J. A. Murphy, J. Garnier, S. R. Park, F. Schoenebeck, S. Zhou and A. T. Turner, Super-electron donors: bis-pyridinylidene formation by base treatment of pyridinium salts, Org. Lett., 2008, 10, 1227–1230 CrossRef CAS PubMed.
- S. S. Hanson, N. A. Richard and C. A. Dyker, Powerful Bispyridinylidene Organic Reducing Agents with Iminophosphorano π-Donor Substituents, Chem. – Eur. J., 2015, 21, 8052–8055 CrossRef CAS PubMed.
- S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Pushing the limits of neutral organic electron donors: a tetra(iminophosphorano)-substituted bispyridinylidene, Angew. Chem., Int. Ed., 2015, 54, 11236–11239 CrossRef CAS PubMed.
- B. Eberle, E. Kaifer and H.-J. Himmel, A Stable Hexakis(guanidino)benzene: Realization of the Strongest Neutral Organic Four-Electron Donor, Angew. Chem., Int. Ed., 2017, 56, 3360–3363 CrossRef CAS PubMed.
- M. M. Burgoyne, T. M. MacDougall, Z. N. Haines, J. W. Conrad, L. A. Calhoun, A. Decken and C. A. Dyker, A strong organic electron donor incorporating highly π-donating triphenylphosphonium ylidyl substituents, Org. Biomol. Chem., 2019, 17, 9726–9733 RSC.
- P. W. Antoni, C. Golz and M. M. Hansmann, Organic Four-Electron Redox Systems Based on Bipyridine and Phenanthroline Carbene Architectures, Angew. Chem., Int. Ed., 2022, 61, e202203064 CrossRef CAS PubMed.
- J. Garnier, A. R. Kennedy, L. E. A. Berlouis, A. T. Turner and J. A. Murphy, Structure and reactivity in neutral organic electron donors derived from 4-dimethylaminopyridine, Beilstein J. Org. Chem., 2010, 6 DOI:10.3762/bjoc.6.73.
- J. Broggi, T. Terme and P. Vanelle, Organic electron donors as powerful single-electron reducing agents in organic synthesis, Angew. Chem., Int. Ed., 2014, 53, 384–413 CrossRef CAS PubMed.
- E. Bindewald, R. Lorenz, O. Hübner, D. Brox, D.-P. Herten, E. Kaifer and H.-J. Himmel, Tetraguanidino-functionalized phenazine and fluorene dyes: synthesis, optical properties and metal coordination, Dalton Trans., 2015, 44, 3467–3485 RSC.
- R. Lorenz, E. Kaifer, H. Wadepohl and H.-J. Himmel, Di- and tetranuclear transition metal complexes of a tetrakisguanidino-substituted phenazine dye by stepwise coordination, Dalton Trans., 2018, 47, 11016–11029 RSC.
- J. Osterbrink, F. Dos Santos, E. Kaifer and H.-J. Himmel, Redox Reactivity Control Through Electromerism, Eur. J. Inorg. Chem., 2024, 27, e202400070 CrossRef CAS.
- A. Maronna, O. Hübner, M. Enders, E. Kaifer and H.-J. Himmel, Bisguanidines with biphenyl, binaphthyl, and bipyridyl cores: proton-sponge properties and coordination chemistry, Chem. – Eur. J., 2013, 19, 8958–8977 CrossRef CAS PubMed.
- P. Roquette, A. Maronna, A. Peters, E. Kaifer, H.-J. Himmel, C. Hauf, V. Herz, E.-W. Scheidt and W. Scherer, On the electronic structure of Ni(II) complexes that feature chelating bisguanidine ligands, Chem. – Eur. J., 2010, 16, 1336–1350 CrossRef CAS PubMed.
