
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,3-Dipolar cycloaddition reactions of triflatophosphanes to afford functionalized azaphospholium salts and azaphospholes†
Jannis
Fidelius
 a,
Kai
Schwedtmann
a,
Sebastian
Schellhammer
b,
Rongjuan
Huang
b,
Felix
Hennersdorf
a,
Moritz
Fink
a,
Jan
Haberstroh
a,
Antonio
Bauzá
c,
Antonio
Frontera
c,
Sebastian
Reineke
b and
Jan J.
Weigand
*a
a,
Kai
Schwedtmann
a,
Sebastian
Schellhammer
b,
Rongjuan
Huang
b,
Felix
Hennersdorf
a,
Moritz
Fink
a,
Jan
Haberstroh
a,
Antonio
Bauzá
c,
Antonio
Frontera
c,
Sebastian
Reineke
b and
Jan J.
Weigand
*a
aFaculty of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany. E-mail: jan.weigand@tu-dresden.de
bDresden Integrated Center for Applied Physics and Photonic Materials (IAPP) and Institute of Applied Physics, Technische Universität Dresden, 01069 Dresden, Germany
cDepartment of Chemistry, Universitat de Illes Balears, 07122 Palma de Mallorca, Spain
First published on 5th March 2025
Abstract
The advent of 1,3-dipolar cycloadditions in organic chemistry has introduced a powerful tool for constructing novel ring systems. However, methods for synthesizing inorganic, phosphorus-containing rings via 1,3-dipolar cycloadditions have remained scarce and are largely limited to the functionalization of phosphaalkynes. In this contribution, we describe the synthesis of 1,3-dipolar triflatophosphanes, demonstrating their ability to engage in (3 + 2)-cycloadditions with a variety of dipolarophiles. In addition to nitriles, (thio)-cyanates, iso-(thio)-cyanates, and (thio)-ketones, phosphaalkenes also exhibit reactivity, yielding a wide range of heteroatom-functionalized di- and triazaphospholium compounds. Density Functional Theory (DFT) calculations provide insight into the likely stepwise mechanism. Furthermore, the reduction of synthesized diazaphospholium salts affords neutral diazaphospholes, offering a novel route to this class of compounds. Importantly, we explored the photophysical properties of selected diazaphospholes that exhibit strong fluorescence with photoluminescence quantum yields of up to 37%, making them attractive candidates for applications in optoelectronics.
Introduction
1,3-Dipolar cycloaddition reactions are versatile and powerful tools in organic chemistry for the construction of heterocyclic ring systems with diverse structural motifs. These reactions involve the combination of a 1,3-dipolar compound with a suitable dipolarophile, resulting in the formation of novel ring structures. Among these, the (3 + 2)-cycloaddition, popularized by Huisgen1 and extensively explored by many others, has gathered significant attention.2 Two prominent variants of these “click” reactions are those of nitriles or alkynes with azides, leading to the formation of tri- or tetrazole derivatives.3 Beyond azides, numerous other dipolarophiles, such as nitrile oxides1 and diazoalkanes,4 have been studied, resulting in a wide array of heterocyclic ring systems. A specific subfield of interest is the synthesis of heterocyclic compounds containing phosphorus atoms, which has attracted considerable attention due to their wide-ranging applications in fields such as materials science5 and coordination chemistry.6 However, only a limited number of (3 + 2)-cycloaddition reactions have been reported for the synthesis of phosphorus-containing heterocycles (see Scheme 1). In line with the classical click reactions of nitriles and azides (Scheme 1, I), Regitz first described the functionalization of phosphaalkynes 1 with azides to afford triazaphospholes 2 – a method that has been further explored by Müller and colleagues (Scheme 1, II).7 Similarly, reactions with diazomethane derivatives yield diazaphospholes 3.8 Later, the same group demonstrated that diazaphospholes 4 could be obtained through analogue cycloadditions with azomethine ylides.9 Nishibayashi described a copper-catalyzed variation of these cycloadditions, where phosphaalkynes 1 react with isocyanoacetates to form azaphospholes 5.10 Recently, Wolf and Hansmann showed that diazaphospholes and diazadiphospholes could be obtained from the reaction of diazoalkenes with either phosphaalkynes 1 or P4.11 | ||
| Scheme 1 Synthetic approaches towards various heterocyclic ring systems; dipolar compounds are marked in red, dipolarophiles in blue. (I) Classical click reactions of alkynes or nitriles with azides; (II) diverse reactions of phosphaalkynes 1 to P/N-containing ring systems; (III) our previous work: dipolar imidazoliumyl-substituted triflatophosphanes reacting with alkynes to form azaphospholium salts, followed by subsequent reduction to 1,3-azaphospholes; (IV) the present work: expansion of our dipolar cycloaddition methodology with different compounds featuring carbon–heteroatom multiple bonds. | ||
In a remarkable reaction, the Cummins group demonstrated that in situ-formed phosphaethyne (H–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P) reacts with sodium azide to form the unsubstituted [HCPN3]− anion.12 In contrast to these results, few examples have been reported where the 1,3-dipolar compound, rather than the dipolarophile, features the phosphorus atom. One such example was recently described by our group, wherein 1,3-dipolar fluorophosphonium compounds react with small molecules to form novel ring systems.13 Additionally, Streubel reported on the in situ preparation of nitrilium phosphaneylide tungsten complexes, which react with alkynes to afford 1,2-azaphosphole tungsten complexes.14
P) reacts with sodium azide to form the unsubstituted [HCPN3]− anion.12 In contrast to these results, few examples have been reported where the 1,3-dipolar compound, rather than the dipolarophile, features the phosphorus atom. One such example was recently described by our group, wherein 1,3-dipolar fluorophosphonium compounds react with small molecules to form novel ring systems.13 Additionally, Streubel reported on the in situ preparation of nitrilium phosphaneylide tungsten complexes, which react with alkynes to afford 1,2-azaphosphole tungsten complexes.14
Recently, our group has reported on dipolar cycloadditions involving imidazoliumyl-substituted triflatophosphanes 6[OTf], which react with alkynes to form azaphospholium salts 7[OTf]2. These salts can be readily reduced to yield neutral azaphospholes 8 (Scheme 1, III), providing a novel approach for accessing this class of compounds.15 With this background in mind, we sought to explore the potential of our cycloaddition method with other dipolarophiles.
