
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trioxanes as photodegradable motifs for additive manufacturing†
Florian
Mayer
 a,
Dominik
Laa
b,
Thomas
Koch
b,
Jürgen
Stampfl
b,
Robert
Liska
a and
Katharina
Ehrmann
*a
a,
Dominik
Laa
b,
Thomas
Koch
b,
Jürgen
Stampfl
b,
Robert
Liska
a and
Katharina
Ehrmann
*a
aInstitute of Applied Synthetic Chemistry, Technische Universität Wien, Vienna, Austria. E-mail: katharina.ehrmann@tuwien.ac.at
bInstitute of Materials Science and Technology, Technische Universität Wien, Vienna, Austria
First published on 7th July 2025
Abstract
We introduce trioxanes as a new motif for on-demand photolysis of 3D printed polymer networks via an orthogonal light source using photoacid generators (PAGs). The acid-induced degradation of otherwise very stable trioxanes in combination with the versatility of generating trioxane-containing monomers makes the motif particularly interesting. Herein, we demonstrate the use of the motif as a trifunctional ene-crosslinker in combination with a trifunctional alicyclic thiol. Thereby, a latent PAG (UVI 6976, absorption ≤ 400 nm) is included into the photopolymerizable formulation and its latency during photocuring at longer wavelengths (≥400 nm) is proven. Upon photopolymerization, the PAG-containing material exhibits equal (thermo)mechanical performance to a reference network excluding PAG. Upon UV light irradiation, degradation of trixoanes is proven via NMR, DSC and solubility tests. Finally, facile printing of the trioxane-containing formulation has been shown.
Introduction
Additive manufacturing techniques for polymers can be divided into two major groups. Extrusion-based methods (fused deposition modelling, FDM) utilize thermoplastic polymers as filaments, which are molten and deposited onto the platform, while photopolymer-based techniques are based on in situ formation of crosslinked polymer networks from a liquid resin in a vat, utilizing light as the trigger to obtain spatio-temporal control over the reaction. These photopolymer-based additive manufacturing technologies, often referred to as vat photopolymerization (VP), exhibit better resolution and surface quality compared to FDM methods. However, the resulting parts consist of highly crosslinked polymers that cannot be recycled by melting like it would be the case for thermoplastics utilized in FDM.Therefore, disintegrating the crosslinks of thermosets on demand has become particularly interesting for the VP community. Common triggers for the decomposition of bonds can be heat,1–5 light4,6–8 or pH,9,10 but also ultrasound11,12 and mechanical agitation13 have been investigated as degradation triggers. Light can be a particularly useful trigger as it can also be used in heat sensitive applications, e.g. in living tissue like dental applications.14 However, this approach is limited by the penetration depth of light into the material.15,16
Photocleavable crosslinkers have become particularly important with the ever-increasing popularity of VP14 in combination with the need for mechanical modulation and spatio-temporally controlled degradation of the materials, especially for biological applications. They are utilized for mechanical modulation as well as full degradation of polymer networks. Several intrinsically photoresponsive functional groups have been explored in photocleavable crosslinkers,17–23 as well as catalytically degradable motifs,24–36 which can be degraded through the use of photolatent degradation catalysts. For example, acid-catalyzed depolymerization of polyphthalaldehyde has been demonstrated.37–40 PAGs are particularly interesting photolatent degradation catalysts because all motifs prone to conventional acidic degradation can undergo photodegradation via PAGs. Another major advantage is the orthogonal combination of PAGs with visible light responsive photoinitiators such as TPO-L, as long as the light sources are applied in the right order (UV irradiation second; sequence-dependent orthogonality or semi-orthogonality). This could enable 3D printing of a photocleavable crosslinker containing formulation at longer wavelengths and subsequent degradation in the harsher UV light regime.