- S. Wiesner, A. Wagner, O. Hübner, E. Kaifer and H.-J. Himmel, Thermochromism of Cu(I) Tetrakisguanidine Complexes: Reversible Activation of Metal-to-Ligand Charge-Transfer Bands, Chem. – Eur. J., 2015, 21, 16494–16503 CrossRef CAS PubMed.
- Y. Wei, G. N. Sastry and H. Zipse, Methyl cation affinities of commonly used organocatalysts, J. Am. Chem. Soc., 2008, 130, 3473–3477 CrossRef CAS PubMed.
- C. Lindner, R. Tandon, B. Maryasin, E. Larionov and H. Zipse, Cation affinity numbers of Lewis bases, Beilstein J. Org. Chem., 2012, 8, 1406–1442 CrossRef CAS PubMed.
- P. Hommes, C. Fischer, C. Lindner, H. Zipse and H.-U. Reissig, Unprecedented strong Lewis bases–synthesis and methyl cation affinities of dimethylamino-substituted terpyridines, Angew. Chem., Int. Ed., 2014, 53, 7647–7651 CrossRef CAS PubMed.
- I. Despotović and R. Vianello, Engineering exceptionally strong oxygen superbases with 1,8-diazanaphthalene di-N-oxides, Chem. Commun., 2014, 50, 10941–10944 RSC.
- C. H. Suresh and S. R. Gadre, A Novel Electrostatic Approach to Substituent Constants: Doubly Substituted Benzenes, J. Am. Chem. Soc., 1998, 120, 7049–7055 ( J. Am. Chem. Soc. , 1998 , 120 , 10276 ) CrossRef CAS.
- F. B. Sayyed and C. H. Suresh, An electrostatic scale of substituent resonance effect, Tetrahedron Lett., 2009, 50, 7351–7354 CrossRef CAS.
- G. S. Remya and C. H. Suresh, Quantification and classification of substituent effects in organic chemistry: a theoretical molecular electrostatic potential study, Phys. Chem. Chem. Phys., 2016, 18, 20615–20626 RSC.
- V. U. Krishnapriya and C. H. Suresh, Imidazolin-2-imine and Imidazolin-2-methylidene Substitutions to Benzene, Pyridine, Phosphine, and N -Heterocyclic Carbene Predict Highly Electron-rich Ligands, Organometallics, 2023, 42, 571–580 CrossRef CAS.
- Y. Han, H. V. Huynh and G. K. Tan, Syntheses and Characterizations of Pd(II) Complexes Incorporating a N -Heterocyclic Carbene and Aromatic N -Heterocycles, Organometallics, 2007, 26, 6447–6452 CrossRef CAS.
- H. V. Huynh, Y. Han, R. Jothibasu and J. An Yang, 13C NMR Spectroscopic Determination of Ligand Donor Strengths Using N-Heterocyclic Carbene Complexes of Palladium(II), Organometallics, 2009, 28, 5395–5404 CrossRef CAS.
- Q. Teng and H. V. Huynh, A unified ligand electronic parameter based on 13C NMR spectroscopy of N-heterocyclic carbene complexes, Dalton Trans., 2017, 46, 614–627 RSC.
- J. H. Franzen, L. F. B. Wilm, P. Rotering, K. Wurst, M. Seidl and F. Dielmann, Electron-rich pyridines with para-N-heterocyclic imine substituents: ligand properties and coordination to CO2, SO2, BCl3 and PdII complexes, Dalton Trans., 2024, 53, 11876–11883 RSC.
- J. L. Doran, B. Hon and K. R. Leopold, Rotational spectrum and structure of the pyridine–CO2 van der Waals complex, J. Mol. Struct., 2012, 1019, 191–195 CrossRef CAS.
- K. D. Vogiatzis, A. Mavrandonakis, W. Klopper and G. E. Froudakis, Ab initio study of the interactions between CO(2) and N-containing organic heterocycles, ChemPhysChem, 2009, 10, 374–383 CrossRef CAS PubMed.