In this study, we expand our methodology to construct novel phosphorus-containing ring systems: triflatophosphanes, like 6[OTf], were found to react with nitriles and various other dipolarophiles, resulting in the formation of (poly)-azaphospholium salts of the type 9[OTf]2. These compounds hold great potential for reduction to their neutral phosphole counterparts, as demonstrated by the reduction of a model compound. Additionally, reactions involving compounds with carbon–heteroatom double bonds were found to yield partially saturated heterophospholium salts 10[OTf]2.
Results and discussion
1,3-Dipolar cycloaddition reactions with CN triple bonds
The formation of dipolar triflatophosphane 6[OTf] was achieved by reacting chlorophosphane 11[OTf] with silver triflate (AgOTf).15 Due to the limited stability of 6[OTf], we employed our in situ generation approach for this salt in the presence of the dipolarophile. Upon the addition of AgOTf to 11[OTf] in acetonitrile, a new signal emerged in the 31P NMR spectrum of the reaction solution at δ(P) = 21.6 ppm, indicating a clean cyclization reaction to diazaphospholium salt 9a[OTf]2 (see Scheme 2). | ||
| Scheme 2 Reaction of in situ prepared 6[OTf] with acetonitrile to afford diazaphospholium salt 9a[OTf]2. | ||
After filtration and removal of the solvent, 9a[OTf]2 was isolated in good yield of 81% as an analytically pure, colourless solid. Single crystals obtained from a saturated solution of 9a[OTf]2, were analyzed by single-crystal X-ray diffraction (SC-XRD). The molecular structure of 9a[OTf]2 is depicted in Fig. 1, confirming the structural connectivity. The molecular structure of 9a[OTf]2 reveals the expected distorted pyramidal bonding environment at the phosphorus atom. The P–C bond (1.8145(13) Å) is comparable to other known examples of imidazoliumyl-substituted phosphanes.16 While the imidazoliumyl fragment remains planar, the diazaphospholium moiety exhibits slight distortion, as evidenced by a torsion angle N2–C2–P1–N1 of 13.20(9)°. A limited π-electron delocalization in the diazaphospholium moiety is furthermore indicated by the N1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 bond length (1.2689(19) Å), characteristic of a CN double bond. In subsequent experiments, selected nitriles with different functional groups were used as substrates for the formation of diazaphospholium compounds 9a–f[OTf]2. The reaction yields and 31P NMR shifts are summarized in Scheme 3.
C3 bond length (1.2689(19) Å), characteristic of a CN double bond. In subsequent experiments, selected nitriles with different functional groups were used as substrates for the formation of diazaphospholium compounds 9a–f[OTf]2. The reaction yields and 31P NMR shifts are summarized in Scheme 3.
 | ||
| Fig. 1 Molecular structure of 9a2+ in 9a[OTf]2 (hydrogen atoms and counter ions are omitted for clarity; thermal ellipsoids are displayed at 50% probability). Selected bond lengths in Å and angles in (°): P1–C1 1.8145(13), P1–N1 1.7122(12), P1–C2 1.8199(14), N1–C3 1.2689(19), C3–N2 1.4606(18), N1–P1–C1 105.27(6), N1–P1–C2 91.72(6), C1–P1–C2 107.50(6), N2–C2–P1–N1 13.20(9). | ||
 | ||
| Scheme 3 Syntheses of 9a–e[OTf]2 and 9f[OTf]4 through reactions of in situ prepared 6[OTf] with various nitriles; 31P NMR chemical shift in ppm and yield in %. | ||
By elevating the reaction temperature to 40 °C, the reaction time for derivatives 9a–f[OTf] was significantly reduced. After workup, 9a–f[OTf]2 were obtained as colourless or red (9e[OTf]2) powders in moderate to excellent yields. Notably, the reactions afforded not only the simple methyl-substituted compound 9a[OTf]2 but also electron-deficient (9c[OTf]2) and electron-rich (9e[OTf]2) derivatives, with no significant changes in yields. However, in the reaction with propionitrile (yielding 9b[OTf]2), no increase in the yield could be achieved, likely due to the limited reactivity of propionitrile. Furthermore, when 1,4-phenyldiacetonitrile was used, the bridged derivative 9f[OTf]4 was obtained. The diazaphospholium compounds show 31P NMR chemical shifts between δ(P) = 21.6 to 27.7 ppm. Single crystals of most derivatives were obtained by the slow diffusion of Et2O into saturated MeCN solutions 9a–f[OTf]2. The molecular structures are given in the ESI (section 2.3†).
The bonding parameters were found to be comparable to those observed for 9a2+, regardless of the steric or electronic properties of the substituents.
To further expand the scope of the cycloadditions, 6[OTf] was reacted with phenylcyanate. The 31P NMR spectrum of the reaction solution revealed the formation of a new compound with a resonance at δ(P) = 14.5 ppm, slightly upfield-shifted compared to that of 9[OTf]2. Filtration and solvent evaporation yielded 12[OTf]2 in an excellent yield of 87%. When the same reaction was conducted using ethylthiocyanate, 13[OTf]2 was obtained in 85% yield (see Scheme 4). The 31P chemical shift of 13[OTf]2 (δ(P) = 27.9 ppm) is similar to those of diazaphospholium salts 9[OTf]2 (vide supra).
 | ||
| Scheme 4 Syntheses of 12[OTf]2 and 13[OTf]2 through the cycloaddition of in situ formed 6[OTf]2 with ethylthiocyanate or phenylcyanate. | ||
Single crystals of 12[OTf]2 were obtained by the slow diffusion of Et2O into a solution of 12[OTf]2 in CH2Cl2. The molecular structure of 122+ is depicted in Fig. 2, and in comparison with 9a2+, the C1–P1 (122+: 1.8263(14) Å; 9a2+: 1.8145(13) Å) and the P1–N1 (122+: 1.7125(13) Å; 9a2+: 1.7122(12) Å) bond lengths are similar. However, an elongation of the P1–C2 (122+: 1.8402(14) Å; 9a2+: 1.8199(14) Å) and a contraction of the N2–C3 (122+: 1.4444(18) Å; 9a2+: 1.4606(18) Å) bond lengths are observed. The C3–O1 (1.3212(18) Å) bond length in 122+ is considerably shorter than the O1–C4 (1.4167(17) Å) bond length, indicating delocalization involving the free lone pairs of the oxygen atom, consistent with the high-field shift observed in the 31P NMR spectrum.