Aside from direct depolymerization,41–44 the polymer community has demonstrated a variety of acid labile motifs as degradable crosslinkers in VP, which are particularly useful to degrade polymer networks effectively. However, several popular acid-degradable motifs in polymers have not been examined as photodegradable motifs so far,45–47 and some acid degradable motifs remain unexplored even in the broader realm of polymer chemistry. Interestingly, reported photodegradable polymers found in literature were typically not synthesised by a (sequence-dependent) orthogonal photochemical polymerisation process and are therefore not suitable for Lithography based printing.37–40 Further, some studies have shown that the presence of latent PAGs in the polymer can cause unwanted depolymerisation prior to photoactivation.38
Trioxanes have already been studied in polymer chemistry, not as cleavable crosslinkers but rather as starting material to synthesize polyoxymethylene.48,49 We have recently published a similar polymerization method where we depolymerize paraformaldehyde via acid to form formaldehyde as a crosslinking agent in situ.50 This approach has inspired us to utilize this same reaction for a reverse approach. However, the integration of paraformaldehyde into a polymer network is unfeasible for a number of reasons, most importantly its linear oligomeric molecular structure, which is unsuitable for building up a network. Similarly, poly(phtalaldehyde), which has been utilized as self-immolative polymer51 and even for acidic photodegradation,52 has an intrinsically linear structural motif unsuitable for polymer networks. The trioxane-related paraldehyde, however, offers facile and flexible threefold endgroup integration and is prone to acidic hydrolysis.53
In this work we aim to introduce a new photodegradable motif based on trioxane that can be used in formulations for additive manufacturing (Fig. 1). Thiol–ene chemistry is chosen as a popular polymerisation mechanism for 3D printing due to its lower shrinkage than acrylate-based formulations,54 high reactivity, and tuneable mechanical properties.54 Most importantly, bioderived aldehydes with double bond end groups are readily commercially available, that can be transformed into trioxanes in only one step.55 Based on this trioxane ene-crosslinker and a cycloaliphatic trithiol, we will demonstrate acidic degradation on demand by triggering latent photoacid generators in the 3D printed part. The printed part containing the trioxane units can be degraded on demand either by using light at moderate temperatures, which is particularly interesting for disassembly on demand, e.g. of electrical components, or by heating it to higher temperatures of 160 °C.
 | ||
| Fig. 1 (A) Schematic representation of visible light-induced network formation and subsequent UV light-induced network degradation utilizing the photoinitiator (PI) TPO-L (radical PI) and the photoacid generator (PAG) UVI6976, respectively, which exhibit sequence-dependent orthogonal photoreactivity as indicated in (B) their UV/vis absorption spectra. | ||
Results and discussion
Cleavable crosslinker design and proof-of-principle
To obtain a polymerizable trioxane moiety, ene- or thiol endgroups have to be introduced to the degradation motif. 10-Undodecenal was chosen as the ideal building block to synthesize a trifunctional ene-crosslinker because it is commercially available and a polymerizable trioxane could be yielded in a one-step synthesis in analogy to Arias-Ugarte et al. (Fig. 2A).56 | ||
| Fig. 2 (A) Synthesis of the trioxane-based ene-crosslinker catalyzed by FeCl3 and its degradation catalyzed by UV-activated PAG and (B) 1H-NMRs of this crosslinker containing PAG (UVI6976, triaryl sulfonium hexafluoroantimonate salts in propylene carbonate, 10 wt% with respect to the trioxane) in the presence and absence of UV light compared to the spectra of pure trioxane and the reference degradation compound 10-undecanal to proof the viability of the trioxane crosslinker as degradable crosslinker. | ||
The newly synthesised monomer should be polymerized using visible light-induced thiol–ene polymerization. In a second step, the degradable trioxane moiety should be cleaved using a semi-orthogonal photoacid generator (PAG, UVI6976), which is only active in the UV light range. The requirements for this monomer are therefore stability during visible light-induced radical polymerisation and degradability upon activation of the PAG. These aspects were investigated for the pure trioxane monomer before using it in a polymerization reaction. 1H-NMR spectroscopy was employed for this purpose (Fig. 2B). To prove the stability of the trioxane moiety during the radical polymerization, the monomer was mixed with the latent PAG and its 1H-NMR spectrum was compared to the trioxane monomer without added PAG. To identify potential degradation products, the spectrum of 10-undecanal, which is the expected degradation product of the trioxane, was recorded for reference. The reference aldehyde exhibits a distinct peak at 9.76 ppm, while the trioxane can be identified via the peak at 4.83 ppm. When adding PAG to the trioxane solution, no change in the trioxane or aldehyde peak was visible. Therefore, it can be concluded that the trioxane is stable in the presence of the latent PAG and can act as a crosslinker during polymerization as long as the PAG remains inactive. When activating the PAG using UV-light with the 320 nm–500 nm light for two minutes at full intensity in the UV oven, the trioxane diminished and the expected aldehyde signal emerged, thereby proving successful degradation upon irradiation with UV light. The small residual peak in the region of the trioxane peak can be attributed to a PAG signal that overlaps with the trioxane peak.
