
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Profiting from light-induced metal-to-metal intramolecular electron transfer: towards highly efficient heterodinuclear photosensitizers for photodynamic therapy†
Juan Sanz-Villafruela ab,
Lisa-Marie Servosa,
Nicolás Montesdeocaa,
José V. Cuevas-Vicariob,
Artur J. Moroc,
João C. Limac,
Marta Martínez-Alonsob,
Gustavo Espino*b and
Johannes Karges*a
ab,
Lisa-Marie Servosa,
Nicolás Montesdeocaa,
José V. Cuevas-Vicariob,
Artur J. Moroc,
João C. Limac,
Marta Martínez-Alonsob,
Gustavo Espino*b and
Johannes Karges*a
aFaculty of Chemistry and Biochemistry, Ruhr-University Bochum, Universitätsstrasse 150, 44780 Bochum, Germany. E-mail: johannes.karges@ruhr-uni-bochum.de
bDepartamento de Química, Facultad de Ciencias, Universidad de Burgos, Plaza Misael Bañuelos s/n, 09001 Burgos, Spain. E-mail: gespino@ubu.es
cLAQV-REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia Universidade NOVA de Lisboa, 2829-516 Caparica, Portugal
First published on 7th May 2025
Abstract
Photodynamic therapy is receiving increasing attention due to its versatile application in anticancer therapy. Ru(II) and Pt(II) complexes are among the most investigated compounds as potential photosensitizers for photodynamic therapy based on their outstanding photophysical and biological properties (i.e., strong emission, high intersystem crossing efficiency, large Stokes shift, high (photo-)stability, biological compatibility, good water solubility). While these classes of compounds have been widely studied separately or combined in derivatives that display a dual therapeutic effect, herein, a novel study on the conjugation of Ru(II) and Pt(II) fragments into a single bimetallic conjugate is proposed. It is assumed that this molecular design could undergo an intramolecular electron transfer from the Ru(II) moiety to the Pt(II) moiety upon irradiation to produce a highly efficient excited state. Capitalizing on the presence of both metals, the bimetallic conjugate was found to generate singlet oxygen with an outstanding efficiency in comparison to its individual components. To enhance the pharmacological profile, the bimetallic complex was encapsulated into polymeric nanoparticles. The nanoparticles were demonstrated to eradicate human breast adenocarcinoma monolayer cells as well as multicellular tumor spheroids upon light irradiation at nanomolar concentrations. We are confident that this approach will open new avenues towards the development of novel highly efficient photosensitizers.
Introduction
Cancer has become one of the deadliest diseases worldwide. In 2022, the estimated number of new cases was around 20 million causing 10 million deaths globally.1 During the last decades, chemotherapy has risen as one of the most important therapies to treat cancer. Despite its global use, some cancers are intrinsically resistant to chemotherapy or develop resistance upon a first successful treatment and the majority of chemotherapeutic drugs produce severe side effects (i.e., kidney damage, nausea, vomiting, bone marrow suppression).2,3 Therefore, there is a need for the development of alternative therapies that can overcome the afore-mentioned limitations. Light-activated therapeutics are gaining interest in chemical and biological research due to their ability to offer spatio-temporal control over treatments.4–8 One of the most promising approaches in this field is photodynamic therapy (PDT).9,10 In PDT, a photosensitizer is administered either locally or systemically. The photosensitizer initially disperses non-specifically throughout the body and reaches the target tissue. After an appropriate incubation period, the target tissue is exposed to light, triggering the local, catalytic production of reactive oxygen species (ROS),10–12 that increase oxidative stress and ultimately leads to cell death. Importantly, the therapeutic effect is confined to the tissue containing the photosensitizer at the time of irradiation, enabling precise and selective treatment.13,14Most photosensitizers applied in clinical settings share a tetrapyrrolic core structure (e.g., porphyrin, chlorin, bacteriochlorin, phthalocyanine).11,15,16 Due to this shared structural scaffold, these photosensitizers face common challenges, including low water solubility, limited photostability, tedious synthesis and purification, slow body clearance leading to prolonged photosensitivity, and limited cancer selectivity.17 To mitigate these issues, researchers have explored the incorporation of metal ions. Certain metals can increase the molecule's stability against metabolic breakdown and light degradation.18 Additionally, specific heavy atoms can enhance intersystem crossing efficiency by the heavy atom effect, thus boosting ROS generation.19–22 Ru(II) and Pt(II) complexes are some of the most extensively studied compounds as potential photosensitizers for PDT, owing to their exceptional photophysical and biological characteristics, including strong emission, high intersystem crossing efficiency, large Stokes shift, excellent (photo-)stability, biological compatibility, and good water solubility.23–26 Notably, the Ru(II) polypyridine complex TLD-1433 has progressed to phase II clinical trials for the treatment of non-muscle invasive bladder cancer.27
While Ru(II) and Pt(II) complexes have been widely studied in the literature either independently,23,28–30 or combined in conjugates that exhibit a dual therapeutic effect,31,32 this work aims to combine these two classes of compounds in a single architecture to attain a synergistic PDT effect. Thus, the computational design, chemical synthesis, photophysical evaluation and biological testing of a novel bimetallic Ru(II)–Pt(II) conjugate is reported here. The new bimetallic photosensitizer exhibits a high efficiency in the production of singlet oxygen compared to its individual components and a remarkable potential as photosensitizer for PDT as a result of a light-induced intramolecular electron transfer process between both metal fragments. To improve its pharmacological profile, the bimetallic complex was encapsulated within polymeric nanoparticles. These nanoparticles were found to effectively eradicate human breast adenocarcinoma monolayer cells and multicellular tumor spheroids under light irradiation at nanomolar concentrations.
Results and discussion
Theoretical design
Previous studies have reported on the chemical synthesis of Ru(II)–Pt(II) conjugates linked through a conjugated system.33–35 However, these compounds have rarely been studied for any biological application. The research groups of Chao and Stang have reported on the first example of a tetranuclear Ru(II)–Pt(II) metallacycle, formed through coordination-driven self-assembly, for biological applications. This compound eradicated human pulmonary carcinoma cells upon light exposure.36 The Gasser and Gibson groups have developed a Ru(II)–Pt(IV) bimetallic complex that undergoes dissociation under red light irradiation, which results in a multi-action, multitarget cytotoxic effect in drug-resistant human ovarian cancer cells.37 A similar strategy was followed by Chakravarty et al. using a biotin-Pt(IV)–Ru(II)-BODIPY conjugate.38 Although these initial studies showed promise for multinuclear Ru–Pt complexes in PDT, the Ru and Pt centers acted independently, producing additive therapeutic effects. Following these preliminary studies, herein, we envisioned that the combination of both metal centers into a single conjugate could benefit from a synergistic effect based on the strong light-harvesting properties of the Ru(II) polypyridine fragments and the high catalytic properties of Pt(II)-terpyridine moieties.Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were utilized as the foundation for the chemical design of a novel conjugate. For the generation of a highly efficient bimetallic photosensitizer, an effective electron transfer from the Ru(II) polypyridine moiety to the Pt(II)-terpyridine moiety is essential. In pseudo-octahedral Ru(II) polypyridyl complexes, the highest occupied molecular orbital (HOMO) is usually located on the ruthenium t2g orbitals, while the lowest unoccupied molecular orbital (LUMO) typically corresponds to π* orbitals distributed over the polypyridyl ligands.39 In Pt(II) complexes of the type [Pt(terpy)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–R)]+ (terpy=terpyridine, CC–R= alkynyl ligand), the HOMO is usually spread over the π orbitals of the alkynyl ligand and the Pt center, while the LUMO is associated with π* orbitals on the terpyridine ligand.30,40 To enable the desired electron transfer between these metal fragments, the bridging ligand must provide the necessary electronic communication. Thus, different approaches were considered to conclude that the use of an alkynyl-functionalized β-carboline ligand could be suitable for this purpose. Consequently, the monometallic Ru(II) complex [Ru(bipy)21-(5-ethynylpyridin-2-yl)-9-methyl-β-carboline]2+ (Ru) (bipy=2,2′-bipyridine) was studied by using this theoretical approach. The topology and relative energy values of the MOs were calculated for the ground electronic state (S0) (Fig. S1, S2 and Table S1†). For this complex the HOMO (−5.87 eV) essentially corresponds to the Ru(dπ) orbitals while the LUMO (−2.70 eV) is located over the π* orbitals of the β-carboline ligand. On the other hand, the Pt(II) complex [Pt(terpy)(phenylacetylene)]+ (Pt) (terpy = 2,2′:6′,2′-terpyridine) was also studied. The topology and relative energy values of the MOs calculated for the ground electronic state (Fig. S1, S3 and Table S2†) showed that the HOMO (−5.92 eV) corresponds mainly to a combination of π orbitals located over the alkynyl ligand with a minor participation of the Pt(II) center, while the LUMO (−3.02 eV) is mainly spread over the π* orbitals of the terpyridine ligand and has a small contribution of the Pt(II) metal center. Next, both metal fragments were combined forming the Ru(II)–Pt(II) conjugate [Ru(bipy)2(1-(5-ethynylpyridin-2-yl)-9-methyl-β-carboline)Pt(terpy)]3+ (RuPt). The topology and relative energy values of the MOs calculated for the ground electronic state of RuPt (Fig. S1, S4 and Table S3†) confirmed the participation of the Ru and Pt fragments into HOMO and LUMO, respectively. The HOMO calculated for the heterobimetallic complex is distributed over the Ru center (approx. 71%, Table S4†) with a small contribution of the β-carboline ligand and consequently exhibits a topology (Ru(dπ) orbital) and an energy value virtually identical to that of Ru (Fig. 1). By contrast, the LUMO of RuPt is spread over the Pt(terpy) fragment (approx. 98%, Table S4†) and therefore it displays a similar topology and a comparable energy value to those of Pt as expected due to the similar structure of both moieties (Fig. 1). These results confirmed the participation of both metal fragments in the HOMO → LUMO monoexcitation and suggest an orthogonal nature for these molecular orbitals, meaning that there is no overlap between them. Moreover, the combination of both metal fragments in RuPt leads to a reduction in the HOMO–LUMO band gap with regard to Ru, and in a lesser extent with respect to Pt. The electronic transitions that take place in the calculated UV-Vis absorption spectrum (Fig. S5†) were analyzed for RuPt (Table S5†). For this complex the nature of the lowest singlet excited state (S1) is mainly described as a HOMO–LUMO monoexcitation, corresponding to a spin-allowed metal-to-ligand charge transfer (1MLCT) transition involving the promotion of one electron from a Ru(dπ) orbital to the Pt(II)-terpy environment. This was also observed in NTO (natural transition orbital) hole and electron patterns for the S0 → S1 electronic transition (Fig. 2A). Nonetheless, the straight formation of S1 is restricted since HOMO and LUMO are orthogonal to each other. Indeed, the oscillator strength for S0 → S1 is 0, suggesting that this transition is forbidden. However, S1 can be efficiently reached through internal conversion from higher energetic singlet excited states (S2, S3, etc.), whose population is more favored (e.g.: f = 0.0106 for S2).
C–R)]+ (terpy=terpyridine, CC–R= alkynyl ligand), the HOMO is usually spread over the π orbitals of the alkynyl ligand and the Pt center, while the LUMO is associated with π* orbitals on the terpyridine ligand.30,40 To enable the desired electron transfer between these metal fragments, the bridging ligand must provide the necessary electronic communication. Thus, different approaches were considered to conclude that the use of an alkynyl-functionalized β-carboline ligand could be suitable for this purpose. Consequently, the monometallic Ru(II) complex [Ru(bipy)21-(5-ethynylpyridin-2-yl)-9-methyl-β-carboline]2+ (Ru) (bipy=2,2′-bipyridine) was studied by using this theoretical approach. The topology and relative energy values of the MOs were calculated for the ground electronic state (S0) (Fig. S1, S2 and Table S1†). For this complex the HOMO (−5.87 eV) essentially corresponds to the Ru(dπ) orbitals while the LUMO (−2.70 eV) is located over the π* orbitals of the β-carboline ligand. On the other hand, the Pt(II) complex [Pt(terpy)(phenylacetylene)]+ (Pt) (terpy = 2,2′:6′,2′-terpyridine) was also studied. The topology and relative energy values of the MOs calculated for the ground electronic state (Fig. S1, S3 and Table S2†) showed that the HOMO (−5.92 eV) corresponds mainly to a combination of π orbitals located over the alkynyl ligand with a minor participation of the Pt(II) center, while the LUMO (−3.02 eV) is mainly spread over the π* orbitals of the terpyridine ligand and has a small contribution of the Pt(II) metal center. Next, both metal fragments were combined forming the Ru(II)–Pt(II) conjugate [Ru(bipy)2(1-(5-ethynylpyridin-2-yl)-9-methyl-β-carboline)Pt(terpy)]3+ (RuPt). The topology and relative energy values of the MOs calculated for the ground electronic state of RuPt (Fig. S1, S4 and Table S3†) confirmed the participation of the Ru and Pt fragments into HOMO and LUMO, respectively. The HOMO calculated for the heterobimetallic complex is distributed over the Ru center (approx. 71%, Table S4†) with a small contribution of the β-carboline ligand and consequently exhibits a topology (Ru(dπ) orbital) and an energy value virtually identical to that of Ru (Fig. 1). By contrast, the LUMO of RuPt is spread over the Pt(terpy) fragment (approx. 98%, Table S4†) and therefore it displays a similar topology and a comparable energy value to those of Pt as expected due to the similar structure of both moieties (Fig. 1). These results confirmed the participation of both metal fragments in the HOMO → LUMO monoexcitation and suggest an orthogonal nature for these molecular orbitals, meaning that there is no overlap between them. Moreover, the combination of both metal fragments in RuPt leads to a reduction in the HOMO–LUMO band gap with regard to Ru, and in a lesser extent with respect to Pt. The electronic transitions that take place in the calculated UV-Vis absorption spectrum (Fig. S5†) were analyzed for RuPt (Table S5†). For this complex the nature of the lowest singlet excited state (S1) is mainly described as a HOMO–LUMO monoexcitation, corresponding to a spin-allowed metal-to-ligand charge transfer (1MLCT) transition involving the promotion of one electron from a Ru(dπ) orbital to the Pt(II)-terpy environment. This was also observed in NTO (natural transition orbital) hole and electron patterns for the S0 → S1 electronic transition (Fig. 2A). Nonetheless, the straight formation of S1 is restricted since HOMO and LUMO are orthogonal to each other. Indeed, the oscillator strength for S0 → S1 is 0, suggesting that this transition is forbidden. However, S1 can be efficiently reached through internal conversion from higher energetic singlet excited states (S2, S3, etc.), whose population is more favored (e.g.: f = 0.0106 for S2).
