
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precursor design for additive manufacturing of ceramics through hydrogel infusion†
Natalie S.
Yaw
ab,
Maryline G.
Ferrier
 *a,
R. Joey
Griffiths
a,
Joshua A.
Hammons
a,
Andrew J.
Swift
a,
Silvina A.
Di Pietro
a,
Jean-Baptiste
Forien
a,
Bradley C.
Childs
a,
Aiden A.
Martin
*a,
Kiel S.
Holliday
a,
Xiaofeng
Guo
b and
Jason R.
Jeffries
a
*a,
R. Joey
Griffiths
a,
Joshua A.
Hammons
a,
Andrew J.
Swift
a,
Silvina A.
Di Pietro
a,
Jean-Baptiste
Forien
a,
Bradley C.
Childs
a,
Aiden A.
Martin
*a,
Kiel S.
Holliday
a,
Xiaofeng
Guo
b and
Jason R.
Jeffries
a
aPhysical and Life Sciences Directorate, Lawrence Livermore National Laboratory, 7000 East Avenue, Livermore, CA 94550, USA. E-mail: kerlin4@llnl.gov; martin248@llnl.gov
bDepartment of Chemistry, Washington State University, Pullman, WA 99163, USA
First published on 10th March 2025
Abstract
Hydrogel-infused additive manufacturing (HIAM) is an emerging technique for the additive manufacturing of ceramics and metals. Distinct from slurry- or powder-based techniques, a hydrogel scaffold is obtained in the desired shape, infused with aqueous metal cations, and subsequently calcined to remove all organic components. This study demonstrates that both organic (hydrogel scaffold formulations) and inorganic (metal salts) precursors shape the quality and morphology of the final ceramic piece. Cu, Ce, Zr, and U oxide-ceramic disks were prepared via HIAM and studied using simultaneous thermal analysis, scanning electron microscopy, X-ray computed tomography, and small-angle X-ray scattering. Hydrogel formulations were found to impact the porosity of the resultant ceramics, with concentrated formulations generally yielding ceramics with a less cracked macrostructure. We hypothesize that this is due to the resulting variation in cation infusion into the matrix. The choice of inorganic salts also influences the morphology and porosity, likely due to the specific cation–polymer interactions and the energetic differences in decomposition pathways upon calcination. In general, chloride salts lead to denser microstructures than nitrate salts with some layer or foam-like macrostructures, while oxo-cations yield denser microstructures and macrostructures when compared to bare (monoatomic) cations. These results demonstrate that the HIAM process can be tailored to deliver a wide range of ceramics successfully, provided precursor feedstocks are adequately optimized.
Introduction
Ceramic materials are widely utilized due to their favorable properties, including high thermal stability, chemical inertness, and mechanical hardness.1 The ability to additively manufacture (AM) complex shapes from these materials is highly desirable for applications ranging from dental to aerospace.2,3 However, specific challenges remain with regard to the AM of ceramic materials in comparison with their metal or polymer counterparts.4 Slurry-based techniques suffer from homogeneity, viscosity, and light permeability challenges, while powder-based techniques can suffer from high porosity, thermal distortions, and the need for intensive optimization of processing parameters.5–9 Nevertheless, the commercial demand for these ceramic parts continues to push innovative research in this field.An emerging AM technique that could improve some of these processing concerns is hydrogel-infused additive manufacturing (HIAM). Yee et al. first reported the AM of lithium cobalt oxide structures using a homogeneous photocurable resin that contained metal nitrate salts dissolved in a solution of poly(ethylene glycol) diacrylate (PEGda).10 Upon curing, the metal salts were trapped in the hydrogel in the desired shape, and during the calcination step, they were transformed to the corresponding micro-architected oxide ceramics without any organic residue. Expanding upon this report, Saccone et al. improved the flexibility of HIAM further by first printing the organic polymer in the desired shape and then infusing the metal salts in a separate step, followed by calcination into the corresponding ceramic part.11 In this way, the act of 3D-printing has been separated from the ceramic material, allowing HIAM to sidestep many of the challenges associated with traditional AM techniques, given the well-studied and facile 3D-printing of hydrogel materials.12–14 While several follow-up studies have confirmed the utility and flexibility of the HIAM approach, a systematic study of the impact of chemical precursors on the final printed pieces is still lacking.15–17
To fully exploit HIAM and rationally design complex ceramic parts, a robust understanding of the precursor feedstocks must be developed. The precursors for the HIAM technique can be divided into two broad categories: the organic component (the polymer scaffold composing the hydrogel, such as PEGda) and the inorganic component (the metal salts selected for infusion). Interactions between these two components in the aqueous medium need to be studied, as this will inform how fully and homogeneously the cation of interest will infiltrate the scaffold and thus dictate the quality of the final ceramic piece. Previous studies, particularly in biomedical and environmental fields where PEGda hydrogels are widely utilized, demonstrated that the interactions between metal cations, water, and PEGda are extremely complex. For example, the behavior of adsorbed water is highly dependent on the physical properties, dissolved ions, and pore structure of the hydrogel. Some portion of this absorbed water can coordinate to the polymer, while the remaining ‘bulk’ water remains free in the pores, resulting in two distinct classes of water.18–21 Cation infusion into hydrogel matrices is similarly complex and is not merely controlled by diffusion but is influenced by cation properties, as well as the interactions with water and/or polymer moieties.22–25 These studies have noted the ability of PEGda hydrogels to discriminate between cations, evidencing the interaction between cations and the polymer scaffold.
Herein, we studied the impact of both organic and inorganic precursors on the formation of ceramic oxides using the HIAM approach. Nitrate and chloride salts, including both bare (i.e., monoatomic) and oxo-cations, were compared for various metals, ranging from transition metals to f-block elements. The copper system, an already studied metal in HIAM, is compared with various metals to probe different chemistry types. The physical characteristics, such as viscosity and cross-linking, of the hydrogel were altered by diluting or concentrating the PEGda resin before curing. Scanning electron microscopy (SEM) and X-ray computed tomography (XCT) techniques were used to study the characteristic structures of the resulting ceramic oxides. Simultaneous thermal analysis (STA), consisting of thermogravimetric analysis (TGA) coupled with differential scanning calorimetry (DSC), was used to study the decomposition of hydrogels and inorganic salts in situ. Ultra-small angle X-ray scattering (USAXS) was used to study the pore structure of hydrogels and ceramics and small angle X-ray scattering (SAXS) was used to study the nanoscale structure within the hydrogels. We found that both inorganic and organic precursors impact the final quality of the ceramic piece (i.e., density, robustness, percentage of shape change, etc.), although element-specific effects cannot be discounted and will be further discussed.
Experimental methods
General synthetic considerations
The reagents were purchased from commercial suppliers. Caution: depleted uranium (238U) is a weak α-emitter with a half-life of 4.47 × 109 years. All the manipulations were carried out in a monitored radiation laboratory.Synthesis of hydrogels
Synthesis of hydrogels was adapted from Saccone et al.11Fig. 1 illustrates the components involved and the procedure. In designing the photoresin which would be cured into the hydrogel scaffold, several material constraints had to be considered. The hydrogel needed to be unreactive towards a variety of metal cations, sufficiently porous to allow cation infusion, mechanically stable after curing, and leave no impurities behind in the ceramic after combustion. For this reason, resin components were chosen strategically to include only carbon, nitrogen, and oxygen (Fig. 1), based on the formulation proposed by Saccone et al., which uses a UV-active molecule that does not contain either phosphine or alkali metals.11 A stock solution was generated by weighing Irgacure 379 (also called Omnirad 379, 2-dimethylamino-2-(4-methyl-benzyl)-1-(4-morpholin-4-yl-phenyl)-butan-1-one, 347 mg, photoinitiator), Michler's ketone (bis[4-(dimethylamino)phenyl]methanone, 229 mg, sensitizer), and Sudan I (1-(phenyldiazenyl)naphthalen-2-ol, 10.3 mg, UV blocker) into a Nalgene bottle, followed by DMF (N,N-dimethylformamide, 35 mL, solvent) and PEGda (poly(ethylene glycol) diacrylate, 35 mL, monomer). The bottle was shaken vigorously until a viscous transparent orange solution was obtained, giving the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 polymer:solvent resin. For the more concentrated resin, an aliquot of this standard resin was taken, and mixed with an equal volume of a polymer to obtain a 2:1 polymer:solvent ratio. For the more dilute resin, an aliquot of this standard resin was taken and diluted with an equal volume of DMF, resulting in a 1:2 polymer:solvent ratio.
:1 polymer:solvent resin. For the more concentrated resin, an aliquot of this standard resin was taken, and mixed with an equal volume of a polymer to obtain a 2:1 polymer:solvent ratio. For the more dilute resin, an aliquot of this standard resin was taken and diluted with an equal volume of DMF, resulting in a 1:2 polymer:solvent ratio.
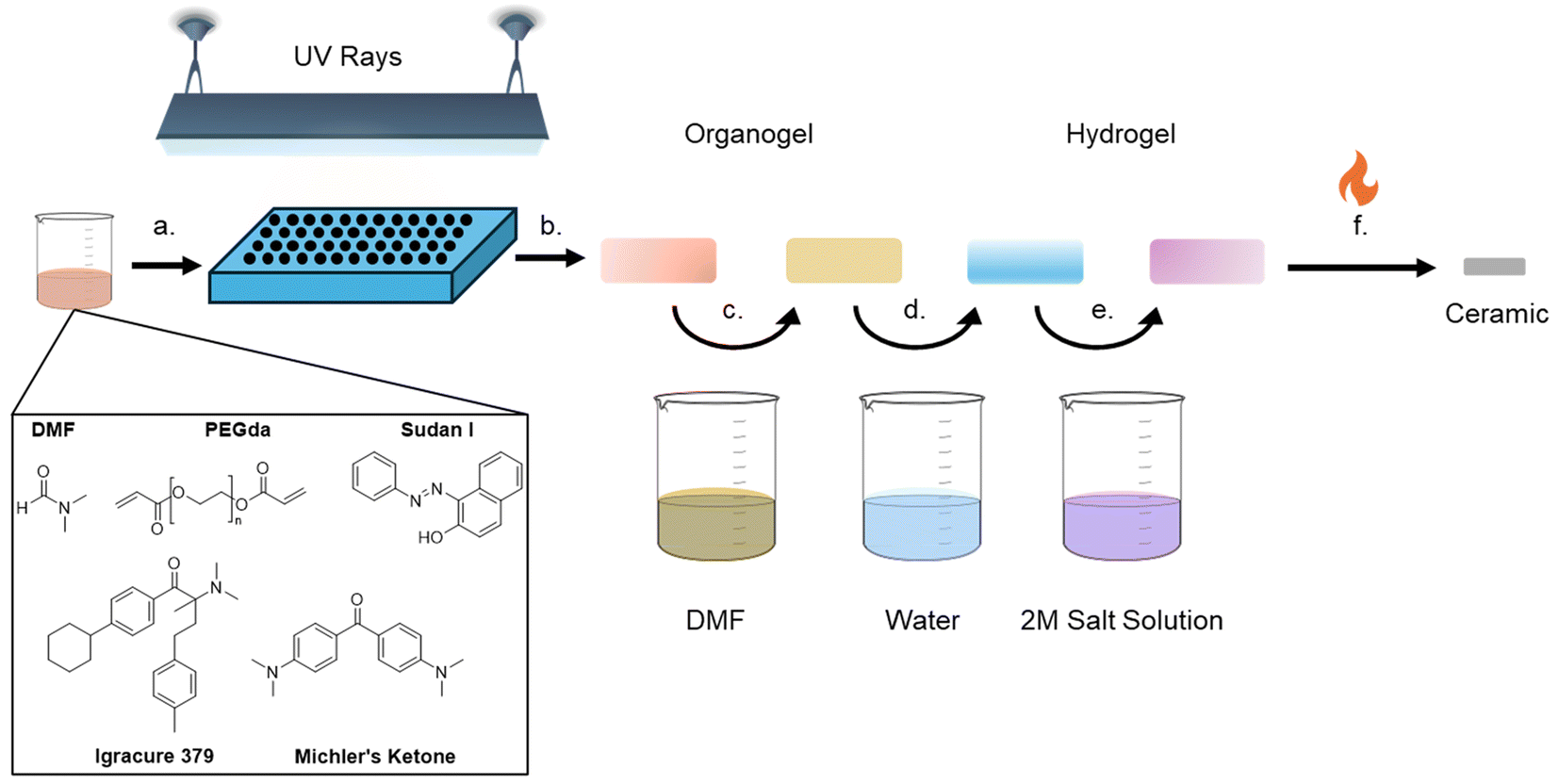 | ||
| Fig. 1 Schematic describing the hydrogel-infused additive manufacturing (HIAM) procedure. (a) Resin is synthesized by mixing various organic components to obtain a viscous bright orange solution. (b) Resin solution is pipetted into molds and then UV-cured to form clear yellow organogel disks. (c) Organogels are rinsed with DMF to remove excess reactants for an hour at 70 °C, and the process was repeated once more. (d) Organogels are transformed into hydrogels by submerging them in water at 70 °C for 1 hour. (e) Hydrogels are infused with the desired metal salt by immersing them in the aqueous solution at 70 °C for 24 hours. (f) Hydrogels are calcined to remove organic components, and the ceramic disks remained in shape. | ||
While dilution of the resin might seem trivial, the viscosity of a resin is an important parameter for many AM technologies.26,27 Therefore, it is important to assess to what extent varying this formulation might impact the integrity of the cured hydrogels and the resultant ceramics, as engineering constraints might necessitate flexibility. One difference from popular recipes is that the UV active molecules are no longer water soluble, thus the formation of an organogel in DMF followed by subsequent transformation into a hydrogel is required. Acetonitrile was explored as an alternative to DMF, given the health hazards of the latter. While altering the original solvent had little impact on the final ceramic piece, the organogels and hydrogels took longer to cure and were far more difficult to handle. Ultimately, DMF was selected for further study as it was easier to handle and widely utilized, but the potential for optimization with safer, greener solvents in the future is possible.
