
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA versatile synthetic strategy for non-symmetric isoindigo polymers via modular sidechain engineering†
Rachael J.
Warner
a,
P. Blake J.
St Onge
a,
Alyssa
Shaw
b,
Tiago C.
Gomes
 a,
Piumi
Kulatunga
a,
Xiaodan
Gu
*b and
Simon
Rondeau-Gagné
*a
a,
Piumi
Kulatunga
a,
Xiaodan
Gu
*b and
Simon
Rondeau-Gagné
*a
aDepartment of Chemistry and Biochemistry, University of Windsor, Ontario, Canada N9B 3P4. E-mail: srondeau@uwindsor.ca
bSchool of Polymer Science and Engineering, The University of Southern Mississippi, Hattiesburg, Mississippi 39406, USA. E-mail: Xiaodan.Gu@usm.edu
First published on 29th April 2025
Abstract
Sidechain engineering is a powerful strategy for tailoring the intrinsic properties of semiconducting polymers, enabling precise modulation of structure–property relationships critical for organic electronic applications. Breaking symmetry via sidechain engineering—by incorporating two non-identical sidechains onto the conjugated backbone—is an emerging approach to control molecular stacking and polymer chain interactions. However, the fundamental impact of sidechain induced non-symmetry remains underexplored, limiting the full tunability and optimization of this design in soft organic electronics. In this work, we investigate the structure–property relationships of isoindigo-thienothiophene-based semiconducting polymers, employing a novel “lego-like” synthetic strategy for non-symmetric condensation of alkylated precursors. This method significantly enhances specificity, customizability, and reproducibility, facilitating the development of fully tunable systems. Using this approach, we synthesized a series of non-symmetric polymers with linear and branched aliphatic sidechains to evaluate their impact on polymer packing motifs and performance in organic field-effect transistors (OFETs). Multimodal characterization revealed that these non-symmetric polymers exhibit electronic properties comparable to their symmetric counterparts while demonstrating reduced crystallinity and low Young's moduli, as shown by atomic force microscopy. Furthermore, increasing the carbon spacer length from 1 to 4 carbons in the branched chain moiety improved charge transport, achieving average hole mobilities of up to 0.12 cm2 V−1 s−1 and 0.10 cm2 V−1 s−1 for P[(iITT)(C1C10C12)(C12)] and P[(iITT)(C4C10C12)(C12)], respectively. Overall, this work establishes a straightforward and highly tunable approach for designing a library of non-symmetric isoindigo derivatives through sidechain engineering, providing precise control over nanoscale polymer structures in thin films and unlocks new pathways for enhancing the performance of organic electronic materials.
Introduction
π-Conjugated semiconducting polymers have gained increasing interest for their applications in organic electronics largely due to their good charge transport properties (comparable to that of amorphous silicon) and tunable optical, electronic, and mechanical properties.1–6 Among the many design elements that can be synthetically fine-tuned to control their performance for improved materials, the sidechains play a particularly important role in the determination of intrinsic properties including molecular stacking, solution processability, and thin film morphology, all of which contribute directly to the optoelectronic and thermomechanical properties of the semiconducting polymer.7–10 In addition to including various functional groups in the sidechains of conjugated polymers, symmetry-breaking via the introduction of non-identical alkyl sidechains or functional groups at both ends of the isoindigo unit through sidechain engineering, is a relatively new design strategy to control the molecular stacking pattern and interactions of the polymer chains.11,12 For almost all high-performance semiconducting polymers, sidechains are symmetric, i.e. they are designed to possess the same length and structure across the entire chain. While this design strategy is efficient, using sidechain engineering to break the symmetry of polymer chains is a promising new design approach for controlling their molecular stacking patterns and interactions. Geometric disparities as a consequence of non-symmetry, has been shown to be beneficial in modulating the solubility, processability, and morphology of semiconducting polymers through various sidechain combinations.Recently, Yen et al. investigated the role of non-symmetry in the modulation of hydrophilicity in isoindigo-based semiconducting polymers using various functionalized sidechains including; oligoether, carbosilane-terminated sidechains, and semifluorinated sidechains.13 Notably the combination of oligoether and carbosilane sidechains resulted in an average hole mobility (μh) of 0.56 cm2 V−1 s−1, measured in organic field-effect transistors (OFETs), which was attributed to the favourable intermolecular interactions of the functionalized sidechains mitigating the steric hindrance between the conjugated backbone and sidechains. The polymer chain stacking orientation, whether edge-on or face-on dominated, was found to be controlled by altering the hydrophilicity through various non-symmetric sidechain combinations. Additionally, the intermolecular interactions between the sidechain combinations influenced the stretchability and overall ductility of the polymers by creating uniformly distributed amorphous domains while maintaining long range crystallinity. In another work, Lin et al. investigated the effect of side chain non-symmetry on the stretchability of semiconducting polymers and related OFET devices using carbosilane and siloxane terminated sidechains in conjunction with backbone fluorination in poly(isoindigo-bithiophene) polymers.14 Each sidechain combination was shown to impart distinct influences on the molecular stacking and orientation with variations in device performances and stretchability owed to the differences in carbon units after the branching point, with average hole mobility maxima of up to 3.49 cm2 V−1 s−1.
Building from these prior works, our group recently investigated a novel family of non-symmetric polyisoindigos bearing aliphatic sidechains combined with a hydroxy-terminated sidechain.15 Notably, the new materials were found to possess favourable electronic and solid-state properties and improved solubility in non-halogenated solvents. A non-symmetric amphiphilic system incorporating oligo(ethylene glycol) in combination with a solubilizing alkyl chain was also investigated for potential applications as mixed ion-electron conductors in organic electrochemical transistors.16,17 However, despite these unique properties, this design strategy possesses significant limitations, suffering from low synthetic yields and difficult control of the non-symmetric synthesis of materials, thus making large-scale production of these novel polymers a challenge.
A ‘one-pot’ alkylation method, where both sidechains are added simultaneously, has commonly been reported as the method of choice for non-symmetric sidechain alkylation.11,14,15,17 Despite successfully obtaining the targeted non-symmetric monomer, this synthetic method has been shown to possess significant limitations, including the generation of multiple products leading to a significantly lower yield of the desired non-symmetric product. Additional problems occur when the sidechains are of similar polarities, for example the use of common branched and linear alkyl chains, increasing the difficulty of purification of the non-symmetric monomer. Considering the need for large quantities of organic semiconducting polymers for large-scale manufacturing, and for exploring new structure–property relationships in electronic materials, this synthetic approach is, therefore, not viable.
