
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpcycling of PET waste: from one polymer to another polymer
Te
Yang
ab,
Zhenjie
Yang
*a,
Yulu
Zhang
ab,
Chenyang
Hu
 a,
Zhenbiao
Xie
ab,
Zhiqiang
Sun
a,
Xuan
Pang
*ab and
Xuesi
Chen
ab
a,
Zhenbiao
Xie
ab,
Zhiqiang
Sun
a,
Xuan
Pang
*ab and
Xuesi
Chen
ab
aState Key Laboratory of Polymer Science and Technology, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: zjyang@ciac.ac.cn; xpang@ciac.ac.cn
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei 230026, China
First published on 26th November 2025
Abstract
Polyethylene terephthalate (PET), a dominant polymer in global plastic production, faces critical recycling challenges due to its persistence in ecosystems and limitations of conventional mechanical/thermal recycling. Upcycling PET waste into value-added polymers represents a transformative approach toward a circular plastics economy. This review systematically examines innovative strategies for chemically converting post-consumer PET into novel polymeric materials, thereby bypassing the performance degradation typically associated with traditional recycling. Key pathways include (1) depolymerization into monomers (terephthalic acid, ethylene glycol) for repolymerization into high-purity PET or advanced polyesters (e.g., biodegradable or bio-based variants), (2) transformation into functional polymers such as polyurethanes, epoxy resins, and ion-exchange membranes via tailored catalytic processes, and (3) copolymerization/blending with biopolymers to enhance material properties. Breakthroughs in catalysts (enzymes, ionic liquids), solvent-free systems, and energy-efficient reactors are highlighted for improving the reaction selectivity and scalability. Despite progress, challenges persist in managing mixed plastic wastes, removing contaminants, and achieving cost parity with virgin polymers. Emerging trends, including enzymatic engineering and AI-guided monomer-to-polymer design, are proposed to address these barriers. By bridging molecular innovation with industrial feasibility, PET upcycling offers dual environmental and economic incentives to close the plastic lifecycle loop.
1. Introduction
Polyethylene terephthalate (PET), a ubiquitous thermoplastic polymer, has become a cornerstone of modern packaging and textile industries due to its exceptional durability, light weight nature, and cost-effectiveness.1 Global PET production exceeds 70 million tons annually, accounting for approximately 18% of total plastic waste.2,3 Alarmingly, only less than 20% of postconsumer PET is effectively recycled, while the majority accumulates in landfills or natural environments, persisting for centuries due to its resistance to hydrolysis and microbial degradation.4,5 The fragmentation of PET waste into microplastics (<5 mm) exacerbates ecological risks, contaminating marine ecosystems and entering the food chain.6–8 A recent study reported that the detection rate of PET microplastics in human blood and tissues has reached up to 50% of all samples.9 Despite being one of the recyclable plastics, PET's recalcitrant aromatic backbone and semi-crystalline structure make conventional waste management strategies insufficient to address its environmental footprint.10–12Traditional PET recycling methods, while widely implemented, face critical technical and economic limitations.12,13 Mechanical recycling—grinding, washing, and remelting PET waste—inevitably degrades molecular weight (Mw) and thermal stability due to chain scission, restricting recycled PET (rPET) to low-value applications such as fibers or non-food containers.14–16 Landfilling and incineration, though prevalent, exacerbate carbon emissions (e.g., 2.7 kg CO2 per kg PET incinerated) and soil contamination. These methods perpetuate a linear “downcycling” model, where the material value diminishes with each cycle.17–19 For instance, mechanically recycled PET bottles typically undergo only 2–3 cycles before becoming unusable, highlighting the unsustainability of current practices.20
Conventional chemical upcycling—via hydrolysis, glycolysis, or alcoholysis to monomers (e.g., terephthalic acid [TPA], bis(2-hydroxyethyl) terephthalate [BHET])—has been explored to bypass mechanical recycling limitations.21–23 However, these routes typically require energy-intensive conditions (>200 °C, 1–4 MPa) and corrosive reagents, leading to high costs, safety hazards, and toxic byproducts.24–27 Currently, much effort has been dedicated to developing milder and efficient methods for upcycling of waste PET to monomers or small molecules. For example, emerging ionic liquid-mediated glycolysis (e.g., [BMIM]Cl with ZnCl2) enhances BHET recovery (>95% at 180 °C) while suppressing side reactions. Electrochemical upcycling further demonstrates the potential to bypass energy-intensive steps and generate significant market profits by converting PET plastic waste into high-value products like hydroxy fatty acids and formamides.28–30 Similarly, bio-upcycling strategies like enzymatic technology produce virgin-quality PET from mixed textile and bottle waste, achieving 97% depolymerization efficiency while reducing energy consumption by 50% compared to conventional methods.27,31 Even with monomer recovery efficiencies >90%, energy-intensive purification (e.g., TPA distillation) and repolymerization into low-value rPET still trap materials in downcycling loops.32
Crucially, emerging “polymer-to-polymer” upcycling strategies now bypass traditional recycling bottlenecks by converting PET waste into high-value polymeric architectures which have drawn much attention in recent years.33,34 These strategies mainly proceed via two core pathways, which are the focus of this review: (i) monomer/oligomer-mediated conversion, where PET is depolymerized into monomers or oligomers like BHET and terephthalate oligomers that are then repolymerized into high-value polymers rather than low-value rPET from conventional monomer recovery. (ii) Direct reconfiguration, where PET chains are restructured in situ into new polymers without isolating monomers. Both strategies can meet the increasing demand for both enhanced performance and energy and cost efficiency. First, by the depolymerization and repolymerization strategy, the PET waste could be upcycled as intermediates for high-value synthesis. Secondly, with direct routes, the energy-intensive monomer purification could be circumvented and the PET structural motifs could be preserved to maintain or enhance mechanical properties. Thirdly, these strategies could enable tailored functionalities via monomer introduction.24,33,35 In this review, we briefly contextualize broader PET upcycling before focusing in detail on these two polymer-to-polymer pathways (Fig. 1). We analyzed the newly reported monomer/oligomer repolymerization routes and direct reconfiguration routes that address challenges in catalyst design, process economics, and lifecycle assessment. Furthermore, outlooks and perspectives on the need for bridging the gap between laboratory-scale innovations and scalable solutions were also proposed. We hope this review could draw more attention to the technologies that redefine waste PET as a new feedstock for high-value products and inspire new ideas addressing the end-of-use issues of waste polymer materials.