 | ||
| Fig. 2 Molecular structure of 122+ in 12[OTf]2 (hydrogen atoms and counter ions are omitted for clarity; thermal ellipsoids are displayed at 50% probability). Selected bond lengths in Å and angles in (°): P1–C1 1.8263(14), P1–N1 1.7125(13), P1–C2 1.8402(14), N1–C3 1.2643(19), C3–N2 1.4444(18), C3–O1 1.3212(18), O1–C4 1.4167(17), N1–P1–C1 104.00(6), N1–P1–C2 91.73(6), C1–P1–C2 107.69(6), N2–C2–P1–N1 12.16(10). | ||
Chemical reductions of the diazaphospholium salts
In our previous work, we demonstrated the reduction of related azaphospholium salts to neutral azaphospholes under elimination of the parent NHC (see Scheme 1).15 Building on this, we investigated the reduction of diazaphospholium salt 9a[OTf]2 as a model compound, aiming to establish a convenient pathway for synthesizing a wide range of diazaphospholes.In order to gain a deeper understanding of its electrochemical reactivity, 9a[OTf]2 was subjected to electrochemical studies, including cyclic voltammetry (CV) and square wave voltammetry (SWV). The CV of 9a[OTf]2 in CH2Cl2 revealed a non-reversible reduction with a peak potential (CV) of Ep = −0.86 V and a SWV formal potential of E = −0.80 V (vs. E1/2(Cp2Fe/Cp2Fe+)). Further details are provided in the ESI.† Subsequently, we investigated the chemical reduction of 9a[OTf]2, using potassium graphite (KC8) as a reducing agent. Upon suspending a mixture of two equivalents of KC8 and 9a[OTf]2 in THF at −78 °C, the clean formation of the neutral diazaphosphole 14 was observed, indicated by a new resonance in the 31P NMR spectrum of the reaction mixture with a chemical shift of δ(P) = 150.4 ppm. After the removal of K[OTf] and NHC 15, 14 was isolated in 62% yield as a yellow crystalline solid (see Scheme 5). In order to gain further understanding of the influence that annulation and substitution have on the properties of these phospholes, we synthesized compounds 16 and 17 from the respective crude diazaphospholium salts 9g[OTf]2 and 31[OTf]2 (see Scheme 5). This demonstrates that, using our synthetic method, a similar range of derivatives is accessible as for the previously described 1,3-azaphospholes 8.
 | ||
| Scheme 5 Reduction of diazaphospholium salts 9a[OTf]2, 9g[OTf]2 and 18[OTf]2 to neutral diazaphosphole 14, with the elimination of NHC 15. | ||
It should be noted, however, that the reductions of 12[OTf]2 and 13[OTf]2 only resulted in the formation of trace amounts of the respective diazaphospholes (>10%); thus, we concluded that the bulk synthesis might not be suitable for this study. Single crystals of 16 were analysed with X-ray diffraction, and the molecular structure is depicted in Fig. 3. As expected, the overall bond lengths in the five-membered diazaphosphole ring are shortened (P1–N1 1.6883(18) Å, P1–C1 1.735(2) Å) compared to 9a2+ (P1–N1 1.7122(12) Å, P1–C2 1.8199(14) Å), reflecting an increased aromatic character. Additionally, the structural parameters of 16 are similar to those observed for the corresponding azaphosphole 8 (R = Ph and R′ = H).15
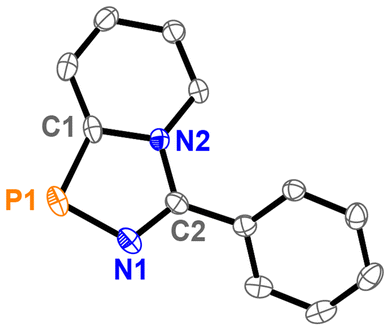 | ||
| Fig. 3 Molecular structure of 16 (hydrogen atoms are omitted for clarity; thermal ellipsoids are displayed at 50% probability). Selected bond lengths in Å and angles in (°): P1–C1 1.735(2), P1–N1 1.6883(18), C1–N2 1.404(2), N1–C2 1.310(3), C2–N2 1.387(3), N1–P1–C1 91.68(10), N1–C2–N2 115.21(16), C2–N1–P1–C1 0.91(19). | ||
Characterization of diazaphospholes 14, 16, and 17
To gain further insights into the electronic structures and photophysical properties of diazaphospholes 14, 16, and 17, we conducted quantum chemical simulations and spectroscopic measurements, similar to our recent studies of azaphospholes 8.15 The key characteristics are summarized in Table S1.† Both molecules 8 (R = Me, R′ = H) and 14 exhibit similar characteristics regarding their frontier molecular orbitals, aromaticity, and absorption/emission properties. Notably, the second nitrogen atom in the diazaphospholes stabilizes both the HOMO and LUMO by approximately 300 meV (Fig. 4a). Both HOMO and LUMO can be slightly shifted towards the gap through pronounced delocalization, as observed for 16, where the substitution of a phenyl group at the C2 position enhances this effect. For 17, benzannulation further increases this delocalization. This latter strategy results in the strongest modification of the aromaticity, as depicted in Fig. S114 in the ESI.† In general, both rings within 14 exhibit substantial aromatic character, as indicated by low nucleus-independent chemical shifts (NICS(1)zz) values of −36.4 for the five-membered ring and −15.6 for the six-membered ring, respectively. These values are similar to those observed for 1,3-azaphospholes 8 and also correlate with the observations made by Dostál et al. for 1,2-azaphospholes.15,17 The high degree of aromaticity in 14 is further supported by the anisotropy of the induced current density (ACID) plot, which reveals the delocalization of electron density across the bicyclic system. Although 16 shows a light decrease in aromaticity due to a pronounced delocalization of the electron density, benzannulation in 17 introduces another aromatic ring (NICS(1)zz = −27.5), though this reduces the aromaticity of the central ring (NICS(1)zz = −7.0). | ||
| Fig. 4 Electronic structure and photophysical properties of 14, 16, and 17 (5 wt% in a PMMA film). (a) Frontier molecular orbitals and the corresponding energies in a vacuum (CAM-B3LYP/6-311G**). (b) Normalized absorption spectra relative to the absorption maximum of 17. (c) Normalized emission spectra (λexc = 340 nm). | ||
Regarding their photophysical properties, the absorption and emission spectra of 14, 16, and 17 were recorded in chloroform (see Fig. S26†) and PMMA films at room temperature (Fig. 4b and c). In PMMA, diazaphosphole 14 shows two absorption peaks at 310 nm and 360 nm. In chloroform, the high-energy absorption peak splits into two absorption features. However, time-dependent density functional theory (TD-DFT, CAM-B3LYP/6-311G**) calculations show that this results from the electronic transition S0 → S2. The simulated spectra depicted in Fig. S115† support the experimental observations well. While the absorption spectra of 16 show strong similarities in both chloroform and PMMA film compared to 14, 17 reveals a pronounced absorption coefficient for the S0 → S1 transition around 350 nm.