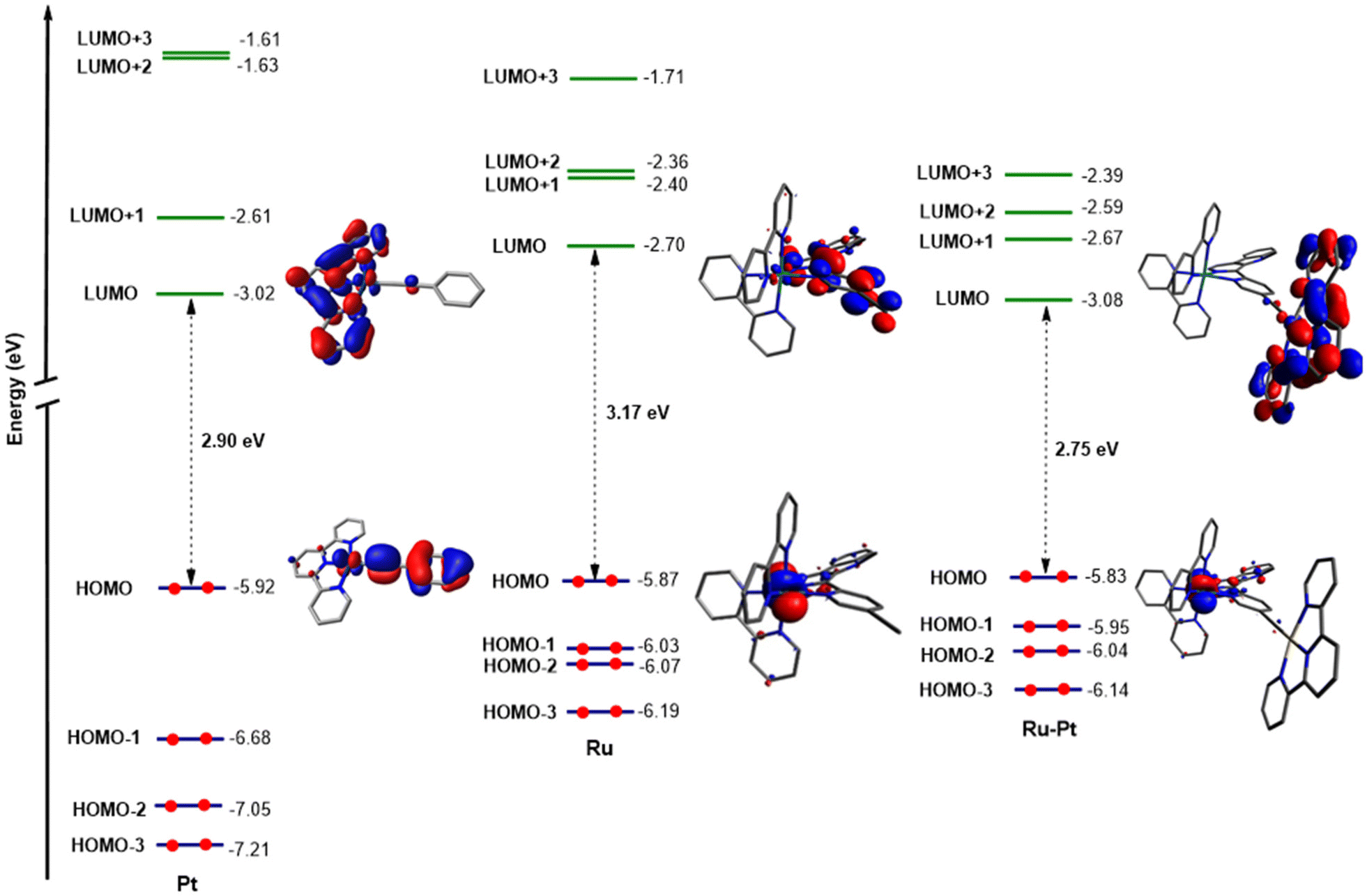 | ||
| Fig. 1 Schematic representation showing the isovalue contour plots calculated for HOMOs and LUMOs of Pt, Ru and RuPt. | ||
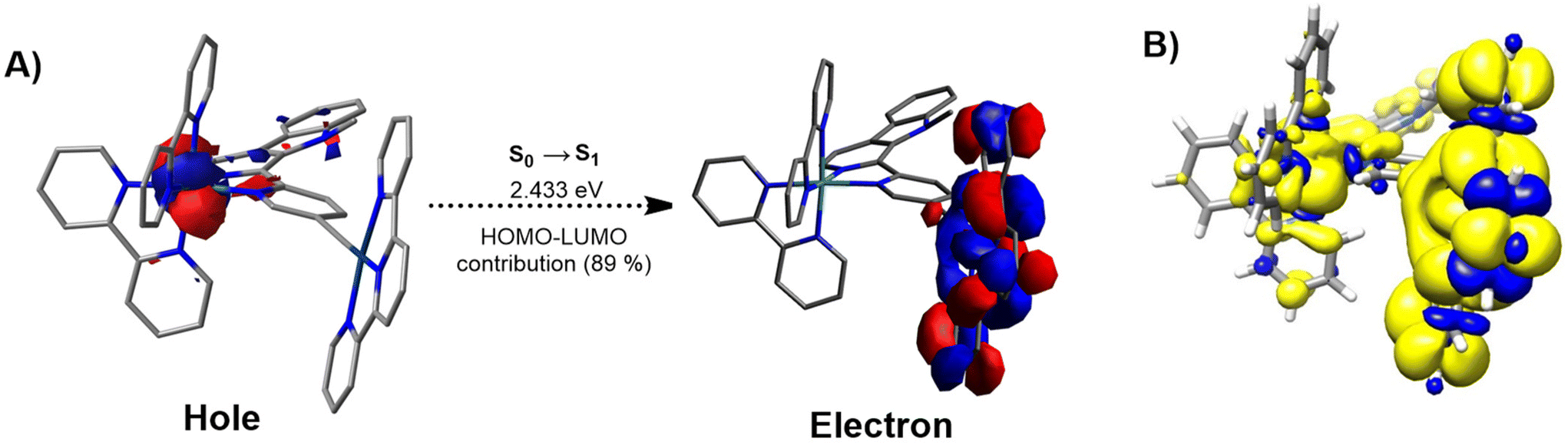 | ||
| Fig. 2 (A) NTO hole and electron patterns for S1 excited state on the optimized S0 geometry of RuPt. (B) Unpaired-electron spin-density contours computed for the fully relaxed triplet excited state (T1) of RuPt. | ||
Moreover, the orthogonal nature of the HOMO and LUMO resulted in a minimal energy difference between S1 and the triplet excited state defined by HOMO–LUMO monoexcitation (Table S5†). The singlet triplet energy gap is given by ΔEST = 2J, where J is the exchange energy. The value of J can be minimized by decreasing the overlap between the HOMO and LUMO orbitals.41 As a result, the singlet and the triplet excited states are almost isoenergetic (2.433 eV and 2.429 eV, respectively). The forbidden nature of S1, which results in a long-lived state, combined with the small ΔEST, facilitates efficient intersystem crossing to the triplet state. For a better comprehension of the nature of the lowest triplet excited state (T1) of RuPt, the geometry of the fully relaxed triplet excited state was optimized (Table S6†) and the unpaired-electron spin-density calculated (Fig. 2B). The unpaired-electron spin-density computed is distributed between the Ru environment and the Pt metal fragment. These findings confirm that the T1 state primarily arises from the HOMO → LUMO monoexcitation and the electron transfer from the Ru(II) center to the Pt(II) metal fragment. The orthogonality between the HOMO and LUMO in this triplet excited state significantly slows electron recombination to the HOMO, hindering decay to S0. This feature adds to the already spin-forbidden nature of the transition, resulting in a highly forbidden T1 → S0 transition and leading to an exceptionally long triplet lifetime (vide infra).
Chemical synthesis
The β-carboline moiety selected as bridging ligand was synthesized in three steps (Fig. 3). Firstly, tryptamine and 5-bromo-2-pyridinecarboxaldehyde were reacted to produce the corresponding tetrahydro-β-carboline (1), which was then oxidized in the presence of activated manganese dioxide to obtain 2 in a one-pot two-steps process.42 Subsequently, the 1-(5-bromopyridin-2-yl)-β-carboline (2) was converted into its methylated form, 1-(5-bromopyridin-2-yl)-9-methyl-β-carboline (3), by using sodium hydride as the base and methyl iodide as a methylating agent. Then, a Sonogashira reaction protocol was followed using 3 and (triisopropylsilyl)acetylene to obtain 9-methyl-1-(5-((triisopropylsilyl)ethynyl)pyridin-2-yl)-β-carboline (4).43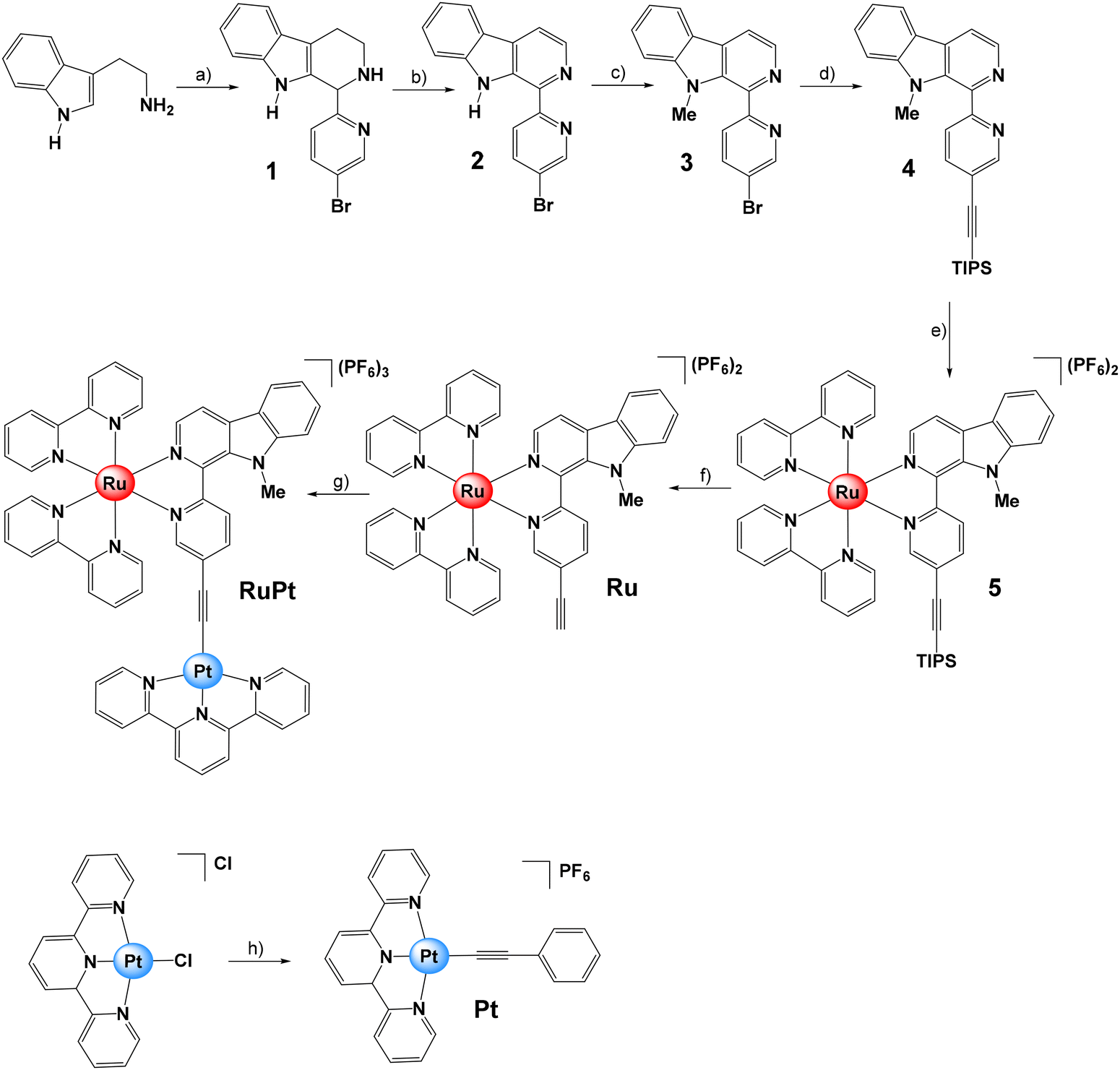 | ||
Fig. 3 Synthesis and molecular structures of β-carboline ligand 4 and Pt, Ru and RuPt investigated in this study. Conditions and reagents: (a) 5-bromo-2-pyridinecarboxaldehyde, anisole, reflux, 3 h; (b) MnO2, reflux, 21 h. (c) NaH, MeI, DMF, 0 °C → rt, overnight; (d) (triisopropylsilyl)acetylene, Pd(PPh3)4, NEt3, CuI, DMF, 65 °C, overnight; (e) Ru(bipy)2Cl2, EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1), pressure flask (120 °C), 24 h; (f) TBAF, AcOH, ACN:THF (2:1), rt, overnight; (g) [Pt(terpy)Cl]Cl, CuI, NEt3, DMF, rt, 72 h; (h) phenylacetylene, CuI, NEt3, DMF, rt, 24 h. :H2O (1:1), pressure flask (120 °C), 24 h; (f) TBAF, AcOH, ACN:THF (2:1), rt, overnight; (g) [Pt(terpy)Cl]Cl, CuI, NEt3, DMF, rt, 72 h; (h) phenylacetylene, CuI, NEt3, DMF, rt, 24 h. | ||