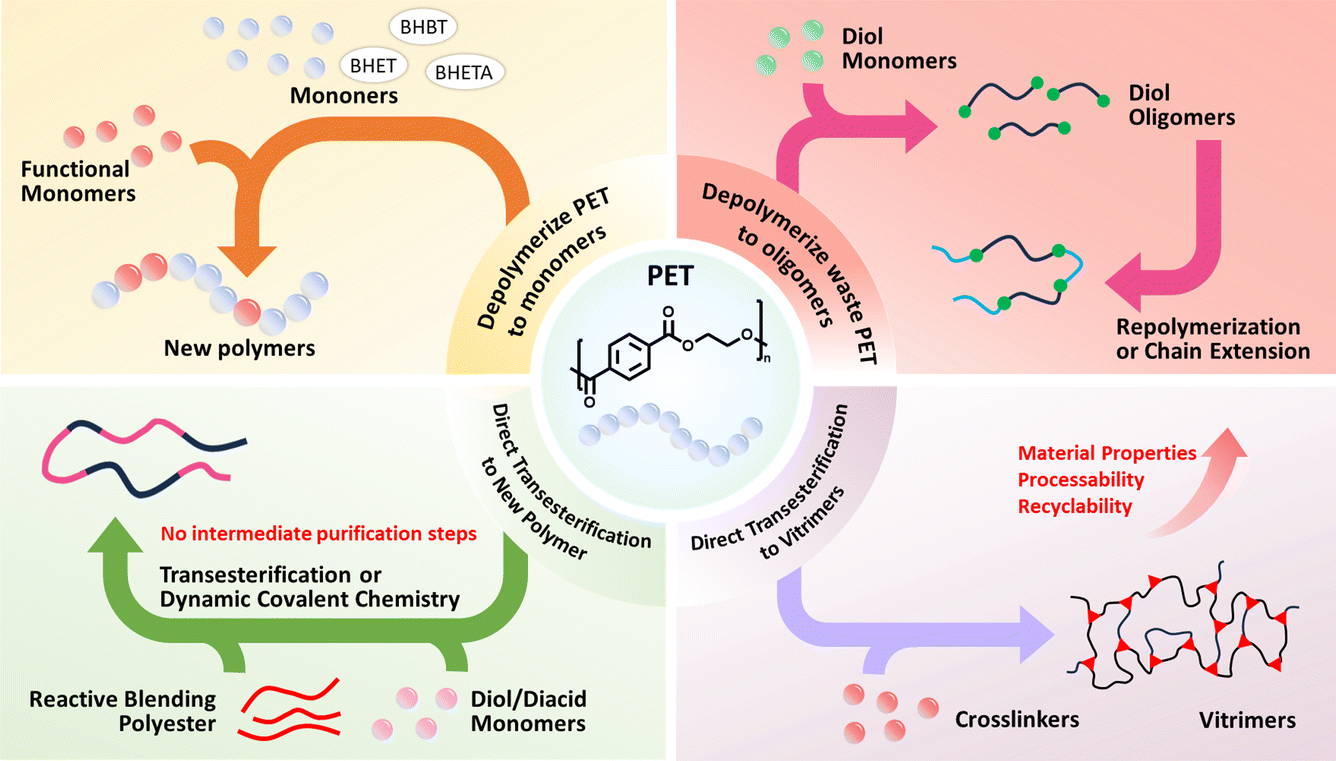 | ||
| Fig. 1 Overview of recycling PET into new polymers. | ||
2. Polymer to polymer upcycling technologies of waste PET
2.1 Depolymerization and repolymerization strategy
Cleaving the ester bonds of PET to obtain small molecules or oligomers has long been a well-established method in PET recycling. In the last few decades, extensive catalyst development has enabled efficient chemical depolymerization of PET, primarily yielding the corresponding monomers and oligomers. Chemists have since leveraged these catalytic approaches for closed-loop repolymerization to synthesize rPET, or alternatively upcycling them into novel polymeric materials. Distinct from conventional monomer recovery, the core of this strategy lies in synergistic catalyst design and process integration to achieve two critical goals: (i) efficient depolymerization of PET waste into high-purity monomers/oligomers under mild conditions; (ii) controlled repolymerization into polymers with tailored performance including enhanced mechanical strength, thermal stability, or functionality for high-value applications.In general, the repolymerization equilibrium is driven forward by efficient removal of the byproduct (EG or H2O) under high temperature (∼270 °C) and vacuum (<1 mbar) conditions, ensuring that the molecular weight and intrinsic viscosity meet industrial standards for PET applications. To reduce the energy cost of this recycling process, advanced catalysts including metal complexes, organic catalysts and ionic liquids have been developed (Table 1).36–40 However, metal catalysts or ionic liquids are difficult to separate from the products, causing the minimization of the adverse impact of catalyst residues on the properties of rPET critical. Recent studies, therefore, have focused on the development of greener and sustainable catalyst systems. Yu et al. employed Ti–Si glycolate during PET glycolysis and repolymerization, yielding rPET with mechanical properties comparable to virgin PET synthesized directly from TPA and EG.41 Additionally, the Si component in the catalyst effectively regulates the catalytic activity of Ti, thereby suppressing yellowing in the rPET product. Zhang et al. developed a supported binuclear Zn catalyst that is readily separable from the reaction system.5 This catalyst enables efficient PET depolymerization under mild conditions, meets industrial depolymerization efficiency requirements, and exhibits excellent recyclability—maintaining high depolymerization activity even after 15 reuse cycles. The bottle-grade rPET produced from the recovered monomers demonstrates performance comparable to commercial PET. To avoid metal residues, Kaiho et al. identified a high-stability organic salt (TBD/p-TSA) that enables efficient depolymerization of PET into monomeric BHET at 180 °C with a conversion rate exceeding 92%.42 The entire process eliminated the need for purification, and the residual catalyst can be directly recycled, thus realizing a “plastic-to-plastic” closed-loop green recycling pathway (Fig. 2a). The development of catalysts that are not required to be removed and are easily separable or recyclable, while balancing the economic viability and environmental sustainability, represents a key trend in future catalyst research.