To overcome these significant limitations and develop an efficient, simple, and fully tunable design strategy for achieving non-symmetric semiconducting polymers, herein, we introduce a novel synthetic method for the development of non-symmetric isoindigo-based polymers. Using this approach, we systematically varied the branching point of one of the sidechains to study its impact on the solid-state, thermomechanical, and electronic properties of the resulting materials. Drawing on the stepwise synthesis of isoindigo, the synthetic approach follows a sequential basic alkylation of the isoindigo monomer, and then is customized further using a non-symmetric condensation of two alkylated precursors in a “lego-like” strategy. The new approach leads to drastically increased overall specificity, customizability, and reproducibility of the synthesis allowing for a fully tunable system. The non-symmetric isoindigo monomers achieved synthetically through this novel method afforded yields ranging between 41% to 76%.
Although non-symmetric designs are attracting attention in current literature, the impact of sidechain non-symmetry at a fundamental level is not fully understood, preventing a complete tunability of the system and its optimization towards more efficient soft organic electronics. Therefore, toward getting further insights into this design and its impact on the properties of the semiconducting polymers, this work investigates a poly(isoindigo-co-thienothiophene) system utilizing only linear and branched aliphatic sidechains to determine how they affect the polymers' packing motifs and overall performance in OFET devices. Overall, increasing the carbon spacer length from 1 to 4 carbons before the branching point was found to have a favourable effect on charge transport with the CnCxCy sidechain series, with average hole mobilities of up to 0.12 cm2 V−1 s−1 and 0.10 cm2 V−1 s−1 obtained for P[(iITT)(C1C10C12)(C12)] and P[(iITT)(C4C10C10)(C12)] respectively. Overall, this work allows for a simple yet completely tunable approach for the development of a library of non-symmetric isoindigo derivatives through sidechain engineering, enabling precise control of nanoscale polymer structure in thin films and enabling new pathways to enhance the performance of these materials in emerging organic electronic devices.
Results and discussion
Isoindigo derivatives are initially formed through the condensation of two precursors: an isatin and oxindole derivative allowing for additional functionalization and customization of the isoindigo monomer through nucleophilic substitution with an alkyl halide.18 This synthetic approach is common in literature, typically achieving alkylated and soluble isoindigo derivatives in moderate yields. As shown in Scheme S1,† a typical one-pot non-symmetric alkylation of isoindigo can generate not only the non-symmetric alkylated monomer, but also two symmetric derivatives. The yield of the desired non-symmetric product is low, primarily due to side-products formation, but also due to difficulty in purification of the crude mixture after alkylation via column chromatography. This is to be expected given the structural similarities between all products, leading to similar polarity and affinity for the stationary phase in column chromatography. This synthetic challenge can be minimized by the utilization of alkyl halides with drastically different polarities; however, this synthetic route still remains challenging and low yielding. As a representative reaction, a one-pot alkylation of the isoindigo monomer with 1-bromododecane and 1-iodo-2-decyltetradecane was performed as shown in Scheme S1.† In addition to the desired product, additional side products including the symmetrically alkylated iI(C12) monomer was also observed in the crude mixture. Notably, separation via both column chromatography on silica gel and preparative thin-layer chromatography on silica were challenging, due to the similar polarities and coelution of each respective product.To circumvent the issues with the commonly used one-pot alkylation route and to improve the overall synthetic route towards non-symmetric semiconducting polymers, a novel “lego-like” approach to synthesize non-symmetric isoindigo derivatives was established (Scheme 1). In this method, 6-bromoisatin and 6-bromooxindole, the precursors to isoindigo, are alkylated with a branched alkyl sidechain and a linear alkyl sidechain, respectively. The process is completed with an aldol condensation, producing the non-symmetric isoindigo monomer with high selectivity. The complete synthetic details are included in ESI.† Briefly, 6-bromooxindole was first reacted with 1-bromodocane and potassium carbonate in dimethylformamide under nitrogen atmosphere. However, after examination by NMR, it was determined that a disubstituted symmetric isoindigo monomer had been formed in one step, instead of an alkylated oxindole derivative. A similar observation was noted by Zhao et al.; an alkylated thiophene fused isoindigo was produced in one step using excess potassium carbonate and 1-bromooctane under aerobic conditions.19 It was proposed that the thiophene-fused oxindole underwent alkylation and oxidation under aerobic basic conditions to produce the alkylated dione. The dione would then condense with an unoxidized oxindole to produce the disubstituted symmetric isoindigo monomer compound 14. An alternative method to synthesize the alkylated oxindole was then utilized as illustrated in Scheme 1. A Wolf–Kishner type reduction was then carried out to reduce the alkylated dione with a dodecyl side chain (compound 1) to an oxindole, (compound 2). The alkylated oxindole was then condensed with an isatin derivative under acidic conditions (compounds 3 to 7), bearing various branched alkyl chains, to produce the non-symmetric iI(CnCxCy)(C12) monomers compounds 8 to 12 in good yields.
