
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of biphenyl groups on the properties of poly(fluorenylidene piperidinium) based anion exchange membranes for applications in water electrolyzers†
Ahmed Mohamed Ahmed
Mahmoud
 ab,
Kenji
Miyatake
*acd,
Fanghua
Liu
a,
Vikrant
Yadav
a,
Fang
Xian
a,
Lin
Guo
a,
Chun Yik
Wong
a,
Toshio
Iwataki
c,
Makoto
Uchida
c and
Katsuyoshi
Kakinuma
ac
ab,
Kenji
Miyatake
*acd,
Fanghua
Liu
a,
Vikrant
Yadav
a,
Fang
Xian
a,
Lin
Guo
a,
Chun Yik
Wong
a,
Toshio
Iwataki
c,
Makoto
Uchida
c and
Katsuyoshi
Kakinuma
ac
aClean Energy Research Center, University of Yamanashi, Kofu, 400-8510, Japan. E-mail: miyatake@yamanashi.ac.jp
bChemistry Department, Faculty of Science, Sohag University, Sohag, 82524, Egypt
cHydrogen and Fuel Cell Nanomaterials Center, University of Yamanashi, Kofu, 400-8510, Japan
dDepartment of Applied Chemistry, Waseda University, Tokyo, 169-8555, Japan
First published on 26th March 2025
Abstract
A series of quaternized poly(arylene fluorenylidene piperidinium)-based copolymers were synthesized using different hydrophobic components, including biphenyl, m- or p-terphenyl, and 9,9-dimethylfluorenyl groups. Among them, the quaternized poly(biphenylene fluorenylidene piperidinium) had the best solvent solubility and membrane formability. Transmission electron microscopy showed that poly(biphenylene fluorenylidene piperidinium) (QBPh-Pip) had a well-interconnected nanoscale phase-separated morphology. The QBPh-Pip membrane with an ion exchange capacity of 1.9 mequiv. g−1 exhibited the most balanced properties, with low water uptake (95% at 80 °C), low swelling (45%), and high hydroxide ion conductivity (160 mS cm−1 at 80 °C). Despite the low water absorption, rapid ion mobility led to high ion conductivity, as calculated using normalized diffusion coefficients. Furthermore, the QBPh-Pip membrane exhibited excellent alkaline stability (91.5% (141 mS cm−1) of the initial conductivity after 1000 h in 8 M potassium hydroxide at 80 °C) and excellent mechanical properties (29.0 MPa of maximum stress and 134% elongation at break). In a water electrolysis cell using a nickel iron oxide anode catalyst, the QBPh-Pip membrane achieved a low cell voltage (1.7 V at 1.0 A cm−2) with 72% efficiency. The QBPh-Pip cell was durable for 1000 h at a constant current density of 1.0 A cm−2 with minor voltage decay of 70 μV h−1.
1. Introduction
Water splitting via electrolysis is expected to play an important role in carbon neutrality, particularly when renewable energy is used to produce green hydrogen; nonetheless, production costs remain a substantial challenge.1 Anion exchange membrane water electrolyzers (AEMWEs) are promising candidates for cost-effective green hydrogen generation because they use nonprecious metal catalysts with inexpensive bipolar plates and current collectors.1,2 In addition to performance, AEMWEs must meet the durability requirements (>50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h at ≥80 °C under high current density and preferably under high pressure of 50–80 bar).3,4 However, to fulfill such criteria, all components, including AEMs, catalysts, and current collectors, must be improved. Among them, AEMs, which function as separators, ion transporters, and catalyst layer binders, should possess multiple chemical, mechanical, and electrochemical functions. Over the past decade, extensive research has focused on improving the properties of AEMs.5 Typical AEMs comprise hydrophobic (co)polymers tethered with cationic head groups, with the polymer main chains responsible for mechanical and thermal stability and the cationic groups dominating ion conduction and alkaline stability.6,7
000 h at ≥80 °C under high current density and preferably under high pressure of 50–80 bar).3,4 However, to fulfill such criteria, all components, including AEMs, catalysts, and current collectors, must be improved. Among them, AEMs, which function as separators, ion transporters, and catalyst layer binders, should possess multiple chemical, mechanical, and electrochemical functions. Over the past decade, extensive research has focused on improving the properties of AEMs.5 Typical AEMs comprise hydrophobic (co)polymers tethered with cationic head groups, with the polymer main chains responsible for mechanical and thermal stability and the cationic groups dominating ion conduction and alkaline stability.6,7
For AEMWE applications, high hydroxide ion (OH−) conductivity (>100 mS cm−1) is essential for high current density operations. Conductivity depends on several factors such as ion exchange capacity (IEC), microphase-separation, structure, and substitution position of cationic head groups.8–15 High IEC (or high ion concentration) directly leads to high OH− conductivity; however, the IEC value should be optimized based on the polymer and cation structures to avoid excessive swelling and mechanical failure of AEMs in a hydrated state. We previously developed partially fluorinated poly(phenylene-co-fluorene)-based AEMs with high IEC (3.0 mequiv. g−1) that achieved high OH− conductivity (168.7 mS cm−1 at 80 °C) with reasonable swelling (37% at room temperature).16 Multi-cations and cross-linking have been used as molecular strategies for reducing swelling in AEMs with medium bulk but locally high IEC values.17–20 For example, Sung et al. developed a cross-linked poly(phenylene oxide) (PPO)-based AEM via multi-cation formation, which exhibited high OH− conductivity (131.96 mS cm−1 at 80 °C) and low dimensional swelling (18%–34% at 80 °C).20 Manipulation of microphase-separated morphology has also been effective in balancing the swelling and conductivity.21–24 He et al. reported PPO-based AEMs with well-defined phase separation using multifunctionalized side chains, achieving high OH− conductivity (69 mS cm−1 at room temperature) and dimensional stability (at 25 °C).21
Another important parameter is alkaline stability, which requires careful design of both cationic head groups and polymer backbones to enable long-term operation of AEMWEs without sacrificing other desirable properties.25–27 The alkaline stability of quaternary ammonium groups, the most common cationic head groups, was thoroughly investigated and found to be in the order of triaryl-1,3-dibutyl imidazoliums > N-spirocyclic piperidiniums > piperidiniums > trimethylalkylammoniums > C2-aryl benzimidazoliums > other imidazoliums.28,29 For the polymer backbones, heteroatom (particularly oxygen ether)-free polymers are preferred to avoid main chain scission caused by nucleophilic attack of OH−, which causes severe mechanical loss in the membrane and premature failure of AEMWEs.30,31 Xiao et al.32,33 reported poly(aryl piperidinium)s functionalized with stable piperidinium head groups, in which the oxygen-free polymer and the ring strain in the piperidinium groups resulted in high resistance to chemical degradation. The membrane maintained its initial conductivity after 2000 h in 1 M potassium hydroxide (KOH) at 100 °C and performed well in AEMWEs with a nickel iron oxide (NiFeO) anode (1.85 V at 1.0 A cm−2 with 0.1 M KOH at 80 °C), achieving improved durability (0.56 mV h−1 voltage increase during 165 h at 0.2 A cm−2). Liu et al.34 investigated two commercial AEMs in operando AEMWEs: Fumasep FAS-50 (polysulfone-functionalized with pendant quaternary ammonium groups) and Sustainion X37-50 (polystyrene-functionalized with methyimidazolium groups). Despite the lower alkaline stability of the methylimidazolium groups, Sustainion maintained 1 A cm−2 at 60 °C for 2000 h with a voltage increase of 5 μV h−1. In contrast, Fumasep suffered from a 200-μV h−1 voltage increase in 200 h, presumably owing to the instability of the polysulfone backbone.
Recently, we developed poly(arylfluorene piperidinium)-based terpolymers35 functionalized with alkaline-stable piperidinium groups linked at the 4-position with a short spacer. The AEM exhibited high alkaline stability (75% remaining conductivity after 1056 h in 8 M KOH at 80 °C), high AEMWE performance (1.64 V at 1.0 A cm−2 with 1.0 M KOH at 80 °C), and excellent durability (28 μV h−1 cell voltage increase for 1000 h at 1.0 A cm−2). Furthermore, superacid-promoted polycondensation produced poly(biphenyl alkylene)s36,37 functionalized with pentyltrimethylammonium groups with excellent alkaline stability (8% IEC loss in 1.0 M sodium hydroxide solution at 95 °C for 60 days), reasonable AEMWE performance (1.75 V at 1.0 A cm−2 with 1.0 M KOH at 60 °C), and excellent durability (6.5 μV h−1 voltage increase for 3500 h at 1.0 A cm−2). The results indicated that tuning both the polymer backbone and cationic head groups is essential for the high performance and long-term durability of AEMWEs.
