
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly isotactic polylactide by binary organocatalyzed polymerization of 1,3-dioxolan-4-ones†
Mengyu
Wang
 ,
Sumin
Lee
,
Hyunhee
Lee
and
Byeong-Su
Kim
*
,
Sumin
Lee
,
Hyunhee
Lee
and
Byeong-Su
Kim
*
Department of Chemistry, Yonsei University, Seoul 03722, Republic of Korea. E-mail: bskim19@yonsei.ac.kr
First published on 10th April 2025
Abstract
Ring-opening polymerization (ROP) of dioxolanones (DOXs) provides access to functional polyesters with properties that are challenging to achieve via traditional polymerization of lactones. However, typical catalysts often induce epimerization in DOXs, limiting the synthesis of isotactic, crystalline polymers. This study reports a binary organocatalytic system that enables controlled ROP of chiral 2,2,5-trimethyl-1,3-dioxolan-4-ones under mild conditions, effectively minimizing epimerization by activating both the monomer and initiator via hydrogen bonding. This strategy facilitates the synthesis of highly isotactic poly(lactic acid) (PLA) with a stereoregularity parameter of 0.92. Moreover, blending enantiomeric PLA chains form a crystalline stereocomplex with a high melting temperature of 195.1 °C. These findings highlight a sustainable and scalable approach for synthesizing stereoregular polyesters, paving the way for advanced material applications.
Introduction
Poly(lactic acid) (PLA) is an important bioplastic that has garnered significant attention for its biodegradability, sustainability, and broad applications in packaging and biomedicine. Derived from renewable resources, PLA offers an environmentally friendly alternative to petroleum-based plastics.1,2 PLA is commonly prepared via ring-opening polymerization (ROP) of cyclic lactide, enabling precise control over molecular weight and dispersity.3,4 Lactide has three diastereomeric forms: L-lactide, D-lactide, and meso-lactide, whose stereochemistry determines the microstructure of the polymer. Achieving high stereoregularity in PLA is essential as it determines crystallinity, mechanical strength, and thermal properties. However, conventional catalysts cause epimerization during polymerization, reducing stereoregularity and hindering crystallinity. Since PLA crystallinity depends on its stereochemical configuration, suppressing epimerization is crucial for obtaining high-performance materials.5–7 Furthermore, the synthesis of functionalized lactide monomers involves intricate procedures, and the relatively modest thermodynamic driving force for lactide polymerization attributed to the relief of six-membered ring strain has spurred the exploration of alternative heterocyclic monomers.8–10O-Carboxy anhydrides (OCAs) have been suggested to increase the driving force for ROP. The polymerization of OCAs is notably fast, owing to the release of CO2 during the reaction.11,12 Additionally, OCAs exhibit excellent functional group tolerance, enabling the incorporation of diverse structural motifs into the polyester backbone.12–14 However, their synthesis typically involves phosgene or its equivalents, increasing cost and toxicity. Moreover, their intrinsic instability complicates long-term storage and handling, thereby restricting their widespread application. These drawbacks have motivated the search for alternative monomeric systems that release small molecules during polymerization to enhance the driving force.
In this context, 1,3-dioxolan-4-ones (DOXs) have emerged as promising monomers because their polymerization eliminates small molecules such as formaldehyde or acetone, thereby improving the thermodynamic driving force for ROP. DOXs can also be derived from sustainable and cost-effective sources, further increasing their value.15,16 Despite these advantages, achieving high-quality and highly isotactic polymers remains a challenge. Previous works on the ROP of DOXs have primarily employed metal-based catalysts such as Salen complexes.16–18 Although these systems have shown some success in producing isotactic PLA, typical side reactions, such as the Tishchenko reaction, often occur under such conditions. Specifically, the Tishchenko reaction results in the retention of acetal or ketal linkages in the polymer backbone, compromising quality and causing uncontrolled polymerization. To mitigate this, metal-catalyzed ROP has been conducted under dynamic vacuum conditions with a condenser to efficiently remove formaldehyde or acetone. Nonetheless, however, this approach requires high temperatures and extended reaction times, limiting scalability and operational feasibility.