 | ||
| Fig. 2 Chemical recycling of PET to BHET for synthesis of new polymers. | ||
| Feedstock | Cat. | Cat. loadinga | Temperature | Time | Depolymerization extent | Product | Ref. |
|---|---|---|---|---|---|---|---|
PET![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEG = 1:10 (mol) :DEG = 1:10 (mol) |
Mn(OAc)2·4H2O | 0.5 wt% | Microwave irradiation 150 W | 2 min | PET depolymerization 100% | Oligoester diol | 37 |
| PET:DEG = 1:10 (mol) |
Mn(OAc)2·4H2O | 0.5 wt% | 180 °C | 5 min | PET depolymerization 100% | Oligoester diol | |
| PET:EG = 1:6 (mol) |
Na2SO4 | 0.5 wt% | 195 °C | 8 h | Yield of BHET 65.72% | BHET | |
| PET:EG = 1:16 (mol) |
DBU | 10 mol% | 160 °C | 20 min | PET depolymerization 100% | BHET | 38 |
| TBD | 10 mol% | 160 °C | 110 min | PET depolymerization 100% | BHET | ||
| PET:EDAc = 1:16 (mol) |
DBU | 5 mol% | 110 °C | 2 h | Yield of BAETA 77% | BAETAd | |
| TBD | 5 mol% | 110 °C | 2 h | Yield of BAETA 89% | BAETA | ||
| PET:EA = 1:12 (mol) |
TBD | 10 mol% | 165 °C | 10 min | Yield of BHETA 90% | BHETA | |
| PET:EG = 1:4 (wt) |
[EMIm]2TPA | 5 wt% | 197 °C | 106 min | PET depolymerization 100% | BHET | 40 |
| Yield of BHET 83.6% |
| Feedstock | Cat. | Cat. loadingb | Temperature | Time | Molecular weight characterization parameters | Product | Ref. |
|---|---|---|---|---|---|---|---|
| a Mol% relative to the repeating unit of PET; wt% relative to the PET mass. b Mol% relative to the monomers listed in the “feedstock” column. c EDA – ethylenediamine. d BAETA – N,N′-bis-(2-amino-ethyl)-terephthalamide. e TPC – terephthaloyl acid chloride, IPC – isophthalic acid chloride, and BPA – bisphenol A. f PTA – p-Phthalic acid. | |||||||
| BHET | KTiOX | 0.02 mol% | 290 °C | 1 h | 0.62 (intrinsic viscosity) | rPET | 36 |
| SnO(nBu)2 | 0.02 mol% | 290 °C | 1 h | 0.57 (intrinsic viscosity) | rPET | ||
| Sb2O3 | 0.02 mol% | 290 °C | 1 h | 0.52 (intrinsic viscosity) | rPET | ||
| Mn-acetate | 0.02 mol% | 290 °C | 1 h | 0.49 (intrinsic viscosity) | rPET | ||
| Zn-acetate | 0.02 mol% | 290 °C | 1 h | 0.44 (intrinsic viscosity) | rPET | ||
| Pb-acetate | 0.02 mol% | 290 °C | 1 h | 0.44 (intrinsic viscosity) | rPET | ||
| No catalyst | 0.02 mol% | 290 °C | 1 h | 0.23 (intrinsic viscosity) | rPET | ||
| TPC:IPC:BPAe = 30:30:60 (mol) |
Butyltrimethylaminium chloride | 0.83 mol% | 15–18 °C | 3–4 h | 1.38–1.77 × 104 (Mn) | PAR | 39 |
| 4.91–6.38 × 104 (Mw) | |||||||
| BHET | Poly(antimony ethylene glycoxide) | — | 260–280 °C | 3.5 h | — | rPET | 40 |
| PTAf | Ti/Si-EG | 0.015 mol% | 260–280 °C | — | 1.10 × 104 (Mn) | rPET | 41 |
| 1.76 × 104 (Mw) | |||||||
| BHET | Ti/Si-EG | 0.015 mol% | 260–280 °C | — | 1.15 × 104 (Mn) | rPET | |
| 1.85 × 104 (Mw) | |||||||
| PTA | Robson-type binuclear zinc catalyst | — | — | — | 1.90 × 104 (Mn) | rPET | 5 |
| 3.89 × 104 (Mw) | |||||||
| BHET | TBD:p-TSA |
5 mol% | 270 °C | 5 h | 4.14 × 104 (Mn) | rPET | 42 |
Beyond the conventional strategy of repolymerizing depolymerized monomers to regenerate rPET, the integration of these monomers with other functionalized molecules has emerged as a transformative pathway to access novel materials with distinct properties from PET. Karanastasis et al. successfully synthesized thermoplastic copolyesters (TPCs) by using bis(2-hydroxyethyl) terephthalate (BHET) as the hard segment and a bulky aliphatic renewable dimer fatty acid (DFA) as the soft segment (Fig. 2b).43 The bis(2-hydroxyethyl) of BHET can react with the diacid functional group of the DFA monomer to form the structural component of the repeating unit of the soft segments. De Dios Caputto et al. reported the reaction of BHET and the inexpensive organic solvent ethylene carbonate (EC) to prepare liquid polyols with different chemical compositions and similar molecular weights (Fig. 2c).44 These reactions could be catalyzed by various organic bases under mild reaction conditions (150 °C, 8 h). It was found that the 1,5,7-triazabicyclo [4.4.0] dec-5-ene (TBD)/methanesulphonic acid (MSA) catalytic system showed the optimal organocatalytic efficiency, with one carbonate group being introduced into the polyol chain for every two EC molecules consumed (Fig. 3).
 | ||
| Fig. 3 Chemical composition and molecular weight of polyols from BHET using organocatalysts. Reproduced from ref. 44 with permission from Elsevier, copyright 2024. | ||
The catalytic depolymerization of waste PET into new small molecules, followed by their reconstruction into high-performance polymers, has also emerged as a promising pathway to break through the limitations of conventional recycling methods. Lyu et al. obtained bis(4-hydroxybutyl) terephthalate (BHBT) using a 1,4-butanediol (1,4-BDO) and chlorobenzene reaction system to depolymerize PET at a low temperature of 120 °C, catalyzed by K2CO3.45 Then they prepared polyurethane (PU) thermosets with closed-loop recycling potential based on the BHBT monomer in combination with the bio-based pentamethylene diisocyanate (PDI) trimer. Distinct from glycolysis or methanolysis, the aminolysis approach yielded terephthalamides with well-defined aromatic–aliphatic structures, which could also be used for the synthesis of novel polyurethane materials (Fig. 4a and d). Tan et al. reported the aminolysis of PET using ethanolamine and organocatalytic TBD and obtained bis(2-hydroxyethyl) terephthalamide (BHETA) as the monomer.46 The resulting BHETA monomer was then copolymerized with poly(ethylene glycol) (PEG) and hexamethylene diisocyanate (HMDI) to form a series of polyurethane materials with different hard and soft segment ratios (Fig. 4b). Interestingly, these PET-derived polyurethanes showed great potential to be used as polymer electrolytes in lithium-ion batteries (Fig. 4e). Moreover, the amide bonds in BHETA could endow the corresponding BHETA-based polymer with hydrophilicity and adhesive ability. Zhang et al. developed a method to convert waste PET into BHETA using acetic acid swelling and mild ethanolamine aminolysis.47 The BHETA-based adhesive was subsequently prepared by crosslinking with diphenylmethane diisocyanate (MDI) (Fig. 4c). Experimental results demonstrated that the adhesive exhibits exceptional bonding performance on wood substrates, achieving a lap shear strength of 2.5 MPa (Fig. 4f), which originates from the synergistic effects of abundant hydrogen bonds and π–π stacking interactions within the material.