- J. W. Keller, Sulfur Dioxide-Pyridine Dimer. FTIR and Theoretical Evidence for a Low-Symmetry Structure, J. Phys. Chem. A, 2015, 119, 10390–10398 CrossRef CAS PubMed.
- H.-J. Himmel, Guanidinyl–Functionalized Aromatic Compounds (GFAs) – Charge and Spin Density Studies as Starting Points for the Development of a New Class of Redox–active Ligands, Z. Anorg. Allg. Chem., 2013, 639, 1940–1952 CrossRef CAS.
- S. Stang, A. Lebkücher, P. Walter, E. Kaifer and H.-J. Himmel, Redox–Active Guanidine Ligands with Pyridine and p –Benzoquinone Backbones, Eur. J. Inorg. Chem., 2012, 2012, 4833–4845 CrossRef CAS.
- L. Lohmeyer, E. Kaifer, H. Wadepohl and H.-J. Himmel, 1,2,5,6-Tetrakis(guanidino)-Naphthalenes: Electron Donors, Fluorescent Probes and Redox-Active Ligands, Chem. – Eur. J., 2020, 26, 5834–5845 CrossRef CAS PubMed.
- F. T. Edelmann, Recent Progress in the Chemistry of Metal Amidinates and Guanidinates, Adv. Organomet. Chem., 2013, 61, 55–374 CrossRef CAS.
- J. Ortmeyer, Y. Vukadinovic, A. Neuba, H. Egold, U. Flörke and G. Henkel, Combining a Phenanthroline Moiety with Two Peralkylated Guanidine Residues: Janus Head Pro-Ligands, Eur. J. Org. Chem., 2017, 6085–6095 CrossRef CAS.
- J. Ortmeyer, Y. Vukadinovic, A. Neuba, U. Flörke and G. Henkel, Combining a Phenanthroline Moiety with Peralkylated Guanidine Residues: Homometallic Cu II, Ni II and Zn II Halide Complexes with Site-Differentiating Janus Head Ligands, Eur. J. Inorg. Chem., 2018, 2018, 5176–5190 CrossRef CAS.
- C. Queffélec, P. B. Pati and Y. Pellegrin, Fifty Shades of Phenanthroline: Synthesis Strategies to Functionalize 1,10-Phenanthroline in All Positions, Chem. Rev., 2024, 124, 6700–6902 CrossRef PubMed.
- P. Alreja and N. Kaur, Recent advances in 1,10-phenanthroline ligands for chemosensing of cations and anions, RSC Adv., 2016, 6, 23169–23217 RSC.
- P. G. Sammes and G. Yahioglu, 1,10-Phenanthroline: a versatile ligand, Chem. Soc. Rev., 1994, 23, 327 RSC.
- W. W. Brandt, F. P. Dwyer and E. D. Gyarfas, Chelate Complexes of 1,10-Phenanthroline and Related Compounds, Chem. Rev., 1954, 54, 959–1017 CrossRef CAS.
- G. Accorsi, A. Listorti, K. Yoosaf and N. Armaroli, 1,10-phenanthrolines: versatile building blocks for luminescent molecules, materials and metal complexes, Chem. Soc. Rev., 2009, 38, 1690–1700 RSC.
- Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M. Kakimoto, Novel bipolar bathophenanthroline containing hosts for highly efficient phosphorescent OLEDs, Org. Lett., 2008, 10, 421–424 CrossRef CAS PubMed.
- Y. Tao, C. Yang and J. Qin, Organic host materials for phosphorescent organic light-emitting diodes, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- A. Buyruk, D. Blätte, M. Günther, M. A. Scheel, N. F. Hartmann, M. Döblinger, A. Weis, A. Hartschuh, P. Müller-Buschbaum, T. Bein and T. Ameri, 1,10-Phenanthroline as an Efficient Bifunctional Passivating Agent for MAPbI3 Perovskite Solar Cells, ACS Appl. Mater. Interfaces, 2021, 13, 32894–32905 CrossRef CAS PubMed.