More recently, we found that the hydrophobic component in the polymer backbone influenced the alkaline stability of the ammonium head groups.16 The fluorinated alkylene groups in poly(phenylene fluorene)-based AEMs were optimized to improve the alkaline stability (68% remaining conductivity; 75.8 mS cm−1 after 810 h in 8 M KOH at 80 °C). The membrane exhibited excellent performance and durability in AEMWEs with a nonprecious metal anode catalyst (1.72 V at 2.0 A cm−2 with a 1.1 μV h−1 voltage increase for 1000 h at 1.0 A cm−2).
The above results prompted us to investigate the effect of nonfluorinated hydrophobic arylene components (biphenylene, m- or p-terphenylenes, and fluorenylidene groups) for poly(arylene fluorene piperidinium)-based AEMs. The effects of hydrophobic arylene groups on membrane formability, as well as the chemical, electrochemical, and mechanical properties of the resulting AEMs, were thoroughly investigated. The optimized AEM with the best-balanced properties was evaluated in an AEMWE cell using a NiFeO anode catalyst.
2. Results and discussion
2.1. Synthesis of monomer and copolymers
To prepare polymers tethered with a piperidinium-based cation at the 4-position, tert-butyl-4-(iodomethyl)piperidine-1-carboxylate (IMP) was prepared according to the literature (Scheme S1†).38 The chemical structure of IMP was confirmed using proton (1H) and carbon-13 (13C) nuclear magnetic resonance (NMR) spectra, which agreed well with the previous report (Fig. S1†).38 Di-tert-butyl-4,4′-((9H-fluorene-9,9-diyl)bis(methylene))bis(piperidine-1-carboxylate) (DTPF) was prepared via a nucleophilic substitution reaction of IMP with fluorene in the presence of aqueous KOH and TBAB as the phase transfer catalyst (Scheme S2†) in reasonable yield (82%). The tert-butyloxycarbonyl (Boc) protecting groups were eliminated via acid-catalyzed hydrolysis in a 50% (% v/v) trifluoroacetic acid (TFA)/dichloromethane (CH2Cl2) mixture to obtain the target 4,4′-((9H-fluorene-9,9-diyl)bis(methylene))bis(piperidin-1-ium)trifluoroacetate (DPF) in high yield (98%). The chemical structure of the DPF monomer was confirmed using 1H, 13C, and fluorine-19 (19F) NMR spectra, in which all peaks were well assigned (Fig. S2†). The resulting DPF monomer was subjected to superacid-promoted Friedel–Crafts polyhydroxyalkylation using various aromatic comonomers, including biphenyl (BPh), p-terphenyl (TPh), m-terphenyl (mTPh), and 9,9-dimethylfluorene (MFl) (Scheme 1). | ||
| Scheme 1 Synthesis of fluorene-based quaternized copolymers from various aromatic comonomers. | ||
A slight excess of ketone (trifluoroacetone, 1.2 equivalent) was required to accelerate the polymerization reaction and increase the molecular weight; however, larger amounts of the ketone should be avoided to prevent unfavorable cross-linking reactions that result in insoluble polymers.39 In addition, the polymerization temperature should be maintained at −5.0 °C to obtain copolymers with solvent solubility. The polymerization of TPh and MFl with DPF monomer produced the TPh-Pip and MFl-Pip copolymers in reasonably high yields (87% and 91%, respectively); however, these copolymers were insoluble in all solvents, even after a short reaction time and at a controlled temperature, likely because of a minor cross-linking reaction. The polymerization of BPh and mTPh with the DPF monomer produced copolymers with good solubility (denoted BPh-Pip and mTPh-Pip) in high yields of 84% and 79%, respectively. The BPh-Pip and mTPh-Pip copolymers were soluble in organic solvents such as tetrahydrofuran (THF), N,N-dimethylformamide (DMF), and dimethylsulfoxide (DMSO) but insoluble in water and lower alcohols such as methanol and ethanol. The chemical structure of the BPh-Pip and mTPh-Pip copolymers was confirmed using 1H NMR spectra (Fig. S3 and 4†), in which the aliphatic protons adjacent to the piperidinium nitrogen atoms were detected at 2.9 and 2.4 ppm, whereas the other protons on the piperidinium groups were detected at 1.0 and 0.6 ppm. The dual methylene protons tethered at the 9-position of the fluorene groups were detected at 2.0 ppm, the protonated amines (+NH2) were detected as a broad peak at 8.2 ppm, and the aromatic protons were detected at 7.1–7.9 ppm. In the 19F NMR spectra, the fluorine peaks related to the –CF3 and CF3–SO3− groups were detected at −69 ppm and −80 ppm, respectively.
The molecular weights of BPh-Pip and mTPh-Pip were estimated using gel permeation chromatography analysis with CHCl3 and 3% trimethylamine (TMA) as the eluent. BPh-Pip exhibited higher molecular weights (weight average molecular weight (Mw) = 97.0–141.1 kDa, number average molecular weight (Mn) = 4.3–28.2 kDa) than mTPh-Pip (Mw = 25.0–79.0 kDa, Mn = 6.5–19.2 kDa) (Table 1). The molecular weight of mTPh-Pip increased as the DPF composition increased (or increasing IEC). The apparent high polydispersity of Bph-Pip is the result of tailing of the GPC curves due presumably to possible interactions with the GPC column (Fig. S5†).
| Copolymer | IECfeed (mequiv. g−1) | IECNMR (mequiv. g−1) | IECtitb (mequiv. g−1) |
M
wa (kDa) |
M
na (kDa) |
PDI | WUb (%) | σ (mS cm−1) |
|---|---|---|---|---|---|---|---|---|
| a Measured in deprotonated form (NH form). b Measured at 30 °C. c Not available. | ||||||||
| QBPh-Pip | 0.85 | 0.90 | 0.80 | 123.54 | 4.38 | 28.18 | 7.00 | 10.50 |
| 1.62 | 1.65 | 1.60 | 118.40 | 28.20 | 4.19 | 25.00 | 62.00 | |
| 2.00 | 2.05 | 1.90 | 141.14 | 18.31 | 7.70 | 45.00 | 97.50 | |
| 2.50 | 2.40 | 2.43 | 97.02 | 7.01 | 13.84 | 150 | NAc | |
| QmTPh-Pip | 0.85 | 0.90 | NAc | 25.00 | 6.70 | 3.70 | NAc | NAc |
| 1.62 | 1.65 | NAc | 41.68 | 6.56 | 6.34 | NAc | NAc | |