In our previous study on 1,3-azaphospholes 8, room temperature phosphorescence (RTP) was observed for several derivatives.15 However, no such features were obtained for diazaphospholes 14, 16, and 17. These compounds show fluorescence with peaks in the range of 450 and 480 nm in PMMA (see Fig. 4c) similar to the fluorescence observed for previous 1,3-azaphospholes 8. The photoluminescence quantum yield (Φ) is significantly increased for diazaphospholes. Compound 14 shows superior results, with a quantum yield of 33.1% in chloroform and 37.0% in PMMA film (see ESI section 4†).
1,3-Dipolar cycloaddition reactions with carbon–heteroatom double bonds
Next, we aimed to explore the potential of reacting triflatophosphane 6[OTf] with various compounds containing carbon-heteroatom or carbon–carbon double bonds (Scheme 6). Initially, we tested reactions of 6[OTf] with alkenes and imines, similar to the other cycloaddition reactions described herein, but no reactions were observed. A comprehensive list of investigated dipolarophiles is provided in ESI section 2.7.† | ||
| Scheme 6 Reactions of in situ prepared 6[OTf] with dipolarophiles containing carbon–heteroatom double bonds. | ||
Upon addition of one equivalent of benzophenone to a solution of in situ prepared 6[OTf], the clean formation of the unusual oxazaphospholium salt 18a[OTf]2 was observed, marked by a broad resonance in the 31P NMR spectrum of the reaction mixture at δ(P) = 65.8 ppm. After filtration and solvent removal, 18a[OTf]2 was isolated in 76% yield as a colourless solid.
When 6[OTf] was reacted with acetone, the oxazaphosphole 18b[OTf]2 was obtained in 25% yield. The relatively low yield of 18b[OTf]2 can be attributed to the formation of side products, which were removed by several recrystallizations. However, when a thioketone such as thiobenzophenone is used, the thiazaphospholium salt 19[OTf]2 was obtained in 87% yield. Notably, reactions with carbonyl and thiocarbonyl compounds proved much faster than cycloadditions with nitriles, reaching full conversion in a matter of minutes. In comparison with 18a,b[OTf]2 (18a[OTf]2: δ(P) = 65.8 ppm; 18b[OTf]2: δ(P) = 71.8 ppm), 19[OTf]2 exhibits a considerably high-field shifted resonance in the 31P NMR spectrum at δ(P) = −15.1 ppm, likely due to the presence of a neighbouring sulfur atom. Using the MesPCPh2 phosphaalkene18 as the dipolarophile, we successfully obtained the unusual azadiphospholium salt 20[OTf]2 in an excellent yield of 88%. The 31P NMR spectrum of 20[OTf]2 shows two resonances as an AM spin system, with chemical shifts of δ(PA) = −51.0 ppm and δ(PM) = 29.7 ppm. The moderate 1JPP coupling constant of −170 Hz is attributed to the trans-orientation of the two neighbouring phosphorus atoms, leading to minimal interaction between their free electron pairs – a phenomenon well-documented in polyphosphanes.19
Surprisingly, we found that 20[OTf]2 is unstable in MeCN solutions, slowly converting into compound 9a[OTf] with the release of MesPCPh2. This slow reaction reached approximately 50% conversion after 10 days, with a reaction rate constant (k′) of 9.29 × 10−7 s−1 (see ESI section 2.7†).
We obtained X-ray quality crystals of compounds 18a[OTf]2, 19[OTf]2, and 20[OTf]2 for SC-XRD analysis. The molecular structures are depicted in Fig. 5. As expected, all three compounds exhibited a distorted pyramidal bonding environment around the central phosphorus atom. The P1–C1 bond lengths (18a2+: 1.8835(15) Å, 192+: 1.8245(12) Å, 202+: 1.8300(18) Å) are within the expected range for imidazoliumyl-substituted phosphanes, similar to those observed for 9a2+. However, 18a2+ shows a slight elongation of the P1–C1 bond, indicative of higher electron density at the phosphorus atom, likely due to a +M effect of the oxygen atom influencing the phosphole ring. The overall planarity of the azaphospholium moiety varied significantly between the compounds. While 18a2+ is near planar (torsion angle N1–C2–P1–O1 10.14(10)°), 192+ (N1–C2–P1–S1 27.41(8)°) and 202+ (N1–C2–P1–P2 30.00(9)) display substantial distortions. The elongation of the P2–C3 bond in 202+ (1.9610(17) Å) is likely due to steric repulsion between the mesityl substituent and the two phenyl groups. For comparison with the diazaphospholium salt 9a[OTf]2, we have also investigated the reduction of 18a[OTf]2, 19[OTf]2 and 20[OTf]2 with two equivalents of KC8, all resulting in the decomposition of the starting material.
