
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced electrocatalytic hydrogen evolution with bimetallic Ru/Pt nanoparticles supported on nitrogen-doped reduced graphene oxide†
Heting
Hou‡
 ab,
Christian
Cerezo-Navarrete‡
c,
Dídac A.
Fenoll
d,
Matilda
Kraft
b,
Carlo
Marini
e,
Luis
Rodríguez-Santiago
d,
Xavier
Solans-Monfort
*d,
Luis M.
Martínez-Prieto
*cf,
Nuria
Romero
*bg,
Jordi
García-Antón
*b and
Xavier
Sala
b
ab,
Christian
Cerezo-Navarrete‡
c,
Dídac A.
Fenoll
d,
Matilda
Kraft
b,
Carlo
Marini
e,
Luis
Rodríguez-Santiago
d,
Xavier
Solans-Monfort
*d,
Luis M.
Martínez-Prieto
*cf,
Nuria
Romero
*bg,
Jordi
García-Antón
*b and
Xavier
Sala
b
aSchool of Chemical and Environmental Engineering, Liaoning University of Technology, Jinzhou, 121001, Liaoning Province, P. R. China
bDepartament de Química, Unitat de Química Inorgànica, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès (Barcelona), Spain. E-mail: Jordi.GarciaAnton@uab.es; nuria.romero@lcc-toulouse.fr
cITQ, Instituto de Tecnología Química, Universitat Politècnica de València (UPV), Av. de los Naranjos S/N, 46022, Valencia, Spain. E-mail: luismiguel.martinez@csic.es
dDepartament de Química, Unitat de Química Física, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès (Barcelona), Spain. E-mail: Xavier.Solans@uab.cat
eALBA Synchrotron Light Facility, Carrer de la Llum 2-26, 08290, Cerdanyola del Valles, Barcelona, Spain
fIIQ, Instituto de Investigaciones Químicas (CSIC-Universidad de Sevilla), Avda. Americo Vespucio 49, 41092, Seville, Spain
gUniv. Toulouse, CNRS, Laboratoire de Chimie de Coordination, Toulouse, France
First published on 12th May 2025
Abstract
The electrocatalytic hydrogen evolution reaction (HER) has been explored using mono- and bimetallic Pt–Ru nanoparticles (NPs) deposited onto nitrogen-doped reduced graphene oxide (NH2-rGO) in acidic media. In this contribution, monometallic and bimetallic nanoparticles with three different Pt/Ru ratios (1/5, 1/1, and 5/1) have been used, yielding five different materials denoted as PtxRuy@NH2-rGO (x = 0, y = 1; x = 1, y = 0; x = 1, y = 5; x = 1, y = 1; x = 5, y = 1). The materials were characterized using a variety of state-of-the-art techniques, including high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM), energy dispersive X-Ray spectroscopy (EDX) and X-Ray absorption spectroscopy (XAS), enabling the investigation of differences in morphology, coordination environment and oxidation state as a function of the metal composition of the graphene-supported NPs. The materials exhibited HER activity and demonstrated competitive overpotentials close to the thermodynamic limit. The initial catalytic activity of the as-synthesized materials enhances as the Pt/Ru ratio increases. Chronopotentiometry cathodic experiments showed that under reductive conditions the electrocatalytic performance is drastically impacted. Ru-rich materials were activated, whereas Pt-rich materials showed poor stability. Upon applying a reducing potential for 58 h, Pt1Ru5@NH2-rGO reached the best catalytic activity with outstanding overpotentials of η0 = 0 mV and η10 = 3 mV and no signs of deactivation even after 12 additional hours of electrolysis. According to DFT calculations, all nanoparticles present surface sites whose hydrogen adsorption energy is optimal for HER. In agreement with the experimental data, the Pt1Ru5 model shows the highest number of highly active sites, especially those involving Ru centres close to the Pt–Ru interface. Combining in-depth characterization and computational modelling, this work reveals that the synergy between the two metals, structural features, and their affinity for the support are responsible for the observed differences in catalytic activities and stabilities.
Introduction
Molecular hydrogen, a zero-emissions fuel when burned and an excellent reservoir of excess green electricity to be fed in fuel cells, is a promising energy vector expected to have a key role in the energy transition and the mitigation of climate change.1–3 Unfortunately, most of the hydrogen produced in large scale nowadays is “grey hydrogen”, produced from gasification or reforming of fossil fuels, which is associated with CO2 emissions. On the contrary, “green hydrogen” produced using renewable electricity and water needs further development.4 The overall water-splitting reaction consists of two half-reactions, called oxygen evolution reaction (OER) and hydrogen evolution reaction (HER). These half-reactions are thermodynamically and kinetically challenging. Sunlight can provide the energy needed to overcome the thermodynamics, but the use of catalysts is required to overcome the kinetics. Focusing on HER, much effort has been devoted in recent years to the development of efficient electrocatalysts. Among all the reported heterogeneous HER catalysts, Pt catalysts are the most efficient ones due to the optimal Gibbs free energy of hydride absorption (ΔGH) associated with the Pt–H bond: it is neither too weak nor too strong, allowing for efficient hydrogen activation and subsequent H2 desorption.5,6 However, the corrosion of the Pt catalyst at neutral/alkaline conditions, together with its scarcity and high price, has limited its large-scale commercial application.7 One way to reduce the amount of material used is to maximize the active catalytic surface by using nanoparticles (NPs). In this respect, Ru NPs with Ru–H bond energies similar to those of Pt–H have recently emerged as promising candidates, demonstrating excellent activity in HER and displaying exceptional stability in both acidic and basic environments.8–15 One possibility is to use Pt in combination with other elements, such as Ru,11,16–19 as the use of bimetallic nanomaterials has often benefits in catalysis by synergistic effects between metals.16–18 For example, some authors have found that Ru combined with Pt solves the corrosion degradation problem of Pt in neutral and basic pH while boosting the activity to overpotentials ranging from 6 to 36 mV. This demonstrates that combining Ru and Pt is a promising strategy for achieving efficient and durable HER electrocatalysts across a wide range of pHs.18 However, these studies do not deeply analyse the atomic structure arrangements, precluding the rational design of catalysts.Different types of bimetallic NPs can be produced depending on the synthetic protocol (Fig. 1), namely alloys, intermetallic, Janus-type or core–shell, with the prevalence of each type being governed by factors such as the metal precursors used and/or the reaction conditions applied.20–23 The preparation of core–shell systems can be relatively straightforward, mainly based on different decomposition rates of the metal precursors.24–26 However, preparing bimetallic nanostructured alloys based on transition metals while maintaining a small particle size is a difficult task. Formation of the intermetallic phases normally needs high temperatures, which can easily lead to the aggregation of the bimetallic NPs.27 Various bimetallic nanoparticles have been obtained by the organometallic approach,28 a wet process consisting of the hydrogenation of organometallic precursors under mild conditions. Examples include RuPt bimetallic nanoparticles used for many different catalytic reactions, as this method allows fine-tuning and clean nanoparticle surface.26,29,30 As stated before, the choice of the metal precursor is crucial in the structure arrangement of the nanoparticles.31
 | ||
| Fig. 1 Types of bimetallic NPs: (A) alloyed, (B) intermetallic, (C) Janus, and (D) core–shell structure. Yellow and grey spheres represent two different metal atoms. | ||
In order to prevent the aggregation of the NPs under turnover conditions and to improve the charge transfer capability between nanocatalysts and the electrode (which in turn increases the conductivity), solid supports are commonly used.10,18 In this regard, conductive supports have been extensively employed to stabilize NPs.32–35 Due to its excellent electronic conductivity (as high as 103 S cm−1) and extremely high surface area (300–700 m2 g−1), reduced graphene oxide (rGO) has become an ideal support for electrocatalysts.36,37 The presence of oxygen-containing functional groups and defects in rGO facilitates the stabilization of metal NPs on its surface. In addition, rGO can be easily doped with heteroatoms such as nitrogen, introducing new active sites and enhancing the electron-transfer properties of the graphene support.7,32,38 For example, it has been recently demonstrated that doping carbon supports with heteroatoms like N and P boost the catalytic performance, reaching such low overpotentials as 2 mV after activation of the Ru NPs on phosphorous-doped reduced graphene oxide.38
The aim of this work is to analyse the effect of different Pt/Ru ratios on the HER electrocatalytic activity of ultrafine bimetallic NPs on nitrogen-doped reduced graphene oxide (NH2-rGO) prepared by the organometallic approach.39 As the structural atomic organization of the nanoparticles is often limited by the analysis techniques, HAADF-STEM, EDX and XAS have been used to determine the chemical composition and the structure of the initial bimetallic NPs as well as their post-catalytic outcome. Additionally, DFT calculations have been performed to gain atomistic insight into the nanomaterials’ catalytic activity.