| 2.00 | 2.05 | NAc | 79.02 | 19.22 | 4.10 | NAc | NAc | |
2.2. Quaternization and membrane formation
The quaternization reaction was performed for the TPh-Pip and MFl-Pip copolymers in a heterogeneous mixture but was unsuccessful, generating only gel-like insoluble products. The quaternization reaction of BPh-Pip and mTPh-Pip was successful using dimethyl sulfate as the quaternization reagent, N-methyl-2-pyrrolidone as the solvent, and potassium carbonate as the promoter. The quaternized QBPh-Pip and QmTPh-Pip copolymers were obtained in reasonable yields of 98% and 96%, respectively. Notably, after quaternization, QBPh-Pip was soluble in chloroform (CHCl3), whereas QmTPh-Pip was only partially soluble. The chemical structures of the quaternized QBPh-Pip and QmTPh-Pip copolymers were confirmed using 1H and 19F NMR spectra. Protonated amine peaks at 8.2 ppm were absent, suggesting a complete quaternization reaction (Fig. 1 and S6†). In addition, new peaks appeared at 2.9 and 3.3 ppm, which were assigned to methyl (N+–CH3) and methylene (N+–CH2) protons adjacent to quaternary ammonium groups respectively, whereas the counterions (CH3OSO3−) were detected at 3.1 ppm. QBPh-Pip and QmTPh-Pip were synthesized with different compositions to obtain target IECs of 0.85, 1.62, and 2.00 mequiv. g−1. The IECs calculated from the NMR integral ratios agreed well with the target IECs (Table 1).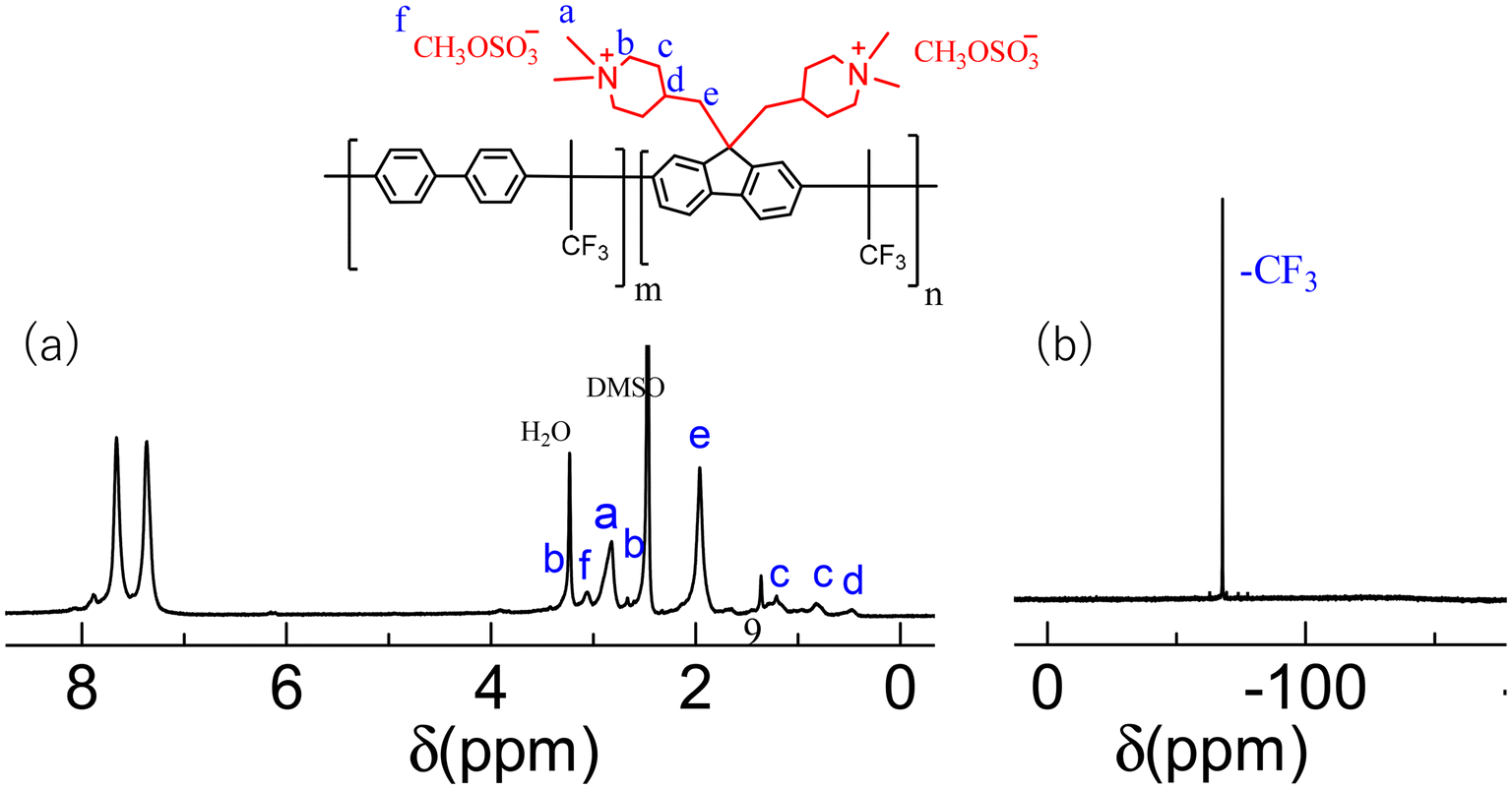 | ||
| Fig. 1 (a) 1H and (b) 19F NMR spectra of quaternized QBPh-Pip copolymer. | ||
QBPh-Pip was cast from DMSO solution to form transparent and flexible QBPh-Pip membranes with 35-μm thicknesses (Fig. S7a–c†). The QBPh-Pip membrane with a higher IEC (∼2.43 mequiv. g−1) was unsuitable for property measurements because of excessive swelling and a gel-like formation when contacted with liquid water, even at room temperature (Fig. S7d†). The titrated IECs of QBPh-Pip agreed well with those determined using NMR integrals (Table 1). For QmTPh-Pip, casting from a DMSO solution resulted in cracked and inflexible membranes for all copolymer compositions (Fig. S8†). The low molecular weight of mTPh-Pip and tortuosity of mTPh appeared to have hindered the membrane formability of QmTPh-Pip.
2.3. Morphology
Well-developed and ordered morphologies are important characteristics of AEMs in terms of properties such as water absorbability, ion conductivity, and mechanical strength. However, several factors influence the AEM morphology, including polymer and ion group structures, as well as ion concentration (i.e., IEC).40 Thus, the morphologies of the QBPh-Pip membranes with different IECs (0.8, 1.6, and 1.9 mequiv. g−1) were investigated using transmission electron microscopy (TEM) images of the dry membranes stained with tetrachloroplatinate ions (Fig. 2). All selected QBPh-Pip membranes possessed an ordered phase-separated morphology, with dark spots and bright spots representing the hydrophilic and hydrophobic domains, respectively. The domain sizes were fitted with histograms, in which the hydrophilic domain size increased slightly with increasing IEC (2.0 ± 1.14 nm for IEC = 0.8 mequiv. g−1, 2.5 ± 0.86 nm for 1.6 mequiv. g−1, and 2.5 ± 0.79 nm for 1.9 mequiv. g−1). The hydrophobic domain size was independent of the ionic content (4.5 ± 2.90 nm for IEC = 0.8 mequiv. g−1, 5.0 ± 2.23 nm for IEC = 1.6 mequiv. g−1, and 4.0 ± 2.37 nm for IEC = 1.9 mequiv. g−1). Compared to our previous poly(fluoroalkyl biphenyl fluorene) membrane functionalized with hexyltrimethylammonium groups (3.0 nm for the hydrophilic domain and 7.0 nm for the hydrophobic domain),41 QBPh-Pip exhibited smaller domain sizes, presumably because of its smaller molecular volume. To support this idea, the molecular volumes of the hydrophilic and hydrophobic components of QBPh-Pip were calculated from the occupied volume using Connolly surface calculations (Fig. S9†). As expected, the results revealed 666.4 Å3 for the hydrophilic component and 268.2 Å3 for the hydrophobic component, which were smaller than our previous 4-QPPAF-TMA41 (806.1 Å3 and 313.2 Å3 for the hydrophilic and hydrophobic components, respectively). | ||
| Fig. 2 TEM images of QBPh-Pip membranes fitted with histograms of hydrophilic and hydrophobic domain sizes. | ||
2.4. Water uptake
The water absorbability of QBPh-Pip with different IECs was measured in liquid water and plotted as a function of temperature (Fig. 3a). The water uptake increased as IEC and temperature increased. Accordingly, the swelling ratio increased in the same manner (Fig. 3b). Compared to our previous 4-QPPAF-TMA membranes41 (water uptake at 80 °C was 200% for IEC = 2.0 mequiv. g−1 and 95% for IEC = 1.7 mequiv. g−1), the QBPh-Pip membranes absorbed less water, possibly because of the rigidity and bulkiness of heterocyclic piperidinium groups compared to trimethylammonium groups. | ||
| Fig. 3 (a) Water uptake and (b) swelling ratio of QBPh-Pip membranes in liquid water as a function of temperature. | ||
To confirm our hypothesis, the number of water molecules per ammonium group (λ) was plotted as a function of IEC (Fig. S10†). The piperidinium groups in QBPh-Pip absorbed less water for hydration than trimethylammonium in 4-QPPAF-TMA, and the effect was more pronounced in membranes with higher IEC (λ = 13.6 for QBPh-Pip with IEC = 1.9 mequiv. g−1 and λ = 26.4 for 4-QPPAF-TMA with IEC = 2.0 mequiv. g−1). The water uptake of the QBPh-Pip membrane (in the CH3OSO3− form) was further investigated under humidified conditions at 60 °C (Fig. 4). The water vapor uptake under humidified conditions was much lower than the water uptake in liquid water, although the trend was similar.