The solution was subsequently added to a mold and UV-cured using a 3D printing wash and cure station (ELGOO Mercury Plus 2 in 1, Version 2 Model). Two sizes of hydrogel disks were generated in this work. For small disks (approx. 4.5 mm diameter, 0.8 mm thick), 30 μL of solution was charged to a silicon mold, slowly to avoid bubble formation, and cured for 60 seconds (only the dilute 1:2 resins required 90 seconds) in the center of the chamber. For larger disks (approximately 8.5 mm in diameter and 2 mm thick), the top was cut from a 2 mL syringe and 100 μL of solution was pipetted in and cured for 60 seconds vertically in the center of the chamber. Across a broad range of resin concentrations and cure times, handleable organogels were produced that could be removed from the mold and rinsed. Successful formulations and curing conditions were judged by whether the organogel changed from a darker orange to a pale yellow upon the first rinse with DMF, indicating that unreacted chemicals were removed. Presumably, a failure to change color indicated a lack of solvent movement in the pores (probably due to overcuring), which may imply that the infiltration of the desired cations would be difficult. On the other hand, under-curing could result in insufficient polymerization and lead to the failure of the ceramic parts to hold their shape. As a result, curing time was selected where the resultant organogels could be easily handled with tweezers but changed colors upon washing, and the same curing conditions were used for all samples and formulations, as it can impact infusion and swelling behavior.28 Successful transformation to the hydrogel and infusion of the metal cations could be observed by witnessing the gel taking on the color of the cation solution (Fig. S1†). The resulting clear yellow organogels were cleaned by fully submerging them in DMF at 70 °C for an hour, to remove the unreacted constituents. The DMF was removed via a pipette and the process was repeated once more with fresh DMF, at which point the organogels were light yellow to colorless. To transform these organogels to hydrogels, the disks were submerged in deionized (DI) water for one hour at 70 °C, three times, whereupon DMF was exchanged for water in the pores of the gel. The hydrogels were stored in DI water until infusion with metal solutions.
Metal infusion and calcination
Hydrogels were submerged in 2 M solutions of the desired metal salts dissolved in DI water (Fig. 1). The metal salts used in this work were CuX2, CeX3, ZrOX2, and UO2(NO3)2 (with X = Cl or NO3). In our set-up, 300 μL was a sufficient volume to completely submerge the part. The closed vials containing the solutions were heated to 70 °C for 24 hours on a hot plate. Hydrogels were removed from the solution immediately prior to calcination. A spatula was used to transfer the hydrogels from the solution to the alumina crucibles. The infused disks were tightly spaced but without any contact with each other. A benchtop box furnace was set, all under an air atmosphere, to ramp up to 700 °C at 0.2 °C min−1, followed by a dwelling time of 3 hours at 700 °C, and then the furnace was cooled by natural convection.Simultaneous thermal analysis (STA)
Analyses were conducted on hydrogels that had been infused with 2 M metal salt solutions. Blank crucible corrections were performed prior to each sample run to baseline-correct the instrument in the same alumina crucible to be used for the sample run. Measurements were performed on a NETZSCH-Geratebau GmbH instrument, classification STA 449F1-Jupiter, simultaneous thermal analyzer. Samples were run under an air atmosphere using laboratory air lines, heated to 700 °C at a constant heating rate of 10 °C min−1, with an air flow of 50 mL min−1 under constant purging. Prior to analysis, the instrument was calibrated over the sampling temperature range (25–700 °C) with a series of standards. Given the highly hydrated nature of the hydrogels, mass loss due to water evaporation occurred while loading the samples into the glovebox as well as into the instrument during purge cycles, leading to underestimation of the mass loss. To correct for this, the mass of each sample post-STA was taken and used to calculate the mass loss that occurred during the heating cycle. The largest mass deviation was observed for the plain hydrogel (1:2). A test from this sample analysed after only 1 pump per purge cycle in the instrument showed a smaller mass deviation, which confirmed the importance to account for water mass loss during the sample transfer.
Scanning electron microscopy (SEM)
Electron microscopy was performed using an Apreo S (Thermo Fisher Scientific, Pleasanton, CA) scanning electron microscope equipped with an EDAX Octane Elite Plus silicon drift detector. The ceramic pieces were placed on conductive carbon tape on an aluminum stub and sputter coated with ∼6 nm of Au–Pd using a Lecia EM ACE600 sputter coating system to avoid or reduce charging of the non-conductive ceramics during imaging.X-ray computed tomography (XCT)
Non-destructive volume characterization of the calcined oxide samples was conducted using an Xradia 510 Versa X-ray microscope (Carl Zeiss X-Ray Microscopy, Inc., Dublin, CA, USA). The X-ray tube voltage and power were set to 100 kV and 9 W, respectively, with no filter used. A ×4 objective lens was employed, and both the source-to-object and object-to-detector distances were set to 17 mm, resulting in a pixel size of 1.67 μm. A total of 1601 projections were collected over a 360° range with a 3-second exposure time. The sample was shifted both vertically and horizontally at each view to minimize ring artifacts. Standard filtered backprojection (FBP) reconstructions were performed using the manufacturer's software.Small angle X-ray scattering (SAXS)
The nano-scale morphology and atomic/molecular structure within the hydrogels and calcined oxides were investigated using a Xeuss 3.0 instrument (Xenocs, Grenoble, France), equipped with a Bonse–Hart setup that can resolve heterogeneities between 0.1 μm and 1 μm, commonly referred to as ultra-small angle X-ray scattering (USAXS). USAXS was only possible on calcined samples (∼0.05 mm thickness) under vacuum using 8 keV X-rays to measure heterogeneity between 1 nm and 1 μm; X-ray diffraction (XRD) was also obtained up to 5 Å−1. Since the wet and dry hydrogels were much thicker (∼1 mm) and filled with a high-Z electrolyte, only heterogeneities between 1 nm and 10 nm were investigated; XRD was obtained up to 3 Å−1 to resolve the polymer and electrolyte structures. Each sample was mounted within a 1 mm thick silicone-gasket cell (Grace Bio-Labs), which was filled with an electrolyte for the wet gels or kept dry with the dry gels or calcined samples. All of the SAXS and USAXS data were measured on an absolute intensity scale using glassy carbon and calibration of the magnitude of the scattering vector (q) was accomplished using AgBeh.29 All 2D images were reduced by subtracting the background scattering of an empty Grace-Bio cell, scaling and azimuthally integrating using the Nika package for Igor Pro.30 The azimuthally averaged SAXS data obtained from the calcined samples were merged together and smeared (qslit = 0.23 Å−1) before merging with the slit-smeared USAXS data using the Irena package for Igor Pro.31 No anisotropy was observed in the 2D images of any of the samples and therefore all USAXS data were modelled using an isotopically slit-smeared model.The pore size distribution for CuO ceramics was modelled using the numerical solution for a spherically symmetric phase as a core–shell with a hollow core and an electron density gradient at the surface; the model was motivated by characteristic surface-gradient scattering observed in the data. The electron density gradient was approximated by the complementary error function and introduced one extra fit parameter, Tshell, which was the thickness of the gradient region. The size distribution of voids is assumed to be log-normal with the scattered intensity equal to:
 | (1) |
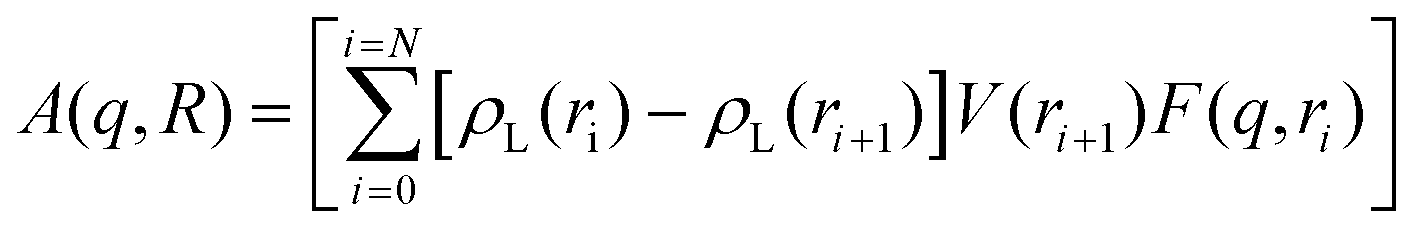 | (2) |
 | (3) |
The nanoscale pore size distribution for Zr and Ce oxides was extracted from these USAXS data using a simple bimodal lognormal distribution of spheres:
 | (4) |
Results
General trends and hydrogel behavior
The fabrication of several metal oxide ceramic disks was attempted in this study, and successful formulations were found to be unique for each metal cation (see Table 1). The identity of the anion (nitrate vs. chloride) and the cation itself both impacted the quality and morphology of the resultant ceramic, as explored in the later sections. Some metal salts (e.g., CuCl2 and ZrOCl2) resulted in handleable ceramics regardless of the resin formulation. In these cases, the best conditions were chosen based on the observed density during microscopy, and in general, the concentrated resin formulation had a denser structure and thus was more attractive. In the event that powdered or friable ceramics were all that survived (e.g. Cu(NO3)2 and CeCl3), the best conditions were chosen to be the ones that occasionally gave a solid ceramic piece that could be manipulated with tweezers. Table 1 shows the variety of metal salts used. While successful conditions were found for most ceramics, some were consistently easier to handle post-calcination (i.e., uranium and zirconium oxides), while others were consistently fragile (i.e., cerium oxide). Pre-calcination, it was observed that some cations significantly shrunk the hydrogels during the infusion process as well as after calcination, notably lanthanides and actinides (Fig. S2 and S3†).:1 resin
| Metal(s) | Cation charge; metal type; oxidation state | Calcined sample physical characteristics for all formulations | Calcination mass loss* |
|---|---|---|---|
| *STA measurements conducted only on 1:1 formulation, except UO2(NO3)2 (2:1). Mass change (%) = [(Initial mass − Final mass)/Initial mass] × 100. Plain hydrogel mass loss is 99.1% (for 1:1) |
|||
| CuCl2·2H2O | +2; transition metal; +II | Solid, easy to handle across all resin conditions | 87.94% |
| Cu(NO3)2·2.5H2O | +2; transition metal; +II | Fragile, porous morphology, often obtained as a powder (concentrated formulations gave more consistent solids) | 82.16% |
| CeCl3·7H2O | +3; f-block; +III | Solid only in dilute resins, otherwise powdery | 80.03% |
| Ce(NO3)3·6H2O | +3; f-block; +III | Fragile, porous morphology, often obtained as a powder (dilute formulations gave more consistent solids) | 81.06% |
| ZrOCl2·8H2O | +2; transition metal; +IV | Solid, easy to handle across resin conditions, very dense morphology across all resin conditions | 58.88% |
| ZrO(NO3)2·6H2O | +2; transition metal; +IV | Solid, easy to handle across all resin conditions | 69.96% |
| UO2(NO3)2·6H2O | +2; f-block; +IV | Solid, easy to handle across resin conditions, low porosity across all resin conditions | 74.94% |
Significant morphological differences across the metals were also observed in SEM with differing grain sizes, shapes, and quality, and these will be described in the later sections.