Results and discussion
The catalysts used in this contribution have been prepared through the organometallic approach, following the synthetic procedure some of us described in a previous publication.39 They consist of mono- and bimetallic ligand-free Pt and/or Ru NPs supported on nitrogen-doped reduced graphene oxide (NH2-rGO), namely Ru@NH2-rGO, Pt@NH2-rGO, Pt1Ru5@NH2-rGO, Pt1Ru1@NH2-rGO and Pt5Ru1@NH2-rGO. The metal content reported for the mono- and bimetallic supported-NPs, determined by inductively coupled plasma (ICP) and X-ray fluorescence (XRF) analyses, is close to 3 wt%. Furthermore, PtxRuy@NH2-rGO catalysts presented particle sizes between 1.6 and 2.1 nm (see TEM micrographs and corresponding size histograms in Fig. S1†), and were characterized by high-resolution transmission electron microscopy (HR-TEM), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS).39 In this work, the bimetallic systems were additionally characterized by point-by-point high-angle annular dark-field scanning transmission electron microscopy coupled with Energy-Dispersive X-ray spectroscopy (HAADF-STEM EDX) (Fig. 2) as well as synchrotron X-ray absorption spectroscopy (XAS), including both X-ray absorption near the edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). | ||
| Fig. 2 TEM and HRTEM micrographs with the corresponding size distribution and HAADF-STEM images with the relative composition determined by EDX of (a), Pt1Ru5@NH2-rGO (b), Pt1Ru1@NH2-rGO (c) and Pt5Ru1@NH2-rGO. | ||
Bimetallic materials characterization
The presence of both metals in the HAADF-STEM EDX spectra for individual particles confirms the bimetallic nature of the nanoparticles (Fig. 2). However, Pt/Ru ratios that significantly deviate from the expected values, such as Pt1Ru99 or Pt95Ru5 for Pt1Ru1@NH2-rGO and Pt5Ru1@NH2-rGO, were also observed (Fig. 2b and c), indicating partial segregation of the bimetallic NPs. The available EDX data for such small nanoparticles did not allow us to fully distinguish between nanoalloys or partially segregated metal clusters on individual nanoparticles due to the limitation of the technique.27,40,41 However, XAS data presented below coupled with this EDX structural information, support the (at least partial) metal segregation, especially for Pt1Ru1@NH2-rGO, without excluding alloyed structure to some extent.According to the structural features given by electron diffraction patterns of nanoparticles, the dominant structure of all nanocatalysts was obtained. Specifically, Ru-rich catalysts, such as Ru@NH2-rGO and Pt1Ru5@NH2-rGO, exhibited a hexagonal close-packed (hcp) structure, whereas Pt-rich catalysts (Pt1Ru1@NH2-rGO, Pt5Ru1@NH2-rGO and Pt@NH2-rGO) presented a face-centered cubic (fcc) structure.39 Moreover, the metal oxidation state and coordination environments were obtained by XANES and FT-EXAFS analysis fitting (shown in Fig. 3, S3, S4, S5, S6† and Table 1). In this way, it was possible to determine the fine structural composition of the nanoparticles. Pt@NH2-rGO exhibits an fcc structure and contains only metallic Pt. On the other hand, Ru@NH2-rGO exhibits an hcp structure, and a mixture of Ru0, RuIII and RuIV oxidation states. It has an average Ru–Ru coordination number (C.N.) of 8.82 (1.66), and an average Ru–O C.N. of 5.03 (0.80). Knowing that the maximum coordination number of a bulk hcp structure is 12, these data suggest the presence of partially oxidized Ru NPs (i.e., Ru0 core with a RuOx shell), as previously observed for other Ru systems.9–13
 | ||
| Fig. 3 XANES spectra of investigated PtxRuy@NH2-rGO (a, Pt L3 edge; b, Ru K edge). | ||
| Catalyst | Ru![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt :Pt |
Cryst. struct. | Oxidation state (%) | Coordination number | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ru0 | RuIII | RuIV | Pt0 | PtIV | Ru–Ru | Ru–Pt | Ru–O | Pt–Pt | Pt–Ru | Pt–O | |||
| Ru@NH 2 -rGO | — | hcp | 11.2 (4.9) | 24.1 (9.3) | 64.7 (5.4) | — | 8.82 (1.66) | — | 5.03 (0.80) | — | — | — | |
| Pt 1 Ru 5 @NH 2 -rGO | 5:1 |
hcp | 23.2 (3.3) | 21.2 (8.4) | 55.6 (9.4) | 81.3 (3.5) | 18.7 (2.9) | 7.84 (1.66) | 1.72 (1.55) | 4.82 (0.95) | 4.99 (0.32) | 0.51 (0.18) | 6.22 (0.58) |
| Pt 1 Ru 1 @NH 2 -rGO | 1:1 |
fcc | 21.8 (4.8) | 25.8 (8.3) | 52.3 (6.5) | 91.3 (1.9) | 8.7 (2.3) | 7.12 (1.77) | 3.41 (2.11) | 5.07 (0.72) | 9.12 (0.61) | 0.21 (0.18) | — |
| Pt 5 Ru 1 @NH 2 -rGO | 1:5 |
fcc | 21.3 (7.7) | 7.7 (8.5) | 71.0 (7.2) | 91.8 (2.9) | 8.2 (2.4) | 8.14 (1.77) | 3.18 (1.98) | 5.04 (0.66) | 8.72 (0.44) | 0.14 (0.09) | — |
| Pt@NH 2 -rGO | — | fcc | — | — | — | 93.3 (2.9) | 6.7 (2.5) | — | — | — | 9.18 (0.54) | — | — |
Concerning bimetallic nanoparticles, Pt1Ru5@NH2-rGO is the only Pt-containing system that retains the typical hcp crystal structure of Ruthenium. Pt1Ru5-catalyst mainly contains metallic Ru and Pt, with an average Ru–Ru C.N. of 7.84 (1.66), and an average Pt–Pt C.N. of 4.99 (0.32). However, Ru–Pt (with a C.N. of 1.72 (1.55)) and Pt–Ru bonding (with a C.N. of 0.51 (0.18)) can also be observed. Surprisingly, Pt–O bonding (with an average coordination number of 6.22 (0.58)) was observed only in this system and not in the other Pt-containing nanoparticles. These results indicate that Pt is mostly located on the surface of these Pt-deficient NPs. This could be related to the fact that Pt1Ru5@NH2-rGO is the only Pt-containing system that shows the hcp crystal structure. This unusual crystal structure of Pt seems to influence its reactivity towards oxidation.42 Thus, it can be suggested that the formation of the hcp-structured Pt in the Ru crystal structures may result in compressive strain throughout the structure to accommodate the interfacial lattice mismatch, thereby altering the surface reactivity (see DFT results below).
On the other hand, Pt1Ru1@NH2-rGO and Pt5Ru1@NH2-rGO show Ru–O bonding and high Ru–Ru C.N. This feature indicates that Ru is likely present at the surface of the nanoparticles and that it is partially oxidized, commonly observed in Ru nanoparticles.10 These Pt-rich samples show the typical fcc structure for Pt and there is no evidence of Pt oxidation.
The Ru–Pt C.N. (3.18 (1.98) for Pt1Ru1@NH2-rGO; 3.41 (2.11) for Pt5Ru1@NH2-rGO) and Pt–Pt C.N. (9.12 (0.61) for Pt1Ru1@NH2-rGO; 8.72 (0.44) for Pt5Ru1@NH2-rGO) are similar for the two compositions and both are higher than for Pt1Ru5@NH2-rGO (only 1.72 (1.55) for Ru–Pt and 4.99 (0.32) for Pt–Pt). Pt-Ru C.N. are low in the two Pt-rich samples (0.14 and 0.21, for Pt1Ru1@NH2-rGO and Pt1Ru5@NH2-rGO, respectively). Such small Ru–Pt C.N., compared to the higher Pt–Pt C.N., seems to indicate that Pt1Ru1@NH2-rGO and Pt1Ru5@NH2-rGO bimetallic particles are partially core–shell segregated, or sub-cluster segregated, with regions of monometallic Pt, regions of monometallic Ru, Pt–Ru interfaces and limited alloy regions, in agreement with the observations of the point-by-point EDX analysis.
Overall, these results show that Pt@NH2-rGO exhibits an fcc structure with only metallic Pt, while Ru@NH2-rGO exhibits an hcp structure with a mixture of Ru0, RuIII and RuIV oxidation states. Pt1Ru5@NH2-rGO retains the hcp structure of Ru, with Pt being partially oxidized and mostly located on the surface of the nanoparticle. In contrast, Pt1Ru1@NH2-rGO and Pt5Ru1@NH2-rGO display typical fcc-Pt0 crystal structures with partial Ru oxidation and possible core–shell or sub-cluster segregation with regions of monometallic Pt or Ru and Pt–Ru interfaces. Different decomposition rates of the metal precursors under hydrogen during the organometallic synthesis could be at the origin of the segregated structures.