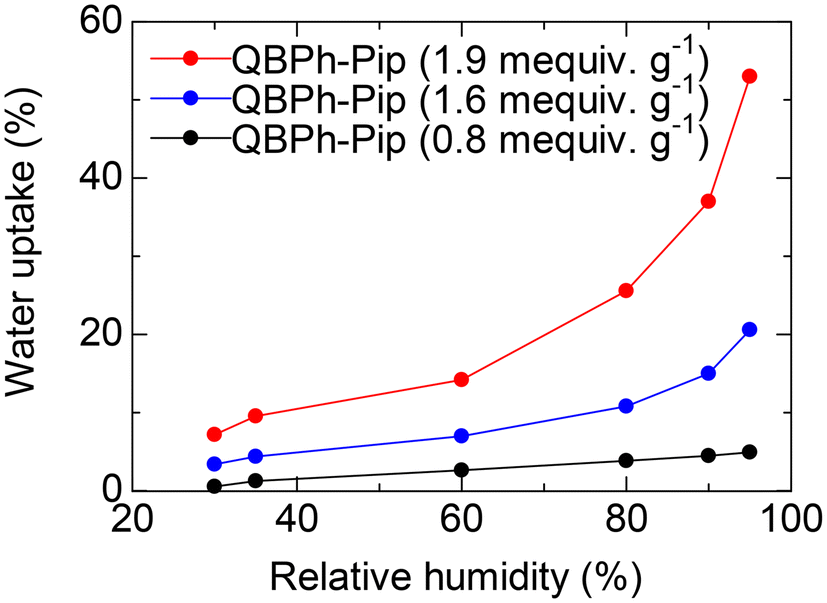 | ||
| Fig. 4 Water vapor sorption at 80 °C as a function of the humidity. | ||
2.5. OH− conductivity
The OH− conductivity of the QBPh-Pip membranes was measured in water at 30 °C and plotted as a function of λ (Fig. 5). The conductivity gradually increased as λ increased owing to the higher IEC. The highest conductivity (97.6 mS cm−1 at λ = 13.6 for IEC = 1.9 mequiv. g−1) was higher than that of 4-QPPAF-TMA41 (80.6 mS cm−1 at λ = 26.4 for IEC = 2.0 mequiv. g−1), suggesting that the compact piperidinium groups utilized water molecules more efficiently for ionic conduction than the trimethylammonium groups. Compared to other reported piperidinium-based AEMs (Fig. S11†), the QBPh-Pip conductivity showed a stronger dependence on λ values, further supporting efficient water utilization for ionic conduction.42–52 | ||
| Fig. 5 OH− conductivity of QBPh-Pip membranes at 30 °C as a function of several water molecules per ammonium group (λ). | ||
The normalized ion diffusion coefficient (D/D0, where D and D0 represent the ion diffusion coefficients of the membranes in the OH− form and dilute hydroxide solution, respectively) of the QBPh-Pip membranes at 30 °C was calculated using the Nernst–Einstein equation. The D/D0 values were 0.08, 0.22, and 0.30 for the membranes with IECs of 0.8, 1.6, and 1.9 mequiv. g−1, respectively (Fig. S12†), which were comparable to our previous AEMs.14,41,53 Compared to the 4-QPPAF-TMA membranes with similar IEC (ca. 2.0 mequiv. g−1), the QBPh-Pip (1.9 mequiv. g−1) membrane was more conductive, because of the higher diffusion coefficient. The results suggest that the ionic species in QBPh-Pip participated more in the migration of hydrated OH−s in the bulk membranes.
The temperature dependence of the OH− conductivity of QBPh-Pip with different IECs was measured (Fig. 6). The conductivity increased as the temperature increased. The QBPh-Pip with the highest IEC (1.9 mequiv. g−1) achieved the highest conductivity of 160 mS cm−1 at 80 °C, followed by QBPh-Pip (1.6 mequiv. g−1, 110 mS cm−1) and QBPh-Pip (0.8 mequiv. g−1, 16.7 mS cm−1). Compared with our previous 4-QPPAF-TMA41 with similar IEC (2.0 mequiv. g−1, 115 mS cm−1 at 80 °C), QBPh-Pip (1.9 mequiv. g−1) was much more conductive. The apparent activation energies (Ea) for the ion conduction of QBPh-Pip were estimated from the Arrhenius plot slopes to be 8.3–10.1 kJ mol−1, which were comparable to 4-QPPAF-TMA41 (8.4–9.0 kJ mol−1). The results indicate that the short-chain piperidinium promoted the migration of OH−s more than the hexyl trimethylammonium groups without affecting the ion conduction mechanism.
 | ||
| Fig. 6 Temperature dependence of OH− conductivity of the different QBPh-Pip membranes. | ||
2.6. Alkaline stability
The alkaline stability of the QBPh-Pip membranes was tested in 8 M KOH at 80 °C for 1000 h. The post-test QBPh-Pip membranes maintained their transparency and bendability, indicating no or only minor degradation in the main polymer chains (Fig. S13†). The OH− conductivity was plotted as a function of testing time (Fig. 7). The membranes with higher IECs (IEC = 0.8, 1.6, and 1.9 mequiv. g−1) showed higher remaining conductivity (10.5 (57.6%), 120 (85.7%), and 141 mS cm−1 (91.5%), respectively). The aged QBPh-Pip membranes were recovered and converted to the chloride ion form for Mohr titration. Compared to the pristine IECs (0.8, 1.6, and 1.9 mequiv. g−1), the post-test titrated IECs were 0.54 (67.5%), 1.43 (89.4%), and 1.78 mequiv. g−1 (93.7%) respectively (Table S1†), which were consistent with the conductivity remaining. | ||
| Fig. 7 Alkaline stability of the different QBPh-Pip membranes in 8 M KOH at 80 °C. | ||
The post-test membranes were further investigated using NMR analyses (Fig. 8). Overall, the three QBPh-Pip membranes exhibited excellent chemical stability, as indicated by minor changes in peaks a and b assigned to the N+–CH3 and N+–CH2 groups of the piperidinium rings, respectively (Fig. 8a). However, in the magnified NMR spectra (Fig. 8b), very minor peaks (g and h) at δ = 3.94 and 6.2 ppm, respectively, were observed in the membrane with the lowest IEC (0.8 mequiv. g−1), indicative of Hofmann β-elimination (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH). The piperidinium cations were susceptible to both nucleophilic substitution and Hofmann elimination,54 and the contribution of the two degradation mechanisms varied with the IEC of the QBPh-Pip membranes. Although the peaks derived from Hofmann elimination were well assigned, the peaks assignable to tertiary amines (N–CH3 or N–CH2) derived from nucleophilic degradation overlapped with the pristine and solvent peaks. The ionic loss of the QBPh-Pip membranes was quantified from the NMR integral ratios and plotted as a function of IEC (Fig. 8c). The ionic loss decreased as the IEC increased, likely because of the steric effect of aggregated piperidinium cations at higher IECs, as well as the increased water absorbability of higher IEC membranes, which is anticipated to solvate OH−s,55 hindering its nucleophilicity and basicity. Specifically, the membrane with IEC = 0.8 mequiv. g−1 underwent an ionic loss of 15%, which was caused by Hofmann elimination. For the higher IEC membranes (1.6 and 1.9 mequiv. g−1), the ionic losses of 8% and 5%, respectively, were caused by nucleophilic substitution. The post-test IECNMR estimated from the NMR integral ratios agreed with those obtained by titration (Table S1†). In the 19F NMR spectra (Fig. 8d), the absence of new peaks suggested the stability of the –CF3 groups. Furthermore, the post-test morphology of the QBPh-Pip membrane with IEC = 1.9 mequiv. g−1 was elucidated using TEM images (Fig. S14†). Compared to the pristine membrane, the hydrophilic domain size slightly decreased from 2.5 to 2.0 nm and the hydrophobic domain size slightly decreased from 4.0 to 3.0 nm. The morphological stability was consistent with the high remaining OH− conductivity and structural stability demonstrated in the NMR analyses discussed above. Compared to the other reported piperidinium-based AEMs prepared via superacid polymerization (Fig. S15†),42–52 QBPh-Pip was among the most stable AEMs in terms of the remaining IEC (and conductivity).
CH). The piperidinium cations were susceptible to both nucleophilic substitution and Hofmann elimination,54 and the contribution of the two degradation mechanisms varied with the IEC of the QBPh-Pip membranes. Although the peaks derived from Hofmann elimination were well assigned, the peaks assignable to tertiary amines (N–CH3 or N–CH2) derived from nucleophilic degradation overlapped with the pristine and solvent peaks. The ionic loss of the QBPh-Pip membranes was quantified from the NMR integral ratios and plotted as a function of IEC (Fig. 8c). The ionic loss decreased as the IEC increased, likely because of the steric effect of aggregated piperidinium cations at higher IECs, as well as the increased water absorbability of higher IEC membranes, which is anticipated to solvate OH−s,55 hindering its nucleophilicity and basicity. Specifically, the membrane with IEC = 0.8 mequiv. g−1 underwent an ionic loss of 15%, which was caused by Hofmann elimination. For the higher IEC membranes (1.6 and 1.9 mequiv. g−1), the ionic losses of 8% and 5%, respectively, were caused by nucleophilic substitution. The post-test IECNMR estimated from the NMR integral ratios agreed with those obtained by titration (Table S1†). In the 19F NMR spectra (Fig. 8d), the absence of new peaks suggested the stability of the –CF3 groups. Furthermore, the post-test morphology of the QBPh-Pip membrane with IEC = 1.9 mequiv. g−1 was elucidated using TEM images (Fig. S14†). Compared to the pristine membrane, the hydrophilic domain size slightly decreased from 2.5 to 2.0 nm and the hydrophobic domain size slightly decreased from 4.0 to 3.0 nm. The morphological stability was consistent with the high remaining OH− conductivity and structural stability demonstrated in the NMR analyses discussed above. Compared to the other reported piperidinium-based AEMs prepared via superacid polymerization (Fig. S15†),42–52 QBPh-Pip was among the most stable AEMs in terms of the remaining IEC (and conductivity).