 | ||
| Fig. 5 Molecular structures of 18a2+ in 18a[OTf]2·CH2Cl2, 192+ in 19[OTf]2 and 202+ in 20[OTf]2·2C2H4Cl2 (hydrogen atoms, solvent molecules, and counter ions are omitted for clarity; thermal ellipsoids are displayed at 50% probability). Selected bond lengths in Å and angles in (°): 18a2+: C1–P1 1.8835(15), P1–O1 1.6651(11), P1–C2 1.8413(14), C2–N1 1.3555(19), N1–C3 1.5230(17), O1–C3 1.4246(16), C1–P1–O1 102.49(6), C1–P1–C2 104.48(6), C2–P1–O1 88.43(6), N1–C2–P1–O1 10.14(10); 192+: C1–P1 1.8245(12), P1–S1 2.1328(5), P1–C2 1.8391(13), C2–N1 1.3553(16), N1–C3 1.5300(15), S1–C3 1.8393(13), C1–P1–S1 104.29(4), C1–P1–C2 98.44(5), C2–P1–S1 90.20(4), N1–C2–P1–S1 27.41(8); 202+: C1–P1 1.8300(18), P1–P2 2.2137(6), P1–C2 1.8322(18), C2–N1 1.358(2), N1–C3 1.514(2), P2–C3 1.9610(17), C1–P1–P2 106.01(6), C1–P1–C2 97.98(8), P2–P1–C2 88.98(6), N1–C2–P1–P2 30.00(9). | ||
Alteration of the N-donor substituent
Given the favourable applicability of our methodology, we sought to synthesize phospholium salts featuring a P–N bond in the nucleophilic moiety, based on previously reported pyrazolyl-substituted derivatives,20,21 enabling post-functionalization. Initially, we encountered challenges in the synthesis of 21[OTf], primarily due to rapid scrambling reactions, which led to the formation of dichloro-substituted compound 22[OTf] and dipyrazolyl-substituted compound 23[OTf], as outlined in Scheme 7. It was observed that an equilibrium between these three compounds rapidly formed in solution (see Fig. S1†). Additional details regarding the proposed mechanism and experiments are provided in the ESI.† Consequently, for the synthesis of the reactive species for 1,3-dipolar cycloaddition reactions, we employed a comproportionation approach. The addition of two equivalents of AgOTf to a mixture of 22[OTf] and 23[OTf] in CH2Cl2 led to the formation of two new resonances in the 31P NMR spectrum at δ(P) = 86.4 ppm and 34.7 ppm, corresponding to a dynamic equilibrium between triflatophosphane 24[OTf] and its dimer, the cyclic compound 25[OTf]4 (Scheme 7).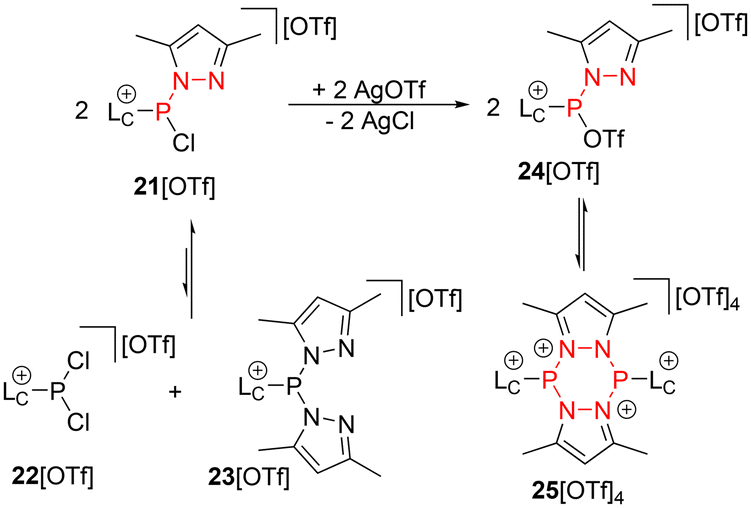 | ||
| Scheme 7 Synthesis of 21[OTf] through a comproportionation reaction of 22[OTf] and 23[OTf], followed by the addition of AgOTf, resulting in an equilibrium between 24[OTf] and its dimeric form, 25[OTf]4. | ||
Variable-temperature 31P NMR experiments revealed a reversible increase in the integration of the low-field resonance at δ(P) = 86.4 ppm, attributed to triflatophosphane 24[OTf] (see ESI section 2†). This finding suggests that the dimerization of 242+ in solution stabilizes the highly electrophilic phosphorus atom, resulting in the formation of dimer 254+, similar to previous observations.15 The equilibrium was confirmed by two correlation peaks observed in the corresponding 31P–31P-EXSY NMR experiment (Fig. S4†).
Single crystals suitable for X-ray analysis were obtained by the slow diffusion of n-pentane into a saturated CH2Cl2 solution of 24[OTf] at −30 °C. The molecular structure of 24+ shows the coordination of one triflate anion to the electrophilic phosphorus atom, confirming the formation of triflatophosphane 24+ (see Fig. 6). A pyramidal bonding environment around the phosphorus atom is observed, with the P–O bond length (P1–O1: 1.7250(11) Å) being significantly shorter than those in other triflatophosphanes, indicating a strong covalent character.22 This is supported by the elongation of the O–S bond (1.5540(11) Å) compared to the free triflate anion (av. 1.4314 Å). The P–C bond (24+: 1.8229(6) Å; 23+: 1.8438(13) Å (ref. 20)) and the P–N bond (24+: 1.6927(13) Å; 23+: 1.7171(11) Å (ref. 20)) are both shortened, indicating the high electrophilicity of the phosphorus atom.
 | ||
| Fig. 6 Molecular structure of cation 24+ in 24[OTf]·CH2Cl2 (hydrogen atoms, solvate molecules, and anions are omitted for clarity; thermal ellipsoids are displayed at 50% probability). Selected bond lengths in Å and angles in (°): P1–C1 1.8229(15), P1–O1 1.7250 (11), P1–N1 1.6927 (13), C1–P1–N1 99.73(6), C1–P1–O1 96.81(6), O1–P1–N1 100.52(6). | ||
Upon dissolving 24[OTf] in acetonitrile, a new resonance of the cycloaddition product, triazaphospholium salt 26a[OTf]2, appeared in the 31P NMR spectrum at δ(P) = 62.7 ppm. After filtration and drying in vacuo, 26a[OTf]2 was isolated in a good yield of 82%. The key to this successful synthesis was to first achieve the in situ formation of 24[OTf] in a three-component reaction from 22[OTf], 23[OTf], and AgOTf, followed by removal of the AgCl by-product and the subsequent addition of acetonitrile. Using this optimized method, we isolated the same range of derivatives previously described for 9[OTf]2 (Scheme 8). The yields were consistent, with most derivatives being isolated in approximately 80% yield. The reaction with propionitrile gave 26b[OTf]2 in lower amounts, due to lower reactivity, as observed previously for 9b[OTf]2. Additionally, both electron-rich (26e[OTf]2) and electron-poor (26c,d[OTf]2) derivatives are accessible.
 | ||
| Scheme 8 Syntheses of triazaphospholes 26a–f[OTf]2 through the cycloaddition of 23[OTf] with various nitriles; 31P NMR chemical shift in ppm, yield in %. | ||
The molecular structures of most of the derivatives were determined via SC-XRD analysis. However, due to similarities in the core unit, we focus here on the discussion of 26a[OTf]2. The molecular structure of 26a2+ (Fig. 7) reveals the expected distorted pyramidal bonding environment around the central phosphorus atom.