Electrocatalytic performance in HER
The as-prepared mono- and bimetallic Pt and/or Ru nanoparticles were tested as catalysts for HER in 1 M H2SO4 aqueous solution by drop-casting their dispersion inks onto a glassy carbon rotating disk electrode (GC-RDE). Cathodic linear sweep voltammetry (LSV) tests were first performed in a typical three-electrode configuration (see Experimental section for further details) to reveal the intrinsic behavior of the catalysts. The experimental results are shown in Fig. 4 and Table 2. From the as-prepared materials, all Pt-containing catalysts showed an onset potential (η0) close to zero. Pt@NH2-rGO requires an overpotential of 12 mV to achieve a current density of −10 mA cm−2 (η10), outperforming Pt5Ru1@NH2-rGO (32 mV), Pt1Ru1@NH2-rGO (33 mV), Pt1Ru5@NH2-rGO (54 mV) and Ru@NH2-rGO (198 mV) catalysts. These results show a clear trend: increasing Ru loading in the catalysts leads to reduced initial hydrogen evolution activity. Therefore, Pt@NH2-rGO exhibits the best performance among all PtxRuy@NH2-rGO catalysts. The Tafel slope values are 39, 48, 42, and 65 mV dec−1 for Pt@NH2-rGO, Pt5Ru1@NH2-rGO, Pt1Ru1@NH2-rGO, and Pt1Ru5@NH2-rGO, respectively, which indicate a Volmer–Heyrovsky mechanism (see Fig. S9†). In contrast, Ru@NH2-rGO displays the highest Tafel slope value (169 mV dec−1), suggesting that the Volmer step is rate-determining.43 | ||
| Fig. 4 (a) Linear sweep voltammetry and (b) their corresponding HER benchmarking overpotentials for all the nanomaterials employed in this work at t = 0 h. (c) Linear sweep voltammetry of Ru@NH2-rGO and Pt1Ru5@NH2-rGO as-synthesized (plane line) and upon applying j = −10 mA cm−2 until reaching an equilibrium for the activation process (dotted line). Experiments were performed in 1 M H2SO4. | ||
| Material | Time of application of j = −10 mA cm−2 | |||
|---|---|---|---|---|
| t = 0 h | Final state | |||
| η 0 (mV) | η 10 (mV) | η 0 (mV) | η 10 (mV) (time of equilibrium) | |
| Pt@NH 2 -rGO | 0 | 12 | 42 | Continuous deactivation |
| Pt 5 Ru 1 @NH 2 -rGO | 0 | 32 | 4 | Continuous deactivation |
| Pt 1 Ru 1 @NH 2 -rGO | 0 | 33 | 21 | Continuous deactivation |
| Pt 1 Ru 5 @NH 2 -rGO | 0 | 54 | 0 | 3 (58 h) |
| Ru@NH 2 -rGO | 61 | 198 | 0 | 20 (5 h) |
When current-controlled bulk electrolysis at j = −10 mA cm−2 were applied to the mono and bimetallic Pt or/and Ru hybrid materials, their catalytic performances were significantly altered. Higher η10 were observed after applying a reductive potential for Pt@NH2-rGO, Pt5Ru1@NH2-rGO and Pt1Ru1@NH2-rGO, showing continuous deactivation over time by observing decreasing potentials during chronoamperometry experiments (Fig. S12a–c† and Table 2). Deactivation is related to the aggregation of the post-operando Pt-rich catalysts (see below). In contrast, the Ru-rich samples Pt1Ru5@NH2-rGO and Ru@NH2-rGO improve their η10 over time, reaching up to 3 mV and 20 mV, after 58 h and 5 h of bulk electrolysis, respectively (Fig. 4(c) and Fig. S12d, e†). These η10 values are comparable to the best catalysts reported in the literature (see Table S3†).
Moreover, these low overpotentials indicate that, upon reduction, Pt1Ru5@NH2-rGO and Ru@NH2-rGO become more active in HER than in their initial state. This behavior has been observed for other passivated Ru nanoparticles,9,10 and it is ascribed to the reduction of the RuO2 shell to metallic Ru0. Another significant difference between catalysts is the time required to reach the most active state of the catalysis, 58 h for Pt1Ru5@NH2-rGO and 5 h for Ru@NH2-rGO (see Fig. 4(c) and Table 2). If we compare Pt1Ru5@NH2-rGO and Ru@NH2-rGO, both are initially partially surface-oxidized, as inferred from XPS39 and XAS data. After applying the reduction potential, the longer time needed to activate the Pt1Ru5@NH2-rGO hybrid material could be attributed to either the slower hydride formation kinetics in its strained crystal structure (vide supra) and/or to the presence of PtOx, which might make the reduction of RuO2 to Ru more difficult. As expected, the Tafel slopes after activation (see Fig. S10†) for Pt1Ru5@NH2-rGO (46 mV dec−1) and Ru@NH2-rGO (38 mV dec−1) are the smallest among all the catalysts, indicating the fastest reaction kinetics. Despite the long activation period, Pt1Ru5@NH2-rGO shows the best activity in HER out of all the mono and bimetallic systems.
In order to study the performance at high current density, LSV measurements were performed on the Pt1Ru5@NH2-rGO catalyst before and after applying chronopotentiometry at −300 mA cm−2 in 1 M H2SO4. The overpotential required to reach a current density of −10 mA cm−2 (η10) decreased significantly over time: starting from an initial value of 130 mV, it dropped to 12 mV after 7 hours, and further decreased to only 2 mV after 19 hours of operation.
TEM analysis was performed for the nanomaterials remaining at the electrodes after bulk electrolysis at j = −10 mA cm−2 (Fig. 5). Pt-rich samples (Pt@NH2-rGO, Pt5Ru1@NH2-rGO, and Pt1Ru1@NH2-rGO) show obvious agglomeration, which may be due to the poor stability of Pt NPs on NH2-rGO in acidic conditions,44 explaining their catalytic activity loss. In contrast, TEM analyses of Pt1Ru5@NH2-rGO and Ru@NH2-rGO indicate that there is no significant change before and after catalysis, as the nanoparticles are uniformly distributed over the NH2-rGO support. It can be concluded that the affinity of Ru for the N-doped support is higher than that of Pt, as the aggregation of Pt samples is more pronounced. In general terms, Ru exhibits greater affinity towards N than Pt, and the N-doping of the support favors Ru anchoring.12,44 The slower activation of Pt1Ru5@NH2-rGO may be due to weaker nanoparticle-support interactions compared to those in monometallic Ru system. As a result, in the bimetallic system, it takes a longer time to transfer the electrons necessary to reduce the oxide passivation layer.
 | ||
| Fig. 5 TEM images of the hybrid materials before (left) and after applying the reduction potentials in 1 M H2SO4 (right). | ||
From the double layer capacitance values (CDL) (Fig. S7†) obtained in a non-faradaic zone of the cyclic voltammetry experiments, the electrochemical active surface area (ECSA) and roughness factor (RF) of all PtxRuy@NH2-rGO catalysts before and after reduction (at maximum activation time) can be estimated. With these data in hand, the specific current density (js) can be calculated before and after activation. These values are summarized in Table 3. Before activation, the ECSA values of Pt@NH2-rGO, Pt5Ru1@NH2-rGO, Pt1Ru1@NH2-rGO, Pt1Ru5@NH2-rGO and Ru@NH2-rGO are ca. 21 cm2, 28 cm2, 31 cm2, 21 cm2, and 7 cm2, respectively. However, after applying the reductive potential, the ECSA significantly changes. In detail, the activated Pt1Ru5@NH2-rGO and Ru@NH2-rGO samples presented an increased ECSA value (from 21 cm2 to 25 cm2 and from 7 cm2 to 47 cm2, respectively) and roughness factor (RF) values (from 105 to 125 and from 33 to 234, respectively). Although values at η = 100 mV are commonly reported in the literature, the current density for the most active materials in this work (reported in Table 3) surpassed the measurable range in our potentiostatic unit. Therefore, for comparison purposes, we analyze the specific current density values at η = 50 mV for all catalysts series. Among them, activated Pt1Ru5@NH2-rGO presents the highest specific current density at η = 50 mV with a value of 0.87 mA cm−2, in agreement with all other benchmarking parameters.
| Entry | Material | Ø (nm) | η 10 (mV) | Tafel slope (b) (mV dec−1) | j 0 (mA cm−2) | ECSA (cm2) | RF | |js|(η=100 mV) (mA cm−2) | j s|(η=50 mV) (mA cm−2) |
|---|---|---|---|---|---|---|---|---|---|
| a Values before/after bulk electrolysis. | |||||||||
| 1 | Pt@NH 2 -rGO | 2.2 ± 0.4 | 12 | 39 | 5.16 | 21 | 106 | − | 0.70 |
| 2 | Pt 5 Ru 1 @NH 2 -rGO | 1.9 ± 0.4 | 32 | 48 | 0.35 | 28 | 142 | 0.63 | 0.16 |
| 3 | Pt 1 Ru 1 @NH 2 -rGO | 1.7 ± 0.4 | 33 | 42 | 2.19 | 31 | 156 | 0.31 | 0.11 |
| 4 | Pt 1 Ru 5 @NH 2 -rGO | 1.7 ± 0.4 | 54/3 | 65/46 | 1.35/0.94 | 21/25 | 105/125 | 0.29 /- | 0.08/0.80 |
| 5 | Ru@NH 2 -rGO | 1.6 ± 0.4 | 198/20 | 169/38 | 0.66/2.86 | 7/47 | 33/234 | 0.07/1.18 | 0.04/0.27 |
Faradaic efficiency experiments are used to quantify the percentage of electrons involved in the desired catalytic reactions (in our case, producing H2). As shown in Fig. S8,† all the PtxRuy@NH2-rGO catalysts display faradaic efficiencies around 97–98% when performing chronoamperometry experiments at j = −10 mA cm−2 for 20 min in 1 M H2SO4, which indicates that virtually all the electrons passing through the system are used in the HER and no side reactions take place.