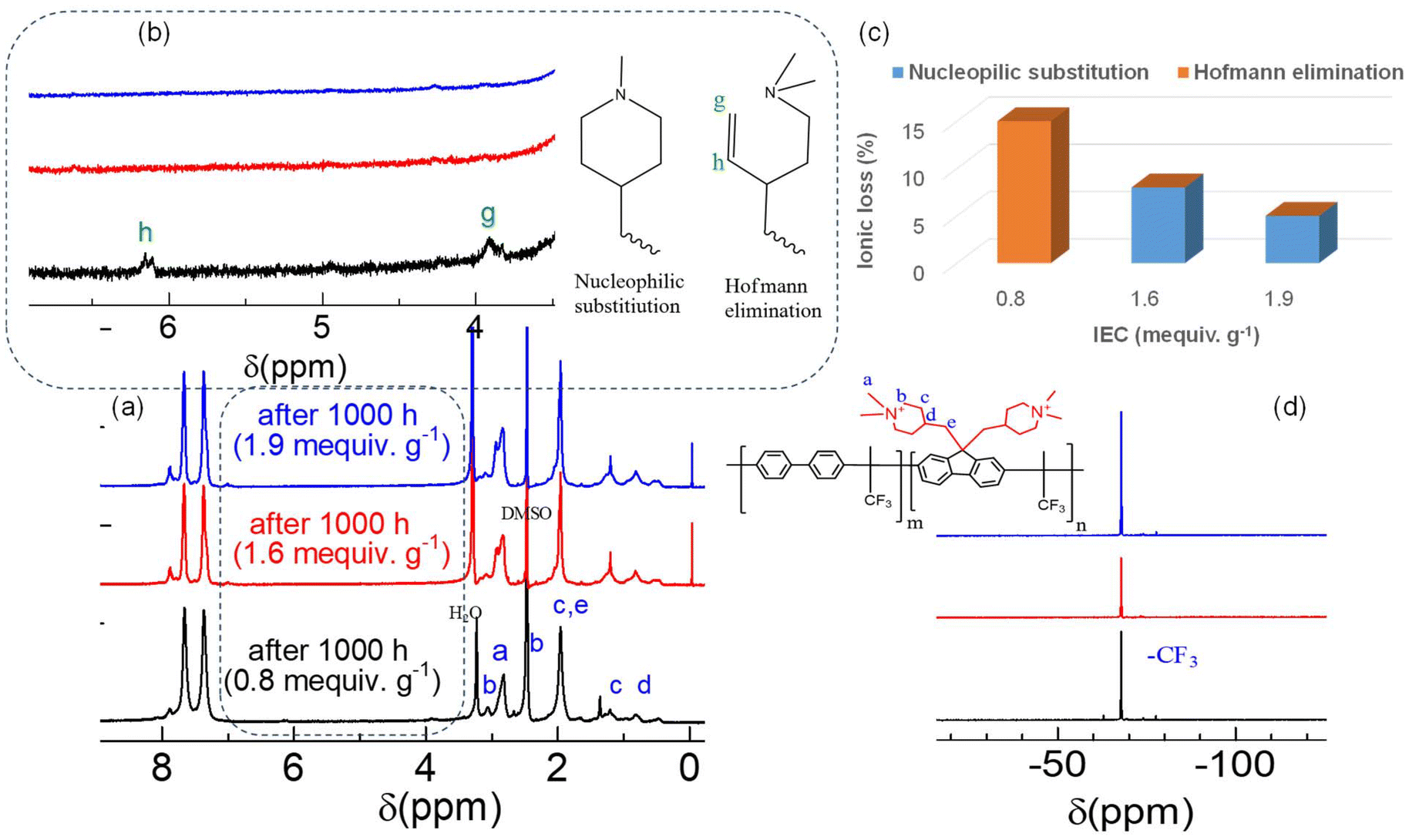 | ||
| Fig. 8 (a) Post-test 1H NMR spectra of QBPh-Pip membranes after 1000 h in 8 M KOH at 80 °C, (b) magnified 1H NMR spectra, (c) IEC loss (%) estimated from the NMR integrals as a function of IEC, and (d) 19F NMR spectra of the post-test QBPh-Pip. | ||
2.7. Mechanical properties
To elucidate the viscoelasticity of the QBPh-Pip membranes, dynamic mechanical analysis (DMA) was performed as a function of temperature at 60% relative humidity (RH) (Fig. 9a). All three QBPh-Pip membranes exhibited nearly constant storage modulus (E′), loss modulus (E′′), and tanδ without glass transition peaks up to 100 °C. The E′ and E′′ moduli were independent of the membrane IEC; however, they showed proportional dependence of Mn (Fig. S16†). In the humidity dependence at 80 °C (Fig. 9b), E′ and E′′ decreased with increasing humidity for the higher IEC membranes, presumably because of the softening effect caused by increased water absorbance, specifically at >80% RH (Fig. 4). For the three membranes, there was no clear evidence for glass transition up to 95% RH.
 | ||
| Fig. 9 DMA of QBPh-Pip membranes as a function of (a) temperature and (b) humidity. | ||
The elongation properties of the QBPh-Pip membranes were evaluated using stress–strain curves (Fig. 10). QBPh-Pip (IEC = 0.8 mequiv. g−1) exhibited the highest maximum stress of 33.0 MPa followed by IEC = 1.6 mequiv. g−1 (29.7 MPa) and IEC = 1.9 mequiv. g−1 (29.0 MPa). QBPh-Pip (IEC = 1.9 mequiv. g−1) exhibited a much higher elongation at break of 134% than the membranes with IECs of 1.6 mequiv. g−1 (22%) and 0.8 mequiv. g−1 (7%). Fig. S17† shows that the elongation properties were related to the copolymer composition, with the elongation at break increasing as the DPF to biphenyl (n/m) ratio increased, whereas the maximum stress slightly decreased as the n/m ratio increased.
 | ||
| Fig. 10 Stress–strain curves of the QBPh-Pip membranes in the CH3OSO3− form at 60% RH and 80 °C. | ||
The elongation properties were further tested for the postalkaline stability test (Fig. S18†). The QBPh-Pip (IEC = 1.9 mequiv. g−1) membrane exhibited 25.1 MPa of the maximum stress and 159% of the elongation at break. The rather minor changes in the elongation properties were consistent with the aforementioned post-test conductivity and NMR analyses.