Alternatively, organocatalytic systems have also been explored, such as using p-toluenesulfonic acid for ROP of DOX.19 However, this approach yielded only low-molecular-weight PLA with poor isotacticity, partly due to catalyst-induced epimerization, similar to traditional catalytic systems. The susceptibility of DOX to epimerization is attributable to both structural and kinetic factors. (1) Structural effects: DOXs contain an additional oxygen in their cyclic structure, which can induce a stronger electron-withdrawing effect, reducing the pKa of the α-hydrogen and making it more susceptible to deprotonation under basic conditions. This promotes epimerization at the chain end (Fig. S1†). (2) Polymerization kinetics: due to its high ring strain, DOX undergoes rapid ring-opening, potentially leading to mismatches in the propagation rate. As a result, the active chain-end remains exposed longer, increasing the likelihood of epimerization. Given these challenges, recent studies show that hydrogen-bonding organocatalysts, particularly thiourea derivatives, effectively activate monomers, regulate polymerization kinetics, and minimize epimerization by forming multiple hydrogen bonds.20,21
In this study, we present a binary organocatalytic system that enables a ROP of DOXs under mild conditions, yielding PLA with controlled molecular weight and minimal epimerization. Specifically, the highly substituted DOXs derivative 2,2,5-trimethyl-1,3-dioxolan-4-one (Me3DOX) was employed as a monomer owing to its high driving force by the release of acetone during polymerization (Scheme 1). This binary organocatalyst, consisting of a thiourea derivative and a base, activates the monomer and initiator through hydrogen bonding, enabling stereochemical control during polymerization. This method provides the efficient synthesis of highly isotactic PLA with a high level of stereoregularity Pm (probability of meso dyad) of 0.92, essential for achieving crystallinity and desirable thermal properties in polyesters. The resulting PLA exhibits crystallinity and blending enantiomeric PLA chains form a stereocomplex with enhanced thermal stability, as evidenced by its high melting temperature (195.1 °C). These findings not only offer a sustainable and scalable method for producing stereoregular polyesters but also advance material applications in biodegradable plastics and drug delivery systems.
 | ||
| Scheme 1 (a) Traditional works for poly(α-hydroxy acids) from the ROP of lactide and OCAs. (b) ROP of Me3DOX using binary organocatalysts: a route to isotactic PLA and its stereocomplexation. | ||
Results and discussion