Simultaneous thermal analysis (STA) was used to interpret the calcining process in situ, to study how the decomposition step affects ceramic quality. First, plain hydrogels (i.e., without any metal infusion) were studied via STA in air to better understand the role of the hydrogel scaffold during calcination, applicable across the metals. Three hydrogels resulting from different resin formulations, as detailed in the Experimental methods section, were tested (Fig. 2). Overall, the behavior of all three hydrogels was quite similar to each other and other literature reports.16 Beginning at approximately 350 °C, all three hydrogels exhibited an extended period of mass loss until approximately no mass remained at 550 °C. Two exothermic events were observed in all three cases, the first near the beginning of this mass loss event and a second distinct exotherm towards the end, at approximately 425 and 500 °C, respectively. The residual mass reached nearly 0% by 525–550 °C, indicating complete combustion of the hydrogel matrix. However, the most dilute hydrogel had an earlier mass loss, a large decrease almost immediately upon heating, likely due to the evaporation of excess water, which is not seen in the two more concentrated hydrogels. In fact, the dilute hydrogel was characterized shortly after removing the disc from water, without an extended air-drying time (e.g., overnight or longer), explaining the rapid and more abrupt mass loss observed around 100 °C. As a result, the dilute hydrogel, due to less crosslinking and thus a bigger pore size, most likely contained more infiltrated water, produced fragile discs difficult to handle and often broke when handling with tweezers. As it will be discussed later, while changing the hydrogel formulation does impact the final ceramic piece, this is unlikely to be due to differences in the decomposition behavior of the hydrogel itself, since all three formulations demonstrated combustion at similar temperatures.
 | ||
| Fig. 2 Simultaneous thermal analysis (STA) results from the plain hydrogels of varying resin formulations. Evolution of temperature-dependent weight % (TGA indicated by dashed lines; left y-axis) and the heat flow rate (DSC indicated by solid lines; right y-axis) as a function of temperature up to 700 °C, with a heating ramp rate of 10 °C min−1. Concentrated (2:1; purple) and normal (1:1; red) hydrogels are very similar, while dilute hydrogels (1:2; black) show an early mass loss around 100 °C, as well as some earlier mass loss and DSC peaks around 400–500 °C. | ||
Ce and Cu salts
The influence of the leaving group (chloride and nitrate) was first studied with bare cations from the d-block and the f-block. Copper salts were the first metal species investigated, due to their well understood oxidation behavior and the bright color of their aqueous solutions, which allowed visual determination of infusion. Prior to calcination, there was no visible difference between the infused Cu hydrogels; both nitrate and chloride appeared to have a homogeneous shade, matching the color of the aqueous solution in which they were soaked (i.e., blue for Cu(NO3)2 and green for CuCl2; see Fig. S1†), and no visible change in size was observed. After calcination, however, there were clear morphological and physical differences between the nitrate and the chloride salts (Fig. S4†). Significant staining marred the crucible containing CuCl2, and the disks were solid and easy to handle. In contrast, there was minimal discoloration in the Cu(NO3)2 crucible and the disks were either powdered or very delicate and difficult to handle. According to XRD, both salts decomposed to CuO over the course of calcination (Fig. S5 and S6†).Investigation of the CuO morphologies using XCT and SEM imaging revealed drastic differences between the ceramics resulting from the nitrate and chloride precursors (Fig. 3 and 4). Macrostructurally, surface imaging of the samples showed a similar appearance with uneven surfaces, a variable topography and many rounded hill-like structures sized in the tens of microns. These are most noticeable in Fig. 4c for nitrates, and in Fig. 4e and f for chlorides, having a blister- or coin-like appearance (Fig. S7a†). Using XCT, however, clear macrostructural differences were observed between the two samples (Fig. 3). The Cu(NO3)2-infused samples had dense regions interspersed with large cracks spanning deep into the sample, while the CuCl2-infused samples had a foam-like macrostructure with large surface grooves. No significant differences were observed when characterizing the 1:1 hydrogels (Fig. S8†).
 | ||
| Fig. 3 XCT reconstruction slices of ceramics resulting from (a) Cu(NO3)2 and (b) CuCl2 infusion into concentrated hydrogels (2:1), along with their corresponding vertical cross-sections (c) and (d). The Cu(NO3)2 sample displays dense regions characterized by large cracks, while the CuCl2 sample reveals a foam-like structure with surface grooves. | ||
 | ||
| Fig. 4 SEM of Cu and Ce ceramics resulting from the HIAM process showing the impact of anions and resin formulations on the final morphology. Results from the infusion of Cu(NO3)2 salt (a–c) and CuCl2 salt (d–f) showed that morphology was impacted by resin condition and anion. Infusion of Ce(NO3)3 (g–i) was only successful in dilute resin conditions, while CeCl3 infusion (j–l) showed morphological changes across resin conditions. | ||
These differences in structure were also evident at the microstructural level, with cracks and fragmentation seen in the lower magnification images (Fig. 4a–c, on the left) for the Cu(NO3)2 infused samples. Examining the higher magnification imaging of the CuCl2-infused samples (Fig. 4d–f) resulted in a condensed microstructure in the imaged surfaces, despite the porous macrostructure. The individual grains had coalesced from the ionic salt infusion and had formed a condensed interconnected structure, akin in appearance to normally pressed and sintered polycrystalline ceramics.32 In contrast, the Cu(NO3)2-infused hydrogels resulted in a discordant microstructure, where individual grains were chained together loosely rather than being densely packed. This limited grain condensation and macroporosity translated into the Cu(NO3)2-infused samples being much more friable and challenging to handle after calcination when compared to the more robust CuCl2-infused samples having a condensed microstructure arranged into a solid foam-like macrostructure (see Fig. S9†).
In addition to the differing coalescence of grains, the two Cu salt infusions varied in the resulting grain size and distribution. While all the samples produced fine grains, a majority with a size of ∼1 μm or less, the grains were typically much finer in the Cu(NO3)2-infused samples, with grains ∼100 nm or less in size being most prevalent. Both salts also gave rise to a bimodal grain size distribution, which was influenced by the resin formulations. The 2:1 concentrated Cu(NO3)2 infusion (Fig. 4a) and the 1:1 CuCl2 infusion (Fig. 4e) resulted in uniform grain sizes; however, all the other samples produced a bimodal structure with patches of extremely fine grains and other patches of larger grains. In the nitrate-based samples, this was typically ∼500 nm:100 nm for the two sets of grain sizes, and ∼1 μm:100 nm in the chloride-based samples.
Ce solutions were colorless, making the judging of a successful infusion challenging. However, post infusion, the gels had shrunk significantly in size (Fig. S4†), giving some indication of infiltration. Upon calcination, most Ce ceramic disks were powdered or crumbled. Ce(NO3)3 yielded only piles of powder except for the most dilute resin conditions, while CeCl3 gave slightly better results and again showed a preference for dilute resin conditions, evidenced by the more cohesive macrostructure (Fig. 4i). XCT reconstruction of the ceramic resulting from 1:1 the hydrogels infused with CeCl3 reflected this observation, showing very thin dense but fragile pieces (Fig. S10c†). Microstructure imaging via SEM revealed that the CeCl3-infused samples produced denser structures when calcining more dilute hydrogels (Fig. 4j–l). While the one successful Ce(NO3)3-infused sample did have a larger grain size (Fig. 4i), these large grains were separated by large voids (Fig. S10a†), explaining the fragility of the sample which ultimately crumbled upon handling. The 1:2 diluted CeCl3 showed a bimodal grain size distribution; the grains were condensed, like that seen in many of the copper-infused samples. In contrast, the nitrate cerium-based infusions showed irregular microstructures with numerous cracks, blisters, and poorly consolidated material (Fig. S7b†). Interestingly, the densest apparent structure of the nitrate sample also suffered from some of the most severe “charging” conditions during electron microscopy, even with sputter coating, which may be due to its large grain size, while the fine-grained chloride counterpart was more easily imaged.
STA analysis was conducted on the Cu and Ce salt-infused hydrogels, to study their transformation into ceramics in situ. The same conditions were used to study the non-infused hydrogels. Fig. 5 shows the STA analyses of the nitrate- and chloride-infused hydrogels for both cations. For both Cu(NO3)2 and CuCl2 salts, the behavior of the metal-infused hydrogels was closer to the one observed for the plain hydrogel than the hydrated salt (Fig. S11†). Overall, Cu(NO3)2 decomposed at a lower temperature, with a large exothermic event and a sharp mass loss at ∼135 to 140 °C. Around the same temperature range, the CuCl2 system displayed only a small mass loss and a small exothermic reaction. Part of the mass loss in both systems can be attributed to the evaporation of bulk water retained in the pores of the hydrogel network. Additional nitrate decomposition was observed with a gradual mass loss from 150–360 °C, while CuCl2 exhibited a somewhat smoother mass loss starting at around the same temperature and ending much later at 560 °C. Compared to the plain hydrogel, which showed 2 exothermic peaks at 420–550 °C, only one broad peak was present in that range for the Cu-infused hydrogels, likely encompassing any final scaffold decomposition as well as metal oxidation. Finally, a confirmation of metal infusion into these hydrogels was the presence of the material in the crucible at the end of calcination that had about 20% mass left from the starting hydrogel. In comparison, 0% mass remained after calcination of the plain hydrogels.