 | ||
| Fig. 7 Molecular structure of cation 26a2+ in 26a[OTf]2·CH2Cl2 (hydrogen atoms, solvate molecules, and anions are omitted for clarity; thermal ellipsoids are displayed at 50% probability). Selected bond lengths in Å and angles in (°): P1–C1 1.8306(16), P1–N1 1.7002(14), P1–N2 1.7757(14), N1–C2 1.277(2), C2–N3 1.427(2), N1–P1–N2 90.10(7), N1–P1–C1 102.66(7), C1–P1–N2 102.60(7), N2–P1–N1–C2 8.46(12). | ||
The P–C bond (1.8306(16) Å) is comparable to other imidazoliumyl-substituted phosphanes.16 While the pyrazolium fragment is planar, the five-membered triazaphosphole ring shows slight distortion (torsion angles in (°): N2–P1–N1–C2 8.46(12)), as is typical for such compounds.23 The low degree of π-electron delocalization in the triazaphosphole moiety is further highlighted by the bond lengths within the ring fragments. The P1–N2 bond (1.7757(14) Å) is elongated compared to 23+ (1.7171(11) Å (ref. 20)), while the pyrazolylium fragment appears aromatic. The N1C2 bond (1.277(2) Å) is consistent with a typical NC double bond. It should be noted that reductions, similar to that of 9a[OTf]2 were unfortunately unsuccessful for 26a[OTf]2.
Mechanistic insights
Given the significant polarization of the CN triple bond in nitriles, we aimed to revisit and broaden our understanding of the reaction mechanism involved in the (3 + 2)-cycloadditions of triflatophosphanes. In a previous study, we proposed that the dipolar cycloaddition reactions with alkynes (cf.Scheme 1, III) leading to compound 8 likely proceed via a concerted pathway.15 However, due to the distinct nucleophilicity of nitriles, attributed to the free electron pair on the terminal nitrogen atom, a step-wise mechanism may be more appropriate. To investigate the energy barriers for the reaction of 24[OTf] with acetonitrile, DFT calculations were performed at the PBE0-D3(COSMO)/def2-TZVP level of theory (Fig. 8). The results showed an overall Gibbs free energy of ΔG* = −17.0 kcal mol−1 for the formation of 26a[OTf]2. Notably, the calculations reveal that the previously proposed concerted transition state pathway involves a high energy barrier through TS′ (P–N 1.88 Å, C–N 2.05 Å). Alternatively, a stepwise mechanism appears more favourable, starting with the formation of an intermediate (INT) viaTS1 through nucleophilic substitution, with a low activation barrier of 2.7 kcal mol−1. This initial step is exergonic by 13.6 kcal mol−1. The subsequent ring closure proceeds through a second transition state (TS2) with a moderate activation barrier of 14.6 kcal mol−1, culminating in the formation of 26[OTf]2. Overall, these computational studies suggest that a stepwise reaction mechanism is energetically preferred when the dipolarophile features a nucleophilic moiety.
 | ||
| Fig. 8 Gibbs free energies (in kcal mol−1 at 298.15 K) of intermediates and transition states predicted for the (3 + 2)-cycloaddition of 24+ with acetonitrile; PBE0-D3/def2-TZVP optimized geometries and energies; distances in Å. | ||
1,3-Dipolar cycloadditions of 24[OTf] with carbonyl compounds and protic substrates
We found that 24[OTf] did not undergo selective reactions with various carbonyl compounds such as ketones and aldehydes, and attempts with alkynes were also unsuccessful (see ESI section 2.7†).However, when 24[OTf] was reacted with substrates like 4-bromphenylisocyanate, successful cycloaddition occurred, leading to the formation of triazaphospholium salt 27[OTf]2. Using naphthylisothiocyanate as the dipolarophile yielded thiatriazaphosphole 28[OTf]2 (Scheme 9). Notably, these reactions showed different regioselectivities as displayed in Scheme 9. In 282+, the isothiocyanate reacted with the CS double bond as the dipolarophile, while in 272+, the CN double bond participates in the ring formation. Single crystals of 27[OTf]2 and 28[OTf]2 were analysed by SC-XRD and the molecular structures of 272+ and 282+ are depicted in Fig. 9. 272+ shows a planar diazaphospholium ring with a comparable P–N bond length (272+: P1–N1 1.735(2)) as in 26a2+ (P1–N1 1.7002(14)). The N1–C2 bond length however is elongated (272+: N1–C2 1.370(3), 26a2+: N1–C2 1.277(2)) due to the limited electron delocalization in 272+. The molecular structure of 282+ is closely comparable to that of 192+, with a similar P–S bond length (282+: P1–S1 2.1212(8), 192+: P1–S1 2.1328(5)).
 | ||
| Scheme 9 Reactions of in situ prepared 24[OTf] with 4-bromphenylisocyanate and naphthylisothiocyanate. | ||
 | ||
| Fig. 9 Molecular structures of 272+ in 27[OTf]2·2CH2Cl2, and 282+ in 28[OTf]2. (hydrogen atoms, solvate molecules, and anions are omitted for clarity; thermal ellipsoids are displayed at 50% probability). Selected bond lengths in Å and angles in (°): 272+: P1–C1 1.819(3), P1–N1 1.735(2), P1–N2 1.765(2), N2–N3 1.363(3), N3–C2 1.433(3), N1–C2 1.370(3), C1–P1–N1 104.52(12), C1–P1–N2 101.74(12), N1–P1–N2 86.60(11), N3–N2–P1–N1 7.85(17); 282+: P1–C1 1.833(3), P1–S1 2.1212(8), P1–N1 1.786(3), N1–N2 1.372(3), N2–C2 1.428(4), C1–S1 1.781(3), C1–P1–N1 101.81(12), C1–P1–S1 106.20(8), N1–P1–S1 90.42(8), N2–N1–P1–S1 3.53(19). | ||
Conclusions
In this study, we successfully expanded our cycloaddition protocol to various dipolarophiles, demonstrating the versatility of our methodology. We showed that in situ generated 1,3-dipolar triflatophosphanes readily react with nitriles, forming dizaphospholium salts 9a–f[OTf]2. These azaphospholium salts can be reduced irreversibly to neutral diazaphospholes, as evidenced by electrochemical examinations. The reduction reactions with KC8 yielded diazaphospholes 14, 16, and 17, offering a novel synthetic pathway to access diazaphospholes. We also explored the photophysical properties of these compounds, comparing them with analogous azaphospholes. Additionally, we demonstrated the selectivity of our approach by employing (thio)ketones as dipolarophiles, leading to the formation of oxazaphospholium salts 18a,b[OTf]2 and the thiazaphospholium salt 19[OTf]2. Remarkably, using phosphaalkene MesPCPh2, we obtained the unusual azadiphospholium salt 20[OTf]2.