The monometallic complexes Pt and Ru were prepared as reference compounds for comparison purposes (Fig. 3). In brief, the Ru(II) complex 5 was obtained by reacting 4 with one equivalent of [Ru(bipy)2Cl2]. Then, the triisopropylsilyl protecting group was removed in the presence of tetrabutylammonium fluoride to yield the Ru(II) complex [Ru(bipy)2(1-(5-ethynylpyridin-2-yl)-9-methyl-β-carboline)][PF6]2 (Ru). The monometallic Pt(II) derivative [Pt(terpy)(phenylacetylene)][PF6] (Pt) was synthesized by reacting the Pt(II) precursor [Pt(terpy)Cl]Cl with phenylacetylene in the presence of copper(I) iodide and triethylamine.30 Finally, [Ru(bipy)2(1-(5-ethynylpyridin-2-yl)-9-methyl-β-carboline)Pt(terpy)][PF6]3 (RuPt) was prepared similarly by reacting Ru with one equivalent of [Pt(terpy)Cl]Cl in the presence of copper(I) iodide and triethylamine. All compounds were analyzed by multinuclear NMR (1H, 13C-NMR) and high-resolution mass spectrometry (HR-MS) (Fig. S6–S36†). The purity of ligands and complexes was verified by elemental analysis (see ESI†).
Absorption and emission
The photophysical properties of the new metal complexes were studied to evaluate their potential use as photosensitizers (Fig. 4, Table 1 and Fig. S37†). In good agreement with the calculated theoretical absorption spectrum (Fig. S5†), a weak absorption profile was observed for Pt in the visible region (Fig. 4). The Pt(II) complex exhibited a low-intensity absorption band around 400 nm, extending into wavelengths beyond 500 nm. | ||
| Fig. 4 Overlaid absorbance spectra of Pt, Ru and RuPt (10−5 M) in H2O:DMSO (99:1, v:v) at room temperature. | ||
:DMSO (99:1, v:v). λabs = absorption maximum; λem = emission maximum; τ = photoluminescence lifetime; ΦPL = photoluminescence quantum yield; ΦΔ = singlet oxygen quantum yield
| Complex | λabs/nm (ε/M−1 cm−1) | λem/nm | τ/ns (contribution/%) | ΦPL/% | ΦΔ/% |
|---|---|---|---|---|---|
| Pt | 262 (48324), 282 (32669), 324 (15206), 403 (5280) |
511 | 507 | 5 | 39 |
| Ru | 288 (75900), 315 (48263), 424 (20414), 486 (14245) |
681 | 307 | 2 | 8 |
| RuPt | 286 (64650), 325 (44367), 428 (25311), 482 (10918) |
676 | 8 (24) | 1 | 88 |
| 311 (76) |
By contrast, Ru showed two main absorption bands centered at 424 nm and around 486 nm with an absorption tail that extends up to 600 nm. The heterobimetallic complex RuPt showed a similar absorption profile compared to that of Ru, with two main absorption bands in the visible region. According to the TD-DFT calculations (Table S5†), the absorption band centered at 428 nm is attributed to the combination of both S11 (2.861 eV, 433.4 nm; f = 0.0961) and S13 (2.931 eV, 423.1 nm; f = 0.1391) excited states while the broad absorption band located around 482 nm that extends up to 600 nm is attributed to the sum of both S8 (2.74 eV, 452.9 nm; f = 0.0551) and S9 (2.74 eV, 452.5 nm; f = 0.0931) excited states. These absorption bands correspond to a mixture of spin-allowed metal-to-ligand charge transfer (1MLCT), ligand-to-ligand charge transfer (1LLCT) and ligand-centered (1LC) transitions.
The emission spectra of the new metal complexes were recorded for aqueous solutions under blue light excitation (λex = 450 nm). Pt displayed a broad emission band with a maximum at 511 nm, while Ru and RuPt featured bands centered at 681 and 676 nm, respectively (Table 1 and Fig. S37†). The lifetime for the observed emission of RuPt exhibited two components, the first one with a value of 8 ns (contributing with a percentage share of 24%) and the second one with a value of 311 ns (percentage share of 76%), indicative of two separate excited states. Moreover, the photoluminescence quantum yield for RuPt was very low (ΦPL = 0.91%). This suggests that non-radiative pathways are very efficient, including the intersystem crossing to non-emissive triplet excited states, which were characterized by transition absorption spectroscopy (vide infra).