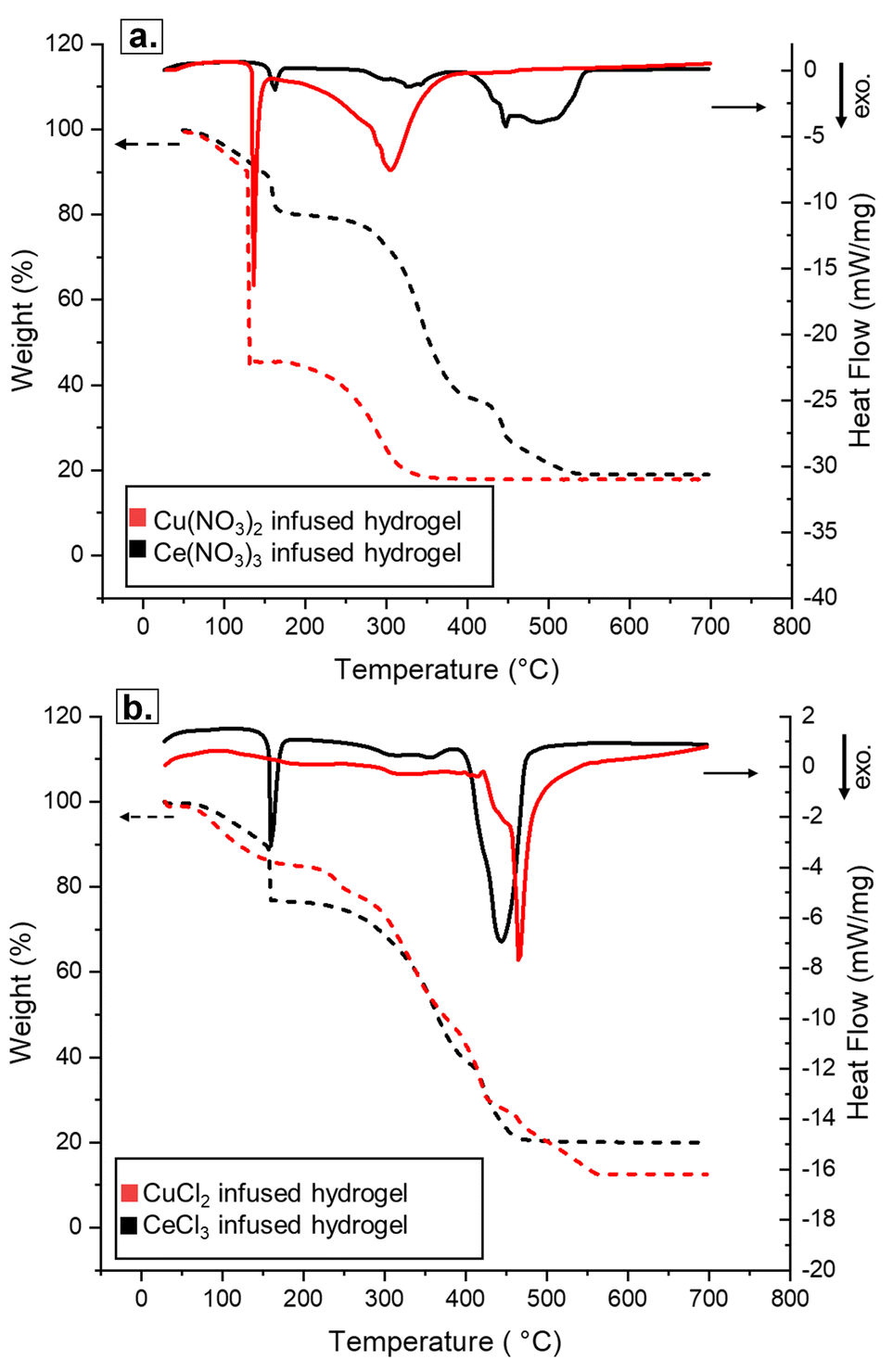 | ||
| Fig. 5 Graphics of a representative experiment illustrating the simultaneous thermal analysis (STA) for Cu (red) and Ce (black) infused 1:1 hydrogels: (a) the nitrate system and (b) the chloride system. Evolution of temperature-dependent mass loss (TGA indicated by dashed lines; left y-axis) and the heat flow rate (DSC indicated by solid lines; right y-axis) as a function of temperature up to 700 °C, with a heating ramp rate of 10 °C min−1. | ||
Ce salt-infused hydrogels also exhibited distinct differences between the nitrate and chloride salts during the STA analysis (Fig. 5). Ce(NO3)3, in contrast to Cu(NO3)2, had only a small early exotherm and mass loss event, at approximately 135 °C, while CeCl3 exhibited a large exothermic event at 135–140 °C. This decomposition is not seen in the analysis of other metal chloride salts studied, and it is worth noting that the CeO2 ceramics were the most challenging to fabricate and handle, even resulting from the chloride precursors, very likely due to this early energetic decomposition. Both Ce salt-infused hydrogels showed a gradual mass loss starting at 250–300 °C and ending at 550 °C for the nitrate and 450 °C for the chloride, accompanied by one large exothermic event near the end of the mass loss region.
ZrO and UO2 salts
Some oxo-salts were tested to determine if they could be successfully used in the HIAM process. ZrOCl2, ZrO(NO3)2 and UO2(NO3)2 were selected for study, mirroring the choice of a transition metal and an f-block element in the previous section. Overall, oxo-cations yielded very dense, handleable ceramics across all resin formulations. Notably, both ZrO(NO3)2 and UO2(NO3)2 yielded handleable ceramics despite the more energetic decomposition of the nitrate salt. SEM analysis (Fig. 6) revealed that both salts showed morphological changes as the hydrogel resin formulations and salts were altered.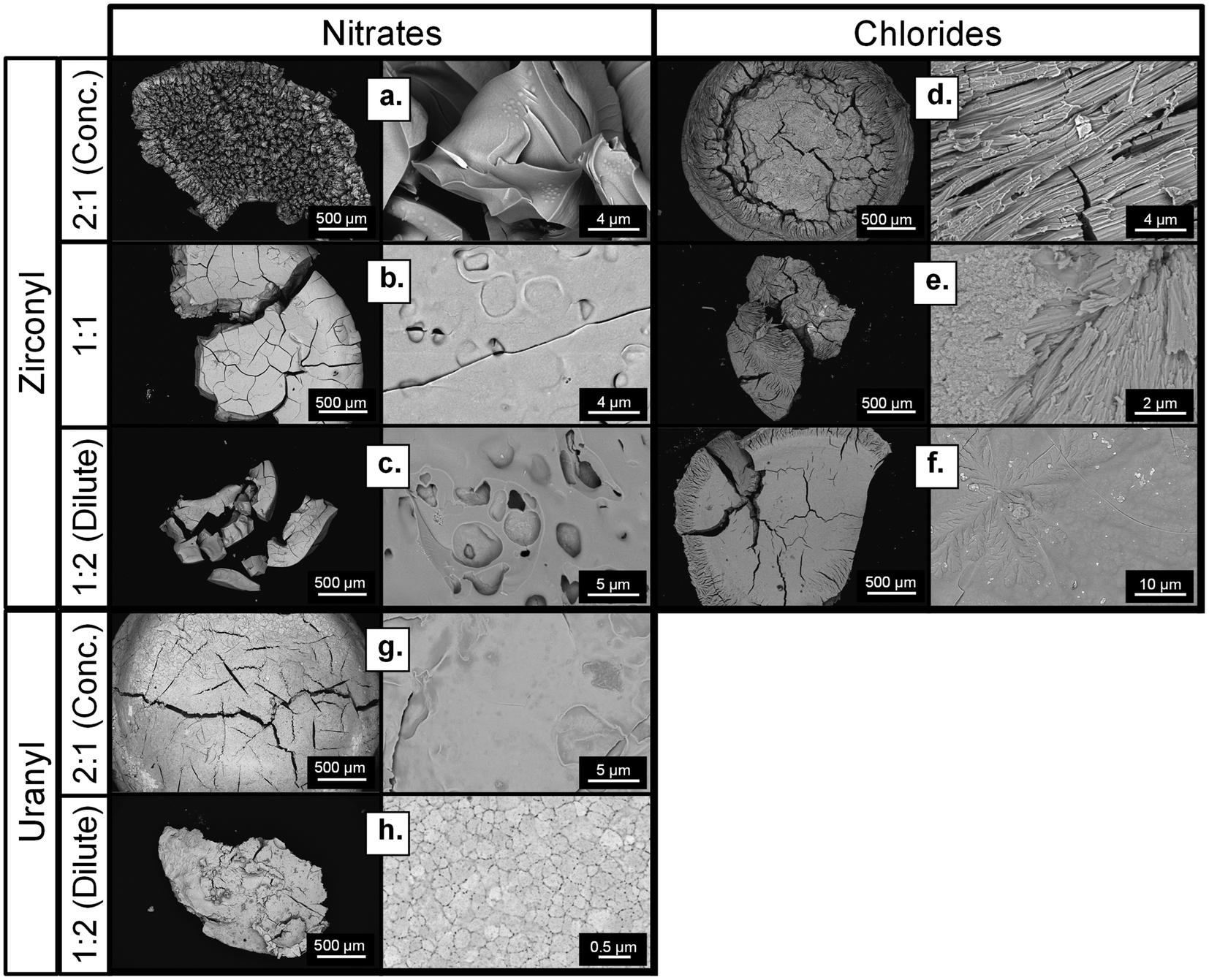 | ||
| Fig. 6 SEM of ceramics resulting from Zr and U oxo-salts. Results from ZrO(NO3)2 salt infusion into different resin conditions is shown in a–c; results from ZrOCl2 salt infusion into different resin conditions is shown in d–f; results from UO2(NO3)2 salt infusion into different resin conditions in shown in g–h. Note: a 1:1 sample with uranium was not fabricated. | ||
Observing the microstructures of the calcined oxo-salt infusions, the microstructures were seen to be typically much denser, with clearly coalesced materials and nearly none of the discordant grain structures. In many cases, it is challenging to provide a description of the specific microstructure due to the poor grain boundary visibility and irregular surface features. In the ZrOCl2-infused samples, the grains were elongated and fibrous in nature, atypical of sintered zirconia polycrystals.33,34 In the dilute ZrOCl2-infused sample, floret-like dendrite structures were seen growing on the surface of the sample as well, which could imply some directionality to the coalescence of solids during the calcination process (Fig. 6d and e). The macrostructure of ZrOCl2-infused samples as studied by XCT reveals a similar trend of dense regions with prominent cracks, forming a more solid piece for the more dilute hydrogel (Fig. S12†). As for the UO2(NO3)2-infused samples, the 1:2 dilute resin formulation also had a clear microstructure, with clearly defined grains typical of polycrystalline uranium oxide (Fig. 6h).35 The 2:1 concentrated UO2(NO3)2 sample shows a similar dense microstructure; however, grain boundaries are not evident to provide a direct comparison (Fig. 6g). The primary features in the high magnification imaging are some cracks and surface topology. These results show that the oxo-salt infusions provide more solids for densification, possibly due to the pre-oxidized state of the oxo-molecules when compared to the monatomic Cu and Ce ions.
To explain the changes in the morphology, STA was used to compare the decomposition pathways of the infused hydrogels (Fig. 7). In the case of zirconyl, STA showed that the nitrate salt-infused hydrogel had energetic events and mass loss at low temperatures (<150 °C), far below the hydrogel decomposition, while the chloride salt-infused hydrogel had only one exothermic event, which coincided with the hydrogel decomposition. This rapid decomposition is likely associated with a physical breakup of the hydrogel disks, resulting in the fragmented pieces observed in the macrostructural images of Fig. 6. Evidently, the zirconyl is effective at delivering materials for coalescence of the solid, but the nitrate is a more aggressive leaving group that results in a break-up of the larger pieces. In general, Zr ceramics were some of the sturdiest, which could be explained by the better infiltration and better intake, since these samples suffered from the least mass loss according to TGA (Fig. 7). Despite UO2(NO3)2 showing the same region of early, rapid mass loss as other nitrate salts via STA, the uranium oxide disks were easy to handle regardless of the resin formulation. This highlights the combined roles of the cation, anion, and resin concentrations on the final structures produced.