Furthermore, by substituting the nucleophilic moiety from pyridine to pyrazole, triflatophosphane 24[OTf] was synthesized and structurally characterized by SC-XRD. This compound exhibits similar reactivity towards nitriles, yielding triazaphospholium salts 26a–f[OTf]2. Quantum chemical calculations revealed that the reaction mechanism is step-wise, in contrast to the concerted mechanism observed with alkynes. While 24[OTf] was unreactive towards (thio)ketones, it readily underwent a cycloaddition reaction with iso(thio)cyanates, demonstrating different regioselectivity depending on the chosen dipolarophile.
In summary, the cycloaddition scheme presented here serves as a strong foundation for further development of 1,3-dipolar cycloaddition reactions using phosphorus-based dipoles, a class with few known examples. This work opens new avenues for the synthesis of diverse heterocyclic ring systems, expanding the synthetic chemistry toolbox.
Author contributions
J. F., K. S., and J. J. W. conceptualized the study; J. F. conducted the experiments and optimized the syntheses, isolations, and purifications; M. F. assisted with the syntheses; J. H. conducted the electrochemical investigation; A. B. and A. F. were responsible for mechanistic studies; S. S. conducted the material simulations; R. H. conducted spectroscopic measurements; J. F., F. H., and J. J. W. were responsible for X-ray data collection and refinement; K. S. and J. J. W. conceived, oversaw, and directed the project; J. F., K. S., and J. J. W. prepared the initial draft of the paper; A. F., S. R., and J. J. W. secured funding. All authors contributed to data analysis, manuscript editing, and discussion.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre under the deposition number 2406453–2406470.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Science Foundation (DFG, WE 4621/6-1) and the European Union (R.H. acknowledges funding through project PNIRED, no. 101068895). A.F. thanks the MICIU/AEI of Spain (project PID2020-115637GB-I00 FEDER funds) for financial support. J.F. thanks the Graduate Academy of TU Dresden for financial support. S.S. thanks the Center for Information Services and High Performance Computing (ZIH) at TU Dresden for the use of computational facilities. Philipp Lange is acknowledged for performing elemental analyses. TU Dresden is also thanked for financial support.References
- R. Huisgen, 1,3-Dipolar Cycloadditions. Past and Future, Angew. Chem., Int. Ed. Engl., 1963, 2, 565 CrossRef.
- (a) M. Breugst and H.-U. Reissig, The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition, Angew. Chem., Int. Ed., 2020, 59, 12293 CrossRef CAS PubMed; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- (a) D. A. Bilodeau, K. D. Margison, M. Serhan and J. P. Pezacki, Bioorthogonal Reactions Utilizing Nitrones as Versitle Dipoles in Cycloaddition Reactions, Chem. Rev., 2021, 121, 6699 CrossRef CAS PubMed; (b) D. Cantillo, B. Gutmann and C. O. Kappe, Mechanistic Insights on Azide-Nitrile Cycloadditions: On the Dialkyltin Oxide-Trimethylsilyl Azide Route and a New Vilsmeier-Haack-Type Organocatalyst, J. Am. Chem. Soc., 2011, 133, 4465 CrossRef CAS PubMed; (c) T. Deb, J. Tu and R. M. Franzini, Mechanisms and Substituent Effects of Metal-Free Bioorthogonal Reactions, Chem. Rev., 2021, 121, 6850 CrossRef CAS PubMed; (d) Z. P. Demko and K. B. Sharpless, A Click Chemistry Approach to Tetrazoles by Huisgen 1,3-Dipolar Cycloaddition: Synthesis of 5-Sulfonyl Tetrazoles from Azides and Sulfonyl Cyanides, Angew. Chem., Int. Ed., 2002, 41, 2110 CrossRef CAS; (e) R. Huisgen, G. Szeimies and L. Möbius, 1.3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Merfachbindungen, Chem. Ber., 1967, 100, 2494 CrossRef CAS; (f) G. S. Kumar and Q. Lin, Light-Triggered Click Chemistry, Chem. Rev., 2021, 121, 6991 CrossRef CAS PubMed; (g) J. C. Jewett and C. R. Bertozzi, Cu-free click cycloaddition reactions in chemical biology, Chem. Soc. Rev., 2010, 39, 1272 RSC; (h) M. Meldal and C. W. Tornøe, Cu-Catalyzed Azide-Alkyne Cycloaddition, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (i) C. G. Neochoritis, T. Zhao and A. Dömling, Tetrazoles via Multicomponent Reactions, Chem. Rev., 2019, 119, 1970 CrossRef CAS PubMed; (j) K. Porte, M. Riomet, C. Figliola, D. Audisio and F. Taran, Click and Bio-Orthogonal Reactions with Mesoionic Compounds, Chem. Rev., 2021, 121, 6718 CrossRef CAS PubMed; (k) K. Li, D. Fong, E. Meichsner and A. Adronov, A Survey of Strain-Promoted Azide-Alkyne Cycloaddition in Polymer Chemistry, Chem. – Eur. J., 2021, 27, 5057 CrossRef CAS PubMed.
- (a) T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, Rhodium-Catalyzed Transannulation of 1,2,3-Triazoles with Nitriles, J. Am. Chem. Soc., 2008, 130, 14972 CrossRef CAS PubMed; (b) H. M. Davies and K. R. Romines, Directs synthesis of furans by 3 + 2 cycloaddtions between rhodium(II) acetate stabilized carbenoids and acetylenes, Tetrahedron, 1988, 44, 3343 CrossRef CAS.
- (a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications, Chem. Rev., 2017, 117, 3479 CrossRef PubMed; (b) M. P. Duffy, W. Delaunay, P.-A. Bouit and M. Hissler, π-Conjugated phospholes and thier incorporation into devices: components with a great deal of potential, Chem. Soc. Rev., 2016, 45, 5296 RSC.
- R. K. Bansal, K. Karaghiosoff and A. Schmidpeter, Anellated heterophospholes, Tetrahedron, 1994, 50, 7675 CrossRef CAS.
- (a) E. Yue, L. Dettling, J. A. W. Sklorz, S. Kaiser, M. Weber and C. Müller, Au(I)-mediated N2-elimination from triazaphospholes: a one-pot synthesis of novel N2P2-heterocycles, Chem. Commun., 2021, 58, 310 RSC; (b) L. Dettling, M. Papke, J. A. W. Sklorz, D. Buzsáki, Z. Kelemen, M. Weber, L. Nyulászi and C. Müller, A new access to diazaphospholes via cycloaddition cycloreversion reactions on triazaphospholes, Chem. Commun., 2022, 58, 7745 RSC.