For the most promising catalysts, Pt1Ru5@NH2-rGO and Ru@NH2-rGO, long-term stability tests were conducted after attaining maximum activation by applying bulk electrolysis (chronopotentiometry) at j = −10 mA cm−2 for 12 h to evaluate their feasibility in practical applications. In Fig. S11,† the LSV curves before and after the bulk electrolysis experiments for Pt1Ru5@NH2-rGO and Ru@NH2-rGO are presented. No significant changes were observed for the η10 overpotential values, indicating that these two catalysts are stable in 1 M H2SO4 in the medium-long term.
The materials were also tested in basic conditions. Chronopotentiometry experiments were conducted in 1 M NaOH (pH 14) at a constant current density of −10 mA cm−2 (see Fig. S15 and S16†). In all cases, the catalysts exhibited higher overpotentials and Tafel slopes under alkaline conditions compared to acidic media. The overpotentials (η10) measured at the stabilized state were: Ru@NH2-rGO (199 mV, stable after 9 hours), Pt1Ru5@NH2-rGO (310 mV, stable after 4 hours), Pt1Ru1@NH2-rGO (288 mV, stable after 4 hours), Pt5Ru1@NH2-rGO (297 mV, stable after 2 hours), and Pt@NH2-rGO (245 mV, stable after 2 hours). This final state was probably reached after an initial deactivation process, during which the NPs with weaker adhesion to the electrode gradually detached until stabilization, as ascertained by the decrease in the ECSA values (Fig. S17–S21†). Under alkaline conditions, monometallic catalysts (especially Ru) behave better than bimetallic catalysts, indicating no synergistic effects in this case. This result is expected as it is well known that Pt is not stable in alkaline media under turnover conditions.10
Atomistic interpretation
With the aim of getting atomistic insights on the catalytic activity of bimetallic Pt1Ru5@NH2-rGO nanoparticles, DFT calculations were performed on three representative nanoparticle models (Pt79, Ru57 and Pt10Ru47) (Fig. 6). The bimetallic model ratio corresponds to that of the most active catalyst, Pt1Ru5@NH2-rGO. The model is constructed maintaining the hcp structure of Ru and a sub-cluster segregated arrangement with Pt–Pt (4.8), Ru–Ru (7.1) and Ru–Pt (0.7) coordination numbers. That is, the experimental structure and coordination numbers were used to build the nanoparticle model (see Computational details for further information). The adsorption energies of one H on different surface sites were used, as defined by Nørskov and co-workers,45 as descriptor for the catalytic activity of the different materials. However, a key point that remains challenging is the determination of the amount of adsorbed H (and their distribution around the nanoparticle) at the reaction conditions. Previous contributions outlined that more than one H atom per surface metal center are adsorbed at working reductive potentials although the exact number is system dependent.9,10,46,47 Consequently, to obtain a fairly representative model, a multistep process was followed consisting of short Ab initio Molecular Dynamics (AIMD) simulations and geometry optimizations at different H coverage until the mean adsorption energy of the last H atoms becomes close to the Nørskov reference adsorption energy. Further details on the model construction can be found in the computational details section. Therefore, the following three representative models were obtained: (i) Pt79H105, (ii) Ru57H61 and (iii) Pt10Ru47H65 (Fig. 6).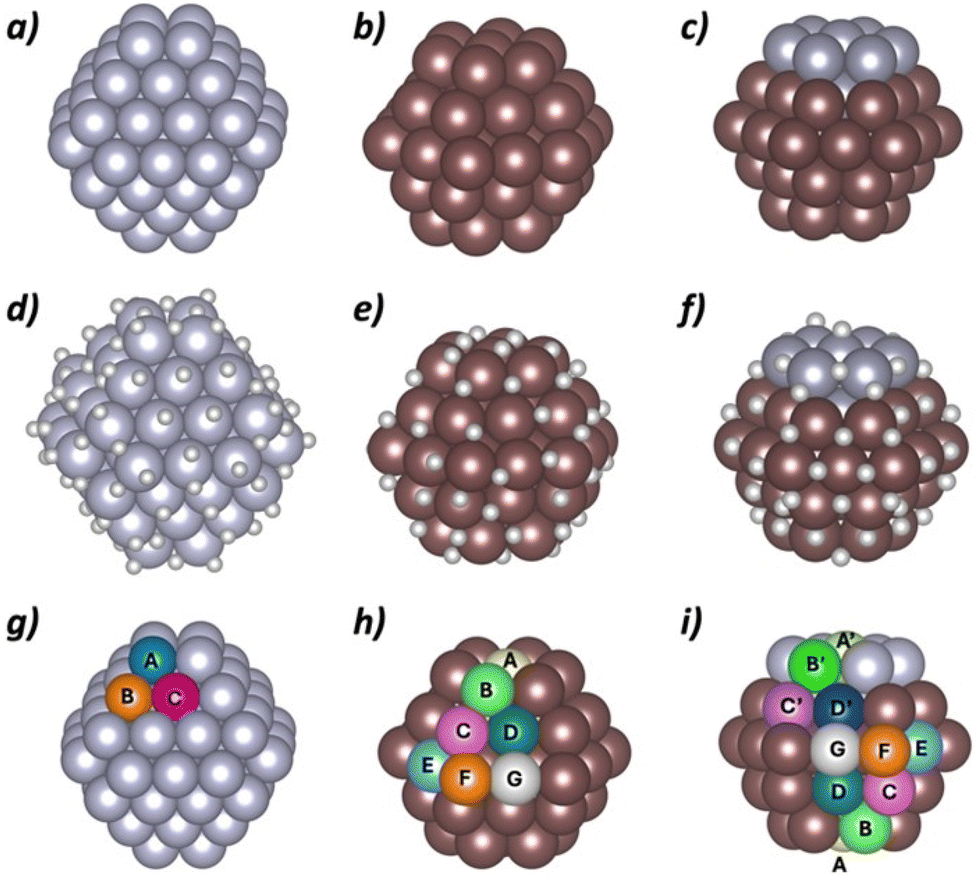 | ||
| Fig. 6 Nanoparticle models used in the DFT simulations and their different sites taking metal environment as criterion: (a) Pt79, (b) Ru57, (c) Pt10Ru47, (d) Pt79H105, (e) Ru57H61, (f) Pt10Ru47H65, (g) sites for Pt79 model, (h) sites for Ru57 model and (i) sites for Pt10Ru47 model. | ||
Different surface sites were identified according to different criteria: (i) surface metal location at the nanoparticle (A–G in Fig. 6g–i); (ii) hydrogen atoms already bonded to the metal center before the adsorption of the additional H; and (iii) presence or absence of an H atom on top of the metal center where the adsorption of the extra hydrogen takes place. All these sites were considered for the adsorption of an H atom either on top of a single center or bridging between two centers. The following nomenclature is used: T(Kia) refers to centers where an H atom is added on top and μ(Kia–Ljb) represents H bridging between two atoms. K and L stand for the metal location at the NP (A to G), i and j specify the number of H already adsorbed (1 to 4), while a and b indicate the presence (Y = yes) or absence (N = no) of a H on top before the adsorption of the additional H atom. As an example, T(B2N) corresponds to the adsorption on top of a metal located in B that already has two H atoms none of them on top.