 | ||
| Fig. 4 (a–c) Utilizing glycols or alkanolamines to depolymerize PET into diols for polymer synthesis. (d) Closed-loop recycling of the BHBT-PU film. Reproduced from ref. 45 with permission from Elsevier, copyright 2024. (e) Room-temperature galvanostatic charge–discharge profiles of the Li-ion cell fabricated with the gel polymer electrolyte (1BHETA: 2PEG + 38% EMI-TFSI), LTO anode, and LFP cathode; the corresponding cycling performance showing the coulombic efficiency and charge/discharge capacity over 150 cycles. Reproduced from ref. 46 with permission from Royal Society of Chemistry, copyright 2022. (f) Lap-shear strength of P-PUa with different PEG contents, and the lap-shear strength of P-PUa60 on the wood surface after multiple cycles. Reproduced from ref. 47 with permission from Elsevier, copyright 2024. | ||
As discussed in the previous section, A. M. Fernandez et al. obtained polyols through the reaction of EC with BHET. They also investigated the direct reaction of PET waste with EC catalyzed by KOH or various organic bases to produce polyols (Fig. 5a).44 The resulting products exhibited stable molecular weights of around 1700 g mol−1, making them ideal raw materials for synthesizing thermoplastic polyurethanes. Notably, the TBD:MSA salt remained inert during PET recycling, which might be attributed to the strong acidity of MSA causing complete protonation of the TBD base. This protonation prevented coordination between TBD and either PET or EC, thereby inhibiting the ring-opening polymerization of EC (Fig. 6). In 2019, Rorrer et al. used EG to depolymerize PET into terminally hydroxylated oligomers catalyzed by titanium butoxide.48 These oligomers were then synthesized by reacting with renewable olefinic acid monomers to obtain unsaturated polyesters (UPEs) or diacrylate polymers (Fig. 5b and c). The resulting polymers were dissolved in a reactive diluent containing a free radical initiator to form a resin, which was coated onto glass fiber mats to produce a series of fibre-reinforced plastics (FRPs).
 | ||
| Fig. 5 (a) Synthesis of polyols via reaction of EC with waste PET. (b) Synthesis of FRPs through crosslinking of UPE obtained via glycolysis of PET followed by the reaction with olefinic diacids. (c) Synthesis of FRPs through crosslinking of the PET-based diacrylic polymer synthesized via glycolysis of PET followed by the reaction with olefinic monoacids. | ||
 | ||
| Fig. 6 Chemical composition and molecular weight of polyols derived from PET waste within 8 h using varying basic organocatalysts and KOH. Reproduced from ref. 44 with permission from Elsevier, copyright 2024. | ||
Beyond conventional diols like EG and propylene glycol for PET depolymerization, specially engineered diols enable precise tuning of polymer properties including flexibility, hydrophilicity/hydrophobicity, and optical characteristics. In 2021, Waskiewicz et al. obtained oligomeric glycol via the polycondensation of diethylene glycol (DEG) and DFAs, which was then used to degrade waste PET and synthesize a new type of oligoesterdiol for use in the development of polyurethane coating binders.49 It is found that the incorporation of DFA could suppress the crystallization tendency during storage and significantly enhance the hydrophobic properties of adhesive coatings (Fig. 7a). These DFA-derived coatings exhibit a performance comparable to those prepared from petroleum-based feedstocks such as adipic acid, DEG, and phthalic anhydride. In 2023, Fan et al. synthesized PG monomers with dual photoresponsive features (fluorescence and photocrosslinking under UV irradiation) containing diphenylacetylene and bis-hydroxyl functional groups and further developed a one-pot “oligomer-scale” in situ chemical upcycling technology.50 After transesterification of the PG monomer with PET to obtain oligomers with end-modified PG and re-polymerization under vacuum conditions, UV-resistant materials with improved mechanical strength were finally obtained (Fig. 7b and d). Liu et al. reported a cardanol-based diol containing a tertiary amine structure and two primary hydroxyl groups, which enabled the glycolysis of PET into diol-type oligomers within 20 minutes.51 Without purification, the resulting diol could be directly reacted with isophorone diisocyanate (IPDI) and pentaerythritol triacrylate (PETA) to produce PET-based PUA UV-curable coatings with excellent mechanical properties (Fig. 7c and e). These oligomer-mediated upcycling strategies circumvent energy-intensive monomer purification challenges and unlock precise control over polymer architectures, enabling tailorable material performance.
 | ||
| Fig. 7 (a–c) Depolymerization of PET into oligomers using functionalized diols followed by upcycling and repolymerization into new polymers. (d) Schematic illustration of the dual photo-response of the diphenylacetylene-containing PET-PG under UV irradiation; tensile strength of the original and UV-irradiated PET and upcycled PET-PG. Reproduced from ref. 50 with permission from John Wiley and Sons, copyright 2023. (e) Mechanical and coating properties of the rPET-PUA coating obtained through UV irradiation curing. Reproduced from ref. 51 with permission from Royal Society of Chemistry, copyright 2023. | ||
2.2 Catalytic direct reconfiguration to new polymers
Except for the monomer- or oligomer-based depolymerization–repolymerization routes, the catalytic direct configuration of PET into new polymers has attracted considerable interest. These pathways directly convert waste polymers into high-value materials in one pot or one step via molecular rearrangement or network reconstruction, using transesterification or dynamic covalent chemistry. Benefiting from advanced catalytic systems, these approaches eliminate intermediate purification steps and avoid the high energy consumption of conventional depolymerization–repolymerization cycles. Consequently, they enable more efficient production of high-value recycled materials. | ||
| Fig. 8 (a) One-pot upcycling of rPET into segmented TPCs using the renewable DFA as the soft building block. (b) Upcycling of PET into antibacterial PET14C using the [C14-EG]5 oligomer. (c) Mechanical properties and stress–strain curves of TPCs. Reproduced from ref. 43 with permission from American Chemical Society, copyright 2022. (d) Antibacterial and thermal properties of the recycled PET14C. Reproduced from ref. 52 with permission from John Wiley and Sons, copyright 2023. | ||
In 2023, Zhang et al. synthesized a quaternary ammonium salt (QAC) antibacterial monomer, 1-tetradecyl-3-methylimidazolium 5-sulfoisophthalate ([C14][SIPA]), featuring enhanced thermostability through the incorporation of a diaromatic ring structure.52 This monomer was pre-polymerized with EG to form the [C14-EG]5 oligomer. Leveraging a solid-state reaction (SSR)-based PET waste upcycling strategy, [C14-EG]5 underwent transesterification with waste PET catalyzed by the intrinsic Sb2O3 catalyst present in PET bottles, achieving molecular weight enhancement without requiring additional catalysts (Fig. 8b). The resulting recycled PET exhibits persistent antibacterial efficacy (>95% activity retention after 30 washing cycles) while maintaining thermal stability comparable to the virgin PET material (Fig. 8d).