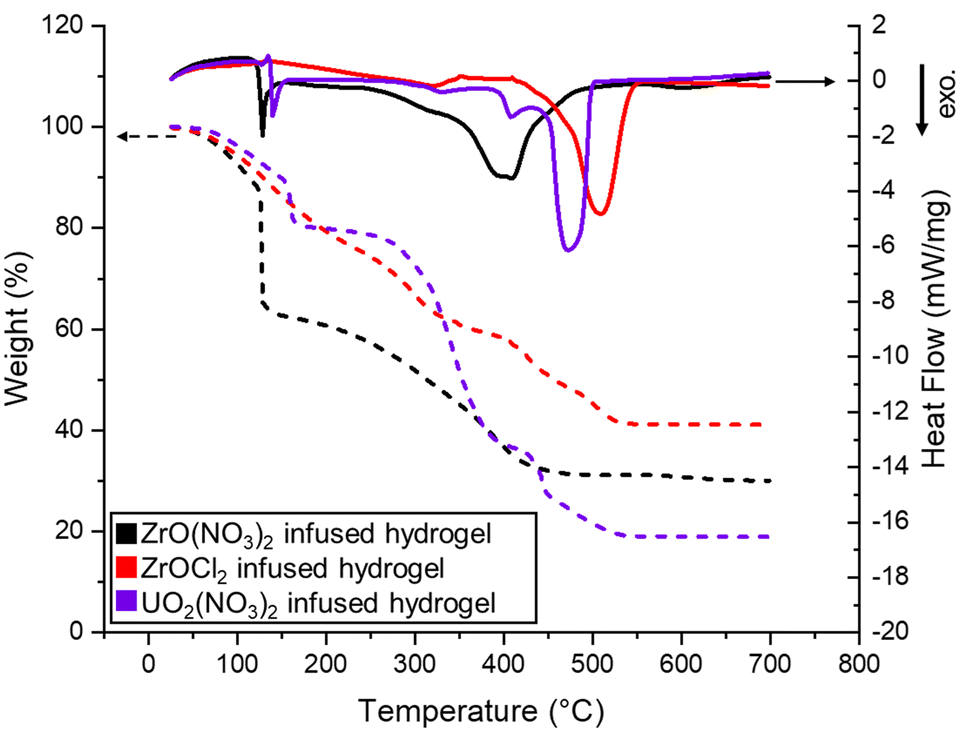 | ||
| Fig. 7 Graphics of a representative experiment illustrating the simultaneous thermal analysis (STA) for the 1:1 hydrogel infused with Zr and U salts. Evolution of temperature-dependent weight % (TGA indicated by dashed lines; left y-axis) and the heat flow rate (DSC indicated by solid lines; right y-axis) as a function of temperature up to 700 °C, with a heating ramp rate of 10 °C min−1. | ||
SAXS and USAXS investigations
Small angle X-ray scattering (SAXS) was used to study how resin formulations and cation choices impacted the infusion step of the HIAM process. Specifically, SAXS was used to study the hydrogels prior to calcination to resolve the size domains of the polymer and electrolyte domains, first in plain hydrogels. In the high-q region, peaks from the polymer (q ≈ 1.48 Å−1) are readily observed in air, but are underneath the primary peak from water (q ≈ 2.06 Å−1) in solution (Fig. 8a). The domain sizes, observed in the low-q region of the hydrated gels, revealed two feature sizes: (1) > 10 nm feature size that decreases in volume with an increasing polymer concentration and (2) a feature size of 1–2 nm that increases in volume with a polymer concentration. Consistent with Waters et al. on PEG hydrogels, the 1–2 nm feature size is attributed to polymer domains, while the larger scatterers are related to the hydrophilic domains.36 Interestingly, the first peak of water shifts at high polymer concentrations (Fig. 8b) and is observed in all of the electrolytes to some degree (Fig. 8a–c). This shifting cannot be explained by a linear combination of the two peaks from the polymer and water and therefore is likely due to an increase in interfacial water that is less dense at high polymer concentrations.37 Therefore, the SAXS data from hydrogels in pure water are consistent with an increase in hydrophobic domains and surface area with increasing polymer.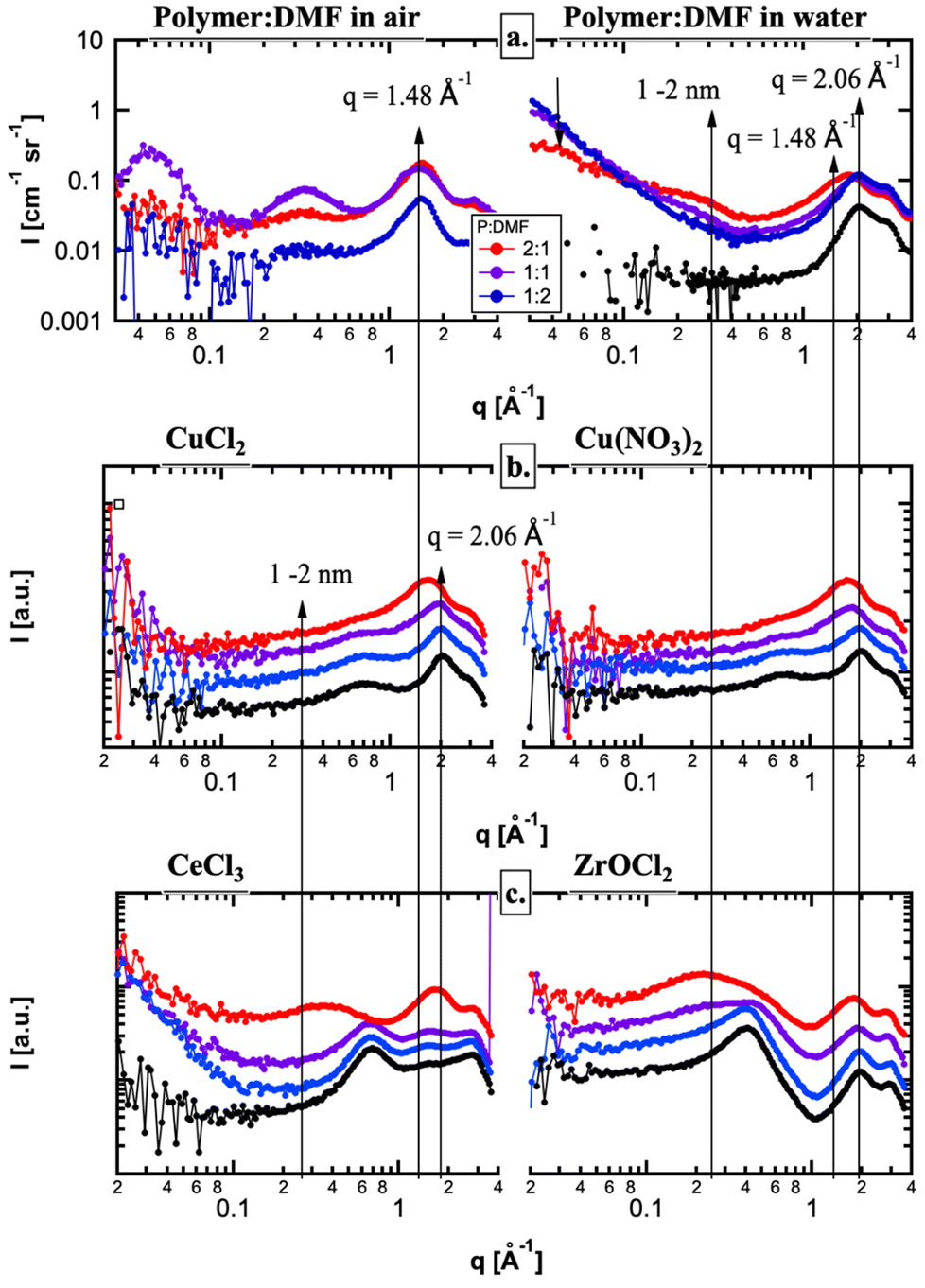 | ||
| Fig. 8 SAXS data showing evidence of hydrogel–cation interactions. The shifting peak indicates a change in the coordination environment around the metal cation when dissolved in water (black) and when infiltrated in hydrogels of various formulations 2:1 (red), 1:1 (purple), and 1:2 (blue). Plain hydrogels are shown in (a), Cu salts in (b), and CeCl3 and ZrOCl2 in (c). | ||
When the hydrogels were submerged in each electrolyte, the small angle scattering (low-q region) changed significantly, as the dissolved ions (primarily, the metal) changed the electron density contrast between the hydrophilic and polymer domains (Fig. 8). SAXS from the Cu(NO3)2 and CuCl2 electrolytes were the same with concentration, suggesting very little contrast between the hydrophilic and hydrophobic domains (Fig. 8b). On the other hand, the polymer domains were very well resolved in the ZrOCl2 system and to some degree in the CeCl3 system (Fig. 8c). The reduced small angle scattering at q ≈ 0.3 Å−1 from the polymer domains in the CuX electrolytes provides direct evidence that the Cu ions infiltrate the polymer domains much differently than the ZrOCl2 ions, as both have the same concentration and a similar number of electrons and should have similar SAXS curves. On the other hand, the Ce ions have significantly more electrons and, for the same concentration, should enhance the contrast between the hydrophilic and polymer domains. However, it is only at the highest polymer concentration that the polymer domains can be observed, suggesting that the Ce ions penetrate the polymer domains to some degree. Therefore, the SAXS data suggest that there is a difference in the way the metal cations interact with the polymer domains within the hydrogel. This confirms that more ZrOCl2 material interacted with the polymer, and as a result more Zr likely infiltrated the hydrogel (as shown per mass left at the end of the STA).
SAXS was also used to study ceramics post-calcination, to further investigate their morphology. Where SEM provides direct observation of pores near the surface, USAXS measurements on the calcined pieces can provide the absolute volume fraction and size of bulk porosity throughout the calcined samples. Guided by SEM imaging and XRD (Fig. 4, 6, S6, and S7†), the small angle scattering is expected to be dominated by voids within the ceramic. Therefore, the USAXS data were modelled as a log-normal size distribution of pores.
The negative deviation from the Porod scattering observed at q > 0.03 Å−1 (as represented from the line depicting the relationship I(q) = Bq−3) for the copper salts suggested that the pore surface in CuO does not meet the Porod condition for a “smooth and well-defined surface”. We can thus infer that an electron density gradient was present at the pore–CuO interface and had to be included to model the high-q data beyond the Guinier region; this feature was not observed in ZrO2 or CeO2 (discussed later). This feature may be associated with Cu infusion into the polymer network, resulting in a surface layer of the pore–CuO interface that has a different electron density from the bulk CuO. Typical values of 4 nm were obtained for Tshell, the thickness of the shell surrounding a pore; the resulting model fits (Fig. 9a) and size distributions are shown in Fig. 9b; the scattering from the samples prepared from the diluted resin (1:2) was weak and is not shown here. There are distinct differences between the pore size distributions between the chloride and the nitrate, with a higher total volume fraction of larger voids being observed in the chloride, also confirmed by XCT. While a clear difference in the void size distributions is qualitatively consistent with a difference in off-gassing during calcination, it is important to note that any voids larger than ∼5 μm cannot be resolved with USAXS and are likely present in the nitrate sample. In both cases, a reduction in polymer domains results in size distributions that are larger in size and broader (Fig. 9b), likely due to larger hydrophilic domains resulting in larger bubbles during calcination.
 | ||
| Fig. 9 Size distribution of pores in calcined ceramics resulting from 1:1 (pale lines) and 2:1 (bold lines) resins. The impact of both salt choice and resin formulation is clear for Cu oxide (a and b). The impact of resin choice is less pronounced for Ce and Zr oxides (c and d), but the results show a clear difference in void space across metals. | ||
Ce and Zr ceramics were also studied via USAXS. For the same anion (Cl−), the two different metal oxides revealed a bi-modal population of much smaller voids (∼10 nm) alongside larger voids similar in size to the CuO samples (Fig. 9b); no evidence of an electron density gradient at the pore-wall boundary was observed (Fig. 9c), since the data followed the angle of the line representing the relationship I(q) = Bq−3. The model fits are shown in Fig. 9c along with the size distributions in Fig. 9d. It is clear from both the USAXS data and size distributions that there is little difference in the nanovoid morphology between ceramics resulting from the 1:1 and 2:1 resin precursors. Moreover, there are significantly smaller voids and fewer large voids on the meso-scale compared with the CuO samples (Fig. 9a and b), suggesting that it is the differences in the Cu infusion in the hydrogel (Fig. 8) that resulted in a different bulk pore morphology.