- N. Kandasamy, N. Keerthi and J. Jeyasekaran, Mixed Ligand Copper(I) Complexes as Emitters Enable Higher OLED Device Performance for Energy-Harvesting Applications, ACS Appl. Electron. Mater., 2023, 5, 4805–4815 CrossRef CAS.
- B. E. Halcrow and W. O. Kermack, Attempts to find new antimalarials; derivatives of o-phenanthroline (7:8:3′:2′-pyridoquinoline), J. Chem. Soc., 1946, 155–157 RSC.
- M. A. de Zwart, H. M. Bastiaans, H. van der Goot and H. Timmerman, Synthesis and copper-dependent antimycoplasmal activity of amides and amidines derived from 2-amino-1,10-phenanthroline, J. Med. Chem., 1991, 34, 1193–1201 CrossRef CAS PubMed.
- S. L. H. Higgins and K. J. Brewer, Designing red-light-activated multifunctional agents for the photodynamic therapy, Angew. Chem., Int. Ed., 2012, 51, 11420–11422 CrossRef CAS PubMed.
- N. M. Gueddouda, M. R. Hurtado, S. Moreau, L. Ronga, R. N. Das, S. Savrimoutou, S. Rubio, A. Marchand, O. Mendoza, M. Marchivie, L. Elmi, A. Chansavang, V. Desplat, V. Gabelica, A. Bourdoncle, J.-L. Mergny and J. Guillon, Design, Synthesis, and Evaluation of 2,9-Bis(substituted-aminomethyl)phenyl-1,10-phenanthroline Derivatives as G-Quadruplex Ligands, ChemMedChem, 2017, 12, 146–160 CrossRef CAS PubMed.
- A. Nano, J. Dai, J. M. Bailis and J. K. Barton, Rhodium Complexes Targeting DNA Mismatches as a Basis for New Therapeutics in Cancers Deficient in Mismatch Repair, Biochemistry, 2021, 60, 2055–2063 CrossRef CAS PubMed.
- J. Guillon, A. Cohen, C. Boudot, S. Monic, S. Savrimoutou, S. Moreau, S. Albenque-Rubio, C. Lafon-Schmaltz, A. Dassonville-Klimpt, J.-L. Mergny, L. Ronga, M. Bernabeu de Maria, J. Lamarche, C. D. Lago, E. Largy, V. Gabelica, S. Moukha, P. Dozolme, P. Agnamey, N. Azas, C. Mullié, B. Courtioux and P. Sonnet, Design, Synthesis, and Antiprotozoal Evaluation of New Promising 2,9-Bis(substituted-aminomethyl)-4,7-phenyl-1,10-phenanthroline Derivatives, a Potential Alternative Scaffold to Drug Efflux, Pathogens, 2022, 11, 1339 CrossRef CAS PubMed.
- F. C. Rix, M. Brookhart and P. S. White, Mechanistic Studies of the Palladium(II)-Catalyzed Copolymerization of Ethylene with Carbon Monoxide, J. Am. Chem. Soc., 1996, 118, 4746–4764 CrossRef CAS.
- J. E. McGarrah, Y. J. Kim, M. Hissler and R. Eisenberg, Toward a molecular photochemical device: a triad for photoinduced charge separation based on a platinum diimine bis(acetylide) chromophore, Inorg. Chem., 2001, 40, 4510–4511 CrossRef CAS PubMed.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Simultaneous Tuning of Activity and Water Solubility of Complex Catalysts by Acid-Base Equilibrium of Ligands for Conversion of Carbon Dioxide, Organometallics, 2007, 26, 702–712 CrossRef CAS.