Inspired by the recent advances in PET acidolysis,53 researchers have leveraged the transesterification principle between carboxylic acids and ester bonds, coupled with sublimation of excess diacid monomers, to achieve polymer-to-polymer upcycling of PET. In 2024, Fang et al. reported a novel PET upcycling strategy using macromolecular carboxyl-ester transesterification.54 This method utilizes waste PET and non-volatile bio-based hydrogenated dimer acid (HDA) as feedstocks. Through melt polymerization under green conditions, it directly synthesizes biodegradable poly(ethylene hydrogenated dimerate-co-terephthalate) (PEHT) (Fig. 9a). Crucially, the process leverages residual antimony (Sb) catalysts inherent in PET waste, without additional catalysts or solvents. Distinct from conventional depolymerization–repolymerization pathways, this strategy enables precise property modulation via controlled reorganization of hard segments (terephthalate units) and soft segments (hydrogenated dimerate units) within PET macromolecular chains. The resulting PEHT materials exhibit mechanical strength comparable to commercial PBAT (tensile strength >30 MPa) (Fig. 9d), significantly enhanced processability (40% increase in melt flow index), and excellent compostability (>95% depolymerization within 60 weeks). Notably, this process demonstrates full compatibility with existing polyester industrial equipment and achieves 100% recovery of the byproduct terephthalic acid (TPA) into the production chain, realizing an atom-efficient circular economy.
 | ||
| Fig. 9 (a) Synthesis of biodegradable PEHT and recovery of high-purity TPA via reaction of HDA with PET. (b) Synthesis of PECT via conventional transesterification between CHDM and PET. (c) Synthesis of PECTOPA via switch-mediated polyesterification with the intervention of OPA. (d) Mechanical properties and processability of PEHTs. Reproduced from ref. 54 with permission from John Wiley and Sons, copyright 2024. (e) Mechanical properties of PECTs and PECTOPA; molecular weight change of PECTOPA under melt polycondensation conditions. Reproduced from ref. 55 with permission from John Wiley and Sons, copyright 2025. | ||
In 2025, Zhang et al. developed an innovative strategy based on a carboxyl-anhydride molecular switch, achieving efficient upcycling of waste PET into high-value, closed-loop recyclable poly(ethylene-co-1,4-cyclohexanedimethanol terephthalate) (PECT) (Fig. 9b).55 By regulating the hydroxyl/carboxyl ratio in polycondensation systems through dynamic ring-closing/opening reactions of ortho-phthalic acid (OPA), this research accomplished cyclic PET depolymerization and PECT repolymerization within a single-reactor melt polycondensation process (Fig. 9c). Experimental validation confirmed that the OPA molecular switch enables controllable molecular weight modulation: during depolymerization (reduced from 51 to 6 kDa within 1 hour) and repolymerization (recovered to 64 kDa), without requiring complex purification steps or excess solvents. The resulting PECT materials exhibit superior mechanical properties (enhanced impact strength versus virgin PET) and solvent processability, with a 24% reduction in recycling costs compared to conventional methods while demonstrating scalability compatible with industrial equipment (Fig. 9e). This work establishes a novel molecular design paradigm for closed-loop recycling of condensation polymers, offering a scientifically sound and engineering-feasible solution to address plastic pollution challenges.
On the other hand, utilizing catalytic transesterification to synthesize novel copolymers through reactive blending of PET with other polyesters also represents a cost-effective strategy. In 2023, Leung et al. successfully achieved random copolymerization of PET and polycaprolactone (PCL) through catalytic transesterification via melt blending at 280 °C.56 The study revealed that titanium-based catalysts (Ti(OBu)4) exhibited optimal catalytic activity, effectively promoting transesterification to form short-block copolymers poly((ET)x-co-(CL)ᵧ) with a structure similar to PBAT. In 2024, building upon previous research, they further elucidated the mechanism of statistical copolymer equilibrium formation in PET/PCL copolymers via catalytic transesterification (Fig. 10).57 The study confirmed that the feedstock ratio serves as the critical parameter for regulating the PET block length, while secondary factors like catalyst loading lose efficacy post-equilibrium. Based on the reaction probability hypothesis, the diminished availability of PCL ester groups was demonstrated to drive equilibrium through reverse reaction dominance. An optimized formulation (65% PET + 1.1–1.5 pph catalyst loading) successfully yielded copolymers with mechanical properties approaching commercial PBAT, accompanied by a proposed chain extension modification strategy to address ductility limitations.
 | ||
| Fig. 10 Synthesis of poly((ET)x-co-(CL)y) via transesterification of PET and PCL through melt blending. | ||
In 2021, Qiu et al. developed continuously reprocessable vitrimer materials by integrating PET with a tertiary amine-containing polyol, 2,2-bis(hydroxymethyl)-2,2′,2″-nitrilotriethanol (BIS-TRIS), and diglycidyl ether of bisphenol A (DGEBA) via reactive extrusion technology (Fig. 11a).59 Dynamic transesterification formed crosslinked networks, significantly enhancing the creep resistance by 2–3 fold versus pure PET (Fig. 11c). The hydroxyl and tertiary amine groups autocatalyze dynamic transesterification during processing. This enables reprocessing via compression molding, extrusion, and injection molding. In 2023, Fabrizio et al. achieved upcycling of low molecular weight PET food trays into vitrimers via melt extrusion-based transesterification using DGEBA and zinc acetylacetonate (Fig. 11b).60 This innovative approach achieved a cross-linking degree of 75% while retaining the material reprocessability. The modified system exhibited breakthrough performance metrics: a three-orders-of-magnitude enhancement in melt strength, doubled tensile strength at room temperature (55 MPa), and retained dimensional stability at 260 °C (Fig. 11d).
 | ||
| Fig. 11 (a) Synthesis of vitrimers from PET via reactive extrusion with BIS-TRIS and DGEBA. (b) Synthesis of vitrimers from PET via reactive extrusion with DGEBA. (c) Comparison of creep curves between PET and the PET vitrimer at 60 °C and 100 °C. Reproduced from ref. 59 with permission from American Chemical Society, copyright 2021. (d) Mechanical properties of PET (P0) and vitrimers (P1D.3Za, P1D.3 Zb, P3D.1Za and P3D.1 Zb). Reproduced from ref. 60 with permission from Elsevier, copyright 2023. | ||
Conventional pathways to PET-derived vitrimers suffer from low reactivity and thus require high temperature and energy cost. Li et al. reported a green and efficient one-pot strategy to convert PET into a covalent adaptable network vitrimer via in situ construction, leveraging the synergistic catalytic effects of neighboring group participation and a tertiary amine structure.61 The vitrimer was synthesized by reacting PET with pyromellitic anhydride (PMDA) and BIS-TRIS in an internal mixer at 270 °C for only 5.5 minutes (Fig. 12a and c). PMDA acts as both a chain extender and internal catalyst via neighboring group participation of its anhydride groups, while BIS-TRIS introduces tertiary amine groups and additional hydroxyl groups to accelerate transesterification. However, such elevated temperatures may also restrict its scale-up and practical application. Besides, it could potentially accelerate undesired side reactions. For example, the excess carboxyl group may promote hydrolytic or acidolytic cleavage of ester bonds in the PET backbone, potentially leading to uncontrolled chain scission or reduced crosslinking efficiency. To further increase the reaction rate, Ng et al. depolymerized PET via a solvent-assisted glycolysis with minimal N-methyl-2-pyrrolidone (NMP) as the solvent (PET/NMP mass ratio = 1:1).35 The reaction used glycerol as the mediator and the ionic liquid 1-ethyl-3-methylimidazolium chloride ([EMIM]Cl) as the catalyst, yielding oligomers bearing reactive end groups. These oligomers were subsequently repolymerized through dynamic transesterification to construct reprocessable covalent adaptable networks without additives or crosslinkers, using only a catalytic amount of Zn(acac)2 (Fig. 12b). The resulting films exhibited superior tensile strength (48 MPa) with a glass transition temperature (Tg) > 80 °C, outperforming commercial thermoplastics and thermosets. After four reprocessing cycles (cold-pressing pelletization, hot-pressing molding), tensile strength retention exceeded 95%, demonstrating the potential to replace virgin plastics (Fig. 12d).