Discussion
The interaction between specific metals and the hydrogel scaffold is difficult to study but nonetheless vital to understand and control the HIAM process. Three precursor choices stand out as clearly impacting the success of the HIAM process and thus merit further discussion: (1) the polymer concentration, (2) the type of metal (bare vs. −yl cations and transition metals vs. f-block), and (3) the anion. The first two choices influence the infusion step of the HIAM process, as the cation and polymer interact with each other. Additionally, the choice of anion and the choice of cation determine the thermal decomposition path during the calcination step. All three proved critical to obtaining handleable ceramic disks.Polymer concentration effect
The change in Vnano with the polymer ratio is small and within errors associated with the method (any voids larger than 5 μm are excluded from Fig. 10). However, SEM results clearly demonstrate that diluting the hydrogel resin impacts the morphology and quality of the final ceramic, albeit not predictably across metals (Fig. 4 and 6). When comparing CuO from Cl− and NO3−, USAXS analysis of nanoscale voids shows lower nanoporosity resulting from concentrated resins only for Cl− (Fig. 10). CuO ceramics exhibited observable differences in the microstructure and porosity when only resin dilution was adjusted, while XCT did not demonstrate any major difference in the global density. Although the macrostructure was similar, with regions of density and regions of large cracks, SEM analysis of the surface of calcined samples showed a shift in the grain size as the resin formulation was changed (Fig. 4 and Fig. S9†). Therefore, it is the larger pores that change with the polymer concentration more than the nanoscale. | ||
| Fig. 10 Plot of the total volume fraction of the nanoporosity (<2 μm) observed by USAXS in the calcined samples. There is a clear difference across metals and slight differences across different resin ratios. | ||
Two competing forces could explain how diluting the PEGda monomer has an impact on the final ceramic and why the impact of dilution is not predictable across metals. Diluting the PEGda monomer concentration means less polymer will be present in the hydrogel, thus less gas will be created when the organic scaffold combusts, potentially creating less porosity in the calcined ceramic. If this was the sole impact of diluting the resin, one would expect a larger grain size and higher handleability for dilute resin formulations, as is the case for Ce ceramics. However, many of the ceramics generated in this study have smaller nanoporosity and better handleability when generated from concentrated resins, like Cu. We hypothesize that altering the resin formulation also has an impact on the infiltration of metal cations, either by altering the size of pores or polymer domains. While more gas might be generated from a concentrated hydrogel, the impact could be tempered if more metal was infiltrated prior to the calcination step.
To study the impact of polymer combustion more closely, thicker hydrogel molds were used to generate larger samples. It could be reasonably assumed that the more energetic decomposition of the nitrate salt necessitates a thinner hydrogel scaffold for off-gassing events to maintain structural integrity. Indeed, the diluted hydrogels were slightly thinner than their concentrated counterparts, 0.7 and 1 mm thick, respectively. Saccone et al. printed hydrogels with even thinner features (200 μm) and reported no challenges handling their ceramics resulting from Cu(NO3)2-infused hydrogels (but presumably fragile, given the fine scale). To test this hypothesis, a thicker hydrogel was prepared using a different mold, approximately 2 mm thick and 8 mm in diameter. Cu(NO3)2 was infused and calcined and a solid ceramic piece was successfully produced, which was handleable and similar to the smaller ceramics resulting from the same formulation. A direct comparison between these larger disks and the small ones cannot be drawn, as engineering differences could have significantly impacted the hydrogel structure. However, it does demonstrate that physically thin hydrogel precursors are not a strict prerequisite for HIAM's success.
Dilution of hydrogel resins has been shown to increase the pore size in hydrogels, due to decreasing entanglements with neighboring strands.38,39 Increased pore size and decreased polymer domains will undoubtedly influence the infusion of metal cations into the hydrogels and vary across cation identities, impacting the quality of the final ceramics. Similarly, increased polymer domains may promote metal infiltration if cations interact with the hydrogel scaffold directly. SAXS analysis of the pre-infused hydrogels showed a decrease in hydrophilic domains (pores) and an increase in polymer domains (pore walls) as the polymer concentration was increased (Fig. 8a). Post-calcination, SEM microstructure analysis demonstrated clear differences in grain size in ceramics resulting from different resin dilutions (Fig. 4 and 6). In particular, the bimodal structure of the CuO ceramics—which vary depending on whether a concentrated or diluted resin is used—likely occurs due to isolated regions of salt-infused hydrogels calcining independently from one another until sufficient shrinkage or surrounding grain growth occurs to bring the different sections into contact. Evidently, the dilution or concentration of the resin influences this behavior, meaning the formulation likely affects the resulting gel polymer networks and the variability and magnitude of salts it can take up, potentially compensating for increased off-gassing from polymer combustion.
The exact pore size of PEGda hydrogels—and the resulting impact on calcined ceramics—is challenging to control and predict due to the high degree of interpenetration and the randomness of radical-initiated chain polymerization reactions, and precise control often requires the use of a solid placeholder.40,41 This is reflected in the USAXS data of calcined pieces, which indeed show a wide size distribution of voids and slight differences across the resin ratios for the same metals (Fig. 10). Combined with the microstructure analysis from SEM, our results demonstrated that dilution of the PEGda monomer can impact the morphology of the calcined ceramic pieces. Metal cation and anion effects prevent clear trends from emerging due to resin dilution (i.e. Ce salts improved in quality with dilute resins, but not Cu), but the formulation of the hydrogel precursor cannot be ignored in designing the HIAM process and will need to be optimized for each ceramic. This study has shown that the morphology of ceramics resulting from HIAM is influenced by a complex combination of factors and cannot be explained by one systematic trend.
Metal identity and interaction with the polymer
The exact nature of the polymer–metal interactions is difficult to study within the hydrogel framework due to the highly disordered polymer strands, the high concentration of the cation in solution both infused and not, and the large excess of water molecules. Nevertheless, interactions between specific cations and the hydrogel polymers must be considered, as each metal had unique results and morphologies, which may be due to their interactions with the hydrogel scaffold and the calcination process. The ability of polyethylene glycol polymers and hydrogels to coordinate with and discriminate between cations is well established in the literature.25,42 Studies have shown that the kinetics of metal adsorption into hydrogels is not diffusion controlled, implying that rather than just diffusing into the hydrogel pores, cations interact with the polymer strands (i.e. the oxygen-containing ether and ester moieties).25Our SAXS data confirmed a clear difference in the nanoscale heterogeneity of the metal infused hydrogels (Fig. 8) and suggested a difference in the metal–polymer interaction. The peak resulting from water, when infiltrated into the hydrogel, indicated a change in the coordination environment. The shift was most dramatic in the concentrated resin hydrogels, regardless of the infiltrated cation, although the magnitude and direction of the shift appear unique between species. While Zr and Cu ions have similar numbers of electrons and thus should have similar SAXS curves, the data looked quite different between the two, directly confirming their different infusion behavior. During Ce infusion, only concentrated resins allowed for polymer domains to be observed, whereas the domains could be faintly observed in the 1:1 formulation with ZrOCl2 (Fig. 8c). This is direct evidence that Ce3+ and ZrO2+ cations infiltrate the hydrogel differently than Cu cations, which may explain the difference in the void morphology between Cu (monomodal) and Zr and Ce (bimodal).
Three potential causes of the observed differences in hydrogel–cation interactions, and thus the resultant ceramic quality, will be discussed: (1) hard/soft acidity, (2) oxo-ligand interaction, and (3) the hydration shell. First, remarkable shrinkage of the hydrogels was observed when f-block elements, Ce(NO3)3 and UO2(NO3)2, were infiltrated (Fig. S2†). Elements from the f-block were hard Lewis acids and capable of forming high coordination number complexes in water, and their aqueous chemistry was distinct from the main group elements.43,44 Furthermore, the ether moieties of the PEGda polymer (Fig. 11), which formed the crosslinking hydrogel scaffold, were hard Lewis bases and thus a hard/hard match between the f-block cations and the ether polymer was achieved. As a result, the f-block cations could form high coordination number complexes with the ether moieties of the hydrogel scaffold, with the hard/hard match making such complexation favorable and strong, which could explain why hydrogel shrinkage was seen upon infusion of f-block elements in contrast to their main-block counterparts. Increased interaction between the cations and the hydrogel could lead to pore collapse and overall hydrogel shrinkage, as neighboring ether moieties crowd around the same metal center (Fig. 11). Elliott et al. demonstrated the ability of the repeating ether moieties of PEGda to crowd metal centers, forming pseudo-crown ethers, and it is possible to extrapolate that such a mechanism, to a lesser extent due to the rigidity of the hydrogel, can occur within the hydrogel matrix.45 Similar complexation has been documented for UO22+ cations when complexed with polyethylene glycol, with neighboring ether moieties crowding the cation in a crown-ether-like complex.46 Furthermore, Elliott et al. noted a preference for hard cations, potentially explaining why drastic shrinkage is only being seen with f-block metals in our system. Our SAXS results corroborate this observation, as the diffraction peaks from CeCl3 and water change significantly at a high polymer concentration, whereas the main block elements Cu and Zr are slightly changed (Fig. 8). While it is impossible to distinguish the nature of this coordination using this technique, it indicated that there are differences between the f-block and main group cations upon interaction with the polymer domains.
 | ||
| Fig. 11 Potential solvent/polymer/cation interactions within the hydrogel matrix, either (i) direct metal–scaffold interaction, hypothesized for Ln/An, or (ii) through the solvation spheres, hypothesized for Cu and Zr. Water molecules are shown in blue. | ||
The existence of oxo-ligands on the cation also substantively improved the ceramic quality, with ZrO2+ and UO22+ cations giving consistently successful HIAM results. The ceramics formed from these precursors were easy to handle regardless of the resin formulation, and the microstructure was significantly larger and possessed denser grain structures compared to the Cu-based infusions (Fig. 6), even when using the energetic nitrate anions. The simplest explanation for the improved microstructure was that the presence of oxygen already coordinated to the metal atom had minimized the disruptive oxidation reactions necessary to form the final ceramic, aiding in retaining the structural integrity of the final pieces. Alternatively, or additionally, the chemistry of the oxo-group could play a role in bolstering infusion into the hydrogel, resulting in improved properties of the final ceramics. The role of the uranyl oxo-group has been extensively studied, due to the preponderance of the uranyl cation in natural and engineered systems. Experimental and theoretical results have shown that the oxo-group is not a spectator in aqueous systems, being able to both exchange with equatorial water ligands and participate in hydrogen bonding with the secondary solvation shell.47,48 Either of these mechanisms could impact the overall infusion of the UO22+ cation into the hydrogel (the solvation shell will be discussed below). As for the oxo-group participating in hydrogen bonding, this offers opportunities for further coordination to the hydrogel or for denser infusion. Fortier and Hayton highlighted many examples of hydrogen bonding in the uranyl oxo-ligand, as well as cation–cation interactions, wherein the oxo-group coordinates to the U of a different uranyl cation.49–51 These chemical interactions may be responsible for improved infusion of UO22+, and by extension ZrO2+, allowing for stronger cation–hydrogel bonding or closer cation–cation packing.
Extending our view beyond the cation core to the larger cation–solvent unit provides another potential mechanism for differences in the infusion behavior. Properties like diffusion and binding strength are often more accurately explained by considering the solvent–ion unit holistically.52,53Fig. 11 illustrates how solvent–ion interactions within the hydrogel matrix could impact interactions within the hydrogel itself. Transition metals and f-block elements behave differently in solution, primarily due to the inability of the f-block valence orbitals to interact strongly with solvent molecules.54 Although some literature studies propose a direct metal–ether interaction in metal-infused hydrogels, it is not clear that this is necessarily the case for all cations.42 The drastic shrinkage of hydrogels infused with f-block elements, but not when infused with main group metals, could stem from the ability of Ln/An cations to interact directly with the hydrogel due to their transient bonding with solvent molecules (Fig. 11 (area i) and Fig. S3†). In contrast, the main group elements tend to have much lower rates of exchange and thus might be primarily interacting with the hydrogel through their solvation sphere (Fig. 11 (area ii)). There is evidence in the literature that hydration shells can influence the cation behavior and infusion within hydrogels. A detailed study of alkali metal infusion into hydrogels suggested that the observed trends, which did not follow hardness or size, were due to the differing nature of the first hydration shell and its ability to either shield interactions with the hydrogel or strengthen them.22 Another study showed that larger metals, not harder ones, were preferentially absorbed into hydrogels and suggested that more diffuse charges and the ability to induce electrostatic charges in the surrounding solution were factors responsible for the behavior observed.55 Given the differing behavior seen in the hydrogels that had been infiltrated by f-block elements, i.e. extreme shrinkage, it is likely that stronger interactions with the solvent shell prevent direct Cu2+ or ZrO2+–hydrogel interactions (small ionic radii), but Ce3+ and UO22+ (large ionic radii) are able to directly associate with the monomeric strands due to the rapid exchange of water, impacting the quality and morphology of the final pieces.