Table 4 reports the adsorption energies of the additional H atoms that are computed about ±10 kJ mol−1 away from the ideal value proposed by Nørskov (−23.4 kJ mol−1). The adsorption of all other computed sites can be found in the ESI (Table S1†). For Pt79H105, the adsorption energies range between −35.0 and +14.7 kJ mol−1. H adsorbed on bridges and top sites that do not present previous H atoms on top present similar adsorption energies, ranging between −35.0 and −10.4 kJ mol−1. For the adsorption on top sites the adsorption energy does not seem to depend much on the nanoparticle metal site environment. Conversely, sites with H atoms already adsorbed on top exhibit very weak or even unfavorable adsorption energies for a second H, indicating that the adsorption of a second top H atom on Pt is unlikely. Overall, the computed adsorption energies suggest that the high initial catalytic activity for HER of the synthesized Pt@NH2-rGO nanoparticles comes mainly from bridge and top sites, whose adsorption energies are close to the ideal value.
| Pt79H105 | Ru57H61 | Pt10Ru47H64 | |||
|---|---|---|---|---|---|
| Site | E ads | Site | E ads | Site | E ads |
| T(B2N) | −32.8 to −30.1 | μ(C3N–D1N) | −30.6 | μ(C′3N–D′1N) | −29.0 |
| μ(A4Y–C1N) | −26.8 | T(F4N) | −26.7 to −24.6 | T(B′3N) | −28.2 to −15.2 |
| μ(A3Y–B2Y) | −26.1 | T(B4N) | −24.1 to −14.7 | μ(C3N–C3N) | −24.6 to −19.6 |
| T(A3N) | −20.4 | T(C4N) | −22.9 to −9.7 | T(F3N) | −21.5 |
| T(C3N) | −16.6 to −10.4 | T(F3N) | −17.4 | T(B4N) | −20.5 to −11.6 |
| T(E4N) | −19.5 | ||||
| T(F4N) | −17.4 | ||||
| T(C′3N) | −15.8 | ||||
| T(D2N) | −15.4 | ||||
Adsorption energies of H atoms exceeding Ru57H61 coverage ranges from −46.7 and +6.8 kJ mol−1. The H atoms adsorbing on bridging sites interact more strongly with the nanoparticle than those adsorbed on top position. The adsorption energies of the bridge sites range between −46.7 and −30.6 kJ mol−1 and thus, the values are too high to suggest that bridge H atoms are involved in HER. The adsorption energies on top sites range between −26.7 and +6.8 kJ mol−1. Interestingly, the adsorption energy is dependent on the Ru surface metal coordination: corner sites (F and B) show stronger adsorption than edges (C, E, G) and face located sites (A and D). Indeed, the least coordinated sites present adsorption energies that are close to the Nørskov ideal value and suggest that they would be responsible for the high catalytic activity of activated (reduced) Ru@NH2-rGO.
The additional H atoms on the Pt10Ru47H64 nanoparticle present adsorption energies between −45.7 and +13.4 kcal mol−1. The adsorption energies of bridge and top H atoms on Pt surface metals are similar to those found for Pt79 nanoparticle thus suggesting that these sites exhibit similar activity to pure metallic nanoparticles. Remarkably, the number of Pt centers without a pre-adsorbed H on top is larger on the Pt–Ru interface, thus indicating a higher concentration of Pt active sites in this region. In addition, the low coordinated Ru surface centers also present adsorption energies close to Nørskov value for very active sites. Finally, some bridge Ru sites of the interface become also active for the bimetallic system. Therefore, according to calculations, the Pt10Ru47H64 nanoparticle is highly active in HER mainly due to the presence of Pt and Ru domains that keep their individual high catalytic activities. In addition, a synergic effect of the bimetallic nanoparticle seems also to be present. The Pt-Ru interface promotes the presence of new active sites: (i) a higher density of available Pt top sites; and (ii) some Ru bridge sites in the interface in which H interacts with the appropriate adsorption energy. Overall, the high performance of activated (reduced) Pt1Ru5@NH2-rGO could be attributed to the activity of the two domains, the synergy between metals and the high stability of the hybrid electrode due to the affinity of ruthenium for the NH2-rGO support.
Conclusion
In this contribution, mono- and bimetallic Pt–Ru NPs supported onto NH2-rGO have been used as electrocatalysts for HER. The NH2-rGO support facilitates the stabilization of ultra-small Pt and/or Ru NPs, which have been synthesized via the organometallic approach. TEM micrographs and XAS analysis confirmed the bimetallic nature of the catalysts and revealed how the nanoparticle morphology varies with the atomic composition, obtaining limited alloy regions together with segregated structures.All PtRu-based catalysts (Pt@NH2-rGO, Pt5Ru1@NH2-rGO, Pt1Ru1@NH2-rGO, Pt1Ru5@NH2-rGO, and Ru@NH2-rGO) were tested as electrocatalysts for the HER in acidic conditions. In the initial state, Pt@NH2-rGO outperformed all the other four catalysts offering the lowest overpotentials (η0 ≈ 0 mV and η10 = 12 mV), and lowest Tafel slope (39 mV dec−1), while Ru@NH2-rGO displayed the highest overpotential (η10 = 198 mV) to achieve a current density of −10 mA cm−2. Upon applying a reductive potential, the performances of all the catalysts changed. Deactivation was observed for the Pt-rich catalysts (Pt@NH2-rGO, Pt5Ru1@NH2-rGO, and Pt1Ru1@NH2-rGO), which agglomerate at the surface of the carbonaceous support due to a weak affinity between the Pt metal and the support. However, activation was observed for the Ru-rich catalysts (Pt1Ru5@NH2-rGO and Ru@NH2-rGO), likely indicating that the passivation shell of RuO2 is reduced to Ru0, as previously reported in the literature.9 Finally, both Pt1Ru5@NH2-rGO (η0 = 0 mV and η10 = 3 mV, Tafel slope of 46 mV dec−1) and Ru@NH2-rGO (η0 = 0 mV and η10 = 20 mV, Tafel slope of 36 mV dec−1) systems have demonstrated outstanding activity and stability for HER, showing no signs of deactivation or nanoparticle aggregation under turnover conditions for 12 h, maintaining η10 values comparable to the best catalysts reported in the literature and paving the way for their real-device application.
DFT calculations suggest that pure Pt and Ru nanoparticles show several active sites that are also found in the monometallic regions of the bimetallic cluster segregated catalyst Pt1Ru5@NH2-rGO. Moreover, some additional sites at the Ru–Pt interface of Pt1Ru5@NH2-rGO are also suggested to be active according to our calculations. Overall, the activity of the Ru-rich bimetallic systems could be attributed to three factors: (i) the high activity of the Ru and Pt domains; (ii) synergistic effects at the Pt–Ru interface with the presence of a higher density of active sites and (iii) the affinity of ruthenium for the carbonaceous support. The significant novelty and impact of this work lie in the combination of advanced synchrotron-based XANES and EXAFS analysis with DFT modelling, which enables a deep, atomic-scale understanding of the structure–activity relationship in Pt–Ru HER catalysts, offering valuable insights for the rational design of future electrocatalysts.
Experimental section
Reagents and materials
All synthesis and reagent handling procedures were performed using a standard Schlenk line, Fisher–Porter tube techniques or glove-box workstation following the procedure described in a previous publication.39 The solvents used (THF, hexane) were purified and treated in solvent stills and then degassed by the freeze–pump–thaw method before use. The precursors [Ru(COD)(COT)] (COD = 1,5-cyclooctadiene; COT = 1,3,5-cyclooctatriene) and [Pt(NBE)3] (NBE = norbornene) were purchased from Nanomeps. Hydrogen gas (Alphagaz) was purchased from Air Liquide. High-purity deionized water was obtained by passing distilled water through a nanopore Milli-Q water purification system.Mono- and bimetallic catalyst (Ru@NH2-rGO, Pt1Ru5@NH2-rGO, Pt1Ru1@NH2-rGO, Pt5Ru1@NH2-rGO and Pt@NH2-rGO) were synthesized according to a previous publication.39
Characterization techniques
 | (1) |
Transmission EXAFS measurements at Ru K-edge (22117 eV) and Pt K-edge (78404 eV) were performed in quick-EXAFS mode at the CLAESS beamline of the Spanish National Light Source, (ALBA Synchrotron, Barcelona, Spain), using a Si (311) double-crystal monochromator.49 The incoming and outgoing photon fluxes were measured by ionization chambers filled with appropriate mixtures of N2 and Kr gases. The second focusing mirror of the beamline has been set to have a focal point of 400 × 300 μm2. Fitting procedure is indeed very similar to the one explained in literature.50 Theoretical parameters like phase and amplitude functions were calculated using FEFF8.2 and experimental factors (S02) derived from reference materials (RuO2, Ru foil and Pt foil) initially fitted apart. As a consequence, a S02 value of 0.79 (0.21) and 0.82 (0.34) has been set for Pt and Ru edges modelling. In the following we consider three main contributions mimicking metal–oxygen (M–O) and metal–metal (M–M) and metal-different metal (M–M1) interactions in the EXAFS fitting. More in detail, we considered an M–O coordination number, M–O bond length, and M–O Debye Waller factor for the first shell. M–M and M–M1 coordination numbers, M–M and M–M1 bond lengths, and M–M and M–M1 Debye Waller factors for the second shell. Correction to edge position E0 has found to be 5.80 (0.26) eV and 5.66 (1.33) eV for Pt L3 and Ru K edges respectively. M–O, M–M, M–M1 Debye Waller factors have been set common to all samples. Finally, all the paths included in the analysis have been weighted by the corresponding metallic and oxide fractions obtained by the LCF of the XANES. The M–O interaction has been considered in the analysis only if its value is found above 20%.
