DOI:
10.1039/D5PY00243E
(Paper)
Polym. Chem., 2025,
16, 2530-2542
Synthesis of butadiene-derived polyolefin graft copolymers and their crystallization behaviors†
Received
8th March 2025
, Accepted 1st May 2025
First published on 2nd May 2025
Abstract
The ability to synthesize polyolefin-based copolymers with controlled chemical composition and chain microstructure provides great opportunities for their use in compatibilizing polyolefin blends, particularly including polyethylene (PE) and polypropylene (PP), to address their waste recycling challenges. In this study, polyolefin graft copolymers comprising semicrystalline PE side chain and amorphous polyethylethylene (PEE) backbone were synthesized by grafting amine-terminated 1,4-polybutadiene (PB) onto carboxyl-grafted 1,2-PB, followed by hydrogenation. A suite of characterization techniques were employed to assess the degree of functionalization, polymer microstructure, and molecular weight of these graft copolymers containing different side chain lengths and graft densities. Moreover, since most commodity polyolefins are semicrystalline, understanding the crystallization behavior of these polyolefin copolymers is important. Specifically, the non-isothermal crystallization behavior of these graft copolymers was investigated using differential scanning calorimetry (DSC) and the Jeziorny-modified Avrami model. Both the graft copolymers and their homopolymer grafts exhibited heterogeneous nucleation with predominantly one-dimensional crystal growth and similar overall crystallization rates. However, PEE-g-PE10k and PEE-g-PE5k showed opposite trends in activation energy, likely due to a competition between nucleation facilitation and crystal growth restriction introduced by the graft structure. A preliminary investigation indicates that the incorporation of these graft copolymers into mixed PE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PP (50:50) blends can lead to improved mechanical performance. This study reveals the impact of graft architecture on the crystallization behavior of polyolefin-based blend compatibilizers.
:PP (50:50) blends can lead to improved mechanical performance. This study reveals the impact of graft architecture on the crystallization behavior of polyolefin-based blend compatibilizers.
Introduction
Polyolefins are among the most widely used polymer materials in the world, contributing over 250 million metric tons annually for diverse applications such as packaging, automotive components, construction materials, and consumer goods.1 Notably, polyethylene (PE) and polypropylene (PP) dominate the market due to their low cost, satisfactory mechanical strength, as well as excellent chemical resistance, which are largely attributed to their semicrystalline microstructures.2,3 The ability to control the crystalline structure and kinetics of polyolefin materials is an important research focus, as these properties are strongly influenced by factors such as polymer architecture, tacticity, chemical identity, and overall composition.4–6 For instance, by controlling the degree of chain branching in PE materials, researchers can significantly modulate their crystallinity, accessing a wide range of elastic moduli and toughness.7–13 Similarly, the tacticity in PP can dictate its crystalline behavior, shifting the material from an amorphous to a highly crystalline state and consequently altering its mechanical properties.14,15
In recent years, polyolefin copolymers with distinct chemical components have attracted considerable interest, particularly as a promising solution for addressing mixed plastic waste, which is a critical sustainability challenge.16,17 These copolymers can serve as compatibilizers for polymer blends by selectively interacting with respective domains, thereby enhancing interfacial adhesion and overall material performance.18 Specifically, PP/PE blends represent a common yet very challenging waste stream. Their similar physical properties complicate sorting and separation processes, while their chemical immiscibility in the melt state leads to macrophase separation and poor interlayer adhesion at domain interfaces.19,20 Consequently, recycled products from mixed PP/PE often exhibit significantly downgraded mechanical performance, limiting their practical applications. To address this challenge, linear and graft block compatibilizers based on polyolefin chemistry have been studied to enhance the compatibility of PP/PE blends.17,18,21–29 Notably, while various copolymer structures have been developed for PP/PE blends, co-crystallization between the compatibilizer and polymer blend remains the predominant compatibilization mechanism in this system.17,28 As such, most copolymers designed for PP/PE systems incorporate both semicrystalline PP and PE blocks, largely relying on organometallic catalysts to achieve controlled chain architecture.17,21,22,27,30–33 A seminal work from Eagan et al. found significant improvements in mechanical properties of PE/iPP (isotactic PP) blends compatibilized with PE-b-iPP multiblock copolymers.21 This superior compatibilization performance was attributed to the trapped entangled mechanism, where the multiblock architecture allowed half of the iPP and PE block to be flanked by thermodynamically immiscible counterparts, leading to the formation of entangled loops at the interface during melt process. As a result, this approach can effectively stitch together the PE and iPP domains upon crystallization during cooling, thereby enhance the mechanical properties of the compatibilized blends.
While most previous studies in this area have focused on leveraging a co-crystallization mechanism and/or physical entanglements between amorphous domains for blend compatibilizers to improve interfacial stability between PE and PP domains, a recent study introduced a new compatibilization mechanism, referred to as ‘threading-the-needle’.34,35 This mechanism attributes the improved mechanical performance of the compatibilized blends to the interfacial topological entanglements of the amorphous block of the compatibilizer with the PP phase, along with the co-crystallization of the semicrystalline block with the PE phase. Specifically, a block copolymer compatibilizer for PP/PE blends, comprising an amorphous PP-like block and a semicrystalline PE-similar block, was prepared by hydrogenating polybutadiene (PB) block copolymer with PB segments of different 1,2- and 1,4-ratios. We note that this approach is especially attractive as it introduces an emerging compatibilization mechanism that presents new opportunities for enhancing PP/PE recycling, while allowing for synthesizing these effective compatibilizers using potentially scalable methods. Since the performance of butadiene-derived copolymers relies on their interaction with distinct crystalline domains, understanding and controlling their crystallization behavior is crucial for enhancing compatibilization performance and informing processing conditions.36–38 Notably, these butadiene-derived polyolefin block copolymers can also enhance the toughness of recycled PE and PP, with their mechanical performance exhibiting a clear dependence on sample cooling rate in processing, further highlighting the need to understand the non-isothermal crystallization kinetics of these materials.35
To date, most studies on PP/PE compatibilization have focused on the synthesis and application of linear block copolymers, while the significant potential of graft copolymers remains underexplored. To investigate the ability of polyolefin-based graft copolymers by leveraging the “threading-the-needle” mechanism for addressing PE/PP blends, this study establishes a synthetic platform to prepare butadiene-derived graft copolymers with chemically dissimilar backbones and grafts by controlling their chain microstructures. Specifically, we synthesized polyolefin graft copolymers with varied chemical compositions by grafting amine-terminated 1,4-polybutadiene (1,4-PB-NH2) onto carboxyl-functionalized 1,2-polybutadiene (1,2-PB-COOH), followed by hydrogenation to obtain semicrystalline polyethylene (PE) side chains and an amorphous polyethylethylene (PEE) backbone. The non-isothermal crystallization kinetics of the graft copolymers and their corresponding homopolymer grafts were investigated to elucidate the impact of molecular architecture on polymer crystallization behavior. Moreover, it is found that the incorporation of these graft copolymers into mixed PP and PE blends can result in improved mechanical performance. This study provides important insights for developing semicrystalline polyolefin graft copolymers from butadiene precursors, which could potentially address the challenges associated with PE/PP recycling and improve the sustainability of polyolefin-based materials.