Salt's thermal path decomposition/anion identity
Investigation through USAXS, XCT, and SEM showed that the anion choice also drives morphological differences in the ceramics. Macrostructural features on the surface of ceramics, namely rounded hill-like structures, almost certainly resulted from off-gassing during thermal treatment, combined with the degree of copper coalescence and densification during calcination (Fig. 4). USAXS data showing the differences in the morphology of nanoscale (<2 μm) voids also confirmed that the anion choice impacts the ceramic morphology (Fig. 6). Importantly, USAXS measurements could not accurately resolve pores larger than 2 μm, which were clearly present in the oxide beneath the surface of the CuO samples, as shown by SEM (Fig. 4 and Fig. S9†). Therefore, the total volume fraction of the nanoscale voids observed by USAXS in CuO resulting from Cu(NO3)2 is actually lower than CuCl2 (Fig. 10), when only accounting for pores smaller than 2 μm. With a much weaker and more porous CuO disk obtained from Cu(NO3)2 (Fig. 3 and 4), it is likely that much larger voids (>2 μm) are present in the bulk, and indeed XCT revealed large cracks penetrating the Cu(NO3)2 sample. The difference in the nanoscale voids between the two CuO samples in addition to markedly different micro- and macrostructures clearly demonstrates the impact of the anion on the final ceramic morphology. On the other hand, the total volume fractions of the nanoscale voids in ZrO2 and CeO2 were far less than those in CuO with the Cl− anion (Fig. 10), indicating that the anion was not solely dictating the pore volume.STA analysis showed that during the calcination step, the anion influenced the decomposition behavior, and correlations between the timing of energetic events and ceramic quality could be drawn. Early energetic events, exhibited by nitrate salts (Fig. 5 and 7), resulted in ceramics that were crumbly and difficult to handle, while those with later decomposition events (chloride salts) that coincided with hydrogel decomposition were consistently of higher quality. The decomposition pathways for Cu salts are presented as an example in Table 2, while Ce, Zr, and U salts followed a similar pattern (Tables S1–S3†). Cu(NO3)2 hydrate decomposed in a complex series of reactions with an onset of 126 °C (Table 2, eqn (5)–(8)), while CuCl2 hydrate was reported to decompose fully in a concerted step with the evolution of Cl2 gas with an onset of 365 °C (Table 2, eqn (9) and (10)).56,57 The resultant nitrate gas was highly reactive and could easily oxidize at high temperatures, causing further energetic reactions with the copper species. The resultant ceramics from Cu(NO3)2 were much more fragile and porous than those resulting from CuCl2, and this trend was also seen in Ce and Zr-oxo salts. The extended period of decomposition and associated off-gassing interfered with the ability of cation clusters to coalesce into a dense structure, producing the discordant microstructures seen in the copper-nitrate samples. In contrast, the off-gassing in the chloride samples was seen to be confined to a singular exothermic event, enabling a more condensed final structure, clearly reflected in the SEM of the final ceramics (Fig. 4).
| Thermal decomposition of Cu(NO3)2·6H2O | Cu(NO3)2·6H2O → CuOH(NO3) + HNO3 + 5 H2O (eqn 5) |
| HNO3 → H2O + 2 NO2 (eqn 6) | |
| CuOH(NO3) + NO2 → CuO(NO2) + HNO3 (eqn 7) | |
| CuO(NO2) → NO + ½O + CuO (eqn 8) | |
| Thermal decomposition of CuCl2·2H2O | CuCl2·2H2O → 2 H2O + CuCl2 (eqn 9) |
| CuCl2 → Cl2 + CuO (eqn 10) |
Conclusions
Hydrogel-infused additive manufacturing (HIAM) is an emerging process for the additive manufacturing of ceramics, in which an organic scaffold is formed first, the desired metals are subsequently infiltrated, and finally, they are calcined to obtain ceramic parts. Overall, the variability in ceramic quality and morphology between the elements tested herein indicates that complex mechanisms between hydrogels, water, metal ions, and salts dictate HIAM outcomes. Different metal salt solutions were rapidly tested with minimal optimization of the photocurable resin.In this study, a PEGda hydrogel acted as the organic scaffold and was subsequently infiltrated with a variety of transition metal and f-block elements, followed by calcination to the respective ceramics. The importance of the individual chemical precursors (hydrogel and inorganic metal salt solutions) was studied using a variety of techniques including SEM, STA, SAXS, and XCT. First, the hydrogel formulation was found to impact the porosity of the resultant ceramics. Concentrated formulations generally yielded low porosity and denser ceramics, which could impact the cation infiltration into the matrix. SAXS measurements showed that Cu, Zr, and Ce ions penetrated differently into the polymer domain. Better ceramics had measurements showing smaller voids and fewer large voids. The inorganic salt choice also impacted morphology and porosity due to specific cation–polymer interactions and the energetic differences in decomposition pathways upon calcination. Metal adsorption into hydrogels is a complex mechanism. Cations can have different hydration shell sizes and will mechanically diffuse into the hydrogel pore, as well as interact with the polymer strands via direct metal bonding (i.e., with the oxygen containing ether and ester moieties) or via hydrogen bonding (i.e., from water molecules). Characteristically, successful parts exhibited high interactions between the cations and the hydrogel or could be described as complexes having diffuse charges. Finally, this study showed that the thermal decomposition mechanism was found to impact the quality of the resultant ceramics. XCT revealed that CuO ceramics had vastly different macrostructures when resulting from nitrate or chloride precursors. The former was characterized by dense materials containing prominent cracks and the latter by a more homogeneous foam-like structure. The XCT study allowed us to obtain a more holistic view of the ceramic structures, which could be observed in a low to medium magnification of the SEM images and be confirmed by USAXS conclusions. Moreover, SEM images at a high magnification could focus on microstructures and grain sizes and provide insight into possible polymer coalescence after metal intake and calcination. STA analysis showed that early energetic events exhibited by the nitrate salts resulted in ceramics that were more porous and fragile, while those with later decomposition events that coincided with hydrogel decomposition (e.g., chloride salts) were consistently of higher quality.
The results here provide a guide for the development of high-density ceramics using the HIAM process. Obtaining high-density ceramics is more likely when (a) a high concentration of hydrogel is employed, (b) a metal precursor which binds strongly to the hydrogel scaffold is chosen, or (c) a salt which undergoes decomposition via mild exothermic pathways is used. An understanding of the various ceramic formation mechanisms associated with the HIAM process and subsequent calcination, as presented here, is critical to the expansion of material systems and the enablement of the production of high-density ceramic components. Given the flexibility of HIAM, this technique can be rapidly exploited without the need for complex print parameter optimization, which is typical of other AM processes.
Author contributions
Natalie S. Yaw: investigation, data curation, formal analysis, visualization, and writing – original draft. Maryline G. Ferrier: conceptualization, methodology, supervision, formal analyses, visualization, and writing – review and editing. R. Joey Griffiths: investigation (SEM analyses) and visualization. Joshua A. Hammons: investigation (SAXS analyses), visualization, and writing review. Andrew J. Swift and Silvina A. Di Pietro: investigation (TGA analyses). Jean-Baptiste Forien: investigation (XCT analyses) and visualization. Bradley C. Childs: methodology and supervision. Aiden A. Martin: conceptualization, project administration, supervision, and writing – review and editing. Kiel S. Holliday: funding acquisition and project administration. Xiaofeng Guo: funding acquisition and supervision. Jason R. Jeffries: funding acquisition and project administration. The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Additional data can be accessed upon reasonable request to the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed under the auspices of the U.S. Department of Energy (DOE) by the Lawrence Livermore National Laboratory (LLNL) under Contract No. DE-AC52-07NA27344. LLNL-JRNL-867681. This material is based upon work supported under a University Nuclear Leadership Program Graduate Fellowship DE_NE0009089.References
- Z. Chen, Z. Li, J. Li, C. Liu, C. Lao, Y. Fu, C. Liu, Y. Li, P. Wang and Y. He, 3D Printing of Ceramics: A Review, J. Eur. Ceram. Soc., 2019, 39(4), 661–687, DOI:10.1016/j.jeurceramsoc.2018.11.013.
- M. Javaid and A. Haleem, Current Status and Applications of Additive Manufacturing in Dentistry: A Literature-Based Review, J. Oral Biol. Craniofacial Res., 2019, 9(3), 179–185, DOI:10.1016/j.jobcr.2019.04.004 , 179–185.
- A. H. Alami, A. G. Olabi, A. Alashkar, S. Alasad, H. Aljaghoub, H. Rezk and M. A. Abdelkareem, Additive Manufacturing in the Aerospace and Automotive Industries: Recent Trends and Role in Achieving Sustainable Development Goals, Ain Shams Eng. J., 2023, 14(11), 102516, DOI:10.1016/j.asej.2023.102516.
- A. Zocca, P. Colombo, C. M. Gomes and J. Günster, Additive Manufacturing of Ceramics: Issues, Potentialities, and Opportunities, J. Am. Ceram. Soc., 2015, 98(7), 1983–2001, DOI:10.1111/jace.13700.
- C. J. Bae, A. Ramachandran and J. W. Halloran, Quantifying Particle Segregation in Sequential Layers Fabricated by Additive Manufacturing, J. Eur. Ceram. Soc., 2018, 38(11), 4082–4088, DOI:10.1016/j.jeurceramsoc.2018.02.008.
- M. L. Griffith and J. W. Halloran, Freeform Fabrication of Ceramics via Stereolithography, J. Am. Ceram. Soc., 1996, 79(10), 2601–2608, DOI:10.1111/j.1151-2916.1996.tb09022.x.
- J. Wilkes, Y. Hagedorn, W. Meiners and K. Wissenbach, Additive Manufacturing of ZrO2–Al2O3 Ceramic Components by Selective Laser Melting, Rapid Prototyping J., 2013, 19(1), 51–57, DOI:10.1108/13552541311292736.
- H. Yves-Christian, W. Jan, M. Wilhelm, W. Konrad and P. Reinhart, Net Shaped High Performance Oxide Ceramic Parts by Selective Laser Melting, Phys. Procedia, 2010, 5, 587–594, DOI:10.1016/j.phpro.2010.08.086.
- P. Mercelis and J. Kruth, Residual Stresses in Selective Laser Sintering and Selective Laser Melting, Rapid Prototyping J., 2006, 12(5), 254–265, DOI:10.1108/13552540610707013.
- D. W. Yee, M. A. Citrin, Z. W. Taylor, M. A. Saccone, V. L. Tovmasyan and J. R. Greer, Hydrogel-Based Additive Manufacturing of Lithium Cobalt Oxide, Adv. Mater. Technol., 2021, 6(2), 2000791, DOI:10.1002/admt.202000791.
- M. A. Saccone, R. A. Gallivan, K. Narita, D. W. Yee and J. R. Greer, Additive Manufacturing of Micro-Architected Metals via Hydrogel Infusion, Nature, 2022, 612(7941), 685–690, DOI:10.1038/s41586-022-05433-2.
- P. Imrie and J. Jin, Multimaterial Hydrogel 3D Printing, Macromol. Mater. Eng., 2024, 309(2), 2300272, DOI:10.1002/mame.202300272.
- G. S. Kaliaraj, D. K. Shanmugam, A. Dasan and K. K. A. Mosas, Hydrogels—A Promising Materials for 3D Printing Technology, Gels, 2023, 9(3), 260, DOI:10.3390/gels9030260.
- Y. A. Alli, H. Anuar, A. Bamisaye, M. R. Manshor, N. O. Etafo, M. O. Bamidele, M. A. Rasheed, S. K. Olatunde, A. S. Akinfenwa and A. Lawal, The Appealing Prospect of Hydrogel in 3D/4D Printing Technology: Overview and Opportunities, Polymer, 2024, 315, 127823, DOI:10.1016/j.polymer.2024.127823.