Fitting results were obtained by using the Artemis program program51 and error bars were estimated with k1/k2/k3 weighted fittings.
Electrochemical measurements
Hydrogen evolution reaction (HER) was carried out using an Origalys potentiostat in a typical three-electrode system with Ag/AgCl (Sat'd) as reference electrode (RE). A rotating disk glassy carbon electrode (RDE-GC, 0.2 cm2 surface area) coated with the catalyst and a Pt rod were used as working electrode and auxiliary electrode, respectively. GC disk electrode was carefully polished before being used. The other two electrodes were rinsed with Milli-Q water and dried prior to the measurements. The speed of the RDE was set to 3000 rpm to remove the hydrogen bubbles produced during this reaction. 1 M H2SO4 solution was prepared by diluting 56.1 mL of 95–97% H2SO4 with 943.9 mL Milli Q water and pH was adjusted to 0.Linear sweep voltammetry (LSV) at a scan rate of 10 mV s−1 were carried out in an Argon-saturated 1 M H2SO4 aqueous solution (pH = 0). Chronopotentiometry was performed by applying a current (i) of −2 mA to reach the current density (j) of −10 mA cm−2. All the potentials in this work were converted to Normal Hydrogen Electrode (NHE) by adding a value of 0.199 V (reference value at 25 °C)
Electrode preparation
The modified GC was prepared as follows: 1 mg of PtxRuy@NH2-rGO was dispersed in 0.5 mL of THF, and then sonicated 5 min to obtain an ink. 15 μL of the ink were loaded by drop casting on the GC, then covered with 5 μL of Nafion® solution. The Nafion® solution was prepared as follows: 40 μL of 5% Nafion®, 7.96 mL Milli-Q water and 2 mL of n-propanol. The working electrode was dried at room temperature with an air flow.In the case of faradaic efficiency measurements, the FTO electrode (1 cm2 surface area) was used as the working electrode. 25 μL of the ink was drop-casted on the surface of the FTO electrode and dried at room temperature with an air flow.
Faradaic efficiency
A two-compartment cell with a permeable membrane in the middle is used. The WE and RE were placed in the same compartment together with the Clark electrode, and the CE was placed in the second compartment. Each of the compartments was filled with 7 ml of 1 M H2SO4 solution and a magnetic stirrer. A Unisense H2-NP Clark was employed to obtain the hydrogen gas produced during the 20 min chronopotentiometry. In the end, the Clark electrode was calibrated by adding different volumes of 99% pure H2, and then the amount of hydrogen was calculated according to the ideal gas law.Double-layer capacitance (CDL) and electrochemically active surface area (ECSA) determination
Double-layer capacitance (CDL) was determined by applying a series of cyclic voltammetry (CV) with different scan rates in a non-faradaic zone. In this region, no redox reaction took place, and the current is due to the double layer charging. Based on this, the charging current (ic) can be calculated. ic equals to the product of the electrochemical double-layer capacitance (CDL) and the scan rate (ν) (as shown in eqn (2)). In this experiment, the scan rates increase from 5 mV s−1 to 500 mV s−1, keeping 10 s the potential vertex at each scan of the CV. Then Plotting ic as a function of ν will yield a straight line whose slope equals to CDL. By using the CDL to divide the specific capacitance (Cs), the electrochemically active surface area (ECSA) can be determined (eqn (3)). As reported, the Cs depends on the nature of material used and solution (for C, in 1 M H2SO4CS = 13–17 μF cm−2). The roughness factor (RF) is the ratio of ECSA to the geometrical surface area (S) of the RDE (eqn (4)).
| i = νCDL | (2) |
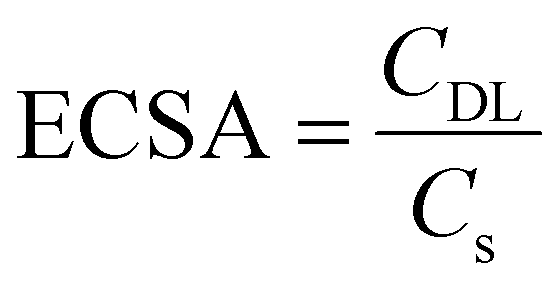
| (3) |
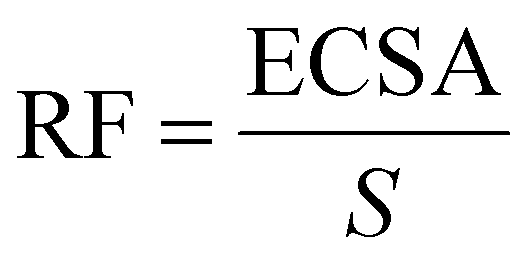
| (4) |
According to ECSA, specific current density (js) can be calculated as follows:
 | (5) |
Computational details
Models
Three nanoparticle models adapted from those existing in the literature9,52–55 were used to represent Pt@NH2-rGO, Ru@NH2-rGO and Pt1Ru5@NH2-rGO (Fig. 6a–c). Pt@NH2-rGO nanoparticles were modeled from the 1.2 nm large, truncated octahedron Pt79 model.54,55 The Ru@NH2-rGO one, with the Ru57 model previously used by some of us, which mainly exhibits (100) and (101) planes.9,52,53 Finally, the Pt1Ru5@NH2-rGO nanoparticles were represented with a cluster segregated Pt10Ru47 model derived from the Ru57 nanoparticle. This model correctly reproduces the experimentally observed nanoparticle size as well as the Pt–Pt, Ru–Ru and Ru–Pt contacts.Both experimental11 and computational9,46 previous contributions showed that more than one hydrogen atom per surface metal center is adsorbed at reaction conditions. However, the exact number of H varies depending on the metal model and the presence or absence of ligands. Therefore, the H content and distribution that should be included in the initial model are unknown. With the aim of determining the appropriate number of H as well as obtaining reasonable structures of the MXHY (MX = Ru57 or Pt79) nanoparticle models, we use a multi-step process consisting of Ab Initio Molecular Dynamics (AIMD) simulations of at least 1.5 ps for each system (see below) and geometry optimization of a representative snapshot of the AIMD simulation. In the first step, we added 1 H per surface metal center (monolayer, θ = 1) and performed the AIMD simulation. Then, we computed the mean adsorption energy of the hydrogen atoms of this monolayer at a representative snapshot of the simulation. Since this adsorption energy is larger than the ideal value suggested by Nørskov and co-workers,45 we increased the coverage (θ) by adding 0.25 H per metal center (Table S2†). We repeated this process several times until the mean adsorption energy of the newly added H atoms became close to the ideal value. It is however worth mentioning that, with the aim of enhancing the precision of our model, once the mean adsorption energy became closer to the ideal value, we also included structures resulting from adding 0.13 H per metal center. The final models in which we computed the hydrogen adsorption energy of additional H atoms are Pt79H105, Ru57H61 and Pt10Ru47H65 (Fig. 6d–f). The Pt79H105 and Ru57H61 models were selected because their H adsorption energy is the closest (but above) to the ideal value,45 while the Pt10Ru47H65 model is constructed by adding the average H atoms per surface Ru and surface Pt found in the two pure metallic systems. Analysis of the metal coordination number as well as the number of H atoms at a given metal center allowed to determine the different surface sites. To all these sites one additional H atom was adsorbed to determine the catalytic activity of each site at reaction conditions.
Since VASP enforces three-dimensional periodicity regardless of the nature of the system, the previously described nanoparticle models were set in a 25 (Ru57 and Pt10Ru47) or 30 (Pt79) Å edge large cubic box.