Transient absorption spectroscopy measurements
Aiming to characterize non-emissive excited states of RuPt and reveal the mechanism involved in the photosensitization of molecular oxygen we performed nanosecond transient absorption spectroscopy (TAS) experiments on this complex. The evolution of the transient absorption spectrum with time, for a degassed aqueous solution at room temperature using a laser excitation wavelength of 355 nm (Fig. 5A) shows a single lifetime over the studied spectral range, corresponding to the decay of T1 and recovery of the ground state absorption. Despite the strong overlap and the existing of a single decay component, the normalized transient absorption spectrum at 0.1 ms, when overlapped with the normalized (and inverted) ground-state absorption spectrum of RuPt allowed us to identify regions of the spectrum that correspond to singlet ground-state depletion and trough spectral decomposition the approximate spectrum of the T1 → Tn transitions (Fig. S38†). Thus, the spectrum exhibits bleaching of the ground state (negative optical density changes) features at 285, 335 and 425 nm, meaning that the ground state absorptions (S0 → Sn) have higher extinction coefficients at these wavelengths than the transient absorptions T1 → Tn. Moreover, two positive transient absorption regions are observed at 305 and 370 nm, where the opposite is true, i.e., the transient absorptions T1 → Tn have higher extinction coefficients than the ground state absorptions. | ||
| Fig. 5 (A) Evolution of the transient absorption spectra of RuPt with time. (B) Recovery of the ground-state after depletion acquired at 280 nm under aerated and degassed conditions; arrows highlight the recovery of the ground state after bleaching (up arrow) and decay of T1 to the ground state (down arrow). | ||
To assess the influence of oxygen on the decay of the RuPt transients, we performed flash photolysis under degassed and aerated conditions over a fresh solution of the complex, by monitoring the ground-state depletion at 280 nm (Fig. 5B). The transients revealed that in the absence of oxygen RuPt has a very long-lived excited state featuring an outstanding lifetime in the hundreds of microseconds scale (0.82 ± 0.20 ms), which is expected for a forbidden triplet excited state. By contrast, this is unusual in the case of metal complexes where the spin–orbit coupling renders T1 → S0 transitions less forbidden, which leads to faster triplet decays. Furthermore, oxygen was proved to be a strong quencher of this triplet excited-state, showing a significant reduction in its lifetime value (0.20 ± 0.05 ms) under aerated conditions. Moreover, the kinetic traces essentially go back to zero in the presence of oxygen, meaning that the ground state is recovered upon the interaction with molecular oxygen. This fact rules out a significant contribution of an electron transfer mechanism to O2, where the kinetic traces would not return to zero. On the contrary, an energy transfer mechanism to produce 1O2 is corroborated.
Then, we determined the energy transfer efficiency from RuPt to O2, ΦET, by using eqn (1) and the values for the lifetimes in the absence (τ0) and in the presence (τ) of oxygen,
 | (1) |
Singlet oxygen generation
The capacity of Pt, Ru and RuPt to generate singlet oxygen (1O2) in aqueous solution was experimentally determined by using 9,10-anthracenediyl-bis(methylene) dimalonic acid (ABDA) as a probe. This probe reacts with a molecule of singlet oxygen, that has been photocatalytically generated by the photosensitizer, forming the corresponding endoperoxide. This reaction was monitored by UV-Vis and the gradual decrease of the absorption band located at 378 nm upon different irradiation times was used to evaluate the ability of Pt, Ru and RuPt to generate 1O2 (Fig. 6 and Fig. S39–S43†). | ||
| Fig. 6 Photobleaching of 9,10-anthracenediyl-bis(methylene) dimalonic acid in the presence of (A) [Ru(bipy)3]Cl2, (B) Pt, (C) Ru and (D) RuPt under blue-light irradiation (460 nm, 24 W) in H2O:DMSO (99:1, v:v). | ||
Upon light irradiation (460 nm, 24 W), a notable decrease of absorbance at 378 nm was observed for all metal complexes, including the reference complex [Ru(bipy)3]Cl2. Indeed, monometallic complexes Ru and Pt displayed similar decomposition rates to that determined for [Ru(bipy)3]Cl2. Strikingly, the heterobimetallic complex RuPt showed an outstanding capacity to generate 1O2, showing the faster photodegradation rate among the evaluated metal complexes. The 1O2 quantum yields (ΦΔ) of our metal complexes were determined using [Ru(bipy)3]Cl2 as a reference photosensitizer (ΦΔ = 0.18 in water).44 The calculated ΦΔ values were 0.39 and 0.08 for the monometallic complexes Pt and Ru, showing a moderate and low efficiency, respectively. By contrast, the heterobimetallic complex RuPt displayed an extraordinary ΦΔ of 0.88 under light irradiation. This exceptional ΦΔ value shown by RuPt could likely be related to the excellent efficiency in the triplet formation and the very long-lived dark triplet excited state. As afore-mentioned, this state is formed as a consequence of the intramolecular electron transfer between both metal fragments, as usually observed in donor-bridge-acceptor organic molecules used in artificial photosynthesis.45
In Fig. 7 we summarize the simplified mechanism for the photophysical process involved in the generation of the triplet excited state of RuPt and the subsequent production of 1O2 through an energy transfer pathway (Fig. 7A). In brief, under light irradiation RuPt is excited to S2 or S3, since the direct excitation to S1 is forbidden owing to the lack of overlapping between HOMO and LUMO (orthogonality). S1 is then populated from S2 by internal conversion thanks to the coupling between the high vibrational states of S1 and the low vibrational states of S2. Next, a triplet state defined by the HOMO–LUMO monoexcitation is formed through an intersystem crossing step, which is very favored due to several factors: (a) the decay from S1 to S0 is highly forbidden; (b) HOMO–LUMO orthogonality leads to a small difference in energy between 1(HOMO–LUMO) and 3(HOMO–LUMO) states, respectively S1 and T3 in the Franck Condon State (Fig. 7B); (c) geometrical relaxation leads to a decrease in the energy of 3(HOMO–LUMO) state, which becomes the new T1 state and (d) the transition from the new T1 state, 3(HOMO–LUMO), to the S0 ground state 1(HOMO–HOMO) is forbidden both by the orthogonality between the HOMO and the LUMO and by the change of spin multiplicity. Finally, the triplet state exhibits a very long lifetime, since electronic recombination to form S0 is forbidden not only because of the spin rule but also due to the lack of overlapping between HOMO and LUMO. Hence, this long lifetime for the triplet excited state of RuPt results from the intramolecular electron transfer between both metal fragments (Fig. 7C), and it is responsible for its high photosensitizing ability of O2 that ultimately generates 1O2 via an energy transfer mechanism.
 | ||
| Fig. 7 (A) Simplified Jablonski diagram for the 1O2 generation by RuPt. (B) Schematic representation of singlet and triplet excited states calculated by TD-DFT approach. (C) Schematic representation for the generation of 3(HOMO–LUMO). | ||
Pharmacological properties
The solubility of the bimetallic conjugate was assessed via dynamic light scattering measurements. A stock solution of RuPt in dimethyl sulfoxide was prepared and subsequently diluted in phosphate-buffered saline to achieve a final dimethyl sulfoxide concentration of 0.5%. Dynamic light scattering measurements were then used to monitor for any precipitate or particle formation. No evidence of particle formation or aggregation was detected, indicating that the conjugate is fully soluble under physiological conditions.Furthermore, the stability of compounds in physiological conditions is an essential feature for biological applications to ensure a safe and effective therapeutic outcome. By contrast, unintended degradation could reduce efficacy and cause side effects. Indeed, Chakravarty et al. have observed the photocleavage of the Pt–C(acetylide) bond under visible light irradiation for complexes of the type [(terpy)Pt–CC–R].46,47 To assess the stability of the bimetallic complex in an aqueous solution, RuPt was incubated in phosphate-buffered saline for 48 h and the absorption profile monitored as well as the identity of the compound analyzed by mass spectrometry. The spectra showed no changes during the incubation time (Fig. S44†), indicating that RuPt remains stable under physiological conditions. Analogously, the photostability of the conjugate in aerated aqueous solutions was studied under continuous blue light irradiation (460 nm, 24 W, 6 h). Within the investigated time interval, no significant changes in the absorption profile of RuPt or any decomposition in the mass spectra were observed (Fig. S45†), corroborating its high photostability.
The lipophilicity of the metal complexes was assessed by determining their distribution coefficient between the phosphate-buffered saline and the octanol phases (logP) using the “shake-flask” method (Table S7†). Ru was predominantly located in the aqueous phase (logP = −0.7 ± 0.1), whereas Pt was found in the octanol phase (logP = +1.1 ± 0.2). Interestingly, RuPt exhibited an amphiphilic character (logP = +0.4 ± 0.2), as expected for a bimetallic derivative that combines features from both monometallic complexes. The cell membrane permeability was evaluated using a parallel artificial membrane permeability assay (Table S8†). The results showed that Ru exhibited poor cell permeability (0.010 ± 0.002 μm s−1), whereas Pt (0.031 ± 0.004 μm s−1) and RuPt (0.024 ± 0.003 μm s−1) displayed moderate cell permeability. Overall, these findings demonstrated that the bimetallic conjugate is soluble and stable under physiological conditions as well as upon irradiation. Furthermore, RuPt showed an amphiphilic character that enables a moderate cell permeability.