Formulation and photoreactivity
In combination with the trioxane ene-crosslinker, 2-[2,4-bis(2-mercaptoethyl) cyclohexyl] ethane thiol (CHTT) was chosen as thiol because it is miscible with the trioxane monomer and, as a trifunctional thiol with a cycloaliphatic group, it is expected to yield less flexible and more stable networks than difunctional and aliphatic thiols (Fig. 3A). TPO-L was used as radical initiator because it can still be activated at 385 nm, which is a wavelength where the employed PAG cannot be activated. This enables the radical polymerization without activating the PAG, i.e. sequence-dependent orthogonality of polymerization and degradation reactions. Butylhydroxytoluene (BHT) was added to the formulation to prevent premature gelation of the formulation when mixing the thiol with the trioxane component. | ||
| Fig. 3 Reactivity of the formulation (A) consisting of the trioxane triene and CHTT triol with and without the active photoacid generator and (B) sample and colour code used for the resulting polymers. (C) Photo-DSC results (tmax, time until maximum heat flow is reached, t95%, time until 95% of conversion is reached) and (D) photorheology results (tgel, time until gel point is reached, t95% time until 95% of conversion is reached). | ||
The reactivity of the system is important because it determines the speed of the 3D printing process. If the reactivity is too low, printing will be slow or even unfeasible: using a standard gap size of 50 μm, it takes 200 layers to build up a structure of 1 cm height. For this reason, every additional second of curing to form stability will multiply by the number of layers.
To test the reactivity, photo-DSC was performed using an LED with a wavelength maximum of 385 nm and an intensity of 50 mW cm−2 at the sample surface, conditions under which the PAG should not cleave – an assumption, which will be investigated in-depth in the next chapter. For the reactivity study, two formulations were measured. One formulation contained latent PAG, i.e. PAG, which was not subjected to UV irradiation (L-PAG), and one formulation did not contain any PAG (N-PAG, Fig. 3B). From the photo-DSC measurements, two important parameters concerning reactivity can be extracted from the recorded exothermic heat flow during the reaction: the time until maximum heat flow is reached (tmax) and the time until 95% of the total heat flow is reached (t95%, Fig. 3C). The tmax of below two seconds indicates very high reactivity, which was expected for a thiol–ene system. A t95% of around 10 s is also very fast, indicating good reactivity for fast 3D printing. In fact, this high reactivity can also cause overpolymerisation and may need to be suppressed using light absorbers during 3D printing later on. As expected, no significant reactivity difference was found between N-PAG and L-PAG formulations.
The t95% determined by complimentary photorheology analysis shows similar trends as the photo-DSC experiment (Fig. 3D, Table 1). Thereby, double bond conversion (DBC) can be tracked via near-infrared (NIR) spectroscopy simultaneously to rheological data during curing with UV light. The light source used in the photorheology experiments was again an LED with its maximum wavelength of 385 nm and an intensity of 50 mW cm−2. The slightly faster t95% can be explained by the thinner layers and better homogenisation of the formulations through the oscillation during the measurement. Additionally, the time until gelation (tgel) was determined from the onset of the normal force decrease during photorheology, since it could not be determined from the intersection of G′ and G′′, which crossed several times in the region of polymerisation (Fig. S3†). In both formulations, DBCs of over 99% were achieved. The DBC at the gel point (DBCgel) is at around 80%, which is consistent with findings from photo-DSC. This is a rather late gel point as expected for a step-growth thiol–ene network.
 , normal force at DBCfinal (FN,final)
, normal force at DBCfinal (FN,final)
| t gel/s | DBCgel/% | t 95%/s | DBCfinal/% |
|
F N,final/N | |
|---|---|---|---|---|---|---|
| No PAG | 2.1 ± 0.1 | 79.1 ± 1.2 | 6.0 ± 1.5 | >99 | 415.5 ± 9.2 | −22.2 ± 0.1 |
| Latent PAG | 2.7 ± 0.5 | 78.6 ± 1.7 | 7.8 ± 1.8 | >99 | 416.1 ± 1.1 | −22.4 ± 0.5 |

Overall, no significant difference between the formulation with and without the PAG could be observed, which indicates that the PAG does not influence the reactivity or the conversion through irradiation at 385 nm. Both formulations have proven to be very reactive, which can be beneficial for 3D printing but may also lead to overpolymerisation.