Level of theory
All calculations were performed with the PBE56 density functional as implemented in VASP.57,58 Dispersion forces are considered by adding Grimme's D2 empirical correction.59 Atomic cores are described with PAW pseudopotentials60,61 and the valence electrons are represented with a plane-wave basis set with an energy cutoff of 500 eV. All calculations were performed at Γ point due to the dimensionality of the model. The energy convergence criteria are fixed to 10−5 and 10−4 eV for electronic and geometry relaxations, respectively. This methodology is equivalent to that previously used by some of us when modeling other Ru-based materials for HER9,46 as well as IrO2 nanoparticles and their electrocatalytic activity.62,63H adsorption energies reported throughout the text are computed with respect to the reference hydrogenated nanoparticle model as proposed by Nørskov and co-workers45,64 as summarized in eqn (6):
| Eads = [EMXH(Y+1)·(EMXHY + 1/2·EH2)] | (6) |
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. S. thanks ICREA for the ICREA Academia Prize 2020. X. S. and J. G.-A. thank MINECO/FEDER (PID2019-104171RB-I00) and MICINN (PID2023-146787OB-I00) for financial support. L. M. M. P. also acknowledges the Junta de Andalucía (ProyExcel_00706) for financial support, as well as Grants PID2021-126080OA-I00, TED2021-132087A-I00 and CNS2023-145078 funded by MICIU/AEI/10.13039/501100011033 and by “ERDF/EU” and “European Union NextGenerationEU/PRTR. L. R.-S. and X. S.-M. acknowledge financial support from MICINN (PID2023-151738NB-I00). X-Ray Absoprtion experiments were performed at CLAESS beamline (experiment 2020084432) with the collaboration of ALBA staff. H. H. acknowledge the China Scholarships Council (No.201908440337) for her PhD grant.References
- N. S. Lewis, Research opportunities to advance solar energy utilization, Science, 2016, 351, aad1920 CrossRef PubMed.
- R. Eisenberg, Addressing the Challenge of Carbon-Free Energy, ACS Energy Lett., 2018, 3, 1521–1522 CrossRef CAS.
- L. Fan, Z. Tu and S. H. Chan, Recent development of hydrogen and fuel cell technologies: A review, Energy Rep., 2021, 7, 8421–8446 CrossRef.
- D. Guan, H. Xu, Q. Zhang, Y.-C. Huang, C. Shi, Y.-C. Chang, X. Xu, J. Tang, Y. Gu, C.-W. Pao, S.-C. Haw, J.-M. Chen, Z. Hu, M. Ni and Z. Shao, Identifying a Universal Activity Descriptor and a Unifying Mechanism Concept on Perovskite Oxides for Green Hydrogen Production, Adv. Mater., 2023, 35, 2305074 CrossRef CAS PubMed.
- S. Trasatti, Work function, electronegativity, and electrochemical behaviour of metals: III. Electrolytic hydrogen evolution in acid solutions, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- Y. Dang, T. Wu, H. Tan, J. Wang, C. Cui, P. Kerns, W. Zhao, L. Posada, L. Wen and S. L. Suib, Partially reduced Ru/RuO2 composites as efficient and pH-universal electrocatalysts for hydrogen evolution, Energy Environ. Sci., 2021, 14, 5433–5443 RSC.
- J. Peng, Y. Chen, K. Wang, Z. Tang and S. Chen, High-performance Ru-based electrocatalyst composed of Ru nanoparticles and Ru single atoms for hydrogen evolution reaction in alkaline solution, Int. J. Hydrogen Energy, 2020, 45, 18840–18849 CrossRef CAS.
- J. Creus, J. De Tovar, N. Romero, J. García-Antón, K. Philippot, R. Bofill and X. Sala, Ruthenium Nanoparticles for Catalytic Water Splitting, ChemSusChem, 2019, 12, 2493–2514 CrossRef CAS PubMed.
- N. Romero, D. A. Fenoll, L. Gil, S. Campos, J. Creus, G. Martí, J. Heras-Domingo, V. Collière, C. A. Mesa, S. Giménez, L. Francàs, L. Rodríguez-Santiago, X. Solans-Monfort, M. Sodupe, R. Bofill, K. Philippot, J. García-Antón and X. Sala, Ru-based nanoparticles supported on carbon nanotubes for electrocatalytic hydrogen evolution: structural and electronic effects, Inorg. Chem. Front., 2023, 10, 5885–5896 RSC.
- J. Creus, S. Drouet, S. Suriñach, P. Lecante, V. Collière, R. Poteau, K. Philippot, J. García-Antón and X. Sala, Ligand-Capped Ru Nanoparticles as Efficient Electrocatalyst for the Hydrogen Evolution Reaction, ACS Catal., 2018, 8, 11094–11102 CrossRef CAS.
- G. Martí, L. Mallón, N. Romero, L. Francàs, R. Bofill, K. Philippot, J. García-Antón and X. Sala, Surface-Functionalized Nanoparticles as Catalysts for Artificial Photosynthesis, Adv. Energy Mater., 2023, 13, 2300282 CrossRef.
- I. Álvarez-Prada, D. Peral, M. Song, J. Muñoz, N. Romero, L. Escriche, A. Acharjya, A. Thomas, R. Schomäcker, M. Schwarze, X. Sala, M. Tasbihi and J. García-Antón, Ruthenium nanoparticles supported on carbon-based nanoallotropes as co-catalyst to enhance the photocatalytic hydrogen evolution activity of carbon nitride, Renewable Energy, 2021, 168, 668–675 CrossRef.
- I. Álvarez-Prada, A. D. Nguyen, N. Romero, H. Hou, E. Benazzi, L. Escriche, A. Acharjya, A. Thomas, M. Schwarze, R. Schomäcker, X. Sala, M. Natali, J. García-Antón and M. Tasbihi, Insights into the light-driven hydrogen evolution reaction of mesoporous graphitic carbon nitride decorated with Pt or Ru nanoparticles, Dalton Trans., 2022, 51, 731–740 RSC.
- J. Creus, L. Mallón, N. Romero, R. Bofill, A. Moya, J. L. G. Fierro, R. Mas-Ballesté, X. Sala, K. Philippot and J. García-Antón, Ruthenium Nanoparticles Supported on Carbon Microfibers for Hydrogen Evolution Electrocatalysis, Eur. J. Inorg. Chem., 2019, 2019, 2071–2077 CrossRef CAS.
- L. Mallón, C. Cerezo-Navarrete, N. Romero, M. Puche, J. García-Antón, R. Bofill, K. Philippot, L. M. Martínez-Prieto and X. Sala, Ru nanoparticles supported on alginate-derived graphene as hybrid electrodes for the hydrogen evolution reaction, New J. Chem., 2022, 46, 49–56 RSC.
- J. Zhang, X. Qu, Y. Han, L. Shen, S. Yin, G. Li, Y. Jiang and S. Sun, Engineering PtRu bimetallic nanoparticles with adjustable alloying degree for methanol electrooxidation: Enhanced catalytic performance, Appl. Catal., B, 2020, 263, 118345 CrossRef CAS.
- P. Lara, T. Ayvalı, M.-J. Casanove, P. Lecante, A. Mayoral, P.-F. Fazzini, K. Philippot and B. Chaudret, On the influence of diphosphine ligands on the chemical order in small RuPt nanoparticles: combined structural and surface reactivity studies, Dalton Trans., 2012, 42, 372–382 RSC.
- J. Yang, J. Feng, Y. Cao, Y. Xiao, L. Qiao, K. An, J. Yang, J. Peng, H. Pan and H.-M. Cheng, Highly Dispersed Ru-Pt Heterogeneous Nanoparticles on Reduced Graphene Oxide for Efficient pH-Universal Hydrogen Evolution, Adv. Funct. Mater., 2024, 34, 2411081 CrossRef CAS.
- M. Tasbihi, F. Fresno, I. Álvarez-Prada, A. Acharjya, A. Thomas, L. Escriche, N. Romero and X. Sala, V. A. de la Peña O'Shea and J. García-Antón, A molecular approach to the synthesis of platinum-decorated mesoporous graphitic carbon nitride as selective CO2 reduction photocatalyst, J. CO2 Util., 2021, 50, 101574 CrossRef CAS.
- G. M. Tomboc, T. Kwon, J. Joo and K. Lee, High entropy alloy electrocatalysts: a critical assessment of fabrication and performance, J. Mater. Chem. A, 2020, 8, 14844–14862 RSC.
- M. Schnedlitz, D. Knez, M. Lasserus, F. Hofer, R. Fernández-Perea, A. W. Hauser, M. Pilar de Lara-Castells and W. E. Ernst, Thermally Induced Diffusion and Restructuring of Iron Triade (Fe, Co, Ni) Nanoparticles Passivated by Several Layers of Gold, J. Phys. Chem. C, 2020, 124, 16680–16688 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Recent Advances in Electrocatalytic Hydrogen Evolution Using Nanoparticles, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- K. Sharma, S. Kaushal, A. Jain, M. H. Sami, S. Kumar, H. Tariq, K. Bano, S. Aggarwal, R. Kumar and P. P. Singh, A comprehensive review on biogenic synthesis of bimetallic nanoparticles and their application as catalytic reduction of 4-nitrophenol, Chem. Pap., 2024, 78, 2757–2782 CrossRef CAS.
- R. Ghosh Chaudhuri and S. Paria, Core/Shell Nanoparticles: Classes, Properties, Synthesis Mechanisms, Characterization, and Applications, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- Q. Shao, P. Wang, S. Liu and X. Huang, Advanced engineering of core/shell nanostructures for electrochemical carbon dioxide reduction, J. Mater. Chem. A, 2019, 7, 20478–20493 RSC.