Encapsulation into nanoparticles
To enhance the pharmacological properties and provide improved tumor selectivity of the bimetallic conjugate, it was encapsulated with the polymer Pluronic F-127/Polaxamer-407 (poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol)) into nanoparticles, hereinafter referred to as RuPt NP. Notably, this polymer has been clinically approved by the US Food and Drug Administration (FDA).48 Dynamic light scattering measurements of the nanoparticles showed that the nanoparticles had a hydrodynamic diameter of approximately 132 nm with a polydispersity of 0.251 (Fig. S46†). The nanoparticles were found with a zeta potential of +27 mV. Using inductively coupled plasma optical emission spectroscopy (ICP-OES), the amount of encapsulated metal complex was quantified. Following concentrations of the nanoparticles refer to the concentration of the metal complex within the polymeric nanoparticles.Biological properties in cancer cells
The cellular uptake of the molecular complex RuPt in comparison to its nanoparticle formulation RuPt NP was evaluated by incubating human breast adenocarcinoma (MCF-7) cells for 4 h, followed by analysis of metal content using ICP-OES. The results indicated that RuPt NP had a cellular uptake approximately 3.3-higher than RuPt (Fig. S47†), confirming that the formulation into a nanomaterial enhanced the internalization into the cancer cells.The therapeutic effect of Ru, Pt, RuPt, RuPt NP was studied in comparison to the clinically applied photosensitizer photofrin and the chemotherapeutic drug cisplatin against cancerous mouse colon carcinoma (CT-26), human breast adenocarcinoma (MCF-7), human hepatocellular carcinoma (HepG2), and non-cancerous human fibroblast (GM-5657) cells. The monometallic complexes Ru and Pt were found to be inactive in the dark as well as upon irradiation (IC50 > 50 μM) against CT-26 cells. The bimetallic complex RuPt was found with a weak therapeutic response (IC50,dark > 50 μM, IC50,light = 26 ± 5 μM) in CT-26 cells, due to the poor cellular uptake. Followingly, the nanoparticles were tested against all cell lines. The nanoparticles displayed a cytotoxic effect upon treatment in the dark in the moderate micromolar range within all tested cell lines (IC50 = 24.81–45.37 μM). Upon irradiation with blue light, the therapeutic effect is strongly enhanced towards the very low micromolar to nanomolar range (IC50 = 0.54–2.64 μM). The strongest therapeutic effect was observed on MCF-7 cells with a phototoxic index of 65. Interestingly, the cytotoxic response for RuPt NP upon irradiation with blue light was approximately 16-times stronger than upon treatment with the clinically approved photosensitizer photofrin under the investigated conditions (Table 2). Notably, the therapeutic effect is strongly dependent on the conditions within the cellular setting. Based on these findings, further experiments were performed using MCF-7 cells.
| MCF-7 | PI | CT-26 | PI | HepG2 | PI | GM-5657 | PI | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dark | Light | Dark | Light | Dark | Light | Dark | Light | |||||
| RuPt NP | 34.86 ± 3.57 | 0.54 ± 0.06 | 64.6 | 24.81 ± 3.85 | 1.37 ± 0.19 | 18.1 | 45.37 ± 4.16 | 1.84 ± 0.23 | 24.7 | 41.68 ± 3.27 | 2.64 ± 0.30 | 15.8 |
| Cisplatin | 21.59 ± 2.48 | n.d. | n.d. | 25.48 ± 1.83 | n.d. | n.d. | 17.36 ± 2.25 | n.d. | n.d. | 33.57 ± 3.64 | n.d. | n.d. |
| Photofrin | >50 | 8.73 ± 0.53 | >5.7 | >50 | 4.31 ± 0.74 | >11.6 | >50 | 7.38 ± 0.61 | >6.8 | >50 | 9.84 ± 0.66 | >5.1 |
The ability of RuPt NP to generate ROS inside the cancer cells was evaluated by fluorescence microscopy using the ROS specific probe 2′,7′-dichlorodihydrofluorescein diacetate. While being non-fluorescent under physiological conditions, the probe is oxidized in the presence of ROS, generating a green-fluorescent dye. As a positive control, hydrogen peroxide was directly added to the cells. Microscopy images of untreated cancer cells and cells treated with RuPt NP in the dark showed no green fluorescence. In contrast, the cells treated with RuPt NP and subsequently irradiated displayed a strong green fluorescent emission, confirming the ability of the nanoparticles to photogenerate ROS (Fig. 8).
 | ||
| Fig. 8 Fluorescence images of MCF-7 cells upon co-incubation of RuPt NP in the dark or upon irradiation (450 nm, power: 20%, 10 min, 1.2 J cm−2) and the ROS specific probe 2′,7′-dichlorodihydrofluorescein diacetate. Untreated cells were used as a negative control and hydrogen peroxide as a positive control. λex = 460–490 nm, λem = 527 nm. Scalebars represent 200 μm. | ||
To gain deeper insights, studies were conducted to identify the specific types of ROS (i.e., ˙OH, 1O2, or ˙O2−) generated by the metal complex. The cancer cells were preincubated with specific scavengers for each ROS type: D-mannitol for ˙OH, sodium azide for 1O2, and 4,5-dihydroxy-1,3-benzenedisulfonic acid disodium salt monohydrate for ˙O2−. Following preincubation in the presence of each scavenger, cells were treated with RuPt NP at its IC50 value (0.54 μM), and cell viability was assessed. Since no change in cell survival was observed with the ˙OH and ˙O2− scavengers, these ROS types were ruled out. On the contrary, preincubation with the 1O2 scavenger significantly increased cell viability (Fig. S48†), suggesting that the cytotoxic mechanism of the nanoparticles primarily involves the generation of 1O2. Complementary, fluorescence microscopy experiments were conducted on the cancer cells treated with RuPt NP in the presence of the specific fluorophores for each ROS: 3′-p-(hydroxyphenyl) fluorescein for ˙OH, singlet oxygen sensor green for 1O2, or dihydroethidium for ˙O2−. No fluorescence was detected from the ˙OH and ˙O2− probes. However, the incubation with the 1O2 probe, produced a strong green emission (Fig. S49†). Overall, these findings indicate that RuPt NP efficiently generated 1O2 in the cancer cells.