- M. G. Pfeffer, B. Schäfer, G. Smolentsev, J. Uhlig, E. Nazarenko, J. Guthmuller, C. Kuhnt, M. Wächtler, B. Dietzek, V. Sundström and S. Rau, Palladium versus platinum: the metal in the catalytic center of a molecular photocatalyst determines the mechanism of the hydrogen production with visible light, Angew. Chem., Int. Ed., 2015, 54, 5044–5048 CrossRef CAS PubMed.
- S. Rau, B. Schäfer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Görls, W. Henry and J. G. Vos, A supramolecular photocatalyst for the production of hydrogen and the selective hydrogenation of tolane, Angew. Chem., Int. Ed., 2006, 45, 6215–6218 CrossRef CAS PubMed.
- L. Jiao, E. Herdtweck and T. Bach, Pd(II)-catalyzed regioselective 2-alkylation of indoles via a norbornene-mediated C-H activation: mechanism and applications, J. Am. Chem. Soc., 2012, 134, 14563–14572 CrossRef CAS PubMed.
- T. Moragas, M. Gaydou and R. Martin, Nickel-Catalyzed Carboxylation of Benzylic C-N Bonds with CO2, Angew. Chem., Int. Ed., 2016, 55, 5053–5057 CrossRef CAS PubMed.
- B. Guan, J. Liu, J. Leng, T. Fan and L. Tong, Hydrogen Evolution Catalysis by Cobalt Complexes of an Aza-bridged Bis-1,10-phenanthroline Ligand Bearing Pendant Basic Sites, ChemCatChem, 2024, 16, e202401284 CrossRef CAS.
- A. Bencini and V. Lippolis, 1,10-Phenanthroline: A versatile building block for the construction of ligands for various purposes, Coord. Chem. Rev., 2010, 254, 2096–2180 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- A. J. Lees, Photophysics of Organometallics, Topics in Organometallic Chemistry 29, Springer: Berlin, Heidelberg, 2010, https://ebookcentral.proquest.com/lib/kxp/detail.action?docID=3064880 Search PubMed.
- S. Quici, M. Cavazzini, G. Marzanni, G. Accorsi, N. Armaroli, B. Ventura and F. Barigelletti, Visible and near-infrared intense luminescence from water-soluble lanthanide Tb(III), Eu(III), Sm(III), Dy(III), Pr(III), Ho(III), Yb(III), Nd(III), Er(III) complexes, Inorg. Chem., 2005, 44, 529–537 CrossRef CAS PubMed.
- S. Quici, C. Scalera, M. Cavazzini, G. Accorsi, M. Bolognesi, L. Armelao and G. Bottaro, Highly Photoluminescent Silica Layers Doped with Efficient Eu(III) and Tb(III) Antenna Complexes, Chem. Mater., 2009, 21, 2941–2949 CrossRef CAS.
- J. Beaudelot, S. Oger, S. Peruško, T.-A. Phan, T. Teunens, C. Moucheron and G. Evano, Photoactive Copper Complexes: Properties and Applications, Chem. Rev., 2022, 122, 16365–16609 CrossRef CAS PubMed.
- V. Balzani, Photochemistry and Photophysics of Coordination Compounds I, Topics in Current Chemistry Ser v.280, Springer, Berlin2007 edn, 2007, https://ebookcentral.proquest.com/lib/kxp/detail.action?docID=7201737 Search PubMed.
- Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Heteroleptic diimine–diphosphine Cu(I) complexes as an alternative towards noble-metal based photosensitizers: Design strategies, photophysical properties and perspective applications, Coord. Chem. Rev., 2018, 356, 127–146 CrossRef CAS.
- R. A. Kunetskiy, S. M. Polyakova, J. Vavřík, I. Císařová, J. Saame, E. R. Nerut, I. Koppel, I. A. Koppel, A. Kütt, I. Leito and I. M. Lyapkalo, A new class of organosuperbases, N-alkyl- and N-aryl-1,3-dialkyl-4,5-dimethylimidazol-2-ylidene amines: synthesis, structure, pK(BH+) measurements, and properties, Chem. – Eur. J., 2012, 18, 3621–3630 CrossRef CAS PubMed.