2.8. Alkaline water electrolysis cell performance
A water electrolysis cell was fabricated using the QBPh-Pip membrane with the most balanced properties (IEC = 1.9 mequiv. g−1, 45 μm thick). The anode catalyst layer comprised a homemade anion conductive ionomer QPAF-4 (IEC = 1.5 mequiv. g−1) as the binder and Ni0.8Fe0.2O as the catalyst. The cathode catalyst comprised QPAF-4 (IEC = 2.0 mequiv. g−1) as the binder and platinum on carbon (Pt/C) as the catalyst. Fig. 11a illustrates the initial current–voltage (I–V) curve at 80 °C fed with 1 M KOH. The onset voltage was 1.44 V, which was comparable to that of the previously reported NiFeO catalyst in aqueous KOH (∼1.45 V measured using the rotating desk electrode method).56 The ohmic resistance of the QBPh-Pip (1.9 mequiv. g−1) cell was 0.103 Ω cm2 at 1.0 A cm−2, which was somewhat higher than that of our previous AEMs (0.055–0.90 Ω cm2 at 1.0 A cm−2) using nickel cobalt oxide (NiCoO) as the anode catalyst.18,38,57 Considering the higher OH− conductivity of QBPh-Pip (1.9 mequiv. g−1) membrane than those AEMs, we speculated that the higher ohmic resistance was caused by the interfacial resistance with the catalyst layer containing NiFeO (Fig. S19a†). The rigidity of QBPh-Pip structure is presumably responsible for the interfacial resistance with the catalyst layer. The cell voltages at current densities of 1.0 and 2.0 A cm−2 were 1.70 and 1.85 V, respectively, which corresponded to 72.4% and 66.5% efficiencies. Fig. S19b† shows a Tafel plot of the internal resistance (IR)-free cell voltage. The Tafel slope of the QBPh-Pip cell was 106.6 mV dec−1 for the linear line at <1.0 A cm−1, which was comparable to that of our previous QPAF-4-based AEMWE cell57 using a NiCoO anode catalyst and the same ionomer binder (99.1 mV dec−1 at current density < 1.0 A cm−2). Because our current and previous AEMWE cells used the same Pt/C and ionomer binder, the comparable Tafel slopes indicated that NiFeO functioned similarly to NiCoO as the anode catalyst. Table S2† summarizes the performance of the reported AEMWEs with the cell voltage at 1.0 A cm−2. The QBPh-Pip (1.9 mequiv. g−1) cell exhibited comparable or higher performance than the previously reported AEMWEs using a NiFeO-based or IrO2 anode catalyst.58–64 | ||
| Fig. 11 (a) IR-included I–V curve of the QBPh-Pip (IEC = 1.9 mequiv. g−1) cell at 80 °C with a 1 M KOH aqueous solution. (b) The durability of the QBPh-Pip (IEC = 1.9 mequiv. g−1) cell at 80 °C with a 1 M KOH aqueous solution. | ||
The cell durability was tested at a constant current density of 1.0 A cm−2 at 80 °C for 1000 h (Fig. 11b). Within the initial 100 h, the cell voltage increased from 1.72 to 1.78 V because of the interfacial structural changes of the ionomers/catalysts under the chronoamperometric conditions. Similar behavior was observed in our previous studies.18,38,57 The cell voltage was much more stable at 1.78 V after 100 h, with an increased rate of 5.0 μV h−1. The ohmic resistance was also stable at ∼0.1 Ω cm2, demonstrating the stability of the QBPh-Pip membrane. The cell experienced forced shutdown/restart because of machine troubles at 624 h, which caused an increase in the cell voltage (to 1.82 V) and ohmic resistance (0.14 Ω cm2). The cell exhibited a faster voltage increase from 624 to 1000 h, reaching 1.82 V at a higher rate of 70 μV h−1 while the ohmic resistance remained constant at 0.14 Ω cm2. The IR-included I–V curves with ohmic resistance and IR-free I–V curves were compared at 0, 624, and 1000 h during the durability test (Fig. S20a and b†). The performance loss was mostly caused by increased ohmic resistance, suggesting some degradation in the membranes and/or its interface with the catalyst layers. The hydrogen (H2) and oxygen (O2) evolution rates were equivalent to the current density and nearly constant for 1000 h (Fig. S20c†).
For the pristine and the post-test QBPh-Pip membranes, cross-sectional SEM images and elemental mapping using energy-dispersive X-ray spectroscopy (EDS) were measured (Fig. 12a and b). The membrane remained free of cracks and pores even after 1000 h of durability testing. The post-test membrane thickness remained comparable to the initial membrane (47 μm) despite compression in the device due to plausible membrane swelling with circulated KOH electrolyte. In addition, the EDS results showed homogeneous distributions of C, N, and F atoms throughout for both pristine and post-test membranes. However, the elemental ratio of N/C decreased from 5.6% to 3.5% (Fig. 12c), indicating a ∼40% loss of the quaternary ammonium groups, which was accountable for the increased ohmic resistance and performance loss. Fig. S21a and b† shows the N atom (Kα) intensity on the EDS line analysis, as quantified by backscattered electrons. The post-test QBPh-Pip membrane attached to the anode side showed a decrease from ∼15% to 3%, with the loss observed up to ∼15 μm from the anode side, suggesting that the membrane degradation was more pronounced at the interface with the NiFeO catalyst (Fig. S20c†). The 1H NMR spectrum was also measured (Fig. 12d) for the recovered QBPh-Pip membrane, which remained soluble in DMSO-d6. The main peak (a) at 2.9 ppm, which corresponded to N+–CH3 protons, remained unchanged; however, the intensity of the peak (c) at 1.3 ppm increased because of product degradation via the SN2 mechanism. The magnified spectra at δ = 4.0–7.0 ppm showed a minor peak at 5.2 ppm caused by the E1 degradation mechanism. The post-test 19F NMR spectrum remained unchanged, revealing the excellent stability of the perfluorinated hydrophobic groups (Fig. 12e). The postdurability IEC was calculated from the NMR integral ratios to be 1.43 mequiv. g−1 (70% of the pristine membrane), which was consistent with the SEM–EDS results.
 | ||
| Fig. 12 SEM–EDS images of (a) initial and (b) postdurability QBPh-Pip (IEC = 1.9 mequiv. g−1) membranes. (c) Elemental analysis (EDS). (d) 1H and (e) 19F NMR spectra of QBPh-Pip (IEC = 1.9 mequiv. g−1) before and after 1000 h of durability testing. | ||
The QBPh-Pip cell was more durable than the reported AEMs (Table S2†), with the cell voltage increase rate exceeding 180 μVh−1, even at lower current density and temperature.59–64 The post-test cell voltage was 1.80 V at 1.0 A cm−2, which was comparable62 or superior61,64 to some of the reported unaged AEMs (Table S2†). Overall, QBPh-Pip is a promising candidate for practical AEMWE applications because of its strong compatibility with nonplatinum group–metal (non-PGM) anode catalysts and ability to achieve reasonable performance and durability.
3. Conclusion
The effect of the hydrophobic components on the properties of quaternized poly(arylene fluorenylidene piperidinium)-based copolymers was investigated. The terphenyl and 9,9-dimethylfluorene-hydrophobic groups were unsuitable for providing high molecular weight and soluble copolymers. In contrast, biphenyl groups provided copolymers with higher molecular weight, better solubility, and membrane formability. The poly(biphenylene fluorenylidene piperidinium) membranes exhibited well-developed phase-separated morphology, as observed in the TEM images, with the hydrophilic domain sizes dependent on the IEC values, whereas the hydrophobic domain sizes were less dependent on the IEC values. The OH− conductivity of the poly(biphenylene fluorenylidene piperidinium) membranes increased as the IEC values increased, achieving the maximum conductivity at IEC = 1.9 mequiv. g−1 (160 mS cm−1 at 80 °C) with balanced water uptake and dimensional swelling. Membranes with an IEC > 1.9 mequiv. g−1 suffered from excessive water absorption and mechanical failure. Similarly, the membranes showed alkaline stability (in 8 M KOH at 80 °C) dependent on the IEC values. In the alkaline stability (in 8 M KOH at 80 °C), membranes with an IEC < 1.0 mequiv. g−1 exhibited faster degradation (32.5% cation loss), which was dominated by the Hofmann elimination mechanism. Membranes with an IEC > 1.5 mequiv. g−1 were less degradable (6.3%–10.6% cation loss), with nucleophilic substitution mechanisms dominating the degradation process. The elongation properties of the poly(biphenylene fluorenylidene piperidinium) membranes were dependent on the copolymer composition rather than the IEC values, whereas the viscoelasticity was related to the Mn of the copolymers and humidity conditions. The poly(biphenylene fluorenylidene piperidinium) membrane with IEC = 1.9 mequiv. g−1 exhibited excellent performance (72.4% efficiency) and reasonable durability for 1000 h in an alkaline water electrolysis cell. The postdurability analysis revealed some degradation in the membrane, specifically at the NiFeO catalyst interface. Overall, poly(biphenylene fluorenylidene piperidinium) is a promising candidate for solid electrolytes used in alkaline water electrolysis cells.4. Experimental section
4.1. Materials
Biphenyl, m-terphenyl, p-terphenyl, 9,9-dimethylfluorene, 1-(tert-butoxycarbonyl)-4-piperidinemethanol, fluorene, dimethyl sulfate, triphenylphosphine, trifluoromethanesulfonic acid (TFSA), and trifluoroacetic acid (TFA) were purchased from TCI Inc. and used as received. Chloroform-d (CDCl3) with 0.03% tetramethylsilane (TMS) and dimethylsulfoxide-d6 (DMSO-d6) with 0.03% TMS were purchased from Acros Organics and used as received. N,N-Dimethylacetamide (DMAc), dimethyl sulfoxide (DMSO), hydrochloric acid, potassium hydroxide, N,N-dimethylformamide (DMF), dichloromethane, and tetrahydrofuran (THF) were purchased from Kanto Chemical Co. and used as received.4.2. Synthesis of tert-butyl-4-(iodomethyl)piperidine-1-carboxylate (Scheme S1†)
A 100 mL round -bottomed flask was charged with tert-butyl 4-(hydroxymethyl)piperidine-1-carboxylate (5.3 g, 25 mmol), triphenylphosphine (15.70 g, 60 mmol) and imidazole (4.08 g, 60 mmol), 60 mL of anhydrous THF under nitrogen atmosphere. The mixture was cooled in ice bath to 0 °C. To the mixture, iodine (7.6 g, 60 mmol) dissolved in 30 mL of THF was added slowly. The mixture was stirred at room temperature overnight. The reaction was quenched by adding excess saturated sodium hydrogen sulfite and sodium chloride aqueous solution. The product was extracted with dichloromethane. The solvent was removed under vacuum and the residue was purified by silica gel column chromatography using dichloromethane and hexane to obtain tert-butyl-4-(iodomethyl)piperidine-1-carboxylate as colorless liquid (8.5 g, 75% yield).4.3. Synthesis of di-tert-butyl-4,4′-((9H-fluorene-9,9-diyl)bis(methylene))bis(piperidine-1-carboxylate) (DTPF) (Scheme S2†)
A 100 mL round-bottomed flask was purged with N2 and charged with tert-butyl-4-(iodomethyl)piperidine-1-carboxylate (4.3 g, 15.5 mmol), fluorene (0.856 g, 5.15 mmol) and TBAB (0.5 g) and 15 mL of DMSO. To the mixture, 4 mL of 50% NaOH aqueous solution was added. The mixture was heated at 70 °C overnight. The reaction was quenched by adding excess water. The product was extracted with ethyl acetate, washed with water and brine. The solvent was removed under vacuum and the residue was purified by silica gel column chromatography using hexane:ethyl acetate (9:1) then (8:1) to obtain di-tert-butyl-4,4′-((9H-fluorene-9,9-diyl)bis(methylene))bis(piperidine-1-carboxylate) (DTPF) as white solid (2.3 g, 82% yield).