 | ||
| Fig. 12 (a) Synthesis of vitrimers from PET via reactive extrusion with BIS-TRIS and PMDA. (b) Synthesis of vitrimers from PET via glycerol glycolysis and Zn(acac)2 catalysis. (c) Creep resistance of PET and vitrimers with various BIS-TRIS contents at different temperatures. Reproduced from ref. 61 with permission from Royal Society of Chemistry, copyright 2022. (d) Comparison of mechanical properties between vitrimers with varying glycerol content and common commercial polymers; ultimate tensile strength of the vRPET1:1 sample after 4 cycles shows little change. Reproduced from ref. 35 with permission from Springer Nature, copyright 2023. | ||
Catalyst-free strategies for upcycling PET into vitrimers have emerged as an attractive method in the upcycling of PET wastes, eliminating the need for exogenous catalysts to streamline processing, reduce production costs, and minimize environmental burdens associated with catalyst recovery and contamination. Wu et al. developed a novel and efficient catalyst-free strategy for preparing PET vitrimers by constructing dynamic covalent networks through melt-blending PET with N,N′-tetraglycidyl-diaminodiphenylmethane (TGDDM) within minutes (Fig. 13a).62 This study utilized TGDDM's multifunctionality: its epoxy groups provided crosslinking sites, while the intrinsic tertiary amine groups enabled concurrent catalysis of the crosslinking reaction and subsequent dynamic transesterification. Controlling the TGDDM content at 0.7–2.0 wt% allowed precise tuning of materials architectures from branched polymers to networks with varying crosslinking densities (Fig. 13b). The resulting vitrimers demonstrated exceptional heat resistance, creep resistance, and reprocessability (Fig. 13c–e). By leveraging the intrinsic chemical reactivity or autocatalytic mechanisms, such approaches are applicable to mixtures of PET products and can enhance the scalability of vitrimer synthesis. Recently, Danielson et al. presented a catalyst-free aminolysis strategy for upcycling post-consumer PET waste into closed-loop recyclable vitrimer plastics and composites, addressing the critical environmental challenge of low PET recycling rates.63 This approach utilizes commercially available amines like Jeffamine T403 to deconstruct diverse PET feedstocks including mixed plastics, textiles, and colored waste into reactive tetramine macromonomers without exogenous catalysts or solvents. The resulting macromonomers are crosslinked with acetoacetates (AcAc) to form dynamic vinylogous urethane networks, yielding vitrimers with tunable thermomechanical properties (Fig. 14a). Notably, the optimized vitrimer (TCD-V) exhibits ultimate tensile strength (84.4 MPa) and Young's modulus (2.73 GPa), surpassing virgin PET by 80% and 150%, respectively, while demonstrating thermal reprocessability and shape memory behavior (Fig. 14b–e). Quantitative monomer recovery and reuse even from fiber-reinforced composites (glass/carbon fibers) could be achieved, with mechanical performance retention exceeding epoxy-based controls by 100% for glass fiber composites. A preliminary technoeconomic analysis confirms cost competitiveness ($5.37 per kg at 6 kg per day scale), highlighting the potential for industrial implementation. This work advances the circular plastic economy by integrating catalyst-free processing, high-performance material recovery, and adaptability to mixed waste streams.
 | ||
| Fig. 13 (a) Synthesis of vitrimers from PET via reactive extrusion with TGDDM. (b) Effect of TGDDM content on the gel fraction of the samples. (c) Comparative heat resistance of samples with varying TGDDM content and pristine PET when subjected to a 200 g load at 150 °C for 20 minutes. (d) Creep curves of PET-0.7 at different temperatures under 10 N stress. (e) Reprocessability of the PET-0.7 sample; storage modulus and tanδ plots of the reprocessed material. Reproduced from ref. 62 with permission from American Chemical Society, copyright 2023. | ||
 | ||
| Fig. 14 (a) Vitrimers based on vinylogous urethane networks are synthesized via the crosslinking of PET macromonomers—efficiently depolymerized from PET through aminolysis—with AcAc crosslinkers. (b) The average tensile stress and Young's modulus for commercial PET and vitrimers incorporating different AcAc crosslinkers. (c) Tensile stress–strain curve of TCD-V vitrimers with different crosslinking densities. (d) Tensile stress–strain curves for the 10:7 TCD-V film after being repressed three times at 200 °C and 500 psi. (e) Consecutive cycles of shape memory behavior acquired from cyclic DMA experiments. Reproduced from ref. 63 with permission from John Wiley and Sons, copyright 2025. | ||
3. Outlook and perspective
The advancement of PET recycling technologies hinges on balancing efficiency, scalability, and material performance to meet the demands of a circular economy. This review highlights the transformative potential of polymer-to-polymer upcycling in redirecting PET waste from landfills to high-value polymeric materials. By preserving PET's macromolecular framework or strategically recombining its building blocks, these approaches, including chemical depolymerization and repolymerization and catalytic reconfiguration, achieve performance parity or superiority over virgin polymers while reducing energy inputs.The chemical depolymerization and repolymerization approaches, which break PET into monomers or oligomers for subsequent repolymerization, have long been the cornerstone of chemical recycling. Their primary strength lies in versatility: purified monomers can be repolymerized into virgin-grade PET or diverted into high-value polymers such as polyurethanes or polyamides. For instance, enzymatic depolymerization under mild conditions achieves high monomer yields with minimal energy input, while chemocatalytic methods like glycolysis and methanolysis enable industrial-scale production of monomers like DMT. However, current methods still face critical limitations: multi-step processing including depolymerization, purification and repolymerization increases energy consumption and cost, and residual impurities often compromise the performance of recycled materials. Future efforts are expected to focus on integrated catalytic systems such as tandem depolymerization–repolymerization catalysts and continuous flow reactors to streamline purification and reduce energy use. Additionally, coupling depolymerization with renewable feedstock integration like bio-based comonomers could further enhance sustainability and expand product portfolios.