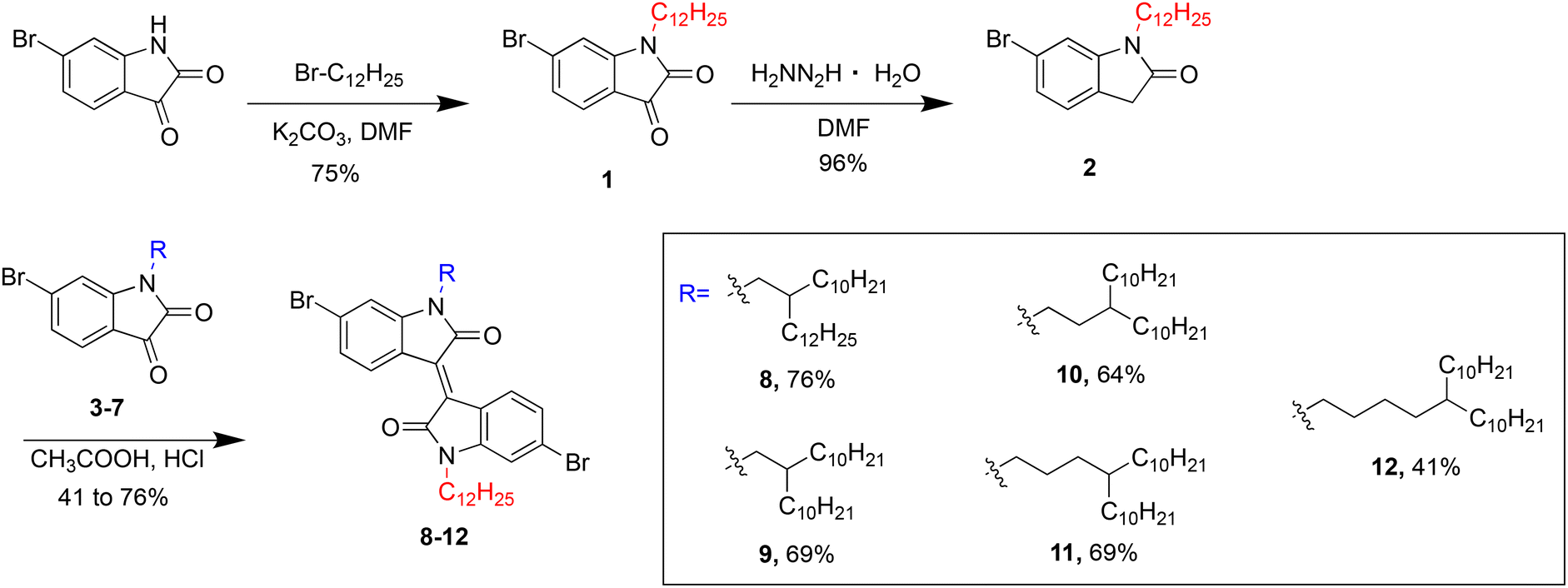 | ||
| Scheme 1 Synthetic pathway to asymmetric isoindigo iI(CnCxCy)(C12) monomers, compounds 8 to 12. | ||
An alternate strategy was also investigated through successive alkylation as shown in Scheme S3.† First, a monoalkylated isoindigo monomer is synthesized, which then undergoes a second alkylation to achieve the desired non-symmetric monomer. This approach was explored for the synthesis of compound 8 as detailed in ESI.† The formation of the monoalkylated monomer compound 13 is performed through acid-catalysed aldol-condensation between compound 3 and the commercially available 6-bromoisatin to afford compound 13 in good yield (54%). The second alkylation was carried out using standard conditions with 1-iodododecane and potassium carbonate in DMF to generate compound 8 in 67% yield, compared to the previous 76% yield obtain in the approach described previously. Notably, while this alternate method increased the specificity of reaction to generate the targeted non-symmetric isoindigo monomer, the purification of the final products was significantly more tedious, and was primarily limited by the solubility of the monoalkylated intermediate. The new “lego-like” synthetic strategy described in Scheme 1 is therefore the most suitable approach to achieve the non-symmetric isoindigo-based monomers with desired synthetic yields and versatility.
Following the preparation of the various non-symmetric isoindigo monomers, co-polymerization with 2,5-bis(trimethylstannyl)thieno[3,2-b]thiophene through palladium-catalyzed Stille cross-coupling afforded the P[(iITT)(CnCxCy)(C12)] series. The crude polymer mixtures were then purified via Soxhlet extractions successively using methanol, acetone, hexanes, and chloroform. The chloroform fractions were collected and precipitated in methanol and then dried under vacuum. The complete synthetic procedure and structural characterization of the new polymers are detailed in ESI.† All polymers were obtained in moderate yields, which may be attributed, at least in part, to the limited solubility of the growing polymer chains. Upon synthesis of the semiconducting polymers, high-temperature gel permeation chromatography in 1,2,4-trichlorobenzene at 180 °C against monodisperse polystyrene standards was performed on the P[(iITT)(CnCxCy)(C12)] series with results summarized in Table 1. All polymers were shown to possess a number-average molecular weight around 15 kDa and degree of polymerization above 10. Only P[(iITT)(C2C10C10)(C12)] showed a smaller molecular weight. UV–vis spectroscopy was subsequently performed to evaluate the optical properties of the polymers, as shown in Fig. 1. The materials were characterized either in solution (CHCl3) or as thin films, which were prepared by drop casting to achieve improved film thickness and spectral quality. In both solution and solid state, the polymers showed dual-band absorption typically associated with donor–acceptor type polymers. The absorption spectra of the polymers in solid state also revealed significant differences in aggregation patterns based on the position of the sidechain branching point. Specifically, a progressive reduction in the (0–1)/(0–0) vibrational peaks ratio was observed as the carbon spacer length before the branch point increased from 1 to 4 carbons.20 This reduction in the (0–0) vibrational peak suggests decreased polymer chain aggregation, attributed to J-type aggregation, as the branching point moves further from the polymer backbone—a trend consistent with previous literature.20–22 The absorption maxima exhibited a hypsochromic shift, from 737 nm for one-carbon spacers to 716 nm for four-carbon spacers, further indicating a reduction in J-aggregates as the spacer length increases. Cyclic voltammetry (CV) was then performed in addition to UV-Vis spectroscopy to determine the optical band gap and highest occupied molecular orbital (HOMO) levels of the polymer series (Fig. S3†). The lowest molecular orbital (LUMO) was then calculated by subtracting the optical bandgap value from the determined HOMO energy level. As indicated in Table 1, all polymers show relatively similar HOMO/LUMO levels, confirming the minimal influence of the sidechain structures on the polymers’ energetic levels. Thermogravimetric analysis (TGA) was then performed in order to determine the thermal stability of the new polymers, with results summarized in Fig. S4.† All polymers show a thermal decomposition (5% weight loss) above 380 °C, thus confirming their high thermal stability.