- L. F. B. Wilm, T. Eder, C. Mück-Lichtenfeld, P. Mehlmann, M. Wünsche, F. Buß and F. Dielmann, Reversible CO2 fixation by N-heterocyclic imines forming water-stable zwitterionic nitrogen-bas-CO2 adducts, Green Chem., 2019, 21, 640–648 RSC.
- M. D. Böhme, T. Eder, M. B. Röthel, P. D. Dutschke, L. F. B. Wilm, F. E. Hahn and F. Dielmann, Synthesis of N-Heterocyclic Carbenes and Their Complexes by Chloronium Ion Abstraction from 2-Chloroazolium Salts Using Electron-Rich Phosphines, Angew. Chem., Int. Ed., 2022, 61, e202202190 CrossRef PubMed.
- L. Maresca, G. Natile, F. P. Fanizzi and F. Stasi, The bisphenanthrolinium ion. An X-ray crystal structure, J. Am. Chem. Soc., 1989, 111, 1492–1493 CrossRef CAS.
- R. V. Andreev, G. I. Borodkin and V. G. Shubin, Structure and Dynamics of the 1,10-Phenanthroline Complex with Nitrosonium Cation, Russ. J. Inorg. Chem., 2001, 37, 144–146 CAS.
- D. F. Azar, H. Audi, S. Farhat, M. El-Sibai, R. J. Abi-Habib and R. S. Khnayzer, Phototoxicity of strained Ru(ii) complexes: is it the metal complex or the dissociating ligand?, Dalton Trans., 2017, 46, 11529–11532 RSC.
- Protonated 1,10-Phenanthroline as a Fluorescent Sensor for Fe(II) at Micro Molar Level Detections, ed. G. Wijesekera, D. T. Arachchi, M. de Costa and R. Senthilnithy, From Innovation to Impact 2020, Colombo, Sri Lanka, 2020 Search PubMed.
- I. Despotović and R. Vianello, Engineering exceptionally strong oxygen superbases with 1,8-diazanaphthalene di-N-oxides, Chem. Commun., 2014, 50, 10941–10944 RSC.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, On the Basicity of Organic Bases in Different Media, Eur. J. Org. Chem., 2019, 6735–6748 CrossRef CAS.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, Extension of the self-consistent spectrophotometric basicity scale in acetonitrile to a full span of 28 pKa units: unification of different basicity scales, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- A. Kütt, S. Selberg, I. Kaljurand, S. Tshepelevitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, pKa values in organic chemistry – Making maximum use of the available data, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef.
- P. Jutzi, C. Müller, A. Stammler and H.-G. Stammler, Synthesis, Crystal Structure, and Application of the Oxonium Acid [H(OEt2)2]+ [B(C6F5)4]−, Organometallics, 2000, 19, 1442–1444 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Molecular single-bond covalent radii for elements 1–118, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- P. Pyykkö and M. Atsumi, Molecular double-bond covalent radii for elements Li-E112, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- J. Ortmeyer, Y. Vukadinovic, A. Neuba, H. Egold, U. Flörke and G. Henkel, CCDC 1545124: Experimental Crystal Structure Determination, 2017.
- H. Ferreira, M. M. Conradie, K. G. von Eschwege and J. Conradie, Electrochemical and DFT study of the reduction of substituted phenanthrolines, Polyhedron, 2017, 122, 147–154 CrossRef CAS.
- B. N. Bandyopadhyay and A. Harriman, Photoreduction of 1,10-phenanthroline, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 663 RSC.