4.4. Synthesis of 4,4′-((9H-fluorene-9,9-diyl)bis(methylene))bis(piperidin-1-ium)trifluoroacetrate (DPF) (Scheme S2†)
A 100 mL round-bottomed flask was charged with 2.3 g of DTPF and mixture of TFA/CH2Cl2 (1:1) (20 mL). The mixture was stirred at room temperature for 4 h. The solvent was removed under vacuum and the residue was washed with diethyl ether to obtain 4,4′-((9H-fluorene-9,9-diyl)bis(methylene))bis(piperidin-1-ium)trifluoroacetrate (DPF) as pale green crystal (2.4 g, 98% yield).
4.5. Synthesis of BPh-Pip copolymer (general procedure)
A general procedure is as follows. In a sealed flask, DPF (0.5 g, 0.89 mmol), biphenyl (0.22 g, 1.45 mmol), trifluoroacetone (0.25 mL, 2.81 mmol), and anhydrous dichloromethane (2 mL) were charged. The mixture was cooled at 0 °C under nitrogen. Trifluoromethanesulfonic acid (2.5 mL, 28.66 mmol) and trifluoroacetic acid (0.86 mL, 11.27 mmol) were added and the polymerization reaction continued for 6 h. The mixture was poured into excess pure water. The precipitate was washed with methanol to obtain 0.8 g, 84% yield of BPh-Pip copolymer as white solid.4.6. Synthesis of mTPh-Pip, TPh-Pip and MFl-Pip copolymers
The mTPh-Pip, TPh-Pip and MFl-Pip copolymers were synthesized in a similar way as mentioned above but the reaction time was shortened to 2 h. The yield was 87% for TPh-Pip, 91% for MFl-Pip and 79% for mTPh-Pip.4.7. Quaternization reaction (general procedure)
In a 100 mL round-bottomed flask, BPh-Pip (0.5 g) was dissolved in 20 mL of NMP. To the solution, potassium carbonate (3.3 equivalent) was added, and the mixture was stirred for 1 h. Dimethyl sulfate (10 equivalents) was added, and the mixture was stirred at room temperature for 48 h. After the reaction, the mixture was poured into excess mixture solvent of hexane:ethanol (1:1) to obtain QBPh-Pip as pale yellow fibrous solid (0.6 g, 98%).
QTPh-Pip, QmTPh-Pip and QMFl-Pip were prepared in a similar way as above.
4.8. Preparation of membranes
A 15wt% solution of QBPh-Pip in DMSO was cast onto a clean, flat glass plate to obtain a transparent and bendable membrane in methyl sulfate ion form with 35 μm thick. QTPh-Pip, QmTPh-Pip and QMFl-Pip copolymers failed to give bendable membranes.4.9. Ion exchange reaction
The IECfeed was calculated from the feed comonomer compositions as follows:
 | (1) |
The IECNMR was calculated from the NMR integral ratios using the eqn (2) and (3):
| 8m + 6n = aromatic integral ratio | (2) |
| 12n = peak (a) integral ratio | (3) |
DFT calculations. Density functional theory (DFT) calculations were performed for the hydrophilic and hydrophobic components of QBPh-Pip copolymers using Dmol3 software (Materials Studio package, Daikin Co.). First, the optimized geometry of the hydrophilic and hydrophobic components was calculated using GGA (PBE) method and DNP Basis set. Then, the Connolly surface and subsequent occupied volume were calculated for the optimized geometry for each component independently.
4.10. Membrane characterization
The experimental procedures for titration (for ion exchange capacity), transmission electron microscopy (TEM), scanning electron microscopy (SEM), electrochemical Impedance spectroscopy (EIS) for hydroxide ion conductivity, viscoelasticity using dynamic mechanical analyses (DMA) and elongation tests using universal stress–strain (S–S) curves, were described in our previous work.14,15,41,53| WUliquid = [(wet weight − dry weight)/dry weight] × 100. |
The water electrolysis cell was operated using 1 M KOH aqueous solution as electrolyte for both anode and cathode sides at a flow rate of 10 mL min−1. The cell was evaluated at 80 °C. Prior to the evaluation, the cell was pre-conditioned by sweeping the current density from 0 to 1.0 A cm−2 twice. The in situ durability was tested for 1000 h at 80 °C under a combined protocol of a constant current density (1.0 A cm−2) and two sweeps of the current density from 0 to 1.0 A cm−2, repeated every 8 h.
Author contributions
Ahmed Mohamed Ahmed Mahmoud: investigation and writing. Kenji Miyatake: supervision, writing, reviewing and editing, funding acquisition, and project administration. Fanghua Liu: reviewing. Vikrant Yadav: reviewing. Fang Xian: reviewing. Lin Guo: reviewing. Chun Yik Wong: reviewing. Toshio Iwataki: investigation. Makoto Uchida: reviewing. Katsuyoshi Kakinuma: investigation and reviewing.Data availability
The data supporting this work has been provided as part of the ESI.†Conflicts of interest
The authors do not have any conflicts of interest to declare.Acknowledgements
This work was partly supported by the New Energy and Industrial Technology Development Organization (NEDO), the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, through Grants-in-Aid for Scientific Research (23H02058) and MEXT Program: Data Creation and Utilization Type Material Research and Development Project (JPMXP1122712807), JST (GteX JPMJGX23H2), and JKA promotion funds from KEIRIN RACE.References
- H. Ishaq, I. Dincer and C. Crawford, Int. J. Hydrogen Energy, 2022, 47, 26238–26264 CrossRef CAS.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed.
- I. Vincent and D. Bessarabov, Renewable Sustainable Energy Rev., 2018, 81, 1690–1704 CAS.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- D. Aili, A. G. Wright, M. R. Kraglund, K. Jankova, S. Holdcroft and J. O. Jensen, J. Mater. Chem. A, 2017, 5, 5055–5066 RSC.
- K. F. L. Hagesteijn, S. Jiang and B. P. Ladewig, J. Mater. Sci., 2018, 53, 11131–11150 CrossRef CAS.
- D. Henkensmeier, M. Najibah, C. Harms, J. Zitka, J. Hnát and K. Bouzek, J. Electrochem. Energy Convers. Storage, 2021, 18, 024001 CrossRef CAS.
- M. Mandal, G. Huang and P. A. Kohl, ACS Appl. Energy Mater., 2019, 2, 2447–2457 CrossRef CAS.
- W. Chen, M. Mandal, G. Huang, X. Wu, G. He and P. A. Kohl, ACS Appl. Energy Mater., 2019, 2, 2458–2468 CrossRef CAS.
- W. You, E. Padgett, S. N. MacMillan, D. A. Muller and G. W. Coates, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9729–9734 CrossRef CAS PubMed.
- Y. Kim, Y. Wang, A. France-Lanord, Y. Wang, Y. C. M. Wu, S. Lin, Y. Li, J. C. Grossman and T. M. Swager, J. Am. Chem. Soc., 2019, 141, 18152–18159 CAS.
- L. Zhu, J. Pan, Y. Wang, J. Han, L. Zhuang and M. A. Hickner, Macromolecules, 2016, 49, 815–824 Search PubMed.
- W. H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6, 566–570 Search PubMed.