 | ||
| Fig. 1 Comparative UV-Vis spectra of P[(iITT)(CnCxCy)(C12)] polymers (a) solution in CHCl3, and (b) drop-casted thin film. | ||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Polymer |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (kDa) a (kDa) |
Đ
wb |
X
nc |
λ
filmmaxd (nm) |
λ
solnmaxd (nm) |
E
optg
(film)e (eV) |
E
optg
(soln)e (eV) |
HOMOf (eV) | LUMOg (eV) |
T
dh (°C) |
| a Number-average molecular weight estimated by high-temperature gel permeation chromatography in 1,2,4-trichlorobenzene at 180 °C using a polystyrene standard. b Dispersity defined as Mw/Mn. c Degree of polymerization defined as Mn/M0, where M0 is the molecular weight of the repeat unit of each polymer. d Absorption maxima. e Calculated by the following equation: gap = 1240/λonset. f Calculated from cyclic voltammetry (potentials vs. Ag/AgCl) using 0.1 M TBAPF6 in CH3CN as electrolyte where EHOMO = −4.38 eV − (OxOnset). g Estimated from calculated Eg and HOMO. h Estimated from thermogravimetry analysis (TGA) at 5% mass loss (Fig. S4†). | ||||||||||
| P[(iITT)(C1C10C12)(C12)] | 17.1 | 2.9 | 19 | 737 | 727 | 1.40 | 1.58 | −5.49 | −4.09 | 397 |
| P[(iITT)(C1C10C10)(C12)] | 12.2 | 2.4 | 14 | 737 | 723 | 1.45 | 1.59 | −5.49 | −4.04 | 382 |
| P[(iITT)(C2C10C10)(C12)] | 7.2 | 2.3 | 8 | 718 | 728 | 1.59 | 1.58 | −5.51 | −3.93 | 389 |
| P[(iITT)(C3C10C10)(C12)] | 16.0 | 2.3 | 18 | 718 | 723 | 1.57 | 1.58 | −5.23 | −3.66 | 387 |
| P[(iITT)(C4C10C10)(C12)] | 24.6 | 3.6 | 23 | 716 | 736 | 1.58 | 1.57 | −5.20 | −3.63 | 393 |

To gain insight into the solid-state properties of these materials in thin films, we investigated the non-symmetric semiconducting polymers using atomic force microscopy (AFM), as shown in Fig. S6 to S9.† Notably, at the nanoscale, all polymers displayed a relatively smooth surface with no distinctive morphological patterns as observed by AFM height images. Surface root mean square roughness (Rq) values ranged from 0.98 to 3.99 nm, consistent with isoindigo-based semiconducting polymers. These findings suggest that side-chain structure has minimal influence on the polymers’ aggregation properties, contributing to their uniform nanoscale morphology in the solid state. AFM measurements of the film height profiles (Fig. S8†) revealed thicknesses ranging from 10 to 36 nm. The variation in polymer thicknesses was primarily attributed to differences in apparent viscosity, resulting from the varying molecular weights of the polymer samples.
Following the characterization of the solid-state morphology, the electronic properties of the P[(iITT)(CnCxCy)(C12)] polymers were investigated through the fabrication of OFETs with a bottom-gate top-contact architecture. The detailed fabrication procedure can be found in ESI.† Briefly, devices were prepared by spin-coating 5 mg mL−1 solutions in chlorobenzene onto octyltrimethoxysilane (OTS) functionalized Si/SiO2.23 Source and drain electrodes were then deposited through physical vapour deposition. All devices showed good output characteristics and transfer behavior. Fig. 2 shows representative output and transfer curves of the devices prepared from P[(iITT)(CnCxCy)(C12)] series using chlorobenzene. Linear fitting of IDS1/2vs. VGS from the transfer curves in the saturation regime was used to extract charge carrier mobility (μsat) using the following equation: IDS (sat) = (WC/2L) μsat(VG − Vth)2, where IDS is drain current, W and L are channel width and length, C is the dielectric constant of SiO2, VG is gate voltage and Vth is threshold voltage. As shown in Table 2, the performance of the materials in OFETs prepared from chlorobenzene solutions was, as expected, significantly influenced by the chemical design of the asymmetric polymer. The polymer P[(iITT)(C1C10C12)(C12)], which combines a linear side chain with an asymmetrical branched side chain, demonstrated an average mobility of 0.12 cm2 V−1 s−1. This value aligns with previously reported mobilities for traditional symmetric isoindigo derivatives, indicating that the presence of a linear chain on one end of the isoindigo moiety does not detrimentally impact charge transport.24 When the C1C10C12 side chain was replaced with a C1C10C10 sidechain in P[(iITT)(C1C10C10)(C12)], the charge mobility decreased by an order of magnitude, with an average value of 0.036 cm2 V−1 s−1. Shifting the branching point progressively further from the π-conjugated backbone led to a gradual increase in average charge mobility, from 0.036 cm2 V−1 s−1 with the C1C10C10 side chain to 0.053 cm2 V−1 s−1 with the C2C10C10 side chain in P[(iITT)(C2C10C10)(C12)]. Interestingly, when the C3C10C10 sidechain was used alongside a linear sidechain in P[(iITT)(C3C10C10)(C12)], the average charge mobility decreased slightly to 0.024 cm2 V−1 s−1. However, extending the branching to the C4 position in P[(iITT)(C4C10C10)(C12)] significantly improved the average mobility to 0.10 cm2 V−1 s−1, comparable to the C1C10C12 non-symmetric counterpart. Several key insights emerged from analyzing these trends. First, shifting from the asymmetric C1C10C12 to the symmetric C1C10C10 sidechain is detrimental to charge mobility, likely due to suboptimal sidechain interdigitation. Additionally, pushing the branching point from C1 spacer to C4 in the C10C10 sidechain series generally increases charge mobility, likely due to reduced steric hindrance along the π-conjugated backbone, which promotes better π–π stacking between polymer chains and enhances charge mobility. This phenomenon has been previously reported for similar semiconducting polymer systems.25–27 Generally, polymers with branched sidechains featuring an even number of carbon spacers exhibited improved charge mobility compared to those with odd-numbered spacers, except in the case of the non-symmetric C1C10C12 which exhibited the overall highest charge mobility, possibly due to the asymmetric branched chain allowing for increased sidechain interdigitation. This observation agrees with the typical odd-even effect seen in such semiconducting polymers, where even spacers usually yield better performance.28,29
 | ||
| Fig. 2 Representative output characteristics (left) and transfer curves (right) of P[(iITT)(CnCxCy)(C12)] series; (a) P[(iITT)(C1C10C12)(C12)], (b) P[(iITT)(C1C10C10)(C12)]; (c) P[(iITT)(C2C10C10)(C12)]; (d) P[(iITT)(C3C10C10)(C12)], and (e) P[(iITT)(C4C10C10)(C12)]. All measurements were performed on thin films prepared via spin coating of a chlorobenzene solution of selected polymer (5 mg ml−1). All OFET devices were fabricated after thermal annealing for 1 hour at hour at 150 °C, VDS = −80 V. | ||
| Polymer | Thicknessa (nm) | W/L | μ aveh/μmaxh [cm2 V−1 s−1] | I ON/IaveOFF | V aveth [V] |
|---|---|---|---|---|---|
| a Thickness confirmed by AFM (Fig. S8†). | |||||
| P(iITT)(C1C10C12)(C12) | 36 | 1000/150 | 0.12 ± 0.02/0.12 | 103 | −15 |
| P(iITT)(C1C10C10)(C12) | 8 | 1000/150 | 0.036 ± 0.005/0.042 | 103 | −9 |
| P(iITT)(C2C10C10)(C12) | 16 | 1000/150 | 0.053 ± 0.005/0.060 | 101 | −11 |