- N. Armaroli, L. de Cola, V. Balzani, J.-P. Sauvage, C. O. Dietrich-Buchecker and J.-M. Kern, Absorption and luminescence properties of 1, 10-phenanthroline, 2, 9-diphenyl-1, 10-phenanthroline, 2,9-dianisyl-1, 10-phenanthroline and their protonated forms in dichloromethane solution, J. Chem. Soc., Faraday Trans., 1992, 88, 553 RSC.
- N. Yoshikawa, S. Yamazaki, Y. Kakimoto, S. Eguchi, R. Yokoyama, N. Kanehisa, N. Tohnai, E. Nakata and H. Takashima, Emission properties of 1,10-phenanthroline derivatives induced by protonation of a nitrogen atom, J. Mol. Struct., 2021, 1242, 130728 CrossRef CAS.
- S. J. Lee, S. S. Lee, M. S. Lah, J.-M. Hong and J. H. Jung, Organic-inorganic hybrid nanomaterial as a new fluorescent chemosensor and adsorbent for copper ion, Chem. Commun., 2006, 4539–4541 RSC.
- B. Zhang, K.-S. Cao, Z.-A. Xu, Z.-Q. Yang, H.-W. Chen, W. Huang, G. Yin and X.-Z. You, Cell–Compatible Fluorescent Chemosensor for Zn 2+ Based on a 3,8-Extended 1,10-Phenanthroline Derivative, Eur. J. Inorg. Chem., 2012, 2012, 3844–3851 CrossRef CAS.
- Z. Aydin, A novel phenanthroline-based colorimetric turn-off optical sensor for Zn2+, Inorg. Chim. Acta, 2021, 517, 120200 CrossRef CAS.
- Y.-Z. Han, G. Tian and Q. Yang, Two colorimetric and ratiometric fluorescence sensors for Zn2+ with 1,10-phenanthroline derivatives, Inorg. Chem. Commun., 2023, 155, 111105 CrossRef CAS.
- C. Bizzarri, C. Strabler, J. Prock, B. Trettenbrein, M. Ruggenthaler, C.-H. Yang, F. Polo, A. Iordache, P. Brüggeller and L. de Cola, Luminescent dinuclear Cu(I) complexes containing rigid tetraphosphine ligands, Inorg. Chem., 2014, 53, 10944–10951 CrossRef CAS PubMed.
- M. S. Henry and M. Z. Hoffman, Photophysics and photochemistry of aromatic nitrogen heterocycles. Fluorescence from 2,2′-bipyridine and 1,10-phenanthroline, J. Phys. Chem., 1979, 83, 618–625 CrossRef CAS.
- N. J. Hestand and F. C. Spano, Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- J. E. Yarnell, J. C. Deaton, C. E. McCusker and F. N. Castellano, Bidirectional “ping-pong” energy transfer and 3000-fold lifetime enhancement in a Re(I) charge transfer complex, Inorg. Chem., 2011, 50, 7820–7830 CrossRef CAS PubMed.
- M. W. Blaskie and D. R. McMillin, Photostudies of copper(I) systems. 6. Room-temperature emission and quenching studies of bis(2,9-dimethyl-1,10-phenanthroline)copper(I), Inorg. Chem., 1980, 19, 3519–3522 CrossRef CAS.
- S. Jensen, K. Tan, W. P. Lustig, D. S. Kilin, J. Li, Y. J. Chabal and T. Thonhauser, Structure-Driven Photoluminescence Enhancement in a Zn-Based Metal–Organic Framework, Chem. Mater., 2019, 31, 7933–7940 CrossRef CAS.
- Y. Li, L. Shi, L.-X. Qin, L.-L. Qu, C. Jing, M. Lan, T. D. James and Y.-T. Long, An OFF-ON fluorescent probe for Zn2+ based on a GFP-inspired imidazolone derivative attached to a 1,10-phenanthroline moiety, Chem. Commun., 2011, 47, 4361–4363 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, mass spectrometry data, crystallographic data, and computational details. CCDC 2420826–2420829. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qi00957j |
| This journal is © the Partner Organisations 2025 |