Thermal stability and photodegradability of trioxane networks
To investigate the cleaving behaviour of the trioxanes in a bulk material, three rectangular specimens were prepared either containing an activated PAG (A-PAG), that was activated via irradiation at 320 nm–500 nm, a latent PAG (L-PAG) or no PAG (N-PAG), and subjected first to thermal stability analysis via differential scanning calorimetry (DSC) on a simultaneous thermal analysis (STA) device and thereafter to photodegradation testing via DSC, melt- and dissolution testing of UV-treated samples. For this purpose, bulk-cured samples were prepared and a part of each sample was used for the different tests (Fig. 4A). | ||
| Fig. 4 Proof of principle for trioxanes as cleavable motifs in a thiol–ene polymer network: (A) experimental design (orange: no PAG; green: latent PAG, blue: irradiated PAG), and melting and dissolution experiments with cured samples after UV irradiation at 320 nm–500 nm; (B) differential scanning calorimetry results (solid lines are polymers, dashed lines are small molecules) and (C) 1H-NMR of the molten sample of the polymer with active PAG compared to the reference aldehyde, which is equivalent to the expected degradation product. | ||
To determine thermal stability of the degradation motif in the polymer network in the absence of a light trigger, DSC curves of samples N-PAG and L-PAG were compared. Both show a peak in the region of 225 °C and 250 °C, which can be attributed to thermal ring opening of the trioxane ring from 225 °C on (Fig. 4B). Since the DSC curves of both samples are similar, L-PAG does not seem to favour degradation compared to N-PAG, indicating thermal stability. As reference to the ring opening of the trioxane ring without incorporation in the network, DSC of the trioxane monomer was conducted as well. In this case, the peak at 225 °C is shifted to 265 °C, which correlates quite well with the boiling point of the degradation product, 10-undecenal (258 °C). Since the peak onset observed during DSC of the monomer coincides with the onset observed during DSC of the polymer, the peak observed during STA of the monomer is considered to include both, the degradation of the trioxane ring and subsequent evaporation of the aldehyde. Additionally, temporal stability over the course of 110 days was proven (Fig. S6†).
Subsequently, DSC was employed to prove photo-triggered degradation of the trioxane motif in the polymer. For this purpose, L-PAG was activated to A-PAG by UV irradiation and subsequent DSC. A signal at 80 °C indicates that acid catalysed ring opening occurs as expected.
Additionally, melting experiments should demonstrate that the crosslinked network is fully degraded upon UV irradiation (Fig. 4A). Therefore, two specimens, A-PAG and N-PAG, were heated up with a heat gun and their temperature was tracked via infra-red thermometer. It is noteworthy that the A-PAG specimen broke when handling it with pincers because it is already mechanically unstable, even before heating it up. Melting was observed at 110 °C for the sample with active PAG, and at approximately 180 °C for the sample without PAG, which aligns well with the DSC results. To confirm that the expected degradation product had formed, a 1H-NMR was measured of the molten A-PAG sample and compared to a reference aldehyde compound, which was synthesised for this purpose (Fig. 7C, Fig. S2†).
The spectra are almost identical with the exception of a peak at 9.25 ppm in the molten sample spectrum (Fig. 4C). This signal is annotated to a common side product that was already observed during trioxane synthesis.
Since the expected linear degradation products should not only be meltable but also soluble, solubility tests were conducted. In both samples, A-PAG and N-PAG, the solid non molten parts were not soluble in CDCl3 but the molten parts were fully soluble. Furthermore, the molten parts did not solidify anymore, even when left overnight, resembling the liquid reference aldehyde. A significant decrease in mechanical stability was again observed when handling the A-PAG sample compared to the N-PAG sample. These observations lead to the conclusion that the activated PAG in the A-PAG sample already causes cleavage of significant amounts of trioxane groups at room temperature as indicated by the model compound NMR study and ATR-IR studies at room temperature, which attested degrees of degradation of 80% (Fig. S6†), but needs higher temperatures to achieve full trioxane cleavage and therefore solubility.
(Thermo-) mechanical analysis of trioxane networks
The thermomechanical properties of the pristine materials, N-PAG and L-PAG, have been investigated using dynamic mechanical thermal analysis (DMTA) and tensile testing (Fig. 5). The DMTA measurements of the pure thiol–ene network without PAG reveal a Tg at around −5 °C, which is expectedly low considering the long aliphatic chains between crosslinking points. The Tg is well defined, judged by the steep decrease in storage modulus and the narrow tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ peak. The elongation at break and tensile strength are as expected for such materials containing only aliphatic components and exhibiting comparably long distances between crosslinking points. To improve the tensile strength, a shorter carbon chain between trioxane and double bound may be beneficial in the future.