Biological properties in multicellular tumor spheroids
Based on the promising biological effects observed in two-dimensional monolayer cancer cells, the therapeutic properties of RuPt NP were further evaluated in multicellular tumor spheroids, a widely used model that closely mimics the pathological conditions of solid tumors.49 Herein, MCF-7 multicellular tumor spheroids with an average diameter of 650 μm were cultivated and treated with RuPt NP in comparison to the chemotherapeutic drug cisplatin and exposed to light irradiation (450 nm, power: 20%, 10 min, 1.2 J cm−2). The tumor spheroids were daily monitored over a period of seven days. While due to the low concentration in the nanomolar range, no significant therapeutic effect for cisplatin was observed, resulting in the further growth of the tumor spheroid, a therapeutic response was monitored upon treatment with RuPt NP (Fig. 9B). To explore the treatment on larger tumors, the therapeutic effects of the compound was further examined in MCF-7 multicellular tumor spheroids with an average diameter of approximately 850 μm. Previous studies have shown that larger multicellular tumor spheroids are considerably more difficult to treat than smaller ones, mainly because of proliferation gradients and hypoxic regions at the tumor core.50 Similarly, to the smaller tumor spheroids, no significant cytotoxic effect for cisplatin was observed. In contrast, the treatment with RuPt NP upon light irradiation resulted in a tumor growth inhibition of the tumor spheroid (Fig. 9C). Light microscopy images of the larger tumor spheroid showed the darkening of the core as well as morphological damage and alterations upon treatment with photoactivated RuPt NP (Fig. 9A). To further investigate the therapeutic effect, multicellular tumor spheroids were treated with RuPt NP under dark and light conditions, followed by staining with specific cellular live (calcein AM) and dead (propidium iodide) dyes. No cytotoxic response was observed when spheroids were treated in the dark; however, a strong therapeutic effect was evident under light exposure. Notably, dead cells were detected even at the spheroid core, a region typically difficult to target, indicating effective nanoparticle penetration (Fig. 9D). Overall, these findings demonstrate the high therapeutic potential of the new bimetallic-based nanoparticles. | ||
| Fig. 9 Evaluation of the therapeutic effect of RuPt NP on MCF-7 multicellular tumor spheroids in the dark or upon irradiation (450 nm, power: 20%, 10 min, 1.2 J cm−2). (A) Light microscopy images of a single tumor spheroid with an initial diameter of 850 μm untreated or treated with RuPt NP and exposure to light. (B) Tumor growth inhibition curves of tumor spheroid with an initial diameter of 650 μm upon treatment with RuPt NP or cisplatin. (C) Tumor growth inhibition curves of tumor spheroid with an initial diameter of 850 μm upon treatment with RuPt NP or cisplatin. (D) Fluorescence microscopy images upon treatment with RuPt NP in the dark or light and incubation with the cell live stain calcein (CAM, λex = 460–490 nm, λem = 515 nm) and cell death stain propidium iodide (PI, λex = 545–580 nm, λem = 617 nm). | ||
Conclusions
In summary, this study presents the computational design, chemical synthesis, photophysical evaluation, and biological testing of a novel bimetallic Ru(II)–Pt(II) conjugate (RuPt). This photosensitizer displays a very high photocatalytic activity in the singlet oxygen generation leveraging an excited state intramolecular electron transfer process from the Ru(II) moiety to the Pt(II) fragment for applications in photodynamic therapy. More specifically, using computational calculations, an orthogonal arrangement was predicted for the HOMO (Ru environment) and LUMO (Pt-terpy environment) of RuPt and the corresponding electron transfer process was characterized. Thanks to this design, the conjugate exhibits an extremely long-lived triplet excited state stemming from the light-induced metal to metal intramolecular electron transfer. This conjugate is capable to produce singlet oxygen upon photoactivation more efficiently that its monometallic congeners, Ru and Pt. To further improve its pharmacological profile, the bimetallic complex was encapsulated within polymeric nanoparticles. The nanoparticles were able to eradicate human breast adenocarcinoma monolayer cells and multicellular tumor spheroids under light irradiation at nanomolar concentrations. We are confident that this approach will open new avenues for highly efficient bimetallic photosensitizers as well as open new perspectives towards its application as PDT agents against cancer.Author contributions
Juan Sanz-Villafruela: writing – original draft, investigation and formal analysis; Lisa-Marie Servos: investigation and formal analysis; Nicolás Montesdeoca: investigation and formal analysis; José V. Cuevas-Vicario: investigation and formal analysis; Artur J. Moro: investigation, formal analysis and data curation; João C. Lima: supervision, formal analysis and writing – review & editing; Marta Martínez-Alonso: supervision; Gustavo Espino: supervision, writing – review & editing, funding acquisition and conceptualization; Johannes Karges: supervision, writing – review & editing, funding acquisition and conceptualization.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. K. gratefully acknowledges the financial support provided by the Liebig fellowship from the Chemical Industry Fund of the German Chemical Industry Association, the Life Sciences Bridge Award from the Aventis Foundation, and the Paul Ehrlich & Ludwig Darmstaedter Early Career Award 2024 – a prize awarded by the Paul Ehrlich Foundation, Germany. G. E. acknowledges the financial support provided by Ministerio de Ciencia e Innovación/Agencia Estatal de Investigación (MCIN/AEI) of Spain (PID2021-127187OB-C21). J. S. V. acknowledges his predoctoral grant to Universidad de Burgos (2019/00002/008/001) and the PhD Exchange Scholarship provided by RUB Research School. J. V. C. V. acknowledges the financial support provided by Ministerio de Ciencia e Innovación/Agencia Estatal de Investigación (MCIN/AEI) of Spain (PID2022-142318NB-I00).References
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2024, 74, 229–263 CrossRef PubMed.
- A. Persidis, Cancer multidrug resistance, Nat. Biotechnol., 1999, 17, 94–95 CrossRef CAS PubMed.
- M. B. Lustberg, N. M. Kuderer, A. Desai, C. Bergerot and G. H. Lyman, Mitigating long-term and delayed adverse events associated with cancer treatment: implications for survivorship, Nat. Rev. Clin. Oncol., 2023, 20, 527–542 CrossRef PubMed.
- C. Imberti, P. Zhang, H. Huang and P. J. Sadler, New Designs for Phototherapeutic Transition Metal Complexes, Angew. Chem., Int. Ed., 2020, 59, 61–73 CrossRef CAS.
- S. Bonnet, Ruthenium-Based Photoactivated Chemotherapy, J. Am. Chem. Soc., 2023, 145, 23397–23415 CrossRef CAS PubMed.
- L. Zhao, X. Zhang, X. Wang, X. Guan, W. Zhang and J. Ma, Recent advances in selective photothermal therapy of tumor, J. Nanobiotechnol., 2021, 19, 335 CrossRef CAS.
- L. Gourdon, K. Cariou and G. Gasser, Phototherapeutic anticancer strategies with first-row transition metal complexes: a critical review, Chem. Soc. Rev., 2022, 51, 1167–1195 RSC.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, A Potent Trans –Diimine Platinum Anticancer Complex Photoactivated by Visible Light, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef CAS PubMed.
- N. Alvarez and A. Sevilla, Current Advances in Photodynamic Therapy (PDT) and the Future Potential of PDT-Combinatorial Cancer Therapies, Int. J. Mol. Sci., 2024, 25, 1023 CrossRef CAS PubMed.
- D. Luo, K. A. Carter, D. Miranda and J. F. Lovell, Chemophototherapy: An Emerging Treatment Option for Solid Tumors, Adv. Sci., 2017, 4, 1600106 CrossRef PubMed.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Clinical development and potential of photothermal and photodynamic therapies for cancer, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef.
- J. H. Correia, J. A. Rodrigues, S. Pimenta, T. Dong and Z. Yang, Photodynamic Therapy Review: Principles, Photosensitizers, Applications, and Future Directions, Pharmaceutics, 2021, 13, 1332 CrossRef CAS PubMed.
- A. Fennes, N. Montesdeoca, Z. Papadopoulos and J. Karges, Rational Design of a Red-Light Absorbing Ruthenium Polypyridine Complex as a Photosensitizer for Photodynamic Therapy, Chem. Commun., 2024, 60, 10724–10727 RSC.
- Z. Chen, T. Feng, J. Shen, J. Karges, C. Jin, Y. Zhao, L. Ji and H. Chao, A mitochondria-localized iridium(III)–chlorin E6 conjugate for synergistic sonodynamic and two-photon photodynamic therapy against melanoma, Inorg. Chem. Front., 2022, 9, 3034–3046 RSC.
- R. Baskaran, J. Lee and S.-G. Yang, Clinical development of photodynamic agents and therapeutic applications, Biomater. Res., 2018, 22, 25 CrossRef PubMed.
- J. Karges, Clinical Development of Metal Complexes as Photosensitizers for Photodynamic Therapy of Cancer, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef CAS PubMed.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Recent progress in photosensitizers for overcoming the challenges of photodynamic therapy: from molecular design to application, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- M. Obata, S. Hirohara, R. Tanaka, I. Kinoshita, K. Ohkubo, S. Fukuzumi, M. Tanihara and S. Yano, In Vitro Heavy-Atom Effect of Palladium(II) and Platinum(II) Complexes of Pyrrolidine-Fused Chlorin in Photodynamic Therapy, J. Med. Chem., 2009, 52, 2747–2753 CrossRef CAS PubMed.
- T. Wang, Y. Hou, Y. Chen, K. Li, X. Cheng, Q. Zhou and X. Wang, Two novel BODIPY–Ru(II) arene dyads enabling effective photo-inactivation against cancer cells, Dalton Trans., 2015, 44, 12726–12734 RSC.
- L. Zhou, F. Wei, J. Xiang, H. Li, C. Li, P. Zhang, C. Liu, P. Gong, L. Cai and K. M.-C. Wong, Enhancing the ROS generation ability of a rhodamine-decorated iridium(III) complex by ligand regulation for endoplasmic reticulum-targeted photodynamic therapy., Chem. Sci., 2020, 11, 12212–12220 RSC.
- A. Linero-Artiaga, L.-M. Servos, V. Rodríguez, J. Ruiz and J. Karges, Rationally Designed Ir(III) Complex with an Exceptionally Strong Binding to Human Serum Albumin for Targeted Photodynamic Therapy, J. Med. Chem., 2025, 68, 7792–7806 CrossRef CAS.
- X. Fan, S. Lv, F. Lv, E. Feng, D. Liu, P. Zhou and F. Song, Type-I Photodynamic Therapy Induced by Pt-Coordination of Type-II Photosensitizers into Supramolecular Complexes, Chem. – Eur. J., 2024, 30, e202304113 CrossRef CAS PubMed.