Results and discussion
Synthesis of polybutadiene grafts and backbone
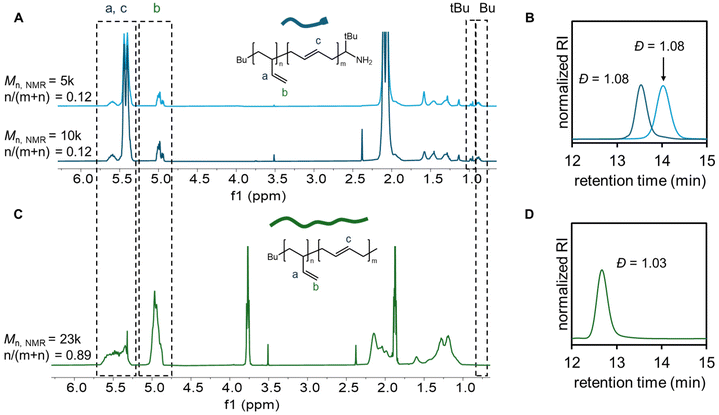
The graft copolymer poly(ethylethylene-ran-ethylene)-graft-polyethylene (PEE-g-PE) was prepared according to the reaction scheme as shown in Fig. 1. Briefly, different types of polybutadiene with a 1,4-dominated microstructure (containing amine end groups) and a 1,2-dominated microstructure were synthesized and employed as grafts and backbones, respectively. Carboxyl groups were introduced onto the 1,2-PB backbone via thiol–ene coupling chemistry, allowing for the chemical attachment of 1,4-PB grafts through amidation reaction. Subsequently, these materials underwent hydrogenation, enabling the saturation of polymer chains. Here, amine-terminated PB (1,4-PB-NH2) was prepared using a previously reported method.39 Specifically, butadiene was polymerized via anionic polymerization using sec-butyllithium as the initiator in toluene, end-capped with pivalonitrile, and subsequently reduced with sodium borohydride (NaBH4) in a protic solvent (Fig. 1A).39 The successful transformation of living polybutadienyllithium into end-capped polybutadiene was confirmed by proton nuclear magnetic resonance spectroscopy (1H-NMR). The disappearance of the allyl proton chain-end signals at 1.65 ppm (ref. 40) and appearance of the new peaks at 1.15 ppm, corresponding to the tert-butyl group, indicate complete end-capping with an end group efficiency reaching 100% (Fig. S1†). Further reduction of the imino group to an amino group upon reaction with NaBH4 was confirmed by the shift of the tert-butyl signals to a higher field (0.98 ppm), which is consistent with previous reports (Fig. S1† and Fig. 2A).39 Moreover, two 1,4-PB-NH2 grafts were prepared, with molar mass of approximately 5 kg mol−1 and 10 kg mol−1, which was determined by 1H-NMR using the signals of sec-butyl as end-group at 0.80 ppm (Fig. 2A). To produce 1,2-dominated PB backbone (1,2-PB), tetrahydrofuran (THF) was added to the standard anionic polymerization of butadiene at a molar concentration of [Li]:[THF] = 1:200 (Fig. 1B),34 yielding 1,2-PB with molar mass of 23 kg mol−1.
 |
| | Fig. 1 Reaction scheme of (A) amine-terminated 1,4-PB, n/(m + n) = 0.12; (B) 1,2-PB, n/(m + n) = 0.89; (C) carboxyl functionalized 1,2-PB, and (D) polyethylethylene-graft-polyethylene. | |
 |
| | Fig. 2 (A) 1H NMR spectra of 1,4-PB-NH2 graft, (B) GPC result of 1,4-PB-NH2 graft, (C) 1H NMR spectrum of 1,2-PB backbone, and (D) GPC result of 1H NMR of 1,2-PB backbone. | |
The chemical structure of the 1,4-PB-NH2 graft and 1,2-PB backbone were characterized by1H-NMR spectroscopy, as shown in Fig. 2A and C, respectively. We note that the peaks in the 4.8–5.6 ppm region are associated with the olefinic protons in the butadiene units. The relative ratio of 1,2-butadiene units was calculated by integrating vinylidene protons at 5.0 ppm (described in ESI†). It was determined that the 1,2-PB backbone contained 89% 1,2-butadiene units, and the 1,4-PB-NH2 grafts contained 12% 1,2-butadiene units. Both 1,2-PB and 1,4-PB samples in this study exhibited a low polydispersity value (Đ), ranging from 1.03–1.08 (also see Table S1†), as shown in the gel permeation chromatography (GPC) traces in Fig. 2B and D, indicating a good control over the polymerization process.
Following the preparation of 1,2-PB polymers, we grafted carboxyl groups onto 1,2-PB using the well-established thiol–ene coupling chemistry to produce the carboxyl functionalized 1,2-PB (1,2-PB-COOH) (Fig. 1C).41,42 The decrease of unsaturated proton in PB at 4.8–5.6 ppm, coupled with the appearance of new peaks at 2.6 and 2.8 ppm (proton adjacent to thioether and carboxyl groups), suggested successful functionalization of 1,2-PB (Fig. S2†).43 GPC results indicated a slightly broader Đ after the thiol–ene coupling, reaching 1.29 over 20 min reaction time, but the number-averaged molecular weight remained similar to that of the starting 1,2-PB (Fig. S3†). We note that the broadening of Đ is primarily due to the radical mechanism of the thiol–ene reaction, which can cause side reactions such as chain scission and coupling/crosslinking, thereby producing both lower and higher molecular weight species. The grafting density of carboxyl groups steadily increased throughout the reaction process, as determined from 1H-NMR spectra. As the vinyl groups exhibit high reactivity with thiol–ene reaction,44 a graft density as high as 70% was achieved within 20 min, as shown in Fig. 3. We note that from NMR results, it was suggested that around 81% of 1,2-unit and less than 0.1% of 1,4-unit were reacted at 20 min, respectively. For the synthesis of PB graft polymer, we selected 1,2-PB with the lowest carboxyl graft density (10%) as the model system for subsequent reactions. We note that a graft density below 10% can be obtained by further reducing the reaction time or using a lower concentration of thiol-containing reagents (3-mercaptopropionic acid in this study).
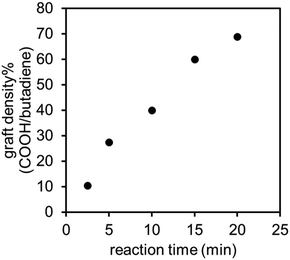 |
| | Fig. 3 Effect of reaction time on the grafting density of the carboxyl functionalization of 1,2-PB. | |
Synthesis of butadiene-derived polyolefin graft copolymers (PEE-g-PE)
The PB-based graft copolymer was then prepared by grafting 1,4-PB-NH2 onto 1,2-PB-COOH via amidation at a 1:1 molar ratio of amine to carboxyl groups, using 4-dimethylaminopyridine (DMAP) as the catalyst and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCl) as the carboxyl activating agent (Fig. 1D).45 The grafting reactions for both 1,4-PB-NH2-5k and 1,4-PB-NH2-10k were monitored by GPC (Fig. S4†), where the appearance of a new polymer fraction (elution peak at ∼11.6 min in Fig. S4A and ∼11.5 min in Fig. S4B†) with a molecular weight higher than that of the backbone and grafts indicated the formation of graft copolymers from the coupling of the 1,2-PB-NH2 graft onto the 1,2-PB-COOH backbone. Specifically, Fig. 4 shows the GPC traces of the homopolymers used as the backbone and grafts, as well as the crude product mixtures at the end of the reaction. The absence of unreacted 1,2-PB in the final product mixtures for both 5k and 10k indicated complete conversion of 1,2-PB-COOH, and the appearance of a new high-molecular-weight fraction evident the formation of PB-based graft copolymers. The reaction progress was determined by the peak area ratio of the graft copolymer to the unreacted graft (Fig. S5†), and the reaction was quenched when this ratio reached a plateau. Notably, the reaction with 1,4-PB-NH2-5k required a longer time to reach completion than that with 1,4-PB-NH2-10k, but ultimately achieved a higher conversion. This difference might be attributed to the impact of graft chain length on their end group reactivity. The shorter 1,4-PB-NH2-5k chain exhibits less steric hindrance and better end group accessibility to reacting with the carboxyl groups on the 1,2-PB-COOH backbone, resulting in a higher conversion. However, the increased steric hindrance from higher grafting density may also slow the coupling process, thereby an extended reaction time might be needed.
 |
| | Fig. 4 GPC traces of the backbone PB (1,2-PB-23k), 1,4-PB grafts (1,4-PB-5k and 1,4-PB-10k), the product from the coupling reaction with 5k graft (PBgp-5k) and 10k graft (PBgp-10k). | |
To further characterize the graft copolymer, PB graft polymers (PBgp) were purified by preparative GPC to remove unreacted 1,4-PB-NH2, which both 1,4-PB-NH2-5k and 10k grafts were successfully isolated, and their 1H NMR and GPC traces were presented in Fig. 5. Effective isolation of graft copolymer was confirmed by the absence of the tert-butyl group peak at 0.9 ppm from 1,2-PB-NH2 in the 1H NMR spectra, as well as the absence of 1,2-PB-NH2 fraction in the GPC traces. The number of grafts was calculated from the ratio of 1,2- and 1,4-butadiene units in PBgp, 1,2-PB-COOH, and 1,4-PB-NH2 (see ESI†). For PBgp-5k, the graft density was approximately 15 grafts per backbone, with a relatively low Đ of 1.11. PBgp-10k had a lower graft density of approximately 10 grafts per backbone and a slightly higher Đ value of 1.43. The lower grafting efficiency in PBgp-10k was consistent with the trends observed in the GPC results (Fig. S5†) and can be attributed to the increased steric hindrance arising from the higher molecular weight of the 1,4-PB-NH2-10k.
 |
| | Fig. 5 (A) 1H NMR of the isolated PBgp-5k, (B) GPC trace of the isolated PBgp-5k, (C) 1H NMR of the isolated PBgp-10k, and (D) GPC trace of the isolated PBgp-10k. | |
The semicrystalline polyolefin graft polymer, PEE-g-PE, was synthesized via hydrogenation of PBgp with the presence of diimide, which was generated through the thermal decomposition of p-toluenesulfonyl hydrazide. The completion of hydrogenation was confirmed by Fourier-transform infrared (FTIR) spectroscopy, as shown in Fig. 6. In PEE-g-PE5k, the appearance of C–H stretch at 2920 cm−1 and the disappearance of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch at 1730 cm−1 and C–H from CHCH at 970 cm−1 indicated complete saturation of PBgp-5k (Fig. 6A).46 Similar FTIR results were observed for PBgp-10k and PEE-g-PE10k, confirming the complete saturation of PBgp-10k as well (Fig. 6B).
C stretch at 1730 cm−1 and C–H from CHCH at 970 cm−1 indicated complete saturation of PBgp-5k (Fig. 6A).46 Similar FTIR results were observed for PBgp-10k and PEE-g-PE10k, confirming the complete saturation of PBgp-10k as well (Fig. 6B).
 |
| | Fig. 6 FTIR of (A) PBgp-5k and the hydrogenated polymer PEE-g-PE5k, and (B) PBgp-10k and the hydrogenated polymer PEE-g-PE10k. | |
Thermal and crystallinity analysis of PEE-g-PE
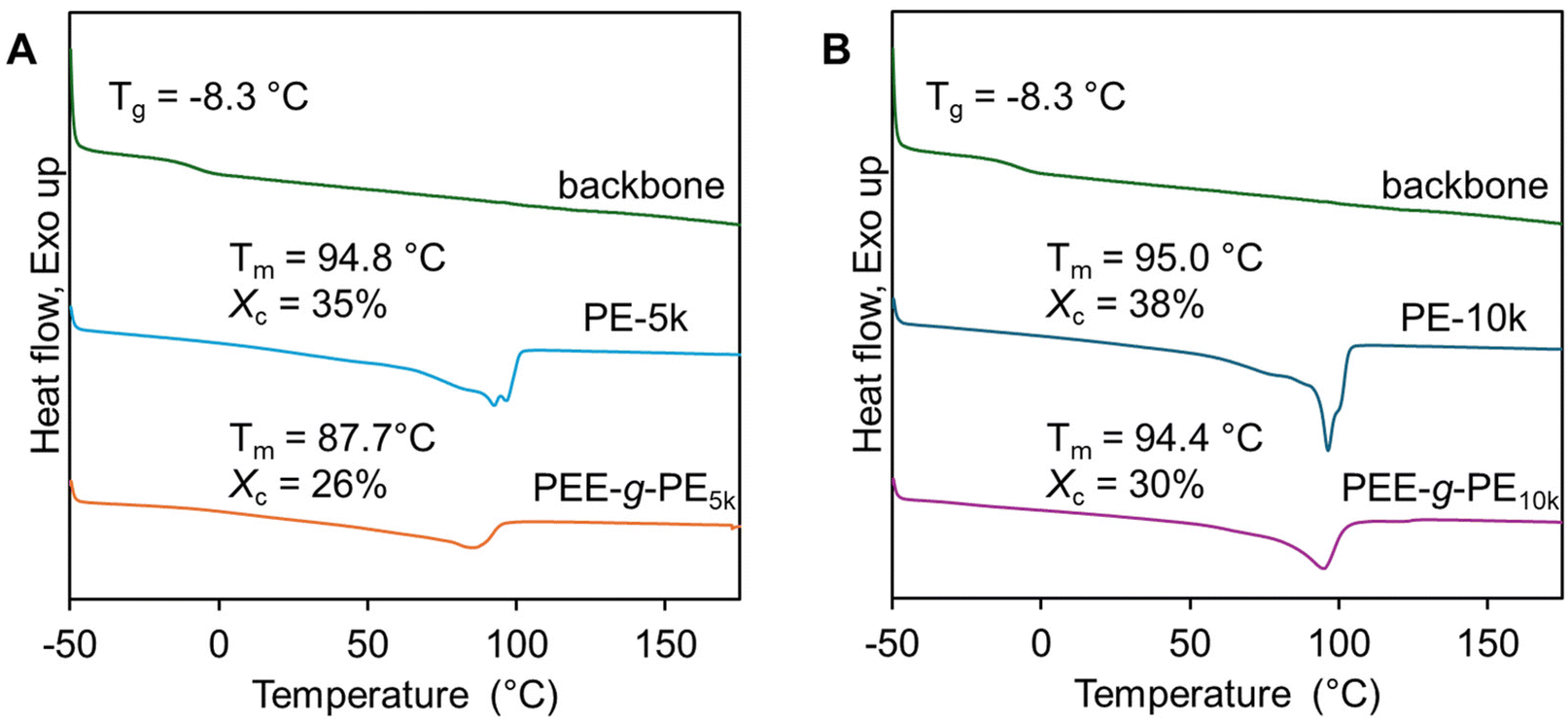
The thermal stability and crystallinity of hydrogenated 1,2-PB-COOH (backbone), hydrogenated 1,4-PB-NH2 (PE-5k and PE-10k), and polyolefin graft copolymers (PEE-g-PE5k and PEE-g-PE10k) were analyzed using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA results revealed that both polyolefin graft copolymers had lower thermal decomposition temperatures (Td5) than their respective homopolymer grafts. Specifically, Td5 decreased from 397 °C for PE-5k to 380 °C for PEE-g-PE5k, and from 396 °C for PE-10k to 378 °C for PEE-g-PE10k (Fig. S6†). This decrease in thermal stability might be attributed to the amide linkages between the backbone and grafts. Despite the decrease in Td5, all samples had decomposition temperatures above typical polymer processing conditions, indicating they are suited for practical applications.
As shown in Fig. 7, the hydrogenated 1,2-PB-COOH was amorphous, exhibiting a glass transition temperature (Tg) of −8.3 °C, which is higher than the typical value of poly(1-butene) (−17 °C).47 This increase may be attributed to restricted chain mobility due to the presence of grafted carboxyl groups.48 Both PEE-g-PE and hydrogenated 1,4-PB-NH2 (PE-5k and PE-10k) were found to be semicrystalline. The PE-5k and PE-10k had similar crystallinity (Xc) of 35%–38% and a melting temperature (Tm) of 95 °C, which are lower than the typical values associated with PE of similar molecular weight (Xc = 73%–68%, Tm = 125–128 °C for PE in the 4k–9k range).49 This reduction in Xc and Tm is likely due to several factors. First, the ethyl branch in PE-5k and PE-10k, generated from the hydrogenation of 1,2-butadiene unit, limits the efficient packing of the polymer chain in the crystalline lattice.50 Additionally, the amine end group of these polymers can also act as defects and strongly associate through hydrogen bonding, further hindering the crystallization by limiting chain mobility.
 |
| | Fig. 7 DSC curves of (A) PEE-g-PE10k, hydrogenated 1,4-PB-NH2 (graft-5k) and hydrogenated 1,2-PB-COOH (backbone), and (B) PEE-g-PE5k, hydrogenated 1,4-PB-NH2 (graft-10k) and hydrogenated 1,2-PB-COOH (backbone), the crystallinity of PEE-g-PE was normalized by the mass ratio of PE over the total mass of the graft polymer. | |
For the graft copolymer PEE-g-PE5k and PEE-g-PE10k, the normalized percent crystallinity is calculated:
| |  | (1) |
where Δ
Hm represents the enthalpy of fusion (293 J g
−1 for 100% crystalline PE), and
Wgraft represents the weight fraction of the graft in the copolymer (see ESI
†). This normalization accounts for the crystallinity of the PE grafts. Both PEE-
g-PE
5k and PEE-
g-PE
10k exhibited lower
Xc and
Tm values than their respective hydrogenated 1,4-PB-NH
2 grafts. Specifically, PEE-
g-PE
5k had a
Tm of 87.7 °C and
Xc of 26%, whereas PEE-
g-PE
10k had a
Tm of 94.4 °C and
Xc of 30%. The lower crystallinity and melting temperature in PEE-
g-PE
5k were likely due to the presence of an increased number of grafts, which impeded polymer chain folding during crystallization and reduced chain mobility.
Non-isothermal crystallization kinetics of PEE-g-PE
As non-isothermal crystallization conditions more closely resemble industrial polymer processing, understanding the crystallization mechanisms and kinetics of these semicrystalline polyolefin polymers is important for informing their processing conditions and performance.51 The non-isothermal crystallization thermograms of the polyolefin grafts and PEE-g-PE at different cooling rates are shown in Fig. 8. As the cooling rate increased, both the onset (Tonset) and peak (Tp) crystallization temperatures shifted to lower values, which are summarized in Fig. S7 and Table S2.† Meanwhile, the exothermic peak became broader with the cooling rate increasing, indicating the formation of imperfect crystal structures upon rapid cooling.52 At the same cooling rate in the range from 5 °C min−1 to 40 °C min−1, the graft copolymers consistently exhibited lower Tp values than their corresponding homopolymers (grafts), suggesting that a greater degree of supercooling was required for crystallization. However, since the crystallization rates obtained from Avrami analysis were similar (which is discussed in the subsequent section), the decrease in Tp is likely due to a dilution effect exerted by the amorphous backbone, which changes the nucleation efficiency rather than significantly reducing the overall crystallization kinetics.53–56 Moreover, Tp of graft copolymer with lower graft density and longer grafts (PEE-g-PE10k) was closer to that of its homopolymer graft (PE-10k) than that of PEE-g-PE5k (Fig. S7†). This result might suggest that the steric constraints in high-graft-density systems hinder chain packing, further delaying crystallization onset. In contrast, the longer grafts in PEE-g-PE10k facilitated chain aggregation, promoting crystallization compared to PEE-g-PE5k.
 |
| | Fig. 8 DSC curves during non-isothermal crystallization at different cooling rates for (A) PE-5k, (B) PEE-g-PE5k, (C) PE-10k, and (D) PEE-g-PE10k. | |
The Avrami model is well known for describing how solids transform from one phase to another at constant temperature and has been widely used for analyzing the isothermal crystallization of polymers.57,58 This method can be extended to describe the kinetics of non-isothermal crystallization under the assumption of constant crystallization temperature.53,54,59–62 In this case, the Avrami equation is expressed as
or
| | | log[−ln(1 − X(t))] = logK + nlogt | (3) |
where
n is the Avrami exponent that is related to the mode of nucleation and crystal growth geometry,
K is the Avrami constant that provides the overall rate of crystallization (nucleation and growth rate parameters), and
X(
t) is the instantaneous relative crystallinity at any time
t, calculated as:
| |  | (4) |
where
Qt is the instantaneous heat flow,
Q∞ is the total heat flow during the crystallization process, and

is the instantaneous enthalpy change rate. In non-isothermal conditions, crystallization time (
t) can be converted from the crystallization temperature (
T) using:
| |  | (5) |
where
ϕ is the cooling rate. Fig. S8
† shows the evolution of the relative crystallinity of PEE-
g-PEs and their corresponding grafts with crystallization temperature. The crystallization half-time (
t1/2) obtained from Fig. S8
† was listed in Table S2.
† As anticipated,
t1/2 decreased with increasing cooling rate for all samples. This trend is attributed to the greater supercooling at higher cooling rates, which enhances the thermodynamic driving force for crystallization and accelerates the crystallization process.
53,62
Fig. 9 presents the Avrami plots of log[−ln(1 − X(t))] versus logt for PEE-g-PEs and their grafts across various cooling rates. All samples exhibited two distinct regions, from which the Avrami parameters (n and K) were extracted from the slope and intercept of the plots, respectively. Considering the non-isothermal character of these measurements, it was established that K should be corrected to obtain the corresponding rate constant (K’, Jeziorny-modified Avrami constant) at a normalized cooling rate as follows:53,62,63
| | 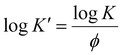 | (6) |
 |
| | Fig. 9 Plots of log[−ln(1 − X(t))] versus logt for non-isothermal crystallization of (A) PE-5k, (B) PEE-g-PE5k, (C) PE-10k, and (D) PEE-g-PE10k. | |
All plots displayed an initial region with anomalously low n values (lower than 1), which indicated a non-ideal crystallization behavior that deviates from the classical Avrami assumptions of constant nucleation rates and isotropic growth.64 This deviation may arise from an inhomogeneous distribution of pre-existing nuclei65 or the constrained growth due to structural defects.66 The low n value was also observed in linear hydrogenated PB by Heeley et al.67 In their study, low molecular weight hydrogenated PB (∼15k g mol−1) had n ∼0.3 at early stages of shear-induced crystallization, which were attributed to a low dimensional dendritic type of growth unit leading to a non-uniform density within the spherulites. Similar observations have also been identified for low molecular weight branched PE.68–70 The calculated values Avrami parameters of the second stage (n2 and  ) were summarized in Fig. 10. The value of n2 for all samples ranged between 1 and 2 across the experimental cooling rates, indicating heterogeneous nucleation with predominantly one-dimensional growth. We noted that some plots possessed a third region near the end of crystallization; however, the Avrami exponent of these regions remained within the range of 1–2, indicating no change in the crystallization mode. Additionally, as shown in Fig. 10B,
) were summarized in Fig. 10. The value of n2 for all samples ranged between 1 and 2 across the experimental cooling rates, indicating heterogeneous nucleation with predominantly one-dimensional growth. We noted that some plots possessed a third region near the end of crystallization; however, the Avrami exponent of these regions remained within the range of 1–2, indicating no change in the crystallization mode. Additionally, as shown in Fig. 10B,  increased with higher cooling rate, consistent with the previous t1/2 observations. The similar
increased with higher cooling rate, consistent with the previous t1/2 observations. The similar  values (0.7–1.1) across all samples at a given cooling rate suggested that the length of the grafts or the graft density does not significantly impact the overall crystallization rate. Similar phenomenon has been reported in a previous study associated with understanding the non-isothermal crystallization of long chain branched PE52 and PP-g-poly(ethylene-co-1-butene) samples.53
values (0.7–1.1) across all samples at a given cooling rate suggested that the length of the grafts or the graft density does not significantly impact the overall crystallization rate. Similar phenomenon has been reported in a previous study associated with understanding the non-isothermal crystallization of long chain branched PE52 and PP-g-poly(ethylene-co-1-butene) samples.53
 |
| | Fig. 10 (A) Avrami exponent and (B) Jeziorny-modified Avrami constant for non-isothermal crystallization of PE-5k, PEE-g-PE5k, PE-10k, and PEE-g-PE10k. | |
To further understand the non-isothermal crystallization process of these semicrystalline polyolefin graft copolymers with distinct graft density and length, the crystallization activation energy (ΔE) was calculated using Kissinger equation:71
| |  | (7) |
where
C is a constant,
R is the gas constant, and
TP is the peak crystallization temperature. The plots of
 versus
versus
for all samples are presented in
Fig. 11. Δ
E was calculated from the slopes of these plots as shown in
Fig. 11. Generally, the Δ
E consists of the transport activation energy (transport molecular segments across the phase boundary to the crystal growth surface) and the nucleation activation energy (the free energy of formation of crystal nuclei with a critical size).
72 We observed an opposite trend in activation energy for the graft polymers with 5k and 10k grafts. PEE-
g-PE
10k (Δ
E = 362.4 kJ mol
−1) had a higher activation energy than its corresponding homopolymer graft (PE-10k, Δ
E = 332.3 kJ mol
−1), whereas PEE-
g-PE
5k had a significantly lower activation energy (186.9 kJ mol
−1) than PE-5k (300.6 kJ mol
−1). This trend consists of their overall crystallization rate without Jeziorny correction (
K, Fig. S8
†). The observed behavior may stem from a competition between nucleation facilitation and crystal growth restriction introduced by the graft structure. While grafting facilitates nucleation by providing additional sites (reducing the activation energy for nucleation), it simultaneously hinders molecular transport to the crystal growth surface (increasing the activation energy of crystal growth). In the case of PEE-
g-PE
5k, its high graft density and low molecular weight of PEE-
g-PE
5k likely promote nucleation while minimizing transport constraints, leading to lower activation energy. In contrast, the longer graft in PEE-
g-PE
10k experiences greater steric hindrance, limiting the ability of crystal growth and resulting in higher activation energy.
 |
| | Fig. 11 Plots of  versus versus for PE-5k, PEE-g-PE5k, PE-10k, and PEE-g-PE10k. for PE-5k, PEE-g-PE5k, PE-10k, and PEE-g-PE10k. | |
Mechanical properties of PP/PE blends with PEE-g-PE
A preliminary investigation of the compatibilization performance of PEE-g-PEs was conducted using LDPE:iPP (50:50; weight ratio), a model blend system. Fig. 12 shows the representative stress–strain curves obtained for the blends with and without 2 wt% PEE-g-PEs. Both LDPE and iPP are well-known for their toughness and ductility.2,34 Blending these two polyolefins together results in a very brittle material with a strain at break (εb) of around 20%, similar to previously reported results.28 The addition of 2 wt% PEE-g-PEs led to more than twofold increase in εb, indicating that the graft copolymer functioned as a compatibilizer in this system. The elastic modulus, strain at break and toughness for the neat blend and the blends with graft copolymers were summarized in Table S3.† Specifically, LDPE/iPP blend with PEE-g-PE10k had a slightly higher strain at break (50%) than the blend with PEE-g-PE5k (46%). The elastic modulus of the neat blend and the blend with 2 wt% PEE-g-PE5k were similar (around 148–150 MPa), while the modulus of PEE-g-PE10k blend was slightly lower (140 MPa).
 |
| | Fig. 12 Representative stress–strain curves for LDPE:iPP (50:50) blends with and without 2 wt% graft copolymer PEE-g-PEs. | |
We note that while the observed improvement in ductility was moderate, several factors may influence the efficacy of this graft copolymer in blend compatibilization performance and suggest future directions for further optimization. First, the chain length of backbone used in this study is relatively low. While the molecular weight exceeds the entanglement molecular weight of iPP (Me = 6300 g mol−1),73 the presence of grafts may hinder entanglement between the PP-like backbone and the amorphous phase of iPP. Moreover, the average molecular weight between the grafting points (around 1400 g mol−1 for PEE-g-PE5k, 2000 g mol−1 for PEE-g-PE10k) is substantially lower than the entanglement molecular weight for iPP, which could further limit the effective entanglement. Second, the unreacted carboxyl group (around 6% per backbond) on the PEE backbone may alter the miscibility of the backbone with iPP, while these groups may also promote loop formation via hydrogen bonding, potentially enhancing interfacial entanglement between iPP and the PEE backbone.74 However, the practical effect of these residual functional groups remains unclear and needs further investigation. Third, prior studies have indicated that “thread-the-needle” compatibilization mechanism and performance can be highly processing condition dependent, with fast cooling promoting co-crystallization between the PE block of the copolymer and the PE homopolymer.34,35 Due to instrument limitations, fast cooling conditions were not explored in this study, but we anticipate that processing conditions will influence the compatibilization efficiency of these graft copolymers as well.
Experimental section
Material
All solvents were purchased from Fisher Scientific and used as received unless otherwise specified. 1,3-Butadiene (20 wt% in toluene) and pivalonitrile, were obtained from Millipore Sigma and dried over activated molecular sieves for around one week before use. sec-Butyllithium (secBuLi) (1.4 M in cyclohexane), anhydrous tetrahydrofuran (THF), azobisisobutyronitrile (AIBN), 4-dimethylaminopyridine (DMAP), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCl), and butylated hydroxytoluene (BHT) were obtained from Millipore Sigma and TCI and used as received.
Synthesis of amine terminated 1,4-enriched polybutadiene (1,4-PB-NH2)
The polymerization was carried out under a nitrogen atmosphere according to a reported procedure.39 For a representative reaction for preparing 1,4-PB-NH2 (with 10 kg mol−1 molar mass), 1,3-butadiene solution (103 mL, 17 g 1,3-butadiene, 310 mmol, 182 eq.) and secBuLi solution (1.2 mL, 1.4 M in cyclohexane, 1.7 mmol, 1 eq.) were placed in an oven-dried glass pressure tube equipped with a magnetic stirrer. The reaction was stirred at 40 °C for 5 h. Before end-capping with pivalonitrile, 1 mL aliquot was withdrawn into methanol to characterize the precursor polymer by GPC. Pivalonitrile (0.2 mL, 1.8 mmol, 1.1 eq.) was added to the reaction mixture under a nitrogen atmosphere at room temperature. The reaction mixture was stirred for 1 h before the addition of NaBH4 (0.14 g, 3.7 mmol, 2.2 eq.)/ethanol (8 mL) solution. The mixture was then stirred at 50 °C for 18 h before precipitated in methanol. The polymer was dried under vacuum at 35 °C for 24 h.
Synthesis of 1,2-enriched polybutadiene (1,2-PB)
For a representative reaction of1,2-PB (with 20 kg mol−1 molar mass), an oven-dried glass pressure tube was equipped with a magnetic stirrer and charged with anhydrous THF (28 mL, 340 mmol, [THF]:[Li] = 200:1), 1,3-butadiene solution (206 mL, 33 g 1,3-butadiene, 620 mmol, 365 eq.) and secBuLi solution (1,2 mL, 1.4 M in cyclohexane, 1.7 mmol, 1 eq.) by sequence under a nitrogen atmosphere. The mixture was stirred at room temperature for 5 h. The polymer was precipitated in methanol and dried under vacuum at 35 °C for 24 h.
Carboxyl functionalization of 1,2-PB (1,2-PB-COOH)
1,2-PB (3 g, 55 mmol double bonds), THF (60 mL), and 3-mercaptopropionic acid (9 g, 82 mmol, 1.5 eq. to double bonds) was introduced into a three-neck round bottom flask. The mixture was sparged with nitrogen for 30 min before heated to 60 °C. AIBN (45 mg, 0.3 mmol, 0.005 eq. to double bonds) was dissolved in 1 mL THF and added to the reaction mixture. The reaction was stirred at 60 °C for the specified time to achieve the target graft density. For this work, 10% graft density was used and the reaction time was 5 min. The reaction was quenched by pouring into a cold BHT/methanol solution. The polymer was precipitated in 10% HCl/methanol solution and dried under vacuum at 35 °C for 24 h. For the kinetic study, 1 mL aliquot was withdrawn into a cold BHT/methanol solution at each time point.
Grafting 1,4-PB-NH2 onto 1,2-PB-COOH (PBgp)
For a representative reaction of PBgp-5k, 1,4-PB-NH2-5k (1 g, 0.2 mmol –NH2, 1 eq.), and 1,2-PB-COOH (0.13 g, 0.2 mmol –COOH, 1 eq.) was dissolved in DCM (40 mL) and placed in a round bottom flask equipped with a magnetic stirrer. DMAP (23 mg, 0.2 mmol, 1 eq.) and EDCl (115 mg, 0.6 mmol, 3 eq.) were added as solids. The mixture was stirred at 40 °C for 72 h before precipitated in methanol. The graft polymer PBgp was separated from the unreacted 1,2-PB-NH2 by preparative GPC system (JAI LaboACE LC5060) equipped with a UV-visible detector. The system was operated at room temperature with a mobile phase of chloroform at a flow rate of 5.0 mL min−1. Preparative GPC traces for the separation of PBgp-5k and PBgp-10k are shown in Fig. S10.† For a representative separation of PBgp-5k, around 1 g of the crude polymer sample was dissolved in 10 mL of chloroform, filtered through a 0.45 μm PTFE syringe filter, and injected to the preparative GPC. As shown in Fig. S10,† the eluted fraction was subjected to GPC recycling for one cycle, and the sub-fraction of PBgp-5k was collected, dried and characterized by analytical GPC and NMR to confirm molecular weight distribution and purity (yield: 0.39 g, 39%).
Poly(ethylethylene-ran-ethylene)-graft-polyethylene (PEE-g-PE)
PBgp (0.5 g, 9 mmol double bonds, 1 eq.), p-toluenesulfonyl hydrazide (5.2 g, 28 mmol, 3.1 eq.), tributylamine (7.5 mL, 31.5 mmol, 3.5 eq.), BHT (10 mg, 0.045 mmol, 0.005 eq.), and xylene (30 mL) were added to a round-bottom flask. The mixture was heated at 140 °C for 24 h. The polymer was precipitated in methanol and washed with methanol and acetone until a white solid was obtained. The isolated white powder was dried under vacuum at 35 °C for 24 h.
General characterization
Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker 400 MHz NMR with sample concentrations of ≈15 mg mL−1 in CDCl3. Gel permeation chromatography (GPC) measurements were performed for the polymers using a TOSOH EcoSEC HLC-8320 GPC with a TSKgel SuperMultiPore HZ-M. The instrument was calibrated with PS standards (PStquick MP-M). All of the experiments were done at 40 °C. The mobile phase was HPLC grade THF using a flow rate of 1.0 mL min−1. The sample concentrations were 2 mg mL−1 in THF with an injection volume of 10 μL. Fourier transfer infrared (FT-IR) spectroscopy was performed on polymer samples using a PerkinElmer FT-IR spectrometer with the universal attenuated total reflectance sampling accessory attached. Scans were taken from 3600 to 600 cm−1 at a resolution of 4 cm−1 and an average of 32 scans. The thermal stability was determined via thermogravimetric analysis (TGA) using a thermogravimetric analyzer Q50 (TA Instruments). In platinum pans, 10–15 mg polymer samples were heated under nitrogen at 10 °C min−1 up to 800 °C. The temperature at which 5% mass loss occurred (Td5) was recorded. The melting and crystallization temperatures (Tm and Tc) of samples were obtained using differential scanning calorimetry (DSC) via a TA Instruments Discovery DSC250. Tzero pans and lids (from TA Instruments) were used, and a heat–cool–heat cycle was employed with a temperature profile ranging from −50 to 180 °C at a ramp rate of 10 °C min−1. Tc and Tm were determined by the temperature at the peak heat flow from the second and third step, respectively. The non-isothermal crystallization kinetics of all polymer samples were studied using a TA Instruments Discovery DSC250 differential scanning calorimeter (DSC). A sample size of around 5 mg was used for each DSC run with Tzero pans and lids (from TA Instruments). The sample was first heated and equilibrated at 130 °C for 3 min to remove any thermal history. The polymer sample was cooled from 130 °C to 0 °C at cooling rates of 5, 10, 20 and 40 °C min−1. The crystallization data were analyzed using the Avrami model,53,57–59 and an activation energy for non-isothermal crystallization was calculated using the Kissinger method.71
Polyolefin blend preparation and tensile test
Blends of 50 wt% LDPE, 50 wt% iPP and 2 wt% (based on the weight of neat LDPE/iPP blends) graft copolymers were prepared by an Xplore MC5 microcompounder at 190 °C at a screw speed of 130 rpm for 5 min. For the tensile testing, the blends were melt pressed into 0.75 mm thick films at 180 °C for a total of 15 min (10 min heating with no pressure and 5 min under 4 MPa) before cooling on an aluminium bench with a steel heat sink on top, holding the mold together for 20 min. Dumbbell shaped tensile bars were prepared with a die cutter (3.7 mm gauge width, 12.7 mm gauge length). All tensile tests were performed on a Mark-10 F105-EM test frame equipped with a series FS05-50 force sensor with a 250 N capacity at a strain rate of 22 mm min−1.
Conclusions
This work reports the synthesis of butadiene-derived polyolefin graft copolymers comprising semicrystalline PE-like side chain and amorphous PP-like backbone. Our method involves preparing polybutadiene with a 1,4-dominated microstructure (containing amine end groups) as grafts and a 1,2-dominated microstructure as backbones, followed by grafting carboxyl groups onto the 1,2-PB backbone via thiol–ene coupling and attaching the 1,4-PB grafts through amidation. The polybutadiene graft polymers were purified via preparative GPC to remove unreacted homopolymers. The shorter graft exhibited higher grafting efficiency due to reduced steric hindrance associated with their lower molecular weight. Two semicrystalline polyolefin-based graft copolymers with different side chain lengths and densities were prepared by hydrogenating polybutadiene graft copolymers. The non-isothermal crystallization of these semicrystalline polyolefin graft copolymers and their homopolymer grafts were investigated, which both samples exhibited heterogeneous nucleation with predominantly one-dimensional growth. Furthermore, the graft copolymers and the homopolymer exhibited similar  and t1/2 at a given cooling rate, suggesting that the grafting architecture had limited influence on the overall crystallization rate. Interestingly, PEE-g-PE10k and PEE-g-PE5k exhibited opposite trend in activation energy, which may stem from a competition between nucleation facilitation and crystal growth restriction introduced by the graft structure. A preliminary investigation of the compatibilization performance with LDPE:
and t1/2 at a given cooling rate, suggesting that the grafting architecture had limited influence on the overall crystallization rate. Interestingly, PEE-g-PE10k and PEE-g-PE5k exhibited opposite trend in activation energy, which may stem from a competition between nucleation facilitation and crystal growth restriction introduced by the graft structure. A preliminary investigation of the compatibilization performance with LDPE:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) iPP (50:50) blends showed the ability of these graft copolymers to improve blend mechanical performance. These findings provide valuable insight into the crystallization behavior of butadiene-derived polyolefin graft copolymers and designing advanced polyolefin materials for practical applications.
iPP (50:50) blends showed the ability of these graft copolymers to improve blend mechanical performance. These findings provide valuable insight into the crystallization behavior of butadiene-derived polyolefin graft copolymers and designing advanced polyolefin materials for practical applications.
Author contributions
Jin Qian: writing – review & editing, writing – original draft, methodology, investigation, formal analysis, data curation, conceptualization. Zhe Qiang: writing – review & editing, methodology, investigation, formal analysis, data curation, conceptualization, funding acquisition.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Science Foundation Office of Integrative Activities #2316351.
References
- A. H. Westlie, E. Y. X. Chen, C. M. Holland, S. S. Stahl, M. Doyle, S. R. Trenor and K. M. Knauer, Macromol. Rapid Commun., 2022, 43, 2200492–2200507 CrossRef CAS PubMed.
- D. W. Sauter, M. Taoufik and C. Boisson, Polymers, 2017, 9, 185–198 CrossRef PubMed.
- P. Galli and G. Vecellio, J. Polym. Sci., Part A:Polym. Chem., 2004, 42, 396–415 CrossRef CAS.
- L. Mandelkern, Biophys. Chem., 2004, 112, 109–116 CrossRef CAS PubMed.
- M. L. Di Lorenzo and C. Silvestre, Prog. Polym. Sci., 1999, 24, 917–950 CrossRef CAS.
- D. Mileva, D. Tranchida and M. Gahleitner, Polym. Cryst., 2018, 1, e10009 Search PubMed.
- J. Liu, F. Zhang and T. He, J. Mater. Sci., 2001, 36, 5345–5349 CrossRef CAS.
- G. Rojas, B. Inci, Y. Wei and K. B. Wagener, J. Am. Chem. Soc., 2009, 131, 17376–17386 CrossRef CAS PubMed.
- I. S. Kolesov, R. Androsch and H.-J. Radusch, Macromolecules, 2005, 38, 445–453 CrossRef CAS.
- R. Gao, X. He, Y. Shao, Y. Hu, H. Zhang, Z. Liu and B. Liu, Macromol. Theory Simul., 2016, 25, 303–311 CrossRef CAS.
- F. Chiu, Q. Fu, Y. Peng and H. Shih, J. Polym. Sci., Part B:Polym. Phys., 2002, 40, 325–337 CrossRef CAS.
- J. I. Iragorri, J. M. Rego, I. Katime, M. T. C. Braña and U. W. Gedde, Polymer, 1992, 33, 461–466 CrossRef CAS.
- R. K. Krishnaswamy, Q. Yang, L. Fernandez-Ballester and J. A. Kornfield, Macromolecules, 2008, 41, 1693–1704 CrossRef CAS.
- P. Maiti, M. Hikosaka, K. Yamada, A. Toda and F. Gu, Macromolecules, 2000, 33, 9069–9075 CrossRef CAS.
- P.-N. Tzounis, D. V. Argyropoulou, S. D. Anogiannakis and D. N. Theodorou, Macromolecules, 2018, 51, 6878–6891 CrossRef CAS.
- A. Graziano, S. Jaffer and M. Sain, J. Elastomers Plastics, 2019, 51, 291–336 CrossRef CAS.
- T.-W. Lin, O. Padilla-Vélez, P. Kaewdeewong, A. M. LaPointe, G. W. Coates and J. M. Eagan, Chem. Rev., 2024, 124, 9609–9632 CrossRef CAS PubMed.
- J. Qian, C. B. Dunn and Z. Qiang, Macromol. Chem. Phys., 2023, 224, 2300291 CrossRef CAS.
- S. Serranti, V. Luciani, G. Bonifazi, B. Hu and P. C. Rem, Waste Manage., 2015, 35, 12–20 CrossRef CAS PubMed.
- S. Yin, R. Tuladhar, F. Shi, R. A. Shanks, M. Combe and T. Collister, Polym. Eng. Sci., 2015, 55, 2899–2909 CrossRef CAS.
- J. M. Eagan, J. Xu, R. Di Girolamo, C. M. Thurber, C. W. Macosko, A. M. La Pointe, F. S. Bates and G. W. Coates, Science, 2017, 355, 814–816 CrossRef CAS PubMed.
- K. Klimovica, S. Pan, T.-W. Lin, X. Peng, C. J. Ellison, A. M. LaPointe, F. S. Bates and G. W. Coates, ACS Macro Lett., 2020, 9, 1161–1166 CrossRef CAS PubMed.
- Y. Lin, G. R. Marchand, A. Hiltner and E. Baer, Polymer, 2011, 52, 1635–1644 CrossRef CAS.
- A. H. Tsou, C. R. López-Barrón, P. Jiang, D. J. Crowther and Y. Zeng, Polymer, 2016, 104, 72–82 CrossRef CAS.
- C. R. López-Barrón and A. H. Tsou, Macromolecules, 2017, 50, 2986–2995 CrossRef.
- Y. Lin, V. Yakovleva, H. Chen, A. Hiltner and E. Baer, J. Appl. Polym. Sci., 2009, 113, 1945–1952 CrossRef CAS.
- J. Xu, J. M. Eagan, S.-S. Kim, S. Pan, B. Lee, K. Klimovica, K. Jin, T.-W. Lin, M. J. Howard, C. J. Ellison, A. M. LaPointe, G. W. Coates and F. S. Bates, Macromolecules, 2018, 51, 8585–8596 CrossRef CAS.
- J. L. Self, A. J. Zervoudakis, X. Peng, W. R. Lenart, C. W. Macosko and C. J. Ellison, JACS Au, 2022, 2, 310–321 CrossRef CAS PubMed.
- T. Zhou, D. Qiu, Z. Wu, S. A. N. Alberti, S. Bag, J. Schneider, J. Meyer, J. A. Gámez, M. Gieler, M. Reithmeier, A. Seidel and F. Müller-Plathe, Macromolecules, 2022, 55, 7893–7907 CrossRef CAS.
- H. Wang, S. Onbulak, S. Weigand, F. S. Bates and M. A. Hillmyer, Polym. Chem., 2021, 12, 2075–2083 RSC.
- F. J. Stadler, B. Arikan-Conley, J. Kaschta, W. Kaminsky and H. Münstedt, Macromolecules, 2011, 44, 5053–5063 CrossRef CAS.
- J. M. Rose, T. H. Mourey, L. A. Slater, I. Keresztes, L. J. Fetters and G. W. Coates, Macromolecules, 2008, 41, 559–567 CrossRef CAS.
- H. Ohtaki, F. Deplace, G. D. Vo, A. M. LaPointe, F. Shimizu, T. Sugano, E. J. Kramer, G. H. Fredrickson and G. W. Coates, Macromolecules, 2015, 48, 7489–7494 CrossRef CAS.
- L. Shen, G. D. Gorbea, E. Danielson, S. Cui, C. J. Ellison and F. S. Bates, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2301352120 CrossRef CAS PubMed.
- G. D. Gorbea, L. Shen, K. Flanigan, C. J. Ellison and F. S. Bates, ACS Appl. Polym. Mater., 2024, 6, 12691–12699 CrossRef CAS.
-
N. F. A. Zainal and C. H. Chan, in Compatibilization of Polymer Blends, Elsevier, 2020, pp. 391–433 Search PubMed.
- W. Phetwarotai and D. Aht-Ong, J. Therm. Anal. Calorim., 2016, 126, 1797–1808 CrossRef CAS.
- Y. Tao and K. Mai, Eur. Polym. J., 2007, 43, 3538–3549 CrossRef CAS.
- S. Nosov, H. Schmalz and A. H. E. Müller, Polymer, 2006, 47, 4245–4250 CrossRef CAS.
- F. Ziaee, M. Ronagh-Baghbani and M. R. Jozaghkar, Polym. Bull., 2020, 77, 2345–2365 CrossRef CAS.
- H. Feng, M. Dolejsi, N. Zhu, P. J. Griffin, G. S. W. Craig, W. Chen, S. J. Rowan and P. F. Nealey, Adv. Funct. Mater., 2022, 32, 2206836 CrossRef CAS.
- W. Zhang, G. Zhang, L. Du, C. Zhang, L. Li, J. Zhu, J. Pei and J. Wu, React. Funct. Polym., 2018, 127, 161–167 CrossRef CAS.
- L. Tian, J. Gu, H. Zhang and B. Dong, RSC Adv., 2020, 10, 42799–42803 RSC.
- J. Xu and C. Boyer, Macromolecules, 2015, 48, 520–529 CrossRef CAS.
- M. Niu, T. Li, R. Xu, X. Gu, D. Yu and Y. Wu, J. Appl. Polym. Sci., 2013, 129, 1833–1844 CrossRef CAS.
- J L. Binder, J. Polym. Sci., Part A:Polym. Chem., 1963, 1, 47–58 CAS.
- T. Luciani, J. Seppala and B. Lofgren, Prog. Polym. Sci., 1988, 13, 37–62 CrossRef.
- P. González Cortes, R. Araya-Hermosilla, K. Wrighton-Araneda, D. Cortés-Arriagada, F. Picchioni, F. Yan, P. Rudolf, R. K. Bose and F. Quero, Mater. Today Chem., 2023, 34, 101771 CrossRef.
- J. Qian and R. J. Comito, Organometallics, 2021, 40, 1817–1821 CrossRef CAS.
- C. De Rosa, R. Di Girolamo, A. Malafronte, M. Scoti, G. Talarico, F. Auriemma and O. Ruiz de Ballesteros, Polymer, 2020, 196, 122423 CrossRef CAS.
- M. L. Di Lorenzo and C. Silvestre, Prog. Polym. Sci., 1999, 24, 917–950 CrossRef CAS.
- B. Yang, M. Yang, W. Wang and S. Zhu, Polym. Eng. Sci., 2012, 52, 21–34 CrossRef CAS.
- L. Wang, D. Wan, J. Qiu and T. Tang, Polymer, 2012, 53, 4737–4757 CrossRef CAS.
- A. Cicolella, M. Scoti, G. Talarico, A. J. Müller, R. Di Girolamo and C. De Rosa, Macromolecules, 2024, 57, 8748–8762 CAS.
- R. V. Castillo, A. J. Müller, J.-M. Raquez and P. Dubois, Macromolecules, 2010, 43, 4149–4160 CrossRef CAS.
- A. J. Müller, A. T. Lorenzo, R. V. Castillo, M. L. Arnal, A. Boschetti-de-Fierro and V. Abetz, Macromol. Symp., 2006, 245246, 154–160 CrossRef.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103–1112 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1941, 9, 177–184 CrossRef CAS.
- S. Gupta, X. Yuan, T. C. M. Chung, M. Cakmak and R. A. Weiss, Polymer, 2014, 55, 924–935 CrossRef CAS.
- A. T. Lorenzo, M. L. Arnal, J. Albuerne and A. J. Müller, Polym. Test., 2007, 26, 222–231 CrossRef CAS.
- H. Q. Hong, H. Y. Zhang, S. Da Jiang, H. He and D. M. Jia, J. Elastomers Plast., 2013, 45, 15–31 CrossRef.
- L. Wang, Z. Jiang, F. Liu, Z. Zhang and T. Tang, Chin. J. Polym. Sci., 2014, 32, 333–349 CrossRef CAS.
- A. Jeziorny, Polymer, 1978, 19, 1142–1144 CrossRef CAS.
- K. Shirzad and C. Viney, J. R. Soc., Interface, 2023, 20, 20230242 CrossRef PubMed.
- N. X. Sun, X. D. Liu and K. Lu, Scr. Mater., 1996, 34, 1201–1207 CrossRef CAS.
- C. De Rosa, R. Di Girolamo, A. Malafronte, M. Scoti, G. Talarico, F. Auriemma and O. Ruiz de Ballesteros, Polymer, 2020, 196, 122423 CrossRef CAS.
- E. L. Heeley, C. M. Fernyhough, R. S. Graham, P. D. Olmsted, N. J. Inkson, J. Embery, D. J. Groves, T. C. B. McLeish, A. C. Morgovan, F. Meneau, W. Bras and A. J. Ryan, Macromolecules, 2006, 39, 5058–5071 CrossRef CAS.
- W. Banks, J. N. Hay, A. Sharples and G. Thomson, Polymer, 1964, 5, 163–175 CrossRef CAS.
- R. Buchdahl, R. L. Miller and S. Newman, J. Polym. Sci., 1959, 36, 215–231 CrossRef CAS.
- E. Maderek and G. R. Strobl, Colloid Polym. Sci., 1983, 261, 471–476 CrossRef CAS.
- H. E. Kissinger, J. Res. Natl. Inst. Stand. Technol., 1956, 57, 217–221 CAS.
- C. Albano, J. Papa, M. Ichazo, J. González and C. Ustariz, Compos. Struct., 2003, 62, 291–302 CrossRef.
- A. Eckstein, J. Suhm, C. Friedrich, R.-D. Maier, J. Sassmannshausen, M. Bochmann and R. Mu, Macromolecules, 1998, 1335–1340 CrossRef CAS.
- C. Zou, J. Chen, M. A. Khan, G. Si and C. Chen, J. Am. Chem. Soc., 2024, 146, 19449–19459 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*









 is the instantaneous enthalpy change rate. In non-isothermal conditions, crystallization time (t) can be converted from the crystallization temperature (T) using:
is the instantaneous enthalpy change rate. In non-isothermal conditions, crystallization time (t) can be converted from the crystallization temperature (T) using:


 ) were summarized in Fig. 10. The value of n2 for all samples ranged between 1 and 2 across the experimental cooling rates, indicating heterogeneous nucleation with predominantly one-dimensional growth. We noted that some plots possessed a third region near the end of crystallization; however, the Avrami exponent of these regions remained within the range of 1–2, indicating no change in the crystallization mode. Additionally, as shown in Fig. 10B,
) were summarized in Fig. 10. The value of n2 for all samples ranged between 1 and 2 across the experimental cooling rates, indicating heterogeneous nucleation with predominantly one-dimensional growth. We noted that some plots possessed a third region near the end of crystallization; however, the Avrami exponent of these regions remained within the range of 1–2, indicating no change in the crystallization mode. Additionally, as shown in Fig. 10B,  increased with higher cooling rate, consistent with the previous t1/2 observations. The similar
increased with higher cooling rate, consistent with the previous t1/2 observations. The similar  values (0.7–1.1) across all samples at a given cooling rate suggested that the length of the grafts or the graft density does not significantly impact the overall crystallization rate. Similar phenomenon has been reported in a previous study associated with understanding the non-isothermal crystallization of long chain branched PE52 and PP-g-poly(ethylene-co-1-butene) samples.53
values (0.7–1.1) across all samples at a given cooling rate suggested that the length of the grafts or the graft density does not significantly impact the overall crystallization rate. Similar phenomenon has been reported in a previous study associated with understanding the non-isothermal crystallization of long chain branched PE52 and PP-g-poly(ethylene-co-1-butene) samples.53

 versus
versus for all samples are presented in Fig. 11. ΔE was calculated from the slopes of these plots as shown in Fig. 11. Generally, the ΔE consists of the transport activation energy (transport molecular segments across the phase boundary to the crystal growth surface) and the nucleation activation energy (the free energy of formation of crystal nuclei with a critical size).72 We observed an opposite trend in activation energy for the graft polymers with 5k and 10k grafts. PEE-g-PE10k (ΔE = 362.4 kJ mol−1) had a higher activation energy than its corresponding homopolymer graft (PE-10k, ΔE = 332.3 kJ mol−1), whereas PEE-g-PE5k had a significantly lower activation energy (186.9 kJ mol−1) than PE-5k (300.6 kJ mol−1). This trend consists of their overall crystallization rate without Jeziorny correction (K, Fig. S8†). The observed behavior may stem from a competition between nucleation facilitation and crystal growth restriction introduced by the graft structure. While grafting facilitates nucleation by providing additional sites (reducing the activation energy for nucleation), it simultaneously hinders molecular transport to the crystal growth surface (increasing the activation energy of crystal growth). In the case of PEE-g-PE5k, its high graft density and low molecular weight of PEE-g-PE5k likely promote nucleation while minimizing transport constraints, leading to lower activation energy. In contrast, the longer graft in PEE-g-PE10k experiences greater steric hindrance, limiting the ability of crystal growth and resulting in higher activation energy.
for all samples are presented in Fig. 11. ΔE was calculated from the slopes of these plots as shown in Fig. 11. Generally, the ΔE consists of the transport activation energy (transport molecular segments across the phase boundary to the crystal growth surface) and the nucleation activation energy (the free energy of formation of crystal nuclei with a critical size).72 We observed an opposite trend in activation energy for the graft polymers with 5k and 10k grafts. PEE-g-PE10k (ΔE = 362.4 kJ mol−1) had a higher activation energy than its corresponding homopolymer graft (PE-10k, ΔE = 332.3 kJ mol−1), whereas PEE-g-PE5k had a significantly lower activation energy (186.9 kJ mol−1) than PE-5k (300.6 kJ mol−1). This trend consists of their overall crystallization rate without Jeziorny correction (K, Fig. S8†). The observed behavior may stem from a competition between nucleation facilitation and crystal growth restriction introduced by the graft structure. While grafting facilitates nucleation by providing additional sites (reducing the activation energy for nucleation), it simultaneously hinders molecular transport to the crystal growth surface (increasing the activation energy of crystal growth). In the case of PEE-g-PE5k, its high graft density and low molecular weight of PEE-g-PE5k likely promote nucleation while minimizing transport constraints, leading to lower activation energy. In contrast, the longer graft in PEE-g-PE10k experiences greater steric hindrance, limiting the ability of crystal growth and resulting in higher activation energy.

 versus
versus for PE-5k, PEE-g-PE5k, PE-10k, and PEE-g-PE10k.
for PE-5k, PEE-g-PE5k, PE-10k, and PEE-g-PE10k.
 and t1/2 at a given cooling rate, suggesting that the grafting architecture had limited influence on the overall crystallization rate. Interestingly, PEE-g-PE10k and PEE-g-PE5k exhibited opposite trend in activation energy, which may stem from a competition between nucleation facilitation and crystal growth restriction introduced by the graft structure. A preliminary investigation of the compatibilization performance with LDPE
and t1/2 at a given cooling rate, suggesting that the grafting architecture had limited influence on the overall crystallization rate. Interestingly, PEE-g-PE10k and PEE-g-PE5k exhibited opposite trend in activation energy, which may stem from a competition between nucleation facilitation and crystal growth restriction introduced by the graft structure. A preliminary investigation of the compatibilization performance with LDPE