- W. Rösch and M. Regitz, [3 + 2]-Cycloaddition Reactions of a Stable Phosphaalkyne-Transition from Singly and Doubly Coordinated Phosphorus, Angew. Chem., Int. Ed. Engl., 1984, 23, 900 CrossRef.
- U. Bergsträßer, A. Hoffmann and M. Regitz, A new access to phosphaindolizines by [3 + 2] cycloaddition of azomethine ylides onto phosphaalkynes, Tetrahedron Lett., 1992, 33, 1049 CrossRef.
- W. Liang, K. Nakajima, K. Sakata and Y. Nishibayashi, Copper-Catalyzed [3 + 2] Cycloaddition Reactions of Isocyanoacetates with Phosphaalkynes to Prepare 1,3-Azaphospholes, Angew. Chem., Int. Ed., 2019, 58, 1168 CrossRef CAS PubMed.
- S. Hauer, J. Reitz, T. Koike, M. M. Hansmann and R. Wolf, Cycloadditions of Diazoalkenes with P4 and tBuCP: Access to Diazaphospholes, Angew. Chem., Int. Ed., 2024, e202410107 Search PubMed.
- W. J. Transue, A. Velian, M. Nava, M.-A. Martin-Drumel, C. C. Womack, J. Jiang, G.-L. Hou, X.-B. Wang, M. C. McCarthy, R. W. Field and C. C. Cummins, A Molecular Precursor to Phosphaethyne and Ist Application in Synthesis of the Aromatic 1,2,3,4-Phostriazolate Anion, J. Am. Chem. Soc., 2016, 138, 6731 CrossRef CAS PubMed.
- C.-X. Guo, K. Schwedtmann, J. Fidelius, F. Hennersdorf, A. Dickschat, A. Bauzá, A. Frontera and J. J. Weigand, Bifunctional Fluorophosphonium Triflates as Intramolecular Frustrated Lewis Pairs: Reversible CO2 Sequestration and Binding of Carbonyls, Nitriles and Acetylenes, Chem. – Eur. J., 2021, 27, 13709 CrossRef CAS PubMed.
- R. Streubel, H. Wilkens, A. Ostrowski, C. Neumann, F. Ruthe and P. G. Jones, Formation of 2H-1,2-Azaphosphole Tungsten Complexes by Trapping Reactions of Nitrilium Phosphane Ylide Complexes, Angew. Chem., Int. Ed. Engl., 1997, 36, 1492 CrossRef CAS.
- J. Fidelius, K. Schwedtmann, S. Schellhammer, J. Haberstroh, S. Schulz, R. Huang, M. C. Klotzsche, A. Bauzá, A. Frontera, S. Reineke and J. J. Weigand, Convenient access to p-conjugated 1,3-azaphospholes from alkynes via [3 + 2]-cycloaddition and reductive aromatization, Chem, 2024, 10, 644 CAS.
- J. J. Weigand, K.-O. Feldmann and F. D. Henne, Carbene-Stabilzed Phosphorus(III)-Centered Cations [LPX2]+ and [L2PX]2+ (L = NHC; X = Cl, CN, N3), J. Am. Chem. Soc., 2010, 132, 16321 CrossRef CAS PubMed.
- V. Kremláček, J. Hyvl, W. Y. Yoshida, A. Růžička, A. L. Rheingold, J. Turek, R. P. Hughes, L. Dostál and M. F. Cain, Heterocycles Derived from Generating Monovalent Pnictogens within NCN Pincers and Bidentate NC Chelates: Hypervalency vs. Bell-Clappers vs. Static Organometallics, Organometallics, 2018, 37, 2481 CrossRef.
- P. Royla, K. Schwedtmann, Z. Han, J. Fidelius, D. P. Gates, R. M. Gomila, A. Frontera and J. J. Weigand, Cationic Phosphinidene as a Versatile P1 Building Block: [LC-P]+ Transfer from Phosphonio-Phosphanides [LC-P-PR3]+ and Subsequent LC Replacement Reactions (LC = N-Heterocyclic Carbene), J. Am. Chem. Soc., 2023, 145, 10364 CrossRef CAS PubMed.
- M. Baudler, G. Reuschenbach, D. Koch and B. Carlsohn, Beiträge zur Chemie des Phosphors, 88. 1,2,3-Triphenyl-1,3-bis(trimethylsilyl)triphosphan, Chem. Ber., 1980, 113, 1264 CrossRef CAS.
- C. Taube, K. Schwedtmann, M. Noikham, E. Somsook, F. Hennersdorf, R. Wolf and J. J. Weigand, P-P Condensation and P-N/P-P Bond Metathesis: Facile Synthesis of Cationic Tri- and Tetraphosphanes, Angew. Chem., Int. Ed., 2020, 59, 3585 CrossRef CAS PubMed.
- K.-O. Feldmann and J. J. Weigand, P-N/P-P bond metathesis for the synthesis of complex polyphosphanes, J. Am. Chem. Soc., 2012, 134, 15443 CrossRef CAS PubMed.
- (a) C. A. Caputo, J. T. Price, M. C. Jennings, R. McDonald and N. D. Jones, N-Heterocyclic phosphenium cations: synthesis and cycloaddition reactions, Dalton Trans., 2008, 3461 RSC; (b) A. Dumitrescu, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, A Crystalline Phosphenium Salt Featuring the Electron-Withdrawing 2,6-Bis(trifluoromethyl)phenyl Group, Eur. J. Inorg. Chem., 2002, 1953 CrossRef CAS; (c) S. Yogendra, F. Hennersdorf, A. Bauzá, A. Frontera, R. Fischer and J. J. Weigand, Carbodiphosphorane mediated synthesis of a triflyloxyphosphonium dication and its reactivity towards nucleophiles, Chem. Commun., 2017, 53, 2954 RSC.
- (a) R. Szűcs, P.-A. Bouit, M. Hissler and L. Nyulászi, Edge modification of PAHs: the effect of embedded heterocycles on the aromaticity pattern, Struct. Chem., 2015, 26, 1351 CrossRef; (b) L. Nyulaszi, T. Veszpremi, J. Reffy, B. Burkhardt and M. Regitz, Electronic structure and aromaticity of azaphospholes, J. Am. Chem. Soc., 1992, 114, 9080 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data and details on calculations. CCDC 2406453–2406470. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qi00427f |
| This journal is © the Partner Organisations 2025 |