| P(iITT)(C3C10C10)(C12) | 12 | 1000/150 | 0.024 ± 0.007/0.033 | 102 | −10 |
| P(iITT)(C4C10C10)(C12) | 20 | 1000/150 | 0.10 ± 0.03/0.13 | 103 | −19 |
Considering the significant impact of thin film conditions and the solvent on polymer chain self-assembly, we also investigated the electronic properties of the new polymers in CHCl3. Due to its lower boiling point, chloroform often produces films with a distinct nanostructure compared to solvents with higher boiling points, especially after thermal annealing. While predicting the effects of solvent substitution is challenging, it provides an opportunity to further assess the influence of chemical design on the electronic properties of the materials. As with the devices prepared from chlorobenzene, all devices fabricated with chloroform exhibited good output and transfer OFET characteristics. Fig. S12 and S13† show representative output and transfer curves for the P[(iITT)(CnCxCy)(C12)] series processed from CHCl3, with key results summarized in Table S2.† Notably, all chloroform-prepared devices demonstrated a lower charge carrier mobility, regardless of the polymer used. Charge mobility decreased by approximately an order of magnitude across all polymers, though a similar trend related to side-chain design and its impact on electronic properties, consistent with that observed in chlorobenzene, was observed. The polymer P[(iITT)(C1C10C12)(C12)], featuring a combination of a linear and an asymmetric branched sidechain, exhibited an average mobility of 0.062 cm2 V−1 s−1. Replacing the C1C10C12 sidechain with a C1C10C10 sidechain in P[(iITT)(C1C10C10)(C12)] slightly reduced the mobility, with an average value of 0.010 cm2 V−1 s−1. Unlike the behavior observed in devices prepared from chlorobenzene, progressively shifting the branching point away from the π-conjugated backbone did not significantly increase the average charge mobility, which remained stable at 0.018 cm2 V−1 s−1 when transitioning from a C1C10C10 to a C2C10C10 side chain in P[(iITT)(C2C10C10)(C12)]. Notably, similar to the trend observed in chlorobenzene, using the C3C10C10 sidechain with a linear sidechain in P[(iITT)(C3C10C10)(C12)] reduced the average charge mobility to 0.0015 cm2 V−1 s−1 for devices prepared from CHCl3. However, extending the carbon spacers to the C4 position in P[(iITT)(C4C10C10)(C12)] markedly improved mobility to 0.050 cm2 V−1 s−1, once again aligning it with the non-symmetric C1C10C12 counterpart. Overall, the results obtained by processing the polymers in CHCl3 confirm that the disruption of symmetry in the isoindigo core, achieved through a combination of linear and branched sidechains, does not significantly alter the polymers electronic properties, which remain consistent with previously reported values for symmetric isoindigo-based polymers.
To further get insights on the impact of sidechain structure, and the non-symmetry of the isoindigo moiety induced by the presence of a linear dodecyl sidechain on electronic properties, characterization of the nanoscale microstructure was performed by grazing incidence wide-angle X-ray scattering (GIWAXS). This analysis was performed directly on thin films prepared from 10 mg mL−1 chlorobenzene solutions of the non-symmetric isoindigo-based polymers, with results summarized in Fig. 3 and Table S1.† Overall, the introduction of asymmetry on the isoindigo core and branched sidechain structure was found to influence the polymer chain packing in the solid-state. GIWAXS analysis revealed long range organization for all polymers with strong out-of-plane diffraction peaks (n00) along the qz plane representing lamellar packing. The presence of strong (400) reflections were observed for the non-symmetric polymer series. In-plane diffraction peaks (010) along the qxy axis represent π–π stacking of the conjugated backbone, and remain consistent for the P[(iITT)(CnCxCy)(C12)] series (q ∼ 1.81 Å−1). Interestingly, the alkyl spacer length before the branching point showed minimal impact on the alkyl packing distance, calculated to be 22.51 Å for P[(iITT)(C1C10C10)(C12)], and 23.00 Å for P[(iITT)(C4C10C10)(C12)]. The alkyl packing distance showed an increasing trend as the carbon spacer was increased from one to three carbons for the CnCxCy series. Similarly, as the branching point was moved progressively further from the backbone, the π–π stacking of the conjugated backbone decreased from 3.47 Å for P[(iITT)(C1C10C12)(C12)] to 3.44 Å for P[(iITT)(C4C10C10)(C12)]. Notably, the π–π stacking distance slightly increased to 3.49 Å for P[(iITT)(C3C10C10)(C12)]. Although the alkyl packing and π–π stacking was not significantly influenced by the branching point of the asymmetric sidechain, 2D GIWAXS analysis indicates variations in polymer ordering within thin films as evidenced by differences in intensity and ordering of the (n00) diffraction peaks. In contrast to traditional symmetric polymers, the presence of a linear dodecyl sidechain most likely plays an important role in molecular packing of the solid-state, thus reducing the impact of the branching point spacer and branched chain interdigitation.
 | ||
| Fig. 3 Grazing incident wide-angle X-ray scattering (GIWAXS) 2D patterns of (a) P[(iITT)(C1C10C12)(C12)]; (b) P[(iITT)(C1C10C10)(C12)]; (c) P[(iITT)(C2C10C10)(C12)]; (d) P[(iITT)(C3C10C10)(C12)], and (e) P[(iITT)(C4C10C10)(C12)]; (f) summary of chain distances (alkyl packing distance and π–π stacking) and hole mobility versus branching point; (g) out-of-plane and (h) in-plane 1D GIWAXS patterns. All measurements have been performed on thin films prepared from chlorobenzene solutions (10 mg mL−1) after thermal annealing at 150 °C. | ||
Following the characterization of the nanostructure of the materials upon non-symmetric sidechain engineering, AFM quantitative nanomechanical mapping analysis was used to probe for the mechanical properties of the new materials by providing insights on the deformation of the thin film upon nanoindentation.17 Derjaguin–Muller–Toporov (DMT) modulus was determined for the P[(iITT)(CnCxCy)(C12)] series and suggested that the branching point of the sidechain did not significantly affect the ductility of the polymers as indicated in Fig. 4. Overall, the elastic moduli for the new polymers ranged between 1.58 GPa for P[(iITT)(C2C10C10)(C12)] to 2.25 GPa for P[(iITT)(C1C10C10)(C12)]. These values agree with our previous work and other reports on similar semiconducting polymers based on diketopyrrolopyrrole.30–32 Notably, the elastic moduli measured are slightly higher than that measured for other isoindigo-based polymers which were reported to range between 0.2–0.8 GPa. This difference can potentially be attributed to the presence of fused ring systems within the conjugated backbone of the polymers, which likely increase the rigidity of the conjugated backbone.33,34 Sidechain interdigitation, directly impacting van der Waals interactions and crystallinity, can also impact elastic moduli.