- P. Lara, M.-J. Casanove, P. Lecante, P.-F. Fazzini, K. Philippot and B. Chaudret, Segregation at a small scale: synthesis of core–shell bimetallic RuPt nanoparticles, characterization and solid state NMR studies, J. Mater. Chem., 2012, 22, 3578–3584 RSC.
- R. Ferrando, J. Jellinek and R. L. Johnston, Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- C. Amiens, D. Ciuculescu-Pradines and K. Philippot, Controlled metal nanostructures: Fertile ground for coordination chemists, Coord. Chem. Rev., 2016, 308, 409–432 CrossRef CAS.
- X. Qi, M. R. Axet, K. Philippot, P. Lecante and P. Serp, Seed-mediated synthesis of bimetallic ruthenium–platinum nanoparticles efficient in cinnamaldehyde selective hydrogenation, Dalton Trans., 2014, 43, 9283–9295 RSC.
- C. Pan, F. Dassenoy, M.-J. Casanove, K. Philippot, C. Amiens, P. Lecante, A. Mosset and B. Chaudret, A New Synthetic Method toward Bimetallic Ruthenium Platinum Nanoparticles; Composition Induced Structural Changes, J. Phys. Chem. B, 1999, 103, 10098–10101 CrossRef CAS.
- M. Lepron, M. Daniel-Bertrand, G. Mencia, B. Chaudret, S. Feuillastre and G. Pieters, Nanocatalyzed Hydrogen Isotope Exchange, Acc. Chem. Res., 2021, 54, 1465–1480 CrossRef CAS PubMed.
- X. Sun, X. Gao, J. Chen, X. Wang, H. Chang, B. Li, D. Song, J. Li, H. Li and N. Wang, Ultrasmall Ru Nanoparticles Highly Dispersed on Sulfur-Doped Graphene for HER with High Electrocatalytic Performance, ACS Appl. Mater. Interfaces, 2020, 12, 48591–48597 CrossRef CAS PubMed.
- Y. Li, Y. Luo, Z. Zhang, Q. Yu, C. Li, Q. Zhang, Z. Zheng, H. Liu, B. Liu and S. Dou, Implanting Ru nanoclusters into N-doped graphene for efficient alkaline hydrogen evolution, Carbon, 2021, 183, 362–367 CrossRef CAS.
- L. M. Martínez-Prieto, M. Puche, C. Cerezo-Navarrete and B. Chaudret, Uniform Ru nanoparticles on N-doped graphene for selective hydrogenation of fatty acids to alcohols, J. Catal., 2019, 377, 429–437 CrossRef.
- A. García-Zaragoza, C. Cerezo-Navarrete, P. Oña-Burgos and L. M. Martínez-Prieto, Boosting the catalytic performance of graphene-supported Pt nanoparticles via decorating with –SnBun: an efficient approach for aqueous hydrogenation of biomass-derived compounds, Nanoscale, 2023, 15, 12319–12332 RSC.
- V. B. Mohan, M. Nieuwoudt, K. Jayaraman and D. Bhattacharyya, Quantification and analysis of Raman spectra of graphene materials, Graphene Technol., 2017, 2, 47–62 CrossRef.
- V. B. Mohan, K. Jayaraman and D. Bhattacharyya, Brunauer–Emmett–Teller (BET) specific surface area analysis of different graphene materials: A comparison to their structural regularity and electrical properties, Solid State Commun., 2020, 320, 114004 CrossRef CAS.
- L. Mallón, J. Navarro-Ruiz, C. Cerezo-Navarrete, N. Romero, I. del Rosal, J. García-Antón, R. Bofill, L. M. Martínez-Prieto, K. Philippot, R. Poteau and X. Sala, Effect of Nitrogen and Phosphorus Doping of Reduced Graphene Oxide in the Hydrogen Evolution Catalytic Activity of Supported Ru Nanoparticles, ACS Appl. Mater. Interfaces, 2025, 17, 6198–6210 CrossRef PubMed.
- C. Cerezo-Navarrete, Y. Mathieu, M. Puche, C. Morales, P. Concepción, L. M. Martínez-Prieto and A. Corma, Controlling the selectivity of bimetallic platinum–ruthenium nanoparticles supported on N-doped graphene by adjusting their metal composition, Catal. Sci. Technol., 2021, 11, 494–505 RSC.
- L. Peng, E. Ringe, R. P. V. Duyne and L. D. Marks, Segregation in bimetallic nanoparticles, Phys. Chem. Chem. Phys., 2015, 17, 27940–27951 RSC.
- T.-Y. Jeon, K.-S. Lee, S. J. Yoo, Y.-H. Cho, S. H. Kang and Y.-E. Sung, Effect of Surface Segregation on the Methanol Oxidation Reaction in Carbon-Supported Pt−Ru Alloy Nanoparticles, Langmuir, 2010, 26, 9123–9129 CrossRef CAS PubMed.
- X. Wang, Y. Zhu, A. Vasileff, Y. Jiao, S. Chen, L. Song, B. Zheng, Y. Zheng and S.-Z. Qiao, Strain Effect in Bimetallic Electrocatalysts in the Hydrogen Evolution Reaction, ACS Energy Lett., 2018, 3, 1198–1204 CrossRef CAS.
- H. Prats and K. Chan, The determination of the HOR/HER reaction mechanism from experimental kinetic data, Phys. Chem. Chem. Phys., 2021, 23, 27150–27158 RSC.
- J. Zhang, Z. Xia and L. Dai, Carbon-based electrocatalysts for advanced energy conversion and storage, Sci. Adv., 2015, 1, e1500564 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- D. A. Fenoll, M. Sodupe and X. Solans-Monfort, Different role of ruthenium and platinum defective sites on the catalytic activity for the hydrogen evolution reaction, Catal. Today, 2024, 442, 114908 CrossRef CAS.
- R. Lipin, A. Ngoipala, R. L. Arevalo and M. Vandichel, Computational surface Pourbaix diagrams to unravel cathodic hydride formation on defective palladium surfaces, Int. J. Hydrogen Energy, 2024, 61, 460–472 CrossRef CAS.
- U. Caudillo-Flores, G. Agostini, C. Marini, A. Kubacka and M. Fernández-García, Hydrogen thermo-photo production using Ru/TiO2: Heat and light synergistic effects, Appl. Catal., B, 2019, 256, 117790 CrossRef.
- L. Simonelli, C. Marini, W. Olszewski, M. Ávila Pérez, N. Ramanan, G. Guilera, V. Cuartero and K. Klementiev, CLÆSS: The hard X-ray absorption beamline of the ALBA CELLS synchrotron, Cogent Phys., 2016, 3, 1231987 Search PubMed.
- D. Guan, G. Ryu, Z. Hu, J. Zhou, C.-L. Dong, Y.-C. Huang, K. Zhang, Y. Zhong, A. C. Komarek, M. Zhu, X. Wu, C.-W. Pao, C.-K. Chang, H.-J. Lin, C.-T. Chen, W. Zhou and Z. Shao, Utilizing ion leaching effects for achieving high oxygen-evolving performance on hybrid nanocomposite with self-optimized behaviors, Nat. Commun., 2020, 11, 3376 CrossRef CAS PubMed.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Moya, J. Creus, N. Romero, J. Alemán, X. Solans-Monfort, K. Philippot, J. García-Antón, X. Sala and R. Mas-Ballesté, Organocatalytic vs. Ru-based electrochemical hydrogenation of nitrobenzene in competition with the hydrogen evolution reaction, Dalton Trans., 2020, 49, 6446–6456 RSC.
- A. Comas-Vives, K. Furman, D. Gajan, M. C. Akatay, A. Lesage, F. H. Ribeiro and C. Copéret, Predictive morphology, stoichiometry and structure of surface species in supported Ru nanoparticles under H2 and CO atmospheres from combined experimental and DFT studies, Phys. Chem. Chem. Phys., 2016, 18, 1969–1979 RSC.
- F. Calle-Vallejo, The ABC of Generalized Coordination Numbers and Their Use as a Descriptor in Electrocatalysis, Adv. Sci., 2023, 10, 2207644 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- D. González, M. Sodupe, L. Rodríguez-Santiago and X. Solans-Monfort, Surface morphology controls water dissociation on hydrated IrO2 nanoparticles, Nanoscale, 2021, 13, 14480–14489 RSC.
- D. González, M. Sodupe, L. Rodríguez-Santiago and X. Solans-Monfort, Metal coordination determines the catalytic activity of IrO2 nanoparticles for the oxygen evolution reaction, J. Catal., 2022, 412, 78–86 CrossRef.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, Electrolysis of water on oxide surfaces, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5qi00451a |
| ‡ Joint first authors with equal contributions. |
| This journal is © the Partner Organisations 2025 |