δ peak. The elongation at break and tensile strength are as expected for such materials containing only aliphatic components and exhibiting comparably long distances between crosslinking points. To improve the tensile strength, a shorter carbon chain between trioxane and double bound may be beneficial in the future.
 | ||
| Fig. 5 (Thermo-) mechanical testing of the trioxane formulation: (A) dynamic mechanical thermal analysis (DMTA) and (B) tensile tests. | ||
Comparing the results of N-PAG and L-PAG, no significant differences were observed in their (thermo-)mechanical behaviour. The Tg is lowered by 5 °C for L-PAG, which is within the expected inter-sample variability, or could alternatively be explained by the additional curing step of N-PAG in a UV oven. The same tendency holds true for the tensile testing results. The best-performing sample of N-PAG specimens slightly outperforms the best-performing L-PAG sample. However, there is no difference in the mean values for stress or strain at break. These measurements confirm that the inclusion of PAG has no significant impact on the polymers’ (thermo-)mechanical properties prior to PAG activation via UV light.
3D printing
For the printing experiments, a custom-made digital light projector (DLP) printer was used. The DLP-light engine is able to irradiate at a maximum intensity of 75 mW cm−2 at 385 nm. An area of 96 mm × 54 mm can be irradiated with a pixel pitch of 50 μm. The printer requires 1.5 mL of formulation for a customized small vat, for the regular larger vat at least 8 mL of material are required for the same fill level. The used building platform for the custom vat allows the creation of objects with a base size of up to 42 × 38 mm. The small dimensions of the printer make it ideal to test new systems in small batches.Quinoline yellow was added to the formulation to prevent overpolymerization. First printing experiments were conducted with an irradiation time of 2 s per layer (total time per layer including irradiation, rising of the building platform and lowering it to a gap size of 50 μm: 9.9 s) and an intensity of 45 mW cm−2, parameters, which were estimated from the photo-DSC and photo-rheology measurements. This led to overpolymerisation despite the use of quinoline yellow. After finetuning the resolution, the final parameters were 2 s at 16 mW cm−2.
Based on these parameters, several parts have been printed, three of which have been chosen for closer investigation via scanning electron microscope (SEM, Fig. 6). The SEM pictures show very defined structures and good resolution of the prints without signs of overpolymerisation.
 | ||
| Fig. 6 Show-prints of the formulation containing the trioxane triene and trithiol CHTT: (A) digital microscope image of hollow ball structure and corresponding SEM images: (B) full structure, (C) close-up, (D) bar. SEM images of hollow pyramid displaying (E) top view and (F) side bar detail. SEM image of benchy displayed (G) in full and (H) from the side at larger resolution. | ||
Experimental
Materials
Iron(III)chloride and 10-undecenal were acquired from Sigma Aldrich. The PAG UVI6976 was purchased from DOW. The radical photoinitiator TPO-L was purchased from BASF. 1,2,4-Trivinylcyclohexane (mixture of isomers, 98%) and thioacetic acid were purchased from TCI. 2,2-Dimethoxy-2 phenylacetophenone (IRGACURE 651), was purchased from Fisher Chemical.Methods