- A. M. A. Mahmoud, A. M. M. Elsaghier, K. Otsuji and K. Miyatake, Macromolecules, 2017, 50, 4256–4266 Search PubMed.
- A. M. A. Mahmoud and K. Miyatake, J. Mater. Chem. A, 2018, 6, 14400–14409 Search PubMed.
- F. Liu, K. Miyatake, A. M. A. Mahmoud, V. Yadav, F. Xian, L. Guo, C. Y. Wong, T. Iwataki, K. Kakinuma and M. Uchida, Adv. Energy Mater., 2024, 202404089 Search PubMed.
- N. Chen, C. Lu, Y. Li, C. Long, Z. Li and H. Zhu, J. Membr. Sci., 2019, 588, 117120–117129 CrossRef CAS.
- A. M. A. Mahmoud, K. Miyatake, F. Liu, V. Yadav, L. Guo, C. Y. Wong, T. Iwataki, K. Kakinuma and M. Uchida, Adv. Energy Sustainability Res., 2024, 5, 2300236 CrossRef CAS.
- T. T. Bai, M. Y. Cong, Y. X. Jia, K. K. Ma and M. Wang, J. Membr. Sci., 2020, 599, 117831 CrossRef CAS.
- S. Sung, T. S. Mayadevi, K. Min, J. Lee, J. E. Chae and T. H. Kim, J. Membr. Sci., 2021, 619, 118774 CAS.
- Y. He, J. Pan, L. Wu, Y. Zhu, X. Ge, J. Ran, Z. J. Yang and T. Xu, Sci. Rep., 2015, 5, 13417 CAS.
- B. Lin, F. Xu, Y. Su, Z. Zhu, Y. Ren, J. Ding and N. Yuan, Ind. Eng. Chem. Res., 2019, 58, 22299–22305 CrossRef CAS.
- L. Liu, D. Li, Y. Xing and N. Li, J. Membr. Sci., 2018, 564, 428–435 Search PubMed.
- X. C. Chen, D. T. Wong, S. Yakovlev, K. M. Beers, K. H. Downing and N. P. Balsara, Nano Lett., 2014, 14, 4058–4064 CAS.
- D. Koronka and K. Miyatake, RSC Adv., 2021, 11, 1030–1038 CAS.
- S. Wierzbicki, J. C. Douglin, A. Kostuch, D. R. Dekel and K. Kruczała, J. Phys. Chem. Lett., 2020, 11, 7630–7636 CrossRef CAS PubMed.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS.
- K. M. Hugar, W. You and G. W. Coates, ACS Energy Lett., 2019, 4, 1681–1686 CrossRef CAS.
- J. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck and D. R. Dekel, Nat. Commun., 2019, 10, 2306 CrossRef PubMed.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS PubMed.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. B. Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- J. Xiao, A. M. Oliveira, L. Wang, Y. Zhao, T. Wang, J. Wang, B. P. Setzler and Y. Yan, ACS Catal., 2021, 11, 264–270 CrossRef CAS.
- Z. Liu, S. D. Sajjad, Y. Gao, H. Yang, J. J. Kaczur and R. I. Masel, Int. J. Hydrogen Energy, 2017, 42, 29661–29665 CrossRef CAS.
- V. Yadav, K. Miyatake, A. M. A. Mahmoud, F. Liu, F. Xian, L. Guo, C. Y. Wong, T. Iwataki, M. Uchida and K. Kakinuma, J. Mater. Chem. A, 2024, 12, 25429–25441 CAS.
- E. J. Park, C. B. Capuano, K. E. Ayers and C. Bae, J. Power Sources, 2018, 375, 367–372 CrossRef CAS.
- Z. Jiang, G. Yi, X. Yao, Y. Ma, X. Su, Q. Liu and Q. Zhang, Chem. Eng. J., 2023, 467, 143442 CAS.
- Y. Ozawa, T. Iwataki, M. Uchida, K. Kakinuma and K. Miyatake, J. Mater. Chem. A, 2023, 11, 19925 CAS.
- M. T. Guzmán-Gutiérrez, D. R. Nieto, S. Fomine, S. L. Morales, M. G. Zolotukhin, M. C. G. Hernandez, H. Kricheldorf and E. S. Wilks, Macromolecules, 2011, 44, 194–202 Search PubMed.
- Z. Zhu and S. J. Paddison, Front. Chem., 2022, 10, 1–24 CAS.
- A. M. A. Mahmoud and K. Miyatake, ACS Appl. Polym. Mater., 2023, 5, 2243–2253 Search PubMed.
- N. Chen, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, C. Hu, J. Y. Bae, E. S. Sim, Y.-C. Chung, J.-H. Jang, S. J. Yoo, Y. Zhuang and Y. M. Lee, Nat. Commun., 2021, 12, 2367 CAS.
- N. Chen, Q. Jiang, F. Song and X. Hu, ACS Energy Lett., 2023, 8, 4043–4051 CAS.
- X. Wu, N. Chen, H. A. Klok, Y. M. Lee and X. Hu, Angew. Chem., Int. Ed., 2022, 61, e202114892 CAS.
- H. Chen, K.-T. Bang, Y. Tian, C. Hu, R. Tao, Y. Yuan, R. Wang, D.-M. Shin, M. Shao, Y. M. Lee and Y. Kim, Angew. Chem., Int. Ed., 2023, 62, e202307690 CAS.
- L. Xu, H. Wang, L. Min, W. Xu, Y. Wang and W. Zhang, Ind. Eng. Chem. Res., 2022, 61, 14232–14241 CAS.
- M. Liu, X. Hu, B. Hu, L. Liu and N. Li, J. Membr. Sci., 2022, 642, 119966 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2018, 6, 16537–16547 RSC.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2019, 7, 15895–15906 RSC.
- T. H. Pham, A. Allushi, J. S. Olsson and P. Jannasch, Polym. Chem., 2020, 11, 6953–6963 RSC.
- C. Hu, N. Y. Kang, H. W. Kang, J. Y. Lee, X. Zhang, Y. J. Lee, S. W. Jung, J. H. Park, M.-G. Kim, S. J. Yoo, S. Y. Lee, C. H. Park and Y. M. Lee, Angew. Chem., Int. Ed., 2024, 63, e2023166 Search PubMed.
- A. M. A. Mahmoud and K. Miyatake, J. Membr. Sci., 2022, 643, 120072 CrossRef.
- A. Allushi, P. M. Bakvand and P. Jannasch, Macromolecules, 2023, 56(3), 1165–1176 Search PubMed.
- D. R. Dekel, M. Amar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29, 4425–4431 CrossRef CAS.
- G. Shi, C. Arata, D. A. Tryk, T. Tano, M. Yamaguchi, A. Iiyama, M. Uchida, K. Iida, S. Watanabe and K. Kakinuma, ACS Omega, 2023, 8, 13068–13077 CrossRef CAS PubMed.
- F. Liu, K. Miyatake, M. Tanabe, A. M. A. Mahmoud, V. Yadav, L. Guo, C. Y. Wong, F. Xian, T. Iwataki, M. Uchida and K. Kakinuma, Adv. Sci., 2024, 2402969 CrossRef CAS PubMed.
- N. Chen, Q. Jiang, F. Song and X. Hu, ACS Energy Lett., 2023, 8, 4043–4051 CrossRef CAS.
- S. C. Zignani, M. L. Faro, S. Trocino and A. S. Aricò, Energies, 2020, 13, 1720 CrossRef CAS.
- A. Caprì, I. Gatto, C. L. Vecchio, S. Trocino, A. Carbone and V. Baglio, ChemElectroChem, 2023, 10, e202201056 CrossRef.
- J. Xiao, A. M. Oliveira, L. Wang, Y. Zhao, T. Wang, J. Wang, B. P. Setzler and Y. Yan, ACS Catal., 2020, 11, 264 Search PubMed.
- J. Wang, C. Liang, X. Ma, P. Liu, W. Pan, H. Zhu, Z. Guo, Y. Sui, H. Liu, L. Liu and C. Yang, Adv. Mater., 2023, 36, 2307925 CrossRef PubMed.
- J. E. Park, S. Y. Kang, S. H. Oh, J. K. Kim, M. S. Lim, C. Y. Ahn, Y. H. Cho and Y. E. Sung, Electrochim. Acta, 2019, 295, 99–106 CAS.
- H. Lim, I. Jeong, J. Choi, G. Shin, J. Kim, T. H. Kim and T. Park, Appl. Surf. Sci., 2023, 610, 155601 CAS.
- H. Ono, T. Kimura, A. Takano, K. Asazawa, J. Miyake, J. Inukai and K. Miyatake, J. Mater. Chem. A, 2017, 5, 24804–24812 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00210a |
| This journal is © The Royal Society of Chemistry 2025 |