In contrast, one-pot reconfiguration strategies could directly construct PET waste into functional materials (e.g., new polymer, vitrimer or composites) without isolating intermediates. These approaches accomplish cyclic PET depolymerization and new PET-derived polymer repolymerization within a single reactor, offering process intensification and reducing carbon footprints. Some catalyst-free systems show the potential to handle mixed, unwashed, or contaminated PET-like dyed textiles and multi-material waste addresses a major barrier in conventional recycling, while in situ crosslinking generates materials with tailored properties including shape memory and self-healing. Despite the advancement in direct upcycling of PET methods, real-world implementation is throttled by inconsistent feedstock quality. There is an urgent need to expand the utilization of dynamic chemistry including transesterification, vinylogous urethanes, and imine bonds for real-life plastic wastes and diversification beyond specific polymer families. For example, future research should explore modular crosslinking systems (e.g., orthogonal dynamic bonds) that enable tunable performance (stiffness, depolymerization rates) and compatibility with diverse waste streams. Scaling such processes will also require advancements in reactor design to handle heterogeneous waste and optimize heat/mass transfer. Another noteworthy priority for PET-to-polymer upcycling is ensuring new polymers do not introduce secondary pollution. This requires designing systems with inherent recyclability: retaining cleavable chemical linkages like esters, urethanes, or dynamic covalent bonds to enable end-of-life chemical recycling, while avoiding permanently crosslinked networks that resist degradation.
A critical yet often overlooked barrier to scaling PET upcycling lies in the heterogeneity of real-world waste streams. Post-consumer plastics rarely exist as pure monomaterials; PET bottles are typically contaminated with polyethylene (PE) caps, polypropylene (PP) labels, adhesives, and residual food/water, while textile waste frequently comprises PET/cotton blends or multi-fiber composites. Such complexity directly challenges the efficiency and selectivity of current recycling technologies, demanding innovations that transcend idealized “clean feedstock” laboratory conditions. To this end, the next generation of PET upcycling will likely merge the strengths of multiple paradigms. For example, hybrid processes coupling mechanical recycling and chemical upcycling could use one-pot and one-step methods to selectively convert PET plastic waste blends into high-purity materials via precision catalysis. As demonstrated by a recent study, Co/Mn-catalyzed autoxidation enables selective depolymerization of mixed polystyrene (PS), high-density polyethylene (HDPE), and PET into benzoic acid, dicarboxylic acids, and terephthalic acid eliminating the need for pre-sorting. This oxygenate mixture is further funneled into single high-value products like β-ketoadipate via precision biocatalysis, exhibiting catalytic synergy in heterogeneous waste streams.64 Similarly, integrating artificial intelligence and machine learning will accelerate the discovery of catalysts and dynamic chemistries tailored to specific waste streams and target applications. Also, a critical unmet need remains end-of-life circularity: while the obtained PET-derived polymer materials enable chemical recycling, ensuring their recyclability at an industrial scale requires standardized protocols for deconstruction and monomer recovery. Moreover, the next step for advancing PET upcycling lies in prioritizing integrated technical-economic analysis (TEA) and life-cycle assessment (LCA) frameworks. While current research has made significant strides in reaction chemistry and material performance, systematic data on cost-effectiveness and environmental impact including carbon footprint and waste generation remain fragmented across upcycling pathways. Future studies should explicitly quantify these metrics to validate whether converting PET into new polymers offers tangible advantages over traditional recycling or virgin polymer production, which would guide policy and investment toward truly sustainable solutions.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review. All information is derived from the previously published literature.Acknowledgements
We appreciate financial support from the Science and Technology Development Plan of Jilin Province-Provincial Natural Science Foundation of Jilin (SKL202302035), the National Natural Science Foundation of China (No. 22293062 52403017), and the International Partnership Program of Chinese Academy of Sciences (029GJHZ2023017MI).References
- N. Yan, Science, 2022, 378, 132–133 CrossRef CAS.
- P. Europe , Plastics—The Fast Facts 2023, https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2023/, (accessed July, 2025).
- Q. Chen, H. Yan, K. Zhao, S. Wang, D. Zhang, Y. Li, R. Fan, J. Li, X. Chen, X. Zhou, Y. Liu, X. Feng, D. Chen and C. Yang, Nat. Commun., 2024, 15, 10732 CrossRef CAS.
- A. Barredo, A. Asueta, I. Amundarain, J. Leivar, R. Miguel-Fernández, S. Arnaiz, E. Epelde, R. López-Fonseca and J. I. Gutiérrez-Ortiz, J. Environ. Chem. Eng., 2023, 11, 109823 CrossRef CAS.
- S. Zhang, Q. Hu, Y.-X. Zhang, H. Guo, Y. Wu, M. Sun, X. Zhu, J. Zhang, S. Gong, P. Liu and Z. Niu, Nat. Sustain., 2023, 6, 965–973 CrossRef.
- S. Moon, L. M. A. Martin, S. Kim, Q. Zhang, R. Zhang, W. Xu and T. Luo, Sci. Adv., 2024, 10, eadh1675 CrossRef CAS PubMed.
- N. P. Ivleva, A. C. Wiesheu and R. Niessner, Angew. Chem., Int. Ed., 2017, 56, 1720–1739 CrossRef CAS PubMed.
- L. Zimmermann, B. Geueke, L. V. Parkinson, C. Schür, M. Wagner and J. Muncke, npj Sci. Food, 2025, 9, 111 CrossRef.
- H. A. Leslie, M. J. M. van Velzen, S. H. Brandsma, A. D. Vethaak, J. J. Garcia-Vallejo and M. H. Lamoree, Environ. Int., 2022, 163, 107199 CrossRef CAS PubMed.
- Y. Zhou, Y. Shan, D. Guan, X. Liang, Y. Cai, J. Liu, W. Xie, J. Xue, Z. Ma and Z. Yang, Nat. Food, 2020, 1, 552–561 CrossRef PubMed.
- T. M. Joseph, S. Azat, Z. Ahmadi, O. Moini Jazani, A. Esmaeili, E. Kianfar, J. Haponiuk and S. Thomas, Case Stud. Chem. Environ. Eng., 2024, 9, 100673 CrossRef.
- C. Pudack, M. Stepanski and P. Fässler, Chem. Ing. Tech., 2020, 92, 452–458 CrossRef CAS.
- K. L. Law and R. Narayan, Nat. Rev. Mater., 2022, 7, 104–116 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- M. del Mar Castro López, A. I. Ares Pernas, M. J. Abad-López, A. Lasagabáster-Latorre, J. M. López-Vilariño and M. V. González-Rodríguez, Mater. Chem. Phys., 2014, 147, 884–894 CrossRef.
- R. K. Foolmaun and T. Ramjeeawon, Environ. Technol., 2012, 33, 563–572 CrossRef CAS.
- T. Abbasi, N. J. Haghighi Fard, F. Madadizadeh, H. Eslami and A. A. Ebrahimi, J. Polym. Environ., 2023, 31, 3493–3508 CrossRef CAS.
- Å. Moberg, G. Finnveden, J. Johansson and P. Lind, J. Cleaner Prod., 2005, 13, 231–240 CrossRef.
- F. P. La Mantia, Handbook of plastics recycling, iSmithers Rapra Publishing, 2002 Search PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- K. Chan and A. Zinchenko, J. Cleaner Prod., 2023, 433, 139828 Search PubMed.