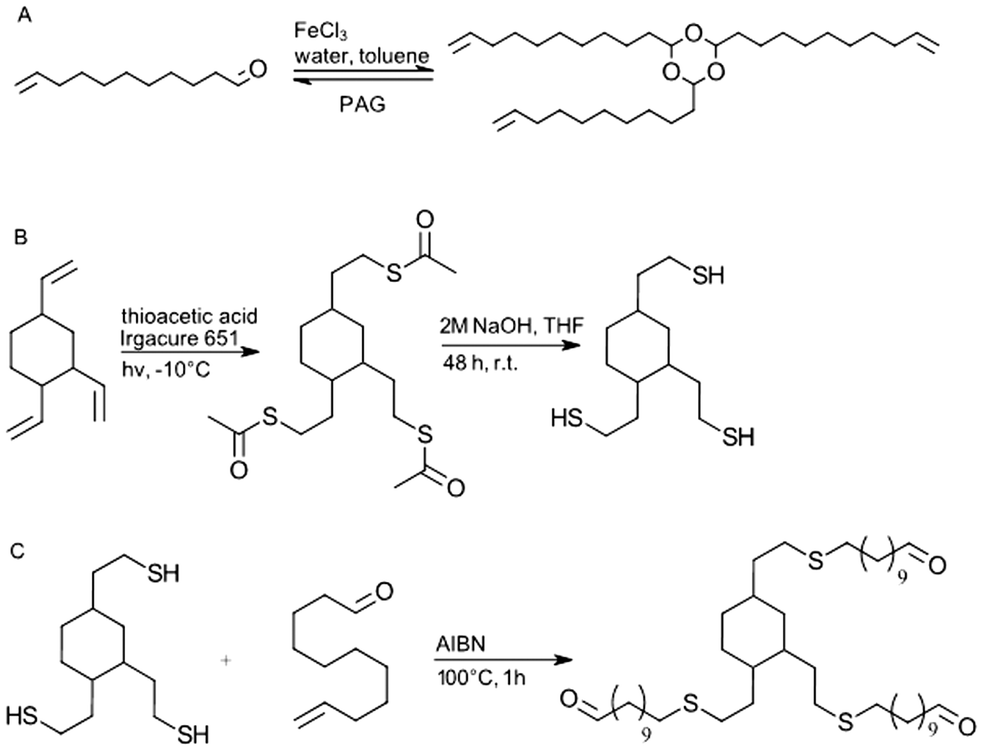 | ||
| Fig. 7 (A) Synthesis of trioxane monomer (B) synthesis of trithiol monomer (C) synthesis of reference aldehyde. | ||
1H-NMR (400 MHz, CDCl3): 5.91–5.70 (m, 3H), 5.09–4.89 (m, 6H), 4.83 (t, 3H, J = 5.3 Hz), 2.12–1.92 (m, 6H, J = 7.8 Hz), 1.72–1.60 (m, 6H, J = 9.9 Hz,) 1.45–1.34 (m, 12H), 1.28 (m, 24H, J = 3.4 Hz) ppm.
13C-NMR: (600 MHz, CDCl3): 139.22, 114.12, 101.70, 34.43, 33.82, 29.16, 23.56 ppm.
1H-NMR (400 MHz, CDCl3) δ 3.04–2.71 (m, 6H), 2.32 (s, 9H), 1.91–0.57 (m, 15H).
1H-NMR (400 MHz, CDCl3) δ = 2.69–2.36 (m, 6H), 1.90–0.57 (m, 18H).
1H-NMR (400 MHz, CDCl3): 9.76 (m, 3H), 2.53–2.38 (m, 16H), 1.72–1.22 (m, 65H) ppm.
δ curve.
Conclusion
We have introduced trioxane as a new photodegradable motif for crosslinkers in polymer networks and tested it in a 3D printable thiol–ene formulation. Its photodegradability is based on the pH-sensitivity of the trioxane, which can be triggered by UV-responsive photoacid generators that can be applied orthogonally to light-based 3D printing at longer wavelengths (385 nm), although only in a sequence-dependent manner due to the simultaneous responsiveness of both photoinitiators under UV light irradiation. We could show that there is no negative impact on the reactivity or the mechanical properties caused by the presence of a latent PAG. When activated, the PAG causes the degradation of the trioxane ring at lower temperatures than if the PAG was not present or activated. The system with latent PAG shows impeccable printability with high resolution and the possibility to print complex shapes. Therefore, trioxane has proven to be a versatile motif for degradable crosslinkers in photopolymer networks exhibiting degradability on demand. In the future, the new degradation motif could also be envisioned in other step-growth polymerizations apart from thiol–ene chemistry.Author contributions
Florian Mayer: data curation, formal analysis, investigation, methodology, project administration, validation, visualization, writing – original draft, writing – review & editing. Dominik Laa: data preparation, formal analysis, investigation, methodology, validation, writing – review & editing. Thomas Koch: data curation, formal analysis, writing – review & editing. Jürgen Stampfl: conceptualization, formal analysis, funding acquisition, methodology, resources, writing – review & editing. Robert Liska: conceptualization, formal analysis, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing. Katharina Ehrmann: conceptualization, data curation, formal analysis, investigation, methodology, project administration, supervision, validation, visualization, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including presented UV/vis, NMR, photo-DSC, photorheological, simultaneous thermal analysis, DMTA, tensile testing and ATR-IR data, are available at TU Wien Research Data at https://doi.org/10.48436/qjcpe-xwq06.References
- Y.-L. Liu, C.-Y. Hsieh and Y.-W. Chen, Polymer, 2006, 47, 2581–2586 CrossRef CAS.
- K. K. Oehlenschlaeger, N. K. Guimard, J. Brandt, J. O. Mueller, C. Y. Lin, S. Hilf, A. Lederer, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Polym. Chem., 2013, 4, 4348–4355 RSC.
- L. F. Hancock and R. A. Minns, US20220056218, 2022.
- C. Heinzmann, S. Coulibaly, A. Roulin, G. L. Fiore and C. Weder, ACS Appl. Mater. Interfaces, 2014, 6, 4713–4719 CrossRef CAS PubMed.
- J. H. Aubert, US20030116272, 2003.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC.
- J. J. Hernandez, G. R. Sama, Y. Hu, S. Soars, C. E. Niemet, C. M. Sanchez and C. N. Bowman, Adv. Funct. Mater., 2024, 34, 2306462 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2693–2697 CrossRef CAS.
- M. C. Parrott, J. C. Luft, J. D. Byrne, J. H. Fain, M. E. Napier and J. M. DeSimone, J. Am. Chem. Soc., 2010, 132, 17928–17932 CrossRef CAS PubMed.
- Z. Xu, Y. Liang, X. Ma, S. Chen, C. Yu, Y. Wang, D. Zhang and M. Miao, Nat. Sustain., 2020, 3, 29–34 CrossRef.
- J. Moon, S. B. Kwak, J. Y. Lee, D. Kim, J. U. Ha and J. S. Oh, Waste Manage., 2019, 85, 557–562 CrossRef CAS PubMed.
- R. Ghanbarinia Firozjah, A. Sadeghi and S. Khoee, ACS Omega, 2020, 5, 27119–27132 CrossRef CAS PubMed.
- S. Soars, J. Kamps, B. Fairbanks and C. Bowman, Macromol. Chem. Phys., 2021, 222, 2100111 CrossRef CAS.
- H. Masai, T. Nakagawa and J. Terao, Polym. J., 2024, 56, 297–307 CrossRef CAS.
- V. Tomeckova and J. W. Halloran, J. Eur. Ceram. Soc., 2010, 30, 2833–2840 CrossRef CAS.
- C. Haslinger, L. P. Leutgeb, M. Haas, S. Baudis and R. Liska, ChemPhotoChem, 2022, 6, e202200108 CrossRef CAS.
- T. Doi, H. Kawai, K. Murayama, H. Kashida and H. Asanuma, Chem. – Eur. J., 2016, 22, 10533–10538 CrossRef CAS PubMed.
- V. X. Truong, F. Li, F. Ercole and J. S. Forsythe, ACS Macro Lett., 2018, 7, 464–469 CrossRef CAS PubMed.
- S. C. Gauci, K. Ehrmann, M. Gernhardt, B. Tuten, E. Blasco, H. Frisch, V. Jayalatharachchi, J. P. Blinco, H. A. Houck and C. Barner-Kowollik, Adv. Mater., 2023, 35, 2300151 CrossRef CAS PubMed.
- M. Gernhardt, V. X. Truong and C. Barner-Kowollik, Adv. Mater., 2022, 34, 2203474 CrossRef CAS PubMed.
- C. Petit, J. Bachmann, L. Michalek, Y. Catel, E. Blasco, J. P. Blinco, A.-N. Unterreiner and C. Barner-Kowollik, Chem. Commun., 2021, 57, 2911–2914 RSC.
- X. Wu, K. Ehrmann, C. T. Gan, B. Leuschel, F. Pashley-Johnson and C. Barner-Kowollik, Adv. Mater., 2025, 37, 2419639 CrossRef CAS PubMed.
- B. Huang, M. Wei, E. Vargo, Y. Qian, T. Xu and F. D. Toste, J. Am. Chem. Soc., 2021, 143, 17920–17925 CrossRef CAS PubMed.
- T. Nakano, H. Iwasa, N. Miyagawa, S. Takahara and T. Yamaoka, J. Photopolym. Sci. Technol., 2000, 13, 715–718 CrossRef CAS.
- T. Sasaki, T. Yoneyama, S. Hashimoto, S. Takemura and Y. Naka, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3873–3880 CrossRef CAS.
- T. Sasaki, T. Kondo, M. Noro, K. Saida, H. Yaguchi and Y. Naka, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1462–1468 CrossRef CAS.
- T. Sasaki and H. Yaguchi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 602–613 CrossRef CAS.
- H. Yaguchi and T. Sasaki, Macromolecules, 2007, 40, 9332–9338 CrossRef CAS.
- H. Okamura and H. Tachi, J. Photopolym. Sci. Technol., 2018, 31, 727–733 CrossRef CAS.
- Y. Kotsuchibashi, M. Ebara, T. Sato, Y. Wang, R. Rajender, D. G. Hall, R. Narain and T. Aoyagi, J. Phys. Chem. B, 2015, 119, 2323–2329 CrossRef CAS PubMed.
- T. J. Spencer and P. A. Kohl, Polym. Degrad. Stab., 2011, 96, 686–702 CrossRef CAS.
- H. Okamura and M. Shirai, J. Photopolym. Sci. Technol., 2011, 24, 561–564 CrossRef CAS.
- M.-S. Jung, J. Hyeon-Lee and T.-L. Choi, J. Appl. Polym. Sci., 2008, 107, 2632–2637 CrossRef CAS.
- M. G. Cupta, P. J. Joseph and P. A. Kohl, J. Appl. Polym. Sci., 2007, 105, 2655–2662 CrossRef CAS.
- M. Hiro and J. M. J. Fréchet, in Irradiation of Polymers, American Chemical Society, 1996, vol. 620, ch. 29, pp. 381–386 Search PubMed.
- M. Shirai, T. Miwa and M. Tsunooka, Chem. Mater., 1995, 7, 642–648 CrossRef CAS.
- Y. Shin, J. M. Schwartz, A. C. Engler, B. Jones, O. Davydovich and P. A. Kohl, ACS Appl. Mater. Interfaces, 2024, 16, 43951–43960 CrossRef CAS PubMed.
- J. Jiang, O. Phillips, A. Engler, M. H. Vong and P. A. Kohl, Polym. Adv. Technol., 2019, 30, 1656–1662 CrossRef CAS.
- K. K. Fu, Z. Wang, J. Dai, M. Carter and L. Hu, Chem. Mater., 2016, 28, 3527–3539 CrossRef CAS.
- O. Phillips, A. Engler, J. M. Schwartz, J. Jiang, C. Tobin, Y. A. Guta and P. A. Kohl, J. Appl. Polym. Sci., 2019, 136, 47141 CrossRef.
- T. Shimizu, R. Whitfield, G. R. Jones, I. O. Raji, D. Konkolewicz, N. P. Truong and A. Anastasaki, Chem. Sci., 2023, 14, 13419–13428 RSC.
- K. Parkatzidis, H. S. Wang and A. Anastasaki, Angew. Chem., Int. Ed., 2024, 63, e202402436 CrossRef CAS PubMed.
- M. Kirstein, C. Lücking, L. Biermann, E. Brepohl, V. Salikov, C. Eichert, M. Paschetag and S. Scholl, Chem. Ing. Tech., 2023, 95, 1290–1296 CrossRef CAS.
- H. S. Wang, M. Agrachev, H. Kim, N. P. Truong, T.-L. Choi, G. Jeschke and A. Anastasaki, Science, 2025, 387, 874–880 CrossRef CAS PubMed.
- Y. Zheng, P. Song, S. Liu, M. Wu, H. Xu, C. Qiao, J. Liu, Z. Gao and Q. Ban, J. Polym. Sci., 2021, 59, 1390–1398 CrossRef CAS.
- L. Sinawehl, R. Wolff, T. Koch, J. Stampfl, R. Liska and S. Baudis, ACS Appl. Polym. Mater., 2023, 5, 5758–5771 CrossRef CAS.
- J. Bachmann, E. Gleis, G. Fruhmann, J. Riedelbauch, S. Schmölzer and O. Hinrichsen, Addit. Manuf., 2021, 37, 101677 CAS.
- M. Iguchi, Polymer, 2019, 168, 255–268 CrossRef CAS.
- J. K. Fink, in Polyoxymethylene Handbook, 2014, pp. 21–51. DOI:10.1002/9781118914458.ch2.
- R. Wolff, K. Ehrmann, P. Knaack, K. Seidler, C. Gorsche, T. Koch, J. Stampfl and R. Liska, Polym. Chem., 2022, 13, 768–777 RSC.
- O. Shelef, S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2021, 143, 21177–21188 CrossRef CAS PubMed.
- J. Jiang, M. Warner, O. Phillips, A. Engler and P. A. Kohl, Polymer, 2019, 176, 206–212 CrossRef CAS.
- R. Cox, Int. J. Mol. Sci., 2011, 12, 8316–8332 CrossRef CAS PubMed.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- V. G. Yadav and S. B. Chandalia, Org. Process Res. Dev., 1997, 1, 226–232 CrossRef CAS.
- R. Arias-Ugarte, F. S. Wekesa and M. Findlater, Tetrahedron Lett., 2015, 56, 2406–2411 CrossRef CAS.
- J. Van Damme, O. van den Berg, J. Brancart, L. Vlaminck, C. Huyck, G. Van Assche, B. Van Mele and F. Du Prez, Macromolecules, 2017, 50, 1930–1938 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00545k |
| This journal is © The Royal Society of Chemistry 2025 |