- Y. Wu, S. Li, Y. Chen, W. He and Z. Guo, Recent advances in noble metal complex based photodynamic therapy, Chem. Sci., 2022, 13, 5085–5106 RSC.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Transition metal complexes as photosensitisers in one- and two-photon photodynamic therapy, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS.
- L.-M. Servos, H. M. Tran, N. Montesdeoca, Z. Papadopoulos, E. Sakong and J. Karges, Functionalization of a Ru(II) Polypyridine Complex with an Aldehyde Group as a Synthetic Precursor for Photodynamic Therapy, Dalton Trans., 2025, 54, 6411–6418 RSC.
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Metal-based photosensitizers for photodynamic therapy: the future of multimodal oncology?, Curr. Opin. Chem. Biol., 2020, 56, 23–27 CrossRef CAS PubMed.
- J. Chen, Q. Tao, J. Wu, M. Wang, Z. Su, Y. Qian, T. Yu, Y. Wang, X. Xue and H.-K. Liu, A lysosome-targeted ruthenium(II) polypyridyl complex as photodynamic anticancer agent, J. Inorg. Biochem., 2020, 210, 111132 CrossRef CAS PubMed.
- E. Ortega-Forte, A. Rovira, M. López-Corrales, A. Hernández-García, F. J. Ballester, E. Izquierdo-García, M. Jordà-Redondo, M. Bosch, S. Nonell, M. D. Santana, J. Ruiz, V. Marchán and G. Gasser, A near-infrared light-activatable Ru(II)-coumarin photosensitizer active under hypoxic conditions, Chem. Sci., 2023, 14, 7170–7184 RSC.
- H. Wang, Y. Lai, D. Li, J. Karges, P. Zhang and H. Huang, Self-Assembly of Erlotinib-Platinum(II) Complexes for Epidermal Growth Factor Receptor-Targeted Photodynamic Therapy, J. Med. Chem., 2024, 67, 1336–1346 CrossRef CAS PubMed.
- J. Zhu, J.Á Rodríguez-Corrales, R. Prussin, Z. Zhao, A. Dominijanni, S. L. Hopkins, B. S. J. Winkel, J. L. Robertson and K. J. Brewer, Exploring the activity of a polyazine bridged Ru(II)–Pt(II) supramolecule in F98 rat malignant glioma cells, Chem. Commun., 2017, 53, 145–148 RSC.
- S. L. H. Higgins, T. A. White, B. S. J. Winkel and K. J. Brewer, Redox, Spectroscopic, and Photophysical Properties of Ru−Pt Mixed-Metal Complexes Incorporating 4,7-Diphenyl-1,10-phenanthroline as Efficient DNA Binding and Photocleaving Agents, Inorg. Chem., 2011, 50, 463–470 CrossRef CAS.
- S. Zhao, S. M. Arachchige, C. Slebodnick and K. J. Brewer, Synthesis and Study of the Spectroscopic and Redox Properties of RuII,PtII Mixed-Metal Complexes Bridged by 2,3,5,6-Tetrakis(2-pyridyl)pyrazine, Inorg. Chem., 2008, 47, 6144–6152 CrossRef CAS PubMed.
- Y. Fan, L.-Y. Zhang, F.-R. Dai, L.-X. Shi and Z.-N. Chen, Preparation, Characterization, and Photophysical Properties of Pt−M (M = Ru, Re) Heteronuclear Complexes with 1,10-Phenanthrolineethynyl Ligands, Inorg. Chem., 2008, 47, 2811–2819 CrossRef CAS.
- K. van der Schilden, F. Garcìa, H. Kooijman, A. L. Spek, J. G. Haasnoot and J. Reedijk, A Highly Flexible Dinuclear Ruthenium(II)–Platinum(II) Complex: Crystal Structure and Binding to 9-Ethylguanine, Angew. Chem., Int. Ed., 2004, 43, 5668–5670 CrossRef CAS PubMed.
- Z. Zhou, J. Liu, T. W. Rees, H. Wang, X. Li, H. Chao and P. J. Stang, Heterometallic Ru–Pt metallacycle for two-photon photodynamic therapy, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5664–5669 CrossRef CAS.
- J. Karges, T. Yempala, M. Tharaud, D. Gibson and G. Gasser, A Multi-action and Multi-target RuII–PtIV Conjugate Combining Cancer-Activated Chemotherapy and Photodynamic Therapy to Overcome Drug Resistant Cancers, Angew. Chem., Int. Ed., 2020, 59, 7069–7075 CrossRef CAS PubMed.
- A. Bera, A. Nepalia, A. Upadhyay, D. K. Saini and A. R. Chakravarty, Biotin–Pt(IV)–Ru(II)–Boron–Dipyrromethene Prodrug as “Platin Bullet” for Targeted Chemo- and Photodynamic Therapy, Inorg. Chem., 2024, 63, 17249–17262 CrossRef CAS.
- A. K. Pal, S. Nag, J. G. Ferreira, V. Brochery, G. La Ganga, A. Santoro, S. Serroni, S. Campagna and G. S. Hanan, Red-Emitting [Ru(bpy) 2 (N-N)] 2+ Photosensitizers: Emission from a Ruthenium(II) to 2,2′-Bipyridine 3 MLCT State in the Presence of Neutral Ancillary “Super Donor” Ligands, Inorg. Chem., 2014, 53, 1679–1689 CrossRef CAS.
- W. H. Lam, E. S.-H. Lam and V. W.-W. Yam, Computational Studies on the Excited States of Luminescent Platinum(II) Alkynyl Systems of Tridentate Pincer Ligands in Radiative and Nonradiative Processes, J. Am. Chem. Soc., 2013, 135, 15135–15143 CrossRef CAS PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Photophysics of thermally activated delayed fluorescence molecules, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- J. Sanz-Villafruela, C. Bermejo-Casadesús, G. Riesco-Llach, M. Iglesias, M. Martínez-Alonso, M. Planas, L. Feliu, G. Espino and A. Massaguer, Bombesin-Targeted Delivery of β-Carboline-Based Ir(III) and Ru(II) Photosensitizers for a Selective Photodynamic Therapy of Prostate Cancer, Inorg. Chem., 2024, 63, 19140–19155 CrossRef CAS PubMed.
- D. V. Kalinin, S. K. Jana, M. Pfafenrot, A. Chakrabarti, J. Melesina, T. B. Shaik, J. Lancelot, R. J. Pierce, W. Sippl, C. Romier, M. Jung and R. Holl, Structure-Based Design, Synthesis, and Biological Evaluation of Triazole-Based smHDAC8 Inhibitors, ChemMedChem, 2020, 15, 571–584 CrossRef CAS.
- X. Cai, K.-N. Wang, W. Ma, Y. Yang, G. Chen, H. Fu, C. Cui, Z. Yu and X. Wang, Multifunctional AIE iridium(III) photosensitizer nanoparticles for two-photon-activated imaging and mitochondria targeting photodynamic therapy, J. Nanobiotechnol., 2021, 19, 254 CrossRef CAS.
- M. T. Colvin, A. B. Ricks, A. M. Scott, A. L. Smeigh, R. Carmieli, T. Miura and M. R. Wasielewski, Magnetic Field-Induced Switching of the Radical-Pair Intersystem Crossing Mechanism in a Donor−Bridge−Acceptor Molecule for Artificial Photosynthesis, J. Am. Chem. Soc., 2011, 133, 1240–1243 CrossRef CAS PubMed.
- K. Mitra, A. Shettar, P. Kondaiah and A. R. Chakravarty, Biotinylated Platinum(II) Ferrocenylterpyridine Complexes for Targeted Photoinduced Cytotoxicity, Inorg. Chem., 2016, 55, 5612–5622 CrossRef CAS.
- V. Ramu, S. Gautam, A. Garai, P. Kondaiah and A. R. Chakravarty, Glucose-Appended Platinum(II)-BODIPY Conjugates for Targeted Photodynamic Therapy in Red Light, Inorg. Chem., 2018, 57, 1717–1726 CrossRef CAS PubMed.
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, A Review of Poloxamer 407 Pharmaceutical and Pharmacological Characteristics, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS.
- J. R. Aguilar Cosme, D. C. Gagui, H. E. Bryant and F. Claeyssens, Morphological Response in Cancer Spheroids for Screening Photodynamic Therapy Parameters, Front. Mol. Biosci., 2021, 8, 1–12 Search PubMed.
- S. J. Han, S. Kwon and K. S. Kim, Challenges of applying multicellular tumor spheroids in preclinical phase, Cancer Cell Int., 2021, 21, 152 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5qi00924c |
| This journal is © the Partner Organisations 2025 |