- J. Mudondo, H.-S. Lee, Y. Jeong, T. H. Kim, S. Kim, B. H. Sung, S.-H. Park, K. Park, H. G. Cha, Y. J. Yeon and H. T. Kim, J. Microbiol. Biotechnol., 2023, 33, 1–14 CrossRef CAS PubMed.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- L. Qin, X. Li, G. Ren, R. Yuan, X. Wang, Z. Hu, C. Ye, Y. Zou, P. Ding and H. Zhang, ChemSusChem, 2024, 17, e202301781 CrossRef CAS.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198 CrossRef CAS.
- S. Zhang, M. Li, Z. Zuo and Z. Niu, Green Chem., 2023, 25, 6949–6970 RSC.
- Q. Shi, Y.-Q. Zhu, X. Liu, B.-J. Yuan, W. Tang, X. Wang, R.-P. Li and H. Duan, J. Am. Chem. Soc., 2025, 147, 14004–14014 CrossRef CAS PubMed.
- G. Wang, Z. Chen, W. Wei and B.-J. Ni, Electron, 2024, 2, e34 CrossRef CAS.
- Q. Shi, W. Tang, K. Kong, X. Liu, Y. Wang and H. Duan, Angew. Chem., Int. Ed., 2024, 63, e202407580 CrossRef CAS PubMed.
- A. Carniel, N. F. dos Santos, F. S. Buarque, J. V. M. Resende, B. D. Ribeiro, I. M. Marrucho, M. A. Z. Coelho and A. M. Castro, Green Chem., 2024, 26, 5708–5743 RSC.
- EPR in the EU Plastics Strategy and the Circular Economy: A focus on plastic packaging, https://ieep.eu/wp-content/uploads/2022/12/Policy-options-brief-EPR-price-modulation-IEEP-Nov-2017-final.pdf, (accessed July, 2025).
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- Z. Guo, J. S. K. Lim, K. W. J. Ng, W. Yan, N. Gupta and X. M. Hu, ACS Sustainable Chem. Eng., 2023, 11, 1394–1404 CrossRef CAS.
- K. W. J. Ng, J. S. K. Lim, N. Gupta, B. X. Dong, C.-P. Hu, J. Hu and X. M. Hu, Commun. Chem., 2023, 6, 158 CrossRef CAS.
- T. H. Shah, J. I. Bhatty, G. A. Gamlen and D. Dollimore, Polymer, 1984, 25, 1333–1336 CrossRef CAS.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- C. Li, G. Yan, Z. Dong, G. Zhang and F. Zhang, Nat. Commun., 2025, 16, 2482 CrossRef CAS PubMed.
- R. Zhang, X. Zheng, X. Yao, K. Song, Q. Zhou, C. Shi, J. Xu, Y. Li, J. Xin, I. E.-T. El Sayed and X. Lu, Ind. Eng. Chem. Res., 2023, 62, 11851–11861 CrossRef CAS.
- Y. Yu, G. Shen, T. J. Xu, R. Wen, Y. C. Qiao, R. C. Cheng and Y. Huo, RSC Adv., 2023, 13, 36337–36345 RSC.
- S. Kaiho, A. A. R. Hmayed, K. R. D. Chiaie, J. C. Worch and A. P. Dove, Macromolecules, 2022, 55, 10628–10639 CrossRef CAS.
- A. A. Karanastasis, V. Safin and L. M. Pitet, Macromolecules, 2022, 55, 1042–1049 CrossRef CAS.
- M. D. de Dios Caputto, R. Navarro, J. López-Valentín and A. Marcos-Fernandez, J. Cleaner Prod., 2024, 454, 142253 CrossRef CAS.
- J. Lyu, S. Lee, H. Jung, Y. I. Park, J. Ahn, Y.-J. Jin, J.-E. Jeong and J. C. Kim, Chem. Eng. J., 2024, 501, 157535 CrossRef CAS.
- M. Y. Tan, L. Goh, D. Safanama, W. W. Loh, N. Ding, S. W. Chien, S. S. Goh, W. Thitsartarn, J. Y. C. Lim and D. W. H. Fam, J. Mater. Chem. A, 2022, 10, 24468–24474 RSC.
- Y. Zhang, F. Tian, C. Liu, X. Liu, Y. He and Z. Wu, J. Cleaner Prod., 2024, 434, 140048 CrossRef CAS.
- N. A. Rorrer, S. Nicholson, A. Carpenter, M. J. Biddy, N. J. Grundl and G. T. Beckham, Joule, 2019, 3, 1006–1027 CrossRef CAS.
- S. Waskiewicz and E. Langer, Polymer, 2021, 227, 123832 CrossRef CAS.
- L.-X. Fan, L. Chen, H.-Y. Zhang, W.-H. Xu, X.-L. Wang, S. Xu and Y.-Z. Wang, Angew. Chem., Int. Ed., 2023, 62, e202314448 CrossRef CAS.
- Z. Liu, H. Zhang, S. Liu and X. Wang, Polym. Chem., 2023, 14, 1110–1116 RSC.
- H. Zhang, T. Fang, X. Yao, X. Li and W. Zhu, Adv. Mater., 2023, 35, 2210758 CrossRef CAS.
- Y. Peng, J. Yang, C. Deng, J. Deng, L. Shen and Y. Fu, Nat. Commun., 2023, 14, 3249 CrossRef CAS.
- T. Fang, W. Jiang, T. Zheng, X. Yao and W. Zhu, Adv. Mater., 2024, 36, e2403728 CrossRef PubMed.
- H. Zhang, M. Fang, S. Niu, M. Wang, M. Gao, Q. Cai, G. Wang, W. Chen and W. Lu, Angew. Chem., Int. Ed., 2025, 64, e202420839 CrossRef CAS PubMed.
- W. H. Leung, E. M. Leitao and C. J. R. Verbeek, Polymer, 2023, 284, 126297 CrossRef CAS.
- W. H. Leung, E. M. Leitao and C. J. R. Verbeek, Polymer, 2024, 308, 127345 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- J. Qiu, S. Ma, S. Wang, Z. Tang, Q. Li, A. Tian, X. Xu, B. Wang, N. Lu and J. Zhu, Macromolecules, 2021, 54, 703–712 CrossRef CAS.
- L. Fabrizio, R. Arrigo, M. T. Scrivani, M. Monti and A. Fina, Polymer, 2023, 266, 125618 CrossRef CAS.
- P. Li, B. Lan, X. Zhang, S. Lei, Q. Yang, P. Gong, C. B. Park and G. Li, Green Chem., 2022, 24, 5490–5501 RSC.
- S. Wu, Y. Li, Z. Hu and J. Zeng, ACS Sustainable Chem. Eng., 2023, 11, 1974–1984 CrossRef CAS.
- M. K. Danielson, C. Gainaru, Z. Demchuk, C. Pan, J. Choi, H.-H. Zhang, J. C. Foster, T. Saito and M. A. Rahman, ChemSusChem, 2025, 18, 2500898 Search PubMed.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |