
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3D printing via polymerization-induced microphase separation using acrylate macromonomers instead of macroRAFT agents†
Maxime
Michelas
,
Nathaniel
Corrigan
 and
Cyrille
Boyer
*
and
Cyrille
Boyer
*
School of Chemical Engineering, UNSW Sydney, Sydney, New South Wales 2052, Australia. E-mail: cboyer@unsw.edu.au
First published on 22nd April 2025
Abstract
Polymerization-induced microphase separation (PIMS) is a versatile technique for manufacturing nanostructured materials. Combining PIMS with 3D printing enables the fabrication of complex objects with nanoscale features, opening possibilities in diverse applications, including nanostructured ceramics, solid polymer electrolytes, and ion-exchange materials. Traditionally, PIMS utilizes polymers synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization (macromolecular Chain Transfer Agents, macroCTAs). However, RAFT-based PIMS can introduce undesirable color and odor into the final materials. To address these limitations, this study explores the use of macromonomers, polymers terminated with acrylate or methacrylate groups, as alternatives to macroCTAs. We synthesized a series of polycaprolactone (PCL) variants with identical molecular weights but differing terminal functionalities: acrylate, methacrylate, trithiocarbonate, and dithiobenzoate. This library enabled a direct comparison of macromonomer and macroCTA approaches for nanostructured material fabrication via PIMS. Small-angle X-ray scattering (SAXS) was employed to determine nanodomain sizes. Notably, both acrylate and trithiocarbonate-terminated PCLs yielded comparable nanodomain sizes. Exploiting PCL degradation, we fabricated nanoporous, 3D-printed objects by selectively etching the PCL from materials formed with both trithiocarbonate and acrylate-terminated PCL. Critically, the acrylate-terminated macromonomer-based PIMS system produced transparent, colorless materials with well-defined microstructures. This work demonstrates the potential of macromonomers to overcome the inherent limitations of RAFT–PIMS, providing a cleaner and more versatile pathway to synthesize advanced nanostructured materials.
Introduction
Additive manufacturing, commonly known as 3D printing, has significantly simplified the fabrication of a wide range of materials with complex shapes and customizable properties.1,2 More recently, advancements in controlling structures at multiple scales, particularly at both the nano- and macroscale, have further expanded its potential.3–9 One notable approach involves polymerization-induced phase separation (PIPS), which generates distinct regions or phases within the material in the microscopic range (from 50 nm to micrometres). This process occurs during polymerization, where phase separation takes place as the polymerization reaction progresses. The key concept behind PIPS is that the growing polymer chains become increasingly incompatible with the surrounding medium (often the monomer or solvent), leading to phase separation.10,11 Levkin and coworkers pioneered the combination of digital light processing (DLP) and PIPS to form 3D-printed hierarchical macro- and mesoporous materials, which can be employed as super-hydrophobic materials, in gas separation, and water repellents for microfluidic applications, as well as superoleophilic materials for the development of oil-absorbent systems.12Inspired by the early work from Seo and Hillmyer on polymerization induced microphase separation (PIMS),13 we introduced a novel 3D printing technology by integrating photoinduced reversible addition–fragmentation chain-transfer (RAFT) with PIMS in photocuring 3D printing, enabling a nanoscopic scale below 100 nm.14 The PIMS methodology relies on an in situ chain-extension reaction of a macromolecular chain transfer agent (macroCTA) with a monomer and a crosslinker, resulting in the formation of a “block” copolymer. This reaction induces microphase separation between the incompatible segments, while simultaneous crosslinking arrests the separation and forms disordered nanostructured materials (Fig. 1a). The resulting nanostructured materials exhibit properties suitable for diverse applications, including in energy storage,15,16 polymer electrolyte membranes,17–20 biomaterials,21 and nanoporous inorganic materials.13,22,23 Extensive research has explored the parameters governing the morphology and nanostructure of PIMS materials, such as the macroCTA chain length and loading,24,25 combinations of different macroCTAs,22,26,27 incorporation of unreactive homopolymers,28 crosslinker's nature,29 reaction conditions,15 macroCTA architecture,22,26,30,31 and choice of RAFT end groups.32 Separately, the thiol–ene coupling reaction has been used for the preparation of bicontinuous networks, providing nanoscale structures using vinyl-terminated polymers (PS and PLA) and a tetra-thiol crosslinker.33
 | ||
| Fig. 1 Illustration of the PIMS mechanism: (a) a reactive polymer undergoes chain extension with acrylic acid (AA) and polyethylene glycol diacrylate (PEGDA) to form block copolymers or (b) in the presence of an acrylate macromonomer the sparse incorporation leads to the formation of a copolymer, which eventually phase-separates, resulting in the formation of distinct domains. (c) Composition of the photocurable resins investigated in this study: functional poly(ε-caprolactone), acrylic acid (AA), and polyethylene glycol diacrylate (PEGDA, Mn = 575 g mol−1). The chemical structures of the four distinct PCL end-chain functionalities are shown: two macroCTAs (represented in orange and purple) and two macromonomers (represented in blue and green). | ||
While PIMS has shown great promise for creating nanostructured materials, the use of RAFT agents often results in objects containing sulfur, resulting in undesirable color (yellowish or pinkish) and odour in the final product. This study explores replacing RAFT end groups with vinyl groups to address these limitations. For the first time, we compare the effects of RAFT-terminated and vinyl-terminated polymers on microphase separation during PIMS.
To achieve this, poly(ε-caprolactone) (PCL–OH) was selected as a model polymer and synthesized via ring-opening polymerization (ROP). The terminal hydroxyl group of PCL was then functionalized with either RAFT agents or acrylate/methacrylate end-groups. The RAFT agents, 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (CPADB) and 2-(((butylthio)carbonothioyl)thio)propanoic acid (BTPA), were reacted with PCL–OH to yield PCL–CPADB and PCL–BTPA macroCTAs (Table 1, Fig. 2, Fig. S8–S15†). Alternatively, the hydroxyl group of PCLs was reacted with 2-isocyanatoethyl methacrylate (IEM) or 2-isocyanatoethyl acrylate (IEA) to produce PCL–methacrylate and PCL–acrylate macromonomers, respectively (Table 1, Fig. 2, Fig. S16–S19†). A range of formulations, achieved by varying PCL, monomer, and/or co-solvent combinations, were screened and characterized using small-angle X-ray scattering (SAXS) to identify those yielding nanodomains. The most promising formulations were then tested for photo-polymerization kinetics and their ability to print complex objects. Finally, inspired by previous work,23,34–37 selective etching of PCL components was performed to demonstrate the potential application of this system for producing nanoporous materials, as evidenced by scanning electron microscopy (SEM). This study showcases the possibility of eliminating RAFT agents in PIMS, resulting in colorless, nanostructured materials, and even nanoporous materials after etching.
 | ||
| Fig. 2 Synthesis and characterization of functionalized PCL polymers. (a) Synthetic route for RAFT-terminated-PCLs (PCL–BTPA and PCL–CPADB) and acrylate/methacrylate-terminated-PCL (PCL–acrylate and PCL–methacrylate) via a two-step process. (b) 1H NMR spectra of the starting PCL–OH and the four modified PCL polymers: PCL–acrylate, PCL–methacrylate, PCL–BTPA and PCL–CPADB (CDCl3 (*), 400 MHz, 298 K). | ||
| Polymer | SEC (RI, DMAc)a | 1H NMR | |||
|---|---|---|---|---|---|
| M n Corr (g mol−1) | Đ |
X
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
M
n
NMRc (g mol−1) |
End group fidelity, fd (%) | |
a Determined by DMAc–SEC analysis using PS standards and applying a correction factor of 0.56.40
b
X
n calculated from the 1H NMR spectrum: Xn = I4.05 ppm/2.
c
M
NMRn = Xn × Mw(CL) + Mw(benzyl alcohol).
d End group fidelity calculated using NMR:  . .
|
|||||
| PCL3k–OH | 3350 | 1.15 | 25 | 3182 | — |
| PCL5.5k–OH | 3800 | 1.17 | 47 | 5643 | — |
| PCL11k–OH | 7380 | 1.06 | 99 | 11805 |
— |
| PCL3k–CPABD | 3890 | 1.17 | 28 | 3788 | 70 |
| PCL3k–BTPA | 4100 | 1.17 | 30 | 3866 | 100 |
| PCL5.5k–BTPA | 4400 | 1.35 | 53 | 6438 | 90 |
| PCL11k–BTPA | 7400 | 1.07 | 97 | 11538 |
100 |
| PCL3k–acrylate | 3600 | 1.13 | 27 | 3323 | 100 |
| PCL5.5k–acrylate | 4500 | 1.27 | 46 | 5600 | 65 |
| PCL11k–acrylate | 8100 | 1.13 | 102 | 11850 |
54 |
| PCL3k–methacrylate | 4400 | 1.36 | 27 | 3389 | 71 |
Results and discussion
To study the effect of the end group on the PIMS process and minimize the influence of macroCTA chain length on both the morphology and nanoscale size, a two-step synthesis approach was employed to ensure consistent molecular weight across all the macroCTAs and macromonomers. In the first step, poly(ε-caprolactone) (PCL–OH) polymers with molecular weights ranging from 3000 to 11000 g mol−1 were synthesized via thermal ring opening polymerization (ROP) following previous procedures.38 Benzyl alcohol (BnOH) was selected as the initiator to facilitate the determination of molecular weight by NMR, and tin(II) 2-ethylhexanoate (Sn(oct)2) acted as the catalyst (Fig. 2a). The ROP method was preferred over polycondensation to achieve a narrow molecular weight distribution (Đ < 1.2).39 Polymerization proceeded to near-complete conversion, as determined by 1H NMR by monitoring the shift of the Hε signal from 4.28 ppm to 4.14 ppm (Fig. S1†). Upon reaching 99% monomer conversion, the resulting polymers were purified by precipitation (Table 1, Fig. S2–S7†). 1H NMR was also employed to calculate the degree of polymerization (Xn) of purified polymers by comparing the polycaprolactone signals at 4.14 ppm (Hε) with that of the CH2 of the benzyl group (Hb) from the initiator. Size exclusion chromatography (SEC) confirmed monodisperse molecular weight distributions for the different PCLs, although a slight discrepancy between the molecular weights determined by NMR and SEC was observed, likely due to the use of polystyrene standards in SEC (Fig. S2–S7†). In the second step, the terminal hydroxyl group of the PCL was functionalized. For RAFT-terminated PCLs, coupling reactions with BTPA and CPADB in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 4-dimethylaminopyridine (DMAP) yielded PCL–BTPA and PCL–CPADB macroCTAs (Table 1, Fig. 2, Fig. S8–S15†). For vinyl-terminated PCL, reactions with 2-isocyanatoethyl methacrylate (IEM) or 2-isocyanatoethyl acrylate (IEA) produced PCL–acrylate and PCL–methacrylate macromonomers, respectively (Table 1, Fig. 2, Fig. S16–S23†).
The resulting PCLs were purified and characterized by 1H NMR and DMAC–SEC (Fig. 2b, Fig. S8–S23†). For PCL–BTPA and PCL–CPADB macroCTAs, 1H NMR spectra (shown in Fig. 2b) confirmed the presence of characteristic signals corresponding to BTPA and CPADB. Specific signals at 1.93 ppm (Hj) and 2.67 ppm (Hi) confirm the presence of CPADB (purple line, Fig. 2b and Fig. S8†), while signals at 0.98 ppm (Hp) and 4.81 ppm (Hn) indicated the presence of BTPA in PCL–BTPA (orange line, Fig. 2b and Fig. S14†). For PCL3k–acrylate, the 1H NMR spectrum exhibited vinyl bond signals (Hu, Ht and Hu′) at 6.44, 6.15 and 5.86 ppm, respectively (blue line, Fig. 2b and Fig. S16†). Similarly, the two characteristic vinyl signals of PCL3k–methacrylate were observed clearly at 6.12 ppm (Hv′) and 5.58 ppm (Hv) (green line, Fig. 2b and Fig. S22†). End-chain fidelity, detailed in Table 1, was determined by comparing these end-group signal integrals to the –CH2 (Hb) at 5.11 ppm from the benzyl initiator fragment. Despite general success, 1H NMR also revealed incomplete end-capping in certain samples, indicated by a residual terminal –CH2OH signal (Hg at 3.64 ppm). This was apparent for PCL–methacrylate and PCL–CPADB (Fig. 2b), and notably more pronounced for higher molecular weight PCL–acrylates (5.5 and 11 kg mol−1; Fig. S18 and S20†). This likely arises from either lower reactivity of the functionalizing agent or sterically hindered access to the hydroxyl terminus on longer chains. Complementary techniques provided further verification. DMAC–SEC equipped with a UV detector (310 nm) was employed to verify the presence of the trithiocarbonate or dithiobenzoate groups on the PCL (Fig. S8–S15†). Fourier-Transform Infrared (FTIR) spectroscopy further confirmed the presence of a double bond at υ = 1529 cm−1 (Fig. S24†).
Photocurable resin formulations containing functionalized PCLs (Mn = 3000 g mol−1), acrylic acid (AA) as the monomer, poly(ethylene glycol) diacrylate (PEGDA, average Mn = 575 g mol−1) as the cross-linker, and diphenyl(2,4,6trimethylbenzoyl) phosphine oxide (TPO) as the photo-initiator were prepared. AA was selected as the resulting PAA block polymer can phase separate, due to the segregation strength. The segregation strength χN is used as a measure of the thermodynamic driving force, where χ is the measure of the incompatibility between the two blocks and N is the total degree of polymerization.41,42 For the PCL-b-P(AA-stat-PEGDA) block copolymer, χ is estimated to be 0.92 at 25 °C indicating that the PCL and P(AA-stat-PEGDA) are thermodynamically incompatible (ESI, Note 1†).43
Components were mixed and sonicated to achieve transparent, homogeneous solutions. Polymerization kinetics were determined via assessment using total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy under violet light irradiation (λmax = 405 nm and I0 = 2.08 mW cm−2) under open-to-air conditions. The molar ratio of [AA]/[PEGDA] and the weight percentages (wt%) of PCL3k–R and TPO were fixed at 9/1, 21 wt%, and 1 wt%, respectively. Vinyl bond conversion was monitored by tracking the decrease in the absorption peak at ≈1620 cm−1, corresponding to the vinylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) stretching mode. As shown in Fig. 3a, resins formulated with acrylate- or methacrylate-functionalized PCL cured rapidly, reaching high vinyl bond conversion (≥90%) within 20 seconds. In contrast, the PCL3k–BTPA containing resin required over 45 seconds to achieve similar conversion (Fig. 3a). This kinetic difference is attributed to the trithiocarbonate group in PCL3k–BTPA, which absorbs light at 405 nm, thereby reducing TPO photoinitiator activation. Furthermore, the PCL3k–CPADB resin showed minimal vinyl bond conversion even after one minute of violet light exposure, consistent with previous reports of CPADB's ineffectiveness under these conditions. This ineffectiveness is due to its low fragmentation efficiency with acrylates and its susceptibility to rapid photodegradation (Fig. S25†).44
stretching mode. As shown in Fig. 3a, resins formulated with acrylate- or methacrylate-functionalized PCL cured rapidly, reaching high vinyl bond conversion (≥90%) within 20 seconds. In contrast, the PCL3k–BTPA containing resin required over 45 seconds to achieve similar conversion (Fig. 3a). This kinetic difference is attributed to the trithiocarbonate group in PCL3k–BTPA, which absorbs light at 405 nm, thereby reducing TPO photoinitiator activation. Furthermore, the PCL3k–CPADB resin showed minimal vinyl bond conversion even after one minute of violet light exposure, consistent with previous reports of CPADB's ineffectiveness under these conditions. This ineffectiveness is due to its low fragmentation efficiency with acrylates and its susceptibility to rapid photodegradation (Fig. S25†).44
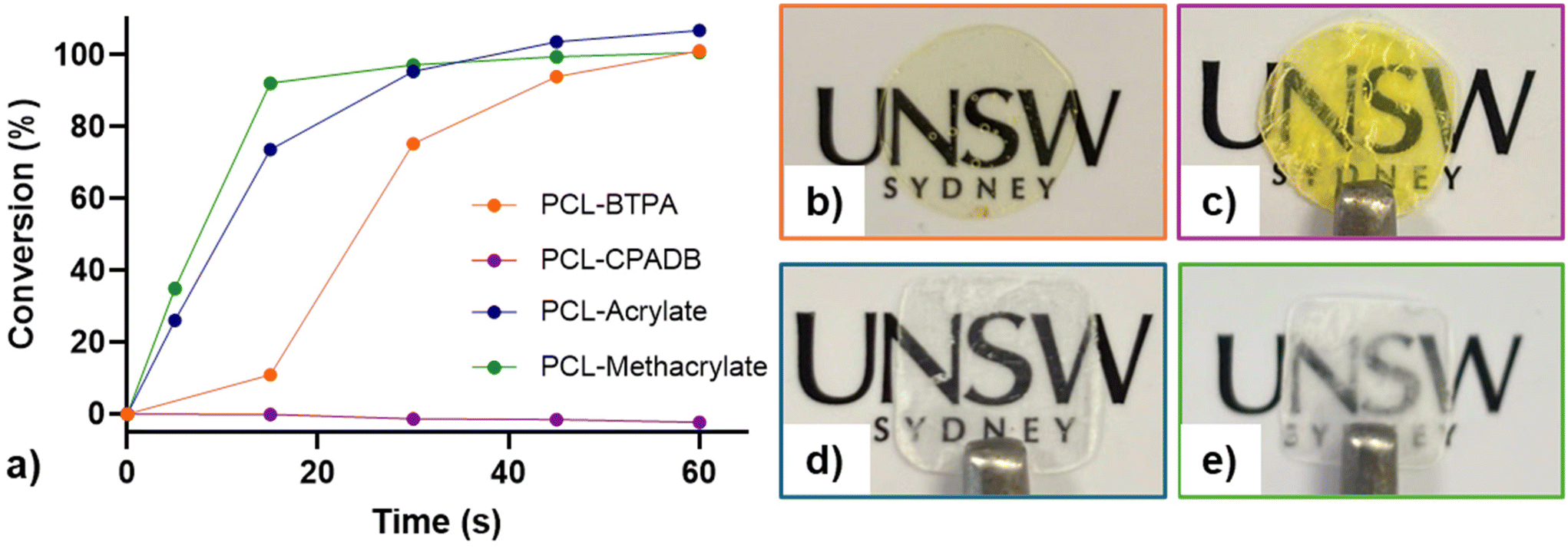 | ||
| Fig. 3 Effect of the PCL end-group on photopolymerization kinetics and 3D printing. (a) Vinyl bond conversion as a function of time for resins containing different PCL polymers. Photographs of the resulting films prepared from the corresponding resins: (b) PCL3k–BTPA, (c) PCL3k–CPADB, (d) PCL3k–acrylate, and (e) PCL3k–methacrylate. Resin formulations: [AA]/[PEGDA] = 9/1 (molar ratio), PCL–R = 21 wt%, and TPO = 1 wt%. Kinetics experiments were performed in triplicate. Double bond conversion was monitored by ATR-FTIR under 2.08 mW cm−2 violet light irradiation (λmax = 405 nm). | ||
Next, the formulated resins were 3D printed using an Anycubic Photon on Mono SE (λmax = 405 nm and I0 = 2.3 mW cm−2) to fabricate simple objects, such as prism or discs (Fig. 3, Fig. S25, Table S1†). For consistency, the [AA]/[PEGDA] molar ratio was fixed at 9/1, and the TPO concentration was 1 wt% for all the formulations. The PCL3k–R content was varied from 15 to 35 wt% (Table S1†). However, the PCL3k–CPADB resin required a higher irradiation intensity (I0 = 20 mW cm−2) to cure the resin, producing a yellow and very brittle object (Fig. 3c). These printed samples were then characterized by small-angle X-ray scattering (SAXS) to investigate their internal nanostructure and phase separation. Initial visual inspection revealed that PCL–BTPA and PCL–acrylate resins yielded transparent samples, while PCL–CPADB and PCL–methacrylate resins produced translucent samples, suggesting the formation of large domains (>200 nm).
Fig. 4 presents the SAXS profiles of 3D-printed objects as a function of macroCTA or macromonomer content. Resins formulated with PCL3k–BTPA and PCL3k–acrylate exhibited broad single scattering peaks across all loadings, indicating the formation of disordered microphase-separated materials (Fig. 4a and c). The scattering peak position shifted to higher q values with an increase in PCL content, consistent with the observations in other similar systems.24 For instance, with 21 wt% loading for PCL3k–BTPA, the peak appeared at 0.439 nm−1, corresponding to a dSAXS value of 14.31 nm, shifting to 0.469 nm−1 (dSAXS of 13.38 nm) at 35 wt% (Fig. 4a). A similar trend was observed for PCL–acrylate, with the peak shifting from 0.432 nm−1 (dSAXS = 14.54 nm) at 20 wt% to 0.488 nm−1 (dSAXS = 12.86 nm) at 35 wt% (Fig. 4c). Furthermore, comparing the two, the PCL3k–BTPA signals were sharper and more intense, suggesting differences in phase separation efficiency; this may reflect more dispersed nanodomains resulting from the free radical polymerization of PCL–acrylate compared to the controlled radical polymerization mechanism of PCL–BTPA. In contrast, the SAXS profiles of objects made with PCL3k–CPADB and PCL3k–methacrylate showed no discernible scattering peaks (Fig. 4b and d). This lack of features strongly indicates macrophase separation, with domain sizes exceeding the typical SAXS detection limit (>∼100 nm). These results are attributed to the inefficiency of CPADB in promoting chain extension for the PCL–CPADB system due to its degradation, and the low reactivity of methacrylate groups during copolymerization with acrylates (AA and PEDGA) for the PCL–methacrylate system.
 | ||
| Fig. 4 SAXS profiles and the corresponding domain spacing (dSAXS) values for materials 3D printed with varying loadings of (a) PCL3k–BTPA, (b) PCL3k–CPADB, (c) PCL3k–acrylate, and (d) PCL3k–methacrylate. Loading: 15 wt% (green), 21 wt% (blue), 25 wt% (red) and 35 wt% (orange). [AA]/[PEGDA] = 9/1 (molar ratio); TPO = 1 wt%. | ||
The observed variations in morphologies can be explained by the reactivity of the PCL end-chains. The trithiocarbonate group in PCL3k–BTPA is a highly efficient chain transfer agent in acrylate polymerization, which promotes effective chain extension, leading to the formation of diblock copolymers that undergo nanoscale phase separation. Conversely, the dithiobenzoate group in PCL3k–CPADB is a less efficient chain transfer agent with acrylates and acrylamides,45,46 resulting in macrophase separation.25,28,29,47 Additionally, this group is more susceptible to photolysis, as evidenced by the color change from reddish to yellowish observed after printing and by the decrease in the characteristic 360 nm absorbance peak of the dithiobenzoate group (Fig. S25 and S26†). Building on these encouraging results, we also investigated resins containing PCLxk–BTPA with molecular weights Mn = 5.5 and 11 kg mol−1. The printed samples from these resins exhibited a single scattering peak that shifted to a lower q value with an increase in Xn. This shift corresponds to an increase in domain spacing, consistent with the formation of higher molecular weight block copolymers (Fig. S27 and S28†). However, the resins formulated with PCL–acrylate (5.5k and 11k) did not produce high-quality printed objects suitable for SAXS analysis.
While these initial results were encouraging, we encountered gelation of the resins within the vat after only a few minutes of 3D printing, preventing the preparation of larger and more complex objects. This effect was particularly prominent with the higher molecular weight PCL (PCL5.5k–BTPA and PCL11k–BTPA) (Fig. S29†). We hypothesized that this issue was due to the low miscibility of PCL in the AA/PEDGA mixture. To mitigate this and improve resin stability for longer print times, we investigated two strategies: replacing AA with alternative monomers or adding a co-solvent to enhance PCL solubility. Initially, we tested several alternative monomers, including methyl acrylate (MA), N,N-dimethylacrylamide (DMA) and isobornyl acrylate (IBoA) (Fig. 5a and Table S2†). SAXS analysis of the resulting materials revealed microstructures in all cases, as evidenced by the broad, single scattering peaks characteristic of disordered microphase-separated systems (Fig. 5a). Interestingly, normalization of the SAXS scattering plots by intensity (I*) revealed that the peak breadth increased with DMA, MA, and IBoA compared to the AA formulation. This suggests that these alternative monomers led to the formation of more polydisperse nanodomains compared to AA, which yielded sharper interfaces. However, despite these promising microstructures, the gelation issue during 3D printing persisted with all tested monomers, hindering their practical application in 3D printing.
 | ||
| Fig. 5 Effect of the monomer and co-solvent on microphase separation. SAXS profiles and the corresponding domain spacing (dSAXS) values of 3D printed materials using (a) PCL3k–BTPA with different co-monomers (AA, MA, DMA, and IBoA), (b) PCL3k–BTPA with AA and toluene, and (c) PCL3k–acrylate with AA and toluene. (d and e) Scaled SAXS data (normalized intensity, I*) comparing peak broadness and position (q) for PCL3k–BTPA with different co-monomers and toluene contents, respectively. Note: [M]/[PEGDA] = 9/1 (molar ratio); TPO = 1 wt%. | ||
As an alternative approach, we investigated the use of a solvent to enhance PCL solubility in our resin formulations. Toluene was selected due to the high solubility of PCL. It was added at 10 wt% and 40 wt% concentrations. Remarkably, 10 wt% toluene alone successfully eliminated gelation, resulting in a stable resin suitable for extended printing (Fig. S29†). Having successfully resolved the gelation problem, we then examined the photopolymerization kinetics of the 10 wt% toluene-containing resins before fabricating complex objects. Subsequent photopolymerization kinetic studies on the 10 wt% toluene-containing resins, maintaining a constant [AA]/[PEGDA] molar ratio (9/1), PCL3k–R weight percentage (19 wt%), and TPO concentration (1 wt%), demonstrated similar kinetic profiles to those of the toluene-free formulations (Fig. S30†). Specifically, resins formulated with PCL–acrylate and PCL–methacrylate achieved near-complete double-bond conversion within 10 seconds, while PCL–BTPA-based resins required approximately 40 seconds to reach a comparable conversion level.
SAXS profiles of the 3D-printed samples revealed that while toluene addition did not significantly alter the broadness of the SAXS signals, it induced a shift in the maximum scattering peak to higher q values, indicating a decrease in dSAXS from 14.11 nm to 13.36 nm as the toluene concentration increased from 10% to 40% (Fig. 5b and e, Table S3†). Moreover, at higher Xn, the SAXS signal became sharper, suggesting the formation of more well-defined nanodomains. A similar trend was noted with PCLXk–acrylate with 10 wt% toluene, with dSAXS values increasing from 12.09 nm (PCL3k–acrylate) to 14.82 nm (PCL11k–acrylate) with a significant change in the SAXS intensity. Interestingly, the SAXS signal intensity of samples prepared with 40 wt% toluene appeared slightly reduced in comparison with the one with 10 wt% toluene (Fig. 5c and Fig. S31†). Conversely, PCL–methacrylate and PCL–CPADB formulations remained opaque/translucent and exhibited no discernible SAXS signals upon toluene addition.
Moreover, the mechanical properties of these formulations were investigated, which revealed that samples prepared with PCL3k–BTPA exhibited a slightly lower storage modulus and a tan δ peak which shifted to a lower temperature compared to those prepared with PCL3k–acrylate, when both used AA and PEGDA (Fig. S32†). Interestingly, the tan δ peak temperature for the PCL3k–BTPA samples shifted significantly higher, from approximately 0 °C to approximately 30 °C, when IBoA and PEGDA were used instead of AA and PEGDA (Fig. S33†).
Subsequently, we assessed the influence of end-chain functionality on printing resolution and macroscopic characteristics by fabricating objects with complex geometries. Resins formulated with PCL3k–BTPA, PCL3k–CPADB, PCL3k–acrylate, and PCL3k–methacrylate (19 wt%) in the presence of 10 wt% toluene and [AA]/[PEGDA] equal to 9/1 were tested alongside a control resin containing unmodified PCL (PCL–OH). Complex snowflake structures (28.9 × 25.7 × 2.0 mm) were successfully printed using a layer thickness of 50 μm, except for the resin based on PCL3k–CPADB which failed to produce acceptable prints. PCL–BTPA and PCL–OH formulations required 30 second exposure time, while the acrylate- and methacrylate-functionalized PCL resins produced high-resolution prints with a shorter 20 second exposure time (Fig. 6).
 | ||
| Fig. 6 3D Printing of complex snowflake structures. (a) Digital model. (b–e) Photographs of 3D-printed snowflakes using resins formulated with (b) PCL3k–BTPA, (c) PCL3k–acrylate, (d) PCL3k–methacrylate, and (e) PCL3k–OH (control). [AA]/[PEGDA] = 9/1 (molar ratio); PCL–R = 19 wt%; toluene = 10 wt%; and TPO = 1 wt%. Layer thickness: 50 μm. Cure times: 30 s (PCL3k–BTPA and PCL3k–OH) and 20 s (PCL–acrylate and PCL3k–methacrylate). | ||
The objects shown in Fig. 6 exhibit notable differences in transparency and color, which can be attributed to variations in phase separation behavior and the presence or absence of the RAFT agent. Objects fabricated with PCL3k–BTPA and PCL3k–acrylate were transparent (Fig. 6b and c). However, the RAFT-based formulation (PCL3k–BTPA) exhibited a yellowish tint, while the PCL3k–acrylate object was colorless. In contrast, objects printed with PCL3k–methacrylate appeared translucent, suggesting the formation of larger domains (>100 nm), consistent with the lack of SAXS signals (Fig. 4d). This effect was even more pronounced in the control formulation (PCL3k–OH, Fig. 6e), where the objects were fully opaque, indicating significant macrophase separation. These results demonstrate that formulations based on PCL–BTPAs enable high-resolution 3D printing of complex objects, with tunable nanodomain sizes (Fig. 6b–d).
Due to the importance of porous materials for a wide range of applications,48,49 the final part of our study focused on creating nanoporous materials by leveraging the degradability of PCL through controlled solvent-based etching, allowing for tunable pore size.23,34–37 Initial etching experiments were performed on 3D-printed objects fabricated from the PCL3k–BTPA formulation. After printing, the objects were immersed in a 0.5 M NaOH aqueous solution at 60 °C for one and two days. SEM images were acquired before etching, after one day, and after two days of etching (Fig. 7a). Using AA as the comonomer resulted in no observable pores after etching, likely due to swelling and collapse of the poly(acrylic acid) (PAA) domains upon freeze-drying. To address this limitation, AA was replaced with a hydrophobic monomer (IBoA) resulting in the formation of water-insoluble domains, preventing pore collapse. Additionally, the PCL Mn was increased from 3000 to 11000 g mol−1 to promote the formation of larger pores. SEM images revealed that samples before etching exhibited no visible pores, but pores emerged after one day of etching (Fig. 7a). After two days, pore coalescence was observed, indicating structural evolution over time. An identical procedure was followed for 3D-printed objects made from PCL11k–acrylate (Fig. 7b), revealing pore formation with broader dispersity, as expected due to the uncontrolled nature of the polymerization mechanism.
 | ||
| Fig. 7 SEM images of freeze-dried 3D-printed object prepared with (a) PCL11k–BTPA and (b) PCL11k–acrylate in the presence of IBoA, PEGDA and 10 wt% toluene, following selective etching of the PCL domains. Images show the control sample before etching (top), after 1 day of treatment (middle), and after 2 days of treatment (bottom) in a 0.5 M NaOH aqueous solution at 60 °C. | ||
To verify the successful etching of PCL, weight loss measurements were conducted (Table S4†), complemented by ATR-FTIR analysis. The weight loss data showed different kinetics depending on the formulation. PCL11k–BTPA samples exhibited a 21.7 wt% loss after one day of etching, which corresponds well with the theoretical PCL content targeted for removal (e.g., ∼29 wt% in that formulation). In contrast, PCL11k–acrylate samples required two days to achieve a comparable weight loss (25 wt%), suggesting slower degradation, potentially due to network differences influencing the etching process. Additionally, ATR-FTIR analysis (Fig. S34†) supported successful PCL degradation. Spectra acquired after etching showed a marked decrease in the ester CO peak (∼1730 cm−1) and the appearance of features indicative of hydrolysis products: specifically, the broad O–H stretching of carboxylic acids (∼3300–2500 cm−1) and the carboxylate anion (COO−) absorption near 1570 cm−1.
Conclusion
In conclusion, this study successfully developed 3D-printable resins containing both RAFT-terminated and vinyl-terminated PCLs for the fabrication of nanostructured materials via PIMS. A systematic comparison of these resins revealed distinct nanodomain structures, significantly influencing the materials’ optical properties. Notably, the use of PCL3k–acrylate enabled the fabrication of high-resolution, colorless nanostructured materials, overcoming the limitations of traditional RAFT systems, which often yield yellowish products. Furthermore, the addition of toluene (up to 40 wt%) improved resin stability, facilitating extended print durations and the creation of complex 3D-printed objects. Finally, selective etching of the PCL component under basic conditions successfully produced nanoporous materials, demonstrating the versatility of this approach. These findings open new avenues for advanced applications in diverse fields such as filtration, catalysis, and tissue engineering.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
CB thanks the Australian Research Council for his Australian Laureate Fellowship (FL220100016).References
- J.-Y. Lee, J. An and C. K. Chua, Fundamentals and applications of 3D printing for novel materials, Appl. Mater. Today, 2017, 7, 120–133 CrossRef.
- J.-T. Lin, J. Lalevee and D.-C. Cheng, A Critical Review for Synergic Kinetics and Strategies for Enhanced Photopolymerizations for 3D-Printing and Additive Manufacturing, Polymers, 2021, 13, 2325 CrossRef CAS.
- K. Lee, N. Corrigan and C. Boyer, Polymerization Induced Microphase Separation for the Fabrication of Nanostructured Materials, Angew. Chem., Int. Ed., 2023, 62(44), e202307329 CrossRef CAS.
- A. Priester, J. Yeng, Y. Zhang, R. Wang and A. J. Convertine, 3D printing soluble solids via PISA, Polym. Chem., 2023, 14(20), 2452–2456 RSC.
- Z. Zhao and M. Huo, Polymerization-induced self-assembly for the construction of nanostructured hydrogels, Polym. Chem., 2024, 15(16), 1577–1590 RSC.
- B. Zhao, J. Li, X. Pan, Z. Zhang, G. Jin and J. Zhu, Photoinduced Free Radical Promoted Cationic RAFT Polymerization toward “Living” 3D Printing, ACS Macro Lett., 2021, 10(10), 1315–1320 CrossRef CAS PubMed.
- A. Priester, J. Yeng, Y. Zhang, K. Hilmas, R. Wang and A. J. Convertine, PISA Printing Microneedles with Controllable Aqueous Dissolution Kinetics, ACS Appl. Polym. Mater., 2024, 6(3), 1944–1950 CrossRef CAS.
- B. Zhao, J. Li, X. Yang, S. He, X. Pan and J. Zhu, Degradable 3D Printed Objects with Tunable Mechanical Properties via Photoinduced Free Radical Promoted Cationic RAFT Polymerization, ACS Appl. Polym. Mater., 2024, 6(2), 1584–1591 CrossRef CAS.
- S. He, X. Yang, Y. Zhao, Y. Liu, B. Zhao, X. Pan, J. Li, J. Zhu and N. Li, Radical promoted cationic RAFT polymerization by photo electron transfer reaction, Polym. Chem., 2024, 15(38), 3847–3853 RSC.
- P. K. Chan and A. D. Rey, Polymerization-Induced Phase Separation. 1. Droplet Size Selection Mechanism, Macromolecules, 1996, 29, 8934–8941 CrossRef CAS.
- P. K. Chan and A. D. Rey, Polymerization-Induced Phase Separation. 2. Morphological Analysis, Macromolecules, 1997, 30, 2135–2143 CrossRef CAS.
- Z. Dong, M. Vuckovac, W. Cui, Q. Zhou, R. H. A. Ras and P. A. Levkin, 3D Printing of Superhydrophobic Objects with Bulk Nanostructure, Adv. Mater., 2021, 33(45), 2106068 CrossRef CAS PubMed.
- M. Seo and M. A. Hillmyer, Reticulated Nanoporous Polymers by Controlled Polymerization-Induced Microphase Separation, Science, 2012, 336, 1422–1425 CrossRef CAS PubMed.
- V. A. Bobrin, K. Lee, J. Zhang, N. Corrigan and C. Boyer, Nanostructure Control in 3D Printed Materials, Adv. Mater., 2022, 34(4), e2107643 CrossRef.
- K. Lee, Y. Shang, V. A. Bobrin, R. Kuchel, D. Kundu, N. Corrigan and C. Boyer, 3D Printing Nanostructured Solid Polymer Electrolytes with High Modulus and Conductivity, Adv. Mater., 2022, 34(42), e2204816 CrossRef PubMed.
- D. Melodia, A. Bhadra, K. Lee, R. Kuchel, D. Kundu, N. Corrigan and C. Boyer, 3D Printed Solid Polymer Electrolytes with Bicontinuous Nanoscopic Domains for Ionic Liquid Conduction and Energy Storage, Small, 2023, 19(50), e2206639 CrossRef.
- C. Jeon, C. Choi, H.-T. Kim and M. Seo, Achieving Fast Proton Transport and High Vanadium Ion Rejection with Uniformly Mesoporous Composite Membranes for High-Efficiency Vanadium Redox Flow Batteries, ACS Appl. Energy Mater., 2020, 3(6), 5874–5881 CrossRef CAS.
- M. W. Schulze, L. D. McIntosh, M. A. Hillmyer and T. P. Lodge, High-modulus, high-conductivity nanostructured polymer electrolyte membranes via polymerization-induced phase separation, Nano Lett., 2014, 14(1), 122–126 CrossRef CAS PubMed.
- S. A. Chopade, J. G. Au, Z. Li, P. W. Schmidt, M. A. Hillmyer and T. P. Lodge, Robust Polymer Electrolyte Membranes with High Ambient-Temperature Lithium-Ion Conductivity via Polymerization-Induced Microphase Separation, ACS Appl. Mater. Interfaces, 2017, 9(17), 14561–14565 CrossRef CAS.
- S. A. Chopade, S. So, M. A. Hillmyer and T. P. Lodge, Anhydrous Proton Conducting Polymer Electrolyte Membranes via Polymerization-Induced Microphase Separation, ACS Appl. Mater. Interfaces, 2016, 8(9), 6200–6210 CrossRef CAS PubMed.
- Y. Xie and M. A. Hillmyer, Nanostructured Polymer Monoliths for Biomedical Delivery Applications, ACS Appl. Bio Mater., 2020, 3(5), 3236–3247 CrossRef CAS PubMed.
- S. A. Saba, B. Lee and M. A. Hillmyer, Tricontinuous Nanostructured Polymers via Polymerization-Induced Microphase Separation, ACS Macro Lett., 2017, 6(11), 1232–1236 CrossRef CAS.
- J. Oh and M. Seo, Photoinitiated Polymerization-Induced Microphase Separation for the Preparation of Nanoporous Polymer Films, ACS Macro Lett., 2015, 4(11), 1244–1248 CrossRef CAS PubMed.
- V. A. Bobrin, K. Lee, J. Zhang, N. Corrigan and C. Boyer, Nanostructure Control in 3D Printed Materials, Adv. Mater., 2022, 34(4), 2107643 CrossRef CAS PubMed.
- X. Shi, V. A. Bobrin, Y. Yao, J. Zhang, N. Corrigan and C. Boyer, Designing Nanostructured 3D Printed Materials by Controlling Macromolecular Architecture, Angew. Chem., Int. Ed., 2022, 61(35), e202206272 CrossRef CAS PubMed.
- Y. Xiu, V. A. Bobrin, N. Corrigan, Y. Yao, J. Zhang and C. Boyer, Engineering internal nanostructure in 3D-printed materials via polymer molecular weight distribution, J. Polym. Sci., 2023, 62, 766–776 CrossRef.