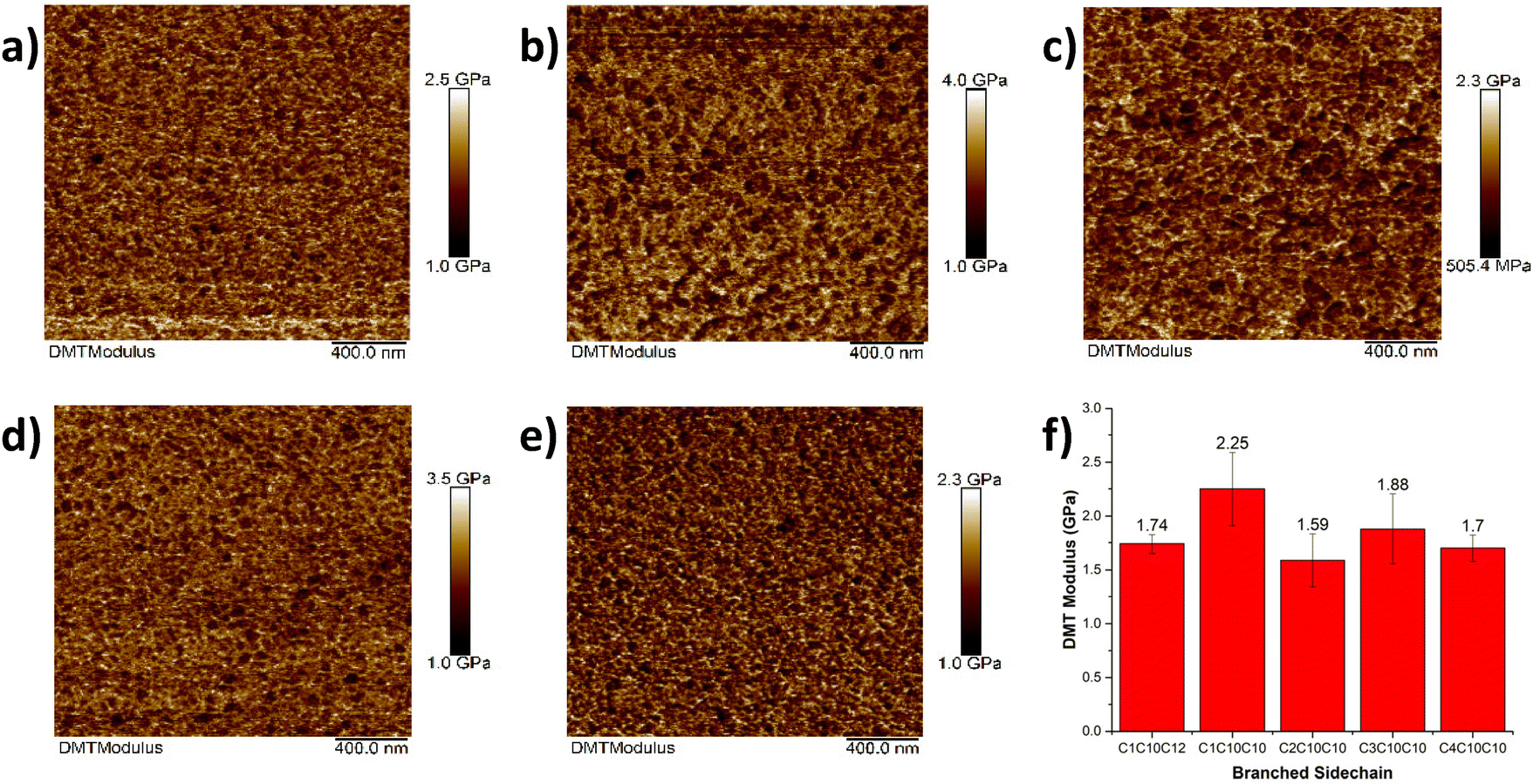 | ||
| Fig. 4 Influence of sidechains on elastic modulus. Nanomechanical mapping of (a) P[(iITT)(C1C10C12)(C12)]; (b) P[(iITT)(C1C10C10)(C12)]; (c) P[(iITT)(C2C10C10)(C12)]; (d) P[(iITT)(C3C10C10)(C12)], and (e) P[(iITT)(C4C10C10)(C12)]; (f) DMT moduli of P[(iITT)(CnCxCy)(C12)] taken using nanoindentation, scale bar of 400 nm. All samples are thin films prepared from chlorobenzene solutions (5 mg mL−1) after thermal annealing at 150 °C. | ||
Conclusion
A novel synthetic strategy was developed to access isoindigo-based semiconducting polymers with two non-identical sidechains, so-called non-symmetric polymers. This modular, “lego-like” approach overcame key challenges associated with traditional one-pot alkylation methods used to prepare these materials, such as low yields and difficult purification, providing a tunable and efficient pathway to non-symmetric semiconducting polymers. Using this method, a series of five non-symmetric isoindigo-based polymers with the general structure P[(iITT)(CnCxCy)(C12)] were synthesized and systematically characterized. A combination of optical spectroscopy, X-ray scattering, and AFM analyses revealed the effects of branching point asymmetry on their mechanical and optoelectronic properties. Additionally, top-contact, bottom-gate OFET devices were fabricated to explore how non-symmetry influenced charge transport in organic electronics. Notably, fixing one sidechain as linear dodecyl and varying the branching point from a C1 spacer to a C4 spacer in the branched sidechain moieties resulted in an increase in charge mobility. P[(iITT)(C4C10C10)(C12)] exhibited an average mobility of 0.10 cm2 V−1 s−1, comparable to its symmetric counterparts. Furthermore, P[(iITT)(C1C10C12)(C12)], containing an asymmetric C1C10C12 branched chain, achieved an average mobility of 0.12 cm2 V−1 s−1, attributed to more optimal sidechain interdigitation. Interestingly, while the branching point had a minimal effect on optical bandgaps and thin-film morphology, as evidenced by AFM, nanomechanical analysis, and GIWAXS, the linear dodecyl sidechain likely played a crucial role in molecular packing within the solid-state. This reduced the influence of the branching point spacer and interdigitation on overall polymer performance, distinguishing these non-symmetric polymers from their symmetric counterparts. These findings highlight the potential of sidechain non-symmetry as a design parameter to tune polymer properties without compromising solid-state packing or charge transport. This approach not only expands the toolkit for polymer design but also paves the way for the development of next-generation materials for organic electronic applications.Author contributions
RW performed the synthesis of all polymers and precursors, assisted by PBJSO. RW performed the UV−vis, CV, FTIR, and NMR measurements and analyzed the data. RW fabricated and characterized all OFETs. XG and AS characterized the materials by GIWAXS. TCG and PK characterized the materials by AFM. SRG contributed to formal analysis, funding acquisition, project administration, supervision, writing-original draft, writing/review & editing. RW and SRG conceived the hypothesis, wrote and edited all version of the manuscript, with input from all co-authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by NSERC through Discovery Grants (SRG: RGPIN-2022-04428). S. R-G. thanks the Canadian Fund for Innovation (CFI) for supporting the infrastructure used in this work. R. J. W. thanks NSERC for financial support through a Canada Graduate Scholarship – Masters scholarship. X. G. acknowledges the support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Science under award no. DE-SC0022050. A. S. acknowledges the support by the National Science Foundation Graduate Research Fellowship under grant no. 2234694. The authors would like to acknowledge the support and access to resources provided by the McMaster Centre for Advanced Light Microscopy (CALM) at McMaster University, Hamilton, Canada.References
- M. U. Ocheje, B. P. Charron, A. Nyayachavadi and S. Rondeau-Gagné, Stretchable electronics: recent progress in the preparation of stretchable and self-healing semiconducting conjugated polymers, Flexible Printed Electron., 2017, 2, 043002 CrossRef.
- A. Moliton and R. C. Hiorns, Review of electronic and optical properties of semiconducting π-conjugated polymers: applications in optoelectronics, Polym. Int., 2004, 53, 1397–1412 CrossRef CAS.
- A. Facchetti, π-Conjugated Polymers for Organic Electronics and Photovoltaic Cell Applications, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- P. W. M. Blom, Polymer Electronics: To Be or Not to Be?, Adv. Mater. Technol., 2020, 5, 2000144 CrossRef CAS.
- I. B. Dimov, M. Moser, G. G. Malliaras and I. McCulloch, Semiconducting Polymers for Neural Applications, Chem. Rev., 2022, 122, 4356–4396 CrossRef CAS PubMed.
- R. M. Pankow and B. C. Thompson, The development of conjugated polymers as the cornerstone of organic electronics, Polymer, 2020, 207, 122874 CrossRef CAS.
- Z. Cao, L. Galuska, Z. Qian, S. Zhang, L. Huang, N. Prine, T. Li, Y. He, K. Hong and X. Gu, The effect of side-chain branch position on the thermal properties of poly(3-alkylthiophenes), Polym. Chem., 2020, 11, 517–526 RSC.
- D. L. Meyer, N. Schmidt-Meinzer, C. Matt, S. Rein, F. Lombeck, M. Sommer and T. Biskup, Side-Chain Engineering of Conjugated Polymers: Distinguishing Its Impact on Film Morphology and Electronic Structure, J. Phys. Chem. C, 2019, 123, 20071–20083 CrossRef CAS.
- S. Lv, L. Li, Y. Mu and X. Wan, Side-chain engineering as a powerful tool to tune the properties of polymeric field-effect transistors, Polym. Rev., 2021, 61, 520–552 CrossRef CAS.
- S. Zhang, A. Alesadi, G. T. Mason, K.-L. Chen, G. Freychet, L. Galuska, Y.-H. Cheng, P. B. J. S. Onge, M. U. Ocheje, G. Ma, Z. Qian, S. Dhakal, Z. Ahmad, C. Wang, Y.-C. Chiu, S. Rondeau-Gagné, W. Xia and X. Gu, Molecular Origin of Strain-Induced Chain Alignment in PDPP-Based Semiconducting Polymeric Thin Films, Adv. Funct. Mater., 2021, 31, 2100161 CrossRef CAS.
- Z. Tang, X. Xu, R. Li, L. Yu, L. Meng, Y. Wang, Y. Li and Q. Peng, Asymmetric Siloxane Functional Side Chains Enable High-Performance Donor Copolymers for Photovoltaic Applications, ACS Appl. Mater. Interfaces, 2020, 12, 17760–17768 CrossRef CAS.
- Y. Yuan, F. Zhao, Y. Ding, G. Zhang, X. Wang and L. Qiu, Asymmetric Hybrid Siloxane Side Chains for Enhanced Mobility and Mechanical Properties of Diketopyrrolopyrrole-Based Polymers, Macromol. Rapid Commun., 2022, 43, 2100636 CrossRef CAS PubMed.
- H.-C. Yen, Y.-C. Lin and W.-C. Chen, Modulation of the Hydrophilicity on Asymmetric Side Chains of Isoindigo-Based Polymers for Improving Carrier Mobility–Stretchability Properties, Macromolecules, 2021, 54, 1665–1676 CrossRef CAS.
- Y.-C. Lin, F.-H. Chen, Y.-C. Chiang, C.-C. Chueh and W.-C. Chen, Asymmetric Side-Chain Engineering of Isoindigo-Based Polymers for Improved Stretchability and Applications in Field-Effect Transistors, ACS Appl. Mater. Interfaces, 2019, 11, 34158–34170 CrossRef CAS PubMed.
- M. Mooney, A. Nyayachavadi, A. Awada, E. Iakovidis, Y. Wang, M.-N. Chen, Y. Liu, J. Xu, Y.-C. Chiu, X. Gu and S. Rondeau-Gagné, Asymmetric side-chain engineering in semiconducting polymers: a platform for greener processing and post-functionalization of organic electronics, Polym. Chem., 2023, 14, 562–572 RSC.
- A. Hu, A. Nyayachavadi, M. Weires, G. Garg, S. Wang and S. Rondeau-Gagné, Unravelling the influence of side-chain symmetry on device performance: insights from isoindigo-based polymers in thin-film transistors, RSC Appl. Polym., 2023, 1, 292–303 RSC.
- G.-H. Jiang, C.-Y. Li, S.-W. Su and Y.-C. Lin, Asymmetric side-chain engineering of conjugated polymers with improved performance and stability in organic electrochemical transistors, J. Mater. Chem. C, 2024, 12, 11752–11762 RSC.