- S. Ma, W. Bai, D. Xiong, G. Shan, Z. Zhao, W. Yi and J. Wang, Additive Manufacturing of Micro-Architected Copper Based on an Ion-Exchangeable Hydrogel, Angew. Chem., Int. Ed., 2024, 63(23), e202405135, DOI:10.1002/anie.202405135.
- A. Saigal, S. J. Velling, A. Dhawan, M. A. Baez, M. Nocum and J. R. Greer, Fabricating Machine Elements Using Hydrogel-Infused Additive Manufacturing (HIAM), in Proceedings of the ASME 2023, San Diego, California, 2023.
- A. Agrawal and C. M. Hussain, 3D-Printed Hydrogel for Diverse Applications: A Review, Gels, 2023, 9(12), 960 CrossRef CAS PubMed.
- U. S. K. Madduma-Bandarage and S. V. Madihally, Synthetic Hydrogels: Synthesis, Novel Trends, and Applications, J. Appl. Polym. Sci., 2021, 138(19), 50376, DOI:10.3390/gels9120960.
- A. S. Hoffman, Hydrogels for Biomedical Applications, Ann. NY Acad. Sci., 2001, 944(1), 62–73, DOI:10.1111/j.1749-6632.2001.tb03823.x.
- X. Yang, B. L. Dargaville and D. W. Hutmacher, Elucidating the Molecular Mechanisms for the Interaction of Water with Polyethylene Glycol-Based Hydrogels: Influence of Ionic Strength and Gel Network Structure, Polymers, 2021, 13(6), 845, DOI:10.3390/polym13060845.
- Y. Yuan, Q. Zhang, S. Lin and J. Li, Water: The Soul of Hydrogels, Prog. Mater. Sci., 2025, 148, 101378, DOI:10.1016/j.pmatsci.2024.101378.
- E. S. Jang, J. Kamcev, K. Kobayashi, N. Yan, R. Sujanani, T. J. Dilenschneider, H. B. Park, D. R. Paul and B. D. Freeman, Influence of Water Content on Alkali Metal Chloride Transport in Cross-Linked Poly(Ethylene Glycol) Diacrylate.1. Ion Sorption, Polymer, 2019, 178, 121554, DOI:10.1016/j.polymer.2019.121554.
- J. Luo, S. Ye, T. Li, E. Sarnello, H. Li and T. Liu, Distinctive Trend of Metal Binding Affinity via Hydration Shell Breakage in Nanoconfined Cavity, J. Phys. Chem. C, 2019, 123(23), 14825–14833, DOI:10.1021/acs.jpcc.9b03004.
- H. Kaşgöz, S. Özgümüş and M. Orbay, Modified Polyacrylamide Hydrogels and Their Application in Removal of Heavy Metal Ions, Polymer, 2003, 44(6), 1785–1793, DOI:10.1016/S0032-3861(03)00033-8.
- M. A. H. Badsha, M. Khan, B. Wu, A. Kumar and I. M. C. Lo, Role of Surface Functional Groups of Hydrogels in Metal Adsorption: From Performance to Mechanism, J. Hazard. Mater., 2021, 408, 124463, DOI:10.1016/j.jhazmat.2020.124463.
- Y. L. Cheng and K. C. Huang, Preparation and Characterization of Color Photocurable Resins for Full-Color Material Jetting Additive Manufacturing, Polymers, 2020, 12(3), 650, DOI:10.3390/polym12030650.
- Y. Guo, Z. Ji, Y. Zhang, X. Wang and F. Zhou, Solvent-Free and Photocurable Polyimide Inks for 3D Printing, J. Mater. Chem. A, 2017, 5, 16307–16314, 10.1039/C7TA01952A.
- A. S. Alketbi, Y. Shi, H. Li, A. Raza and T. Zhang, Impact of PEGDA Photopolymerization in Micro-Stereolithography on 3D Printed Hydrogel Structure and Swelling, Soft Matter, 2021, 17(30), 7188–7195, 10.1039/D1SM00483B.
- L. Fan, M. Degen, S. Bendle, N. Grupido and J. Ilavsky, The Absolute Calibration of a Small-Angle Scattering Instrument with a Laboratory X-Ray Source – IOPscience, J. Phys.: Conf. Ser., 2010, 247, 012005, DOI:10.1088/1742-6596/247/1/012005.
- J. Ilavsky, Nika: Software for Two-Dimensional Data Reduction, J. Appl. Crystallogr., 2012, 45(2), 324–328, DOI:10.1107/S0021889812004037.
- J. Ilavsky and P. R. Jemian, Irena: Tool Suite for Modeling and Analysis of Small-Angle Scattering, J. Appl. Crystallogr., 2009, 42(2), 347–353, DOI:10.1107/S0021889809002222.
- U. Täffner, V. Carle, R. Schäfer and M. J. Hoffmann, Preparation and Microstructural Analysis of High-Performance Ceramics, Metallography and Microstructures, ed. G. F. Vander Voort, ASM International, 2004, vol. 9, pp. 1057–1066. DOI:10.31399/asm.hb.v09.a0003795.
- P. N. De Aza, A. H. De Aza and S. De Aza, Crystalline Bioceramic Materials, Bol. Soc. Esp. Ceram. Vidrio, 2005, 44(3), 135–145, DOI:10.1002/chin.200629272.
- R. Nisticò, Zirconium Oxide and the Crystallinity Hallows, J. Aust. Ceram. Soc., 2021, 57(1), 225–236, DOI:10.1007/s41779-020-00529-2.
- R. Winters, C. Doyle, A. Lupercio, A. Nelson and B. Jaques, Effects of Oxide Additives on the Microstructure of Surrogate Nuclear Fuels, 2020 Undergraduate Research Showcase, 2020.
- D. J. Waters, K. Engberg, R. Parke-Houben, L. Hartmann, C. N. Ta, M. F. Toney and C. W. Frank, Morphology of Photopolymerized End-Linked Poly(Ethylene Glycol) Hydrogels by Small-Angle X-Ray Scattering, Macromolecules, 2010, 43(16), 6861–6870, DOI:10.1021/ma101070s.
- B. L. Dargaville and D. W. Hutmacher, Water as the Often Neglected Medium at the Interface between Materials and Biology, Nat. Commun., 2022, 13(1), 4222, DOI:10.1038/s41467-022-31889-x.
- N. C. Padmavathi and P. R. Chatterji, Structural Characteristics and Swelling Behavior of Poly(Ethylene Glycol) Diacrylate Hydrogels, Macromolecules, 1996, 29(6), 1976–1979, DOI:10.1021/ma950827r.
- J. Mercado-Montijo, D. M. Anstine, S. J. Rukmani, C. M. Colina and J. S. Andrew, PEGDA Hydrogel Structure from Semi-Dilute Concentrations: Insights from Experiments and Molecular Simulations, Soft Matter, 2022, 18(18), 3565–3574, 10.1039/D1SM01708J.
- Y. Zhu, Y. Zheng, F. Wang and A. Wang, Monolithic Supermacroporous Hydrogel Prepared from High Internal Phase Emulsions (HIPEs) for Fast Removal of Cu2+ and Pb2+, Chem. Eng. J., 2016, 284, 422–430, DOI:10.1016/j.cej.2015.08.157.
- Y. C. Chiu, M. H. Cheng, H. Engel, S. W. Kao, J. C. Larson, S. Gupta and E. M. Brey, The Role of Pore Size on Vascularization and Tissue Remodeling in PEG Hydrogels, Biomaterials, 2011, 32(26), 6045–6051, DOI:10.1016/j.biomaterials.2011.04.066.
- N. Sari, E. Kahraman, B. Sari and A. Özgün, Synthesis of Some Polymer–Metal Complexes and Elucidation of Their Structures, J. Macromol. Sci., Part A: Pure Appl. Chem., 2006, 43(8), 1227–1235, DOI:10.1080/10601320600737484.
- N. Kaltsoyannis and P. Scott, The f Elements, Oxford Chemistry Primers, Oxford University Press, 1999 Search PubMed.
- S. A. Cotton, Establishing Coordination Numbers for the Lanthanides in Simple Complexes, C. R. Chim., 2005, 8(2), 129–145, DOI:10.1016/j.crci.2004.07.002.
- B. J. Elliott, A. B. Scranton, J. H. Cameron and C. N. Bowman, Characterization and Polymerization of Metal Complexes of Poly(Ethylene Glycol) Diacrylates and the Synthesis of Polymeric Pseudocrown Ethers, Chem. Mater., 2000, 12(3), 633–642, DOI:10.1021/cm990247z.
- R. D. Rogers, M. M. Benning, R. D. Etzenhouser and A. N. Rollins, Novel Unidentate Co-Ordination of a Crown Ether and of a Polyethylene Glycol to Uranium(VI), J. Chem. Soc., Chem. Commun., 1989, 20, 1586, 10.1039/c39890001586.
- I. Farkas, I. Bányai, Z. Szabó, U. Wahlgren and I. Grenthe, Rates and Mechanisms of Water Exchange of UO22+(Aq) and UO2(Oxalate)F(H2O)2-: A Variable-Temperature 17O and 19F NMR Study, Inorg. Chem., 2000, 39(4), 799–805, DOI:10.1021/ic990929o.
- P. Nichols, E. J. Bylaska, G. K. Schenter and W. de Jong, Equatorial and Apical Solvent Shells of the UO22+ Ion, J. Chem. Phys., 2008, 128(12), 124507, DOI:10.1063/1.2884861.
- S. Fortier and T. W. Hayton, Oxo Ligand Functionalization in the Uranyl Ion (UO22+), Coord. Chem. Rev., 2010, 254(3), 197–214, DOI:10.1016/j.ccr.2009.06.003.
- L. A. Watson and B. P. Hay, Role of the Uranyl Oxo Group as a Hydrogen Bond Acceptor, Inorg. Chem., 2011, 50(6), 2599–2605, DOI:10.1021/ic102448q.
- A. Ekstrom, Kinetics and Mechanism of the Disproportionation of Uranium(V), Inorg. Chem., 1974, 13(9), 2237–2241, DOI:10.1021/ic50139a035.
- Y. Marcus and G. Hefter, Ion Pairing, Chem. Rev., 2006, 106(11), 4585–4621, DOI:10.1021/cr040087x.
- Y. Gao, F. Haso, J. E. S. Szymanowski, J. Zhou, L. Hu, P. C. Burns and T. Liu, Selective Permeability of Uranyl Peroxide Nanocages to Different Alkali Ions: Influences from Surface Pores and Hydration Shells, Chem. – Eur. J., 2015, 21(51), 18785–18790, DOI:10.1002/chem.201503773.
- L. Helm and A. E. Merbach, Inorganic and Bioinorganic Solvent Exchange Mechanisms, Chem. Rev., 2005, 105(6), 1923–1960, DOI:10.1021/cr030726o.
- R. Sartori, L. Sepulveda, F. Quina, E. Lissi and E. Abuin, Binding of Electrolytes to Poly(Ethylene Oxide) in Aqueous Solutions, Macromolecules, 1990, 23(17), 3878–3881, DOI:10.1021/ma00219a002.
- M. C. Ball and R. F. M. Coultard, Thermal Studies on Halides and Basic Halides of Copper(II), J. Chem. Soc. A, 1968, 1417, 10.1039/j19680001417.
- Z. Ding, W. Martens and R. L. Frost, Thermal Activation of Copper Nitrate, J. Mater. Sci. Lett., 2002, 21(18), 1415–1417, DOI:10.1023/A:1019962632204.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures (process, Cu, Ce and UO2 infiltrated gels pre- and post-calcination, phase analysis of ceramics resulting from the HIAM process via XRD and SAXS, SEM images of blister appearance in consolidated and non-consolidated systems, SEM and XCT reconstruction of Cu, Ce, and ZrO ceramics, and STA of Cu salts), as well as tables of decomposition pathways of Ce, Zr oxo and UO2(NO3)2 salts (PDF). See DOI: https://doi.org/10.1039/d5qi00139k |
| This journal is © the Partner Organisations 2025 |