- X. Shi, Y. Yao, J. Zhang, N. Corrigan and C. Boyer, Polymerization Induced Microphase Separation of ABC Triblock Copolymers for 3D Printing Nanostructured Materials, Small, 2023, e2305268 Search PubMed.
- J. Park, S. A. Saba, M. A. Hillmyer, D.-C. Kang and M. Seo, Effect of homopolymer in polymerization-induced microphase separation process, Polymer, 2017, 126, 338–351 CrossRef CAS.
- M. W. Schulze and M. A. Hillmyer, Tuning Mesoporosity in Cross-Linked Nanostructured Thermosets via Polymerization-Induced Microphase Separation, Macromolecules, 2017, 50(3), 997–1007 CrossRef CAS.
- D. Wu, V. Dev, V. A. Bobrin, K. Lee and C. Boyer, Nanostructure design of 3D printed materials through macromolecular architecture, Chem. Sci., 2024, 15(46), 19345–19358 RSC.
- C. Yoo and M. Seo, Drive to Asymmetric Lamellar Order by Polymerization-Induced Microphase Separation from the Janus Bottlebrush Architecture, Macromolecules, 2025, 58, 2303–2309 CrossRef CAS.
- Y. Xiu, V. A. Bobrin, N. Corrigan, J. Zhang and C. Boyer, Effect of Macromolecular Structure on Phase Separation Regime in 3D Printed Materials, Macromol. Rapid Commun., 2023, 44(24), e2300236 CrossRef PubMed.
- D. Zeng and R. C. Hayward, Effects of Randomly End-Linked Copolymer Network Parameters on the Formation of Disordered Cocontinuous Phases, Macromolecules, 2019, 52(7), 2642–2650 CrossRef CAS.
- D. A. Olson, L. Chen and M. A. Hillmyer, Templating Nanoporous Polymers with Ordered Block Copolymers, Chem. Mater., 2008, 20, 869–890 CrossRef CAS.
- C. M. Bates, M. J. Maher, D. W. Janes, C. J. Ellison and C. G. Willson, Block Copolymer Lithography, Macromolecules, 2013, 47(1), 2–12 CrossRef.
- C. G. Gamys, J. M. Schumers, C. Mugemana, C. A. Fustin and J. F. Gohy, Pore-functionalized nanoporous materials derived from block copolymers, Macromol. Rapid Commun., 2013, 34(12), 962–982 CrossRef CAS.
- C. H. Peterson, J. R. Werber, H. K. Lee and M. A. Hillmyer, Tailored Mesoporous Microspheres by Polymerization-Induced Microphase Separation in Suspension, ACS Appl. Polym. Mater., 2022, 4(6), 4219–4233 CrossRef CAS.
- A. Quaak, Q. Thijssen and S. Van Vlierberghe, Exploiting the network architecture of thiol–ene photo-crosslinked poly(ε-caprolactone) towards tailorable materials for light-based 3D-printing, Polym. Chem., 2023, 14(29), 3392–3403 RSC.
- M. Degirmenci, G. Hizal and Y. Yagci, Synthesis and Characterization of Macrophotoinitiators of Poly(ε-caprolactone) and Their Use in Block Copolymerization, Macromolecules, 2002, 35, 8265–8270 CrossRef CAS.
- M. Save, M. Schappacher and A. Soum, Controlled Ring-Opening Polymerization of Lactones and Lactides Initiated by Lanthanum Isopropoxide, 1. General Aspects and Kinetics, Macromol. Chem. Phys., 2002, 203(5–6), 889–899 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Block copolymer thermodynamics: theory and experiment, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS.
- P. A. Small, Some factors affecting the solubility of polymers, J. Appl. Chem., 2007, 3(2), 71–80 CrossRef.
- F. S. Bates and G. H. Fredrickson, Block Copolymers—Designer Soft Materials, Phys. Today, 1999, 52(2), 32–38 CrossRef CAS.
- Z. Zhang, N. Corrigan and C. Boyer, Effect of Thiocarbonylthio Compounds on Visible-Light-Mediated 3D Printing, Macromolecules, 2021, 54(3), 1170–1182 CrossRef CAS.
- Y. Lee, C. Boyer and M. S. Kwon, Photocontrolled RAFT polymerization: past, present, and future, Chem. Soc. Rev., 2023, 52(9), 3035–3097 RSC.
- G. Ng, K. Jung, J. Li, C. Wu, L. Zhang and C. Boyer, Screening RAFT agents and photocatalysts to mediate PET-RAFT polymerization using a high throughput approach, Polym. Chem., 2021, 12(45), 6548–6560 RSC.
- K. Lee, J. Mata, V. A. Bobrin, D. Dipan, V. K. Peterson, N. Corrigan and C. Boyer, 3D Printing Highly Efficient Ion-Exchange Materials via a Polyelectrolyte Microphase Separation Strategy, Small Sci., 2024, 4, 2400019 CrossRef CAS PubMed.
- Y. Gao, J. Lalevée and A. Simon-Masseron, An Overview on 3D Printing of Structured Porous Materials and Their Applications, Adv. Mater. Technol., 2023, 8(17), 2300377 CrossRef CAS.
- B. Keitel, S. Dietl, T. Philipp, G. Neusser, C. Kranz, H. Sobek, B. Mizaikoff and M. Dinc, 3D-Printed Highly Porous Functional Materials for the Efficient Removal of Adenovirus, Adv. Mater. Technol., 2025, 10(1), 2401178 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, NMR spectra of purified poly(ε-caprolactone), molecular weight distributions obtained by SEC of modified poly(ε-caprolactone)s, FTIR spectra of modified poly(ε-caprolactone)s, UV-vis spectra, and SAXS profiles (Fig. S1–S34 and Tables S1–S4). See DOI: https://doi.org/10.1039/d5py00226e |
| This journal is © The Royal Society of Chemistry 2025 |