- T. Lei, J.-Y. Wang and J. Pei, Design, Synthesis, and Structure–Property Relationships of Isoindigo-Based Conjugated Polymers, Acc. Chem. Res., 2014, 47, 1117–1126 CrossRef CAS PubMed.
- N. Zhao, L. Qiu, X. Wang, Z. An and X. Wan, Synthesis of a thiophene-fused isoindigo derivative: a potential building block for organic semiconductors, Tetrahedron Lett., 2014, 55, 1040–1044 CrossRef CAS.
- B. C. Schroeder, T. Kurosawa, T. Fu, Y.-C. Chiu, J. Mun, G.-J. N. Wang, X. Gu, L. Shaw, J. W. E. Kneller, T. Kreouzis, M. F. Toney and Z. Bao, Taming Charge Transport in Semiconducting Polymers with Branched Alkyl Side Chains, Adv. Funct. Mater., 2017, 27, 1701973 CrossRef.
- F. Zhang, Y. Hu, T. Schuettfort, C. Di, X. Gao, C. R. McNeill, L. Thomsen, S. C. B. Mannsfeld, W. Yuan, H. Sirringhaus and D. Zhu, Critical Role of Alkyl Chain Branching of Organic Semiconductors in Enabling Solution-Processed N-Channel Organic Thin-Film Transistors with Mobility of up to 3.50 cm2 V–1 s–1, J. Am. Chem. Soc., 2013, 135, 2338–2349 CrossRef CAS PubMed.
- I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo, J. Nelson and I. McCulloch, Photocurrent Enhancement from Diketopyrrolopyrrole Polymer Solar Cells through Alkyl-Chain Branching Point Manipulation, J. Am. Chem. Soc., 2013, 135, 11537–11540 CrossRef CAS PubMed.
- Y. Ito, A. A. Virkar, S. Mannsfeld, J. H. Oh, M. Toney, J. Locklin and Z. Bao, Crystalline Ultrasmooth Self-Assembled Monolayers of Alkylsilanes for Organic Field-Effect Transistors, J. Am. Chem. Soc., 2009, 131, 9396–9404 CrossRef CAS PubMed.
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Systematic Investigation of Isoindigo-Based Polymeric Field-Effect Transistors: Design Strategy and Impact of Polymer Symmetry and Backbone Curvature, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS.
- S. Chen, Y. Meng, Y. Li, B. Qu and D. Zhuo, Effect of the length and branching point of alkyl side chains on DPP-thieno[3,2-b]thiophene copolymers for organic thin-film transistors, Opt. Mater., 2019, 88, 500–507 CrossRef CAS.
- K. Kawabata, M. Saito, N. Takemura, I. Osaka and K. Takimiya, Effects of branching position of alkyl side chains on ordering structure and charge transport property in thienothiophenedione- and quinacridone-based semiconducting polymers, Polym. J., 2017, 49, 169–176 CrossRef CAS.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, Siloxane-Terminated Solubilizing Side Chains: Bringing Conjugated Polymer Backbones Closer and Boosting Hole Mobilities in Thin-Film Transistors, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou and J. Pei, Influence of Alkyl Chain Branching Positions on the Hole Mobilities of Polymer Thin-Film Transistors, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed.
- J.-H. Dou, Y.-Q. Zheng, T. Lei, S.-D. Zhang, Z. Wang, W.-B. Zhang, J.-Y. Wang and J. Pei, Systematic Investigation of Side-Chain Branching Position Effect on Electron Carrier Mobility in Conjugated Polymers, Adv. Funct. Mater., 2014, 24, 6270–6278 CrossRef CAS.
- M. U. Ocheje, M. Selivanova, S. Zhang, T. H. V. Nguyen, B. P. Charron, C.-H. Chuang, Y.-H. Cheng, B. Billet, S. Noori, Y.-C. Chiu, X. Gu and S. Rondeau-Gagné, Influence of amide-containing side chains on the mechanical properties of diketopyrrolopyrrole-based polymers, Polym. Chem., 2018, 9, 5531–5542 RSC.
- A. Nyayachavadi, A. K. Sur, P. Kulatunga, Y. Wang, T. C. Gomes, M. Mooney, G. T. Mason, A. Hu, X. Gu and S. Rondeau-Gagné, Covalent Topochemical Photo-Cross-Linking of Diketopyrrolopyrrole-Based Polymers with Polydiacetylenes toward Tunable Mechanical and Electronic Properties, Chem. Mater., 2023, 35, 9682–9691 CrossRef CAS.
- Y.-C. Lin, C.-C. Shih, Y.-C. Chiang and W.-C. Chen, Intrinsically stretchable isoindigo–bithiophene conjugated copolymers using poly(acrylate amide) side chains for organic field-effect transistors, Polym. Chem., 2019, 10, 5172–5183 RSC.
- S. Savagatrup, A. D. Printz, T. F. O'Connor, A. V. Zaretski, D. Rodriquez, E. J. Sawyer, K. M. Rajan, R. I. Acosta, S. E. Root and D. J. Lipomi, Mechanical degradation and stability of organic solar cells: molecular and microstructural determinants, Energy Environ. Sci., 2014, 8, 55–80 RSC.
- B. Roth, S. Savagatrup, N. V. de los Santos, O. Hagemann, J. E. Carlé, M. Helgesen, F. Livi, E. Bundgaard, R. R. Søndergaard, F. C. Krebs and D. J. Lipomi, Mechanical Properties of a Library of Low-Band-Gap Polymers, Chem. Mater., 2016, 28, 2363–2373 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedure; NMR spectra; FTIR spectra; high temperature GPC of the new polymers P[(iITT)(CnCxCy)(C12)]; UV-Vis spectroscopy of new polymers P[(iITT)(CnCxCy)(C12)]; cyclovotammograms of P[(iITT)(CnCxCy)(C12)]; thermogravimetric analysis of P[(iITT)(CnCxCy)(C12)]; 1D GIWAXS patterns for P[(iITT)(CnCxCy)(C12)]. Atomic force microscopy images (phase and height) of thin films of P[(iITT)(CnCxCy)(C12)]; representative output characteristics of P[(iITT)(CnCxCy)(C12)] series; transfer characteristics of P[(iITT)(CnCxCy)(C12)] series. See DOI: https://doi.org/10.1039/d5py00289c |
| This journal is © The Royal Society of Chemistry 2025 |