Since DOXs often exhibit moderate reactivity in ROP, their polymerization often requires highly active catalysts or structural modifications to increase the driving force for ring-opening. Thus, a chiral (S)-Me3DOX monomer was designed and synthesized in one step with moderate yield. Initially, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was employed for the ROP of chiral (S)-Me3DOX to synthesize the isotactic PLA according to previous studies.22–24 However, DBU alone was ineffective in the ROP of (S)-Me3DOX (entry 1 in Table 1). To improve catalytic performance, we then combined a series of thioureas, such as TU-A, TU-B, and TU-C, to prepare binary organocatalysts and explored their potential for the ROP of (S)-Me3DOX (Table 1).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | [M]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]:[C] :[I]:[C] |
Time (h) | Conv.a (%) |
M
n,theob (g mol−1) |
M
n,c (g mol−1) |
M
wc (g mol−1) |
Đ |
P
md |
| Conditions: benzyl alcohol as an initiator at 25 °C under the bulk condition.a Monomer conversion determined by 1H NMR in CDCl3 using integrals of the characteristic signals.b Calculated using MDOX (72.08 g mol−1) × ([M]0/[I]0) × conversion + MBnOH (108.14 g mol−1).c Obtained from SEC-MALS analysis (THF).d Possibility of meso diad formation from the homonuclear decoupled 1H NMR spectrum and calculated based on CEC mechanism. | |||||||||
| 1 | DBU | 50:1:1 |
24 | — | — | — | — | — | |
| 2 | DBU/TU-A | 50:1:1:1 |
6 | 4 | — | — | — | — | |
| 3 | DBU/TU-B | 50:1:1:1 |
6 | 73 | 2740 | 1400 | 2640 | 1.48 | n.d. |
| 4 | DBU/TU-C | 50:1:1:1 |
6 | 17 | — | — | — | — | |
| 5 | DBU/TU-B | 50:1:1:1.5 |
4 | 74 | 2770 | 2380 | 3080 | 1.30 | 0.77 |
| 6 | DBU/TU-B | 50:1:1:3 |
2 | 71 | 2660 | 2160 | 2700 | 1.25 | 0.92 |
| 7 | DBU/TU-B | 100:1:1:3 |
4 | 68 | 5000 | 2630 | 3980 | 1.51 | 0.93 |
| 8 | DBU/TU-B | 150:1:1:3 |
5 | 57 | 6260 | 3480 | 4770 | 1.51 | 0.85 |
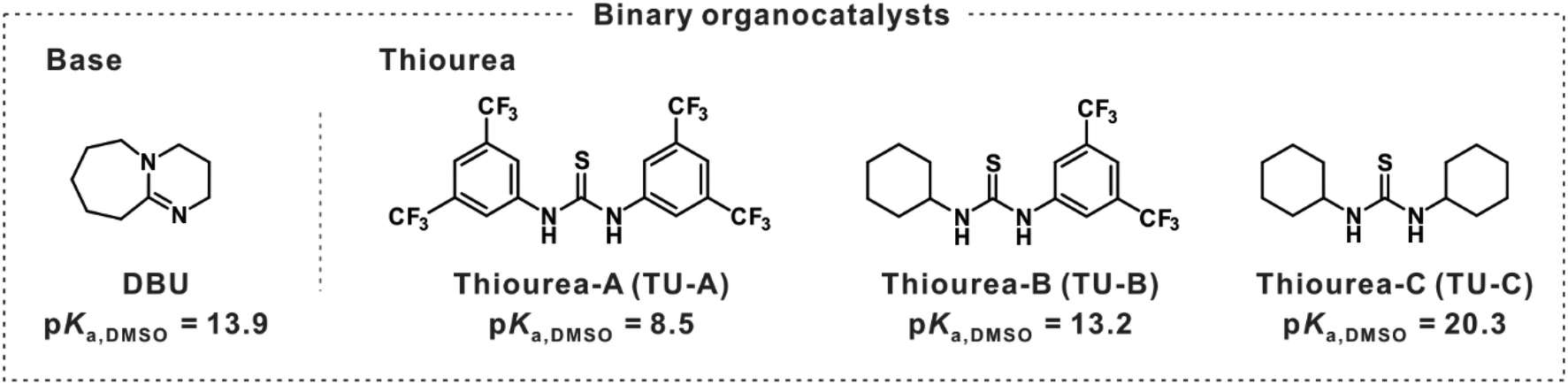
To evaluate the potential of binary organocatalyst systems in (S)-Me3DOX polymerization, we investigated thiourea and DBU catalysis with benzyl alcohol (BnOH) as an initiator. Screening experiments were performed at room temperature (approximately 21 °C) to examine binary organocatalyst systems comprising equimolar amounts of DBU and various thioureas in bulk conditions. The DBU/TU-B combination demonstrated the highest catalytic activity, achieving 73% conversion in 6 h at room temperature, resulting in a poly(s-lactic acid) (P(S-LA)) with a molecular weight of 1400 g mol−1 and a molecular weight dispersity (Đ) of 1.48 (Table 1, entry 3). In contrast, TU-A and TU-C showed poor catalytic activity, with conversions of only 4% and 17%, respectively. TU-A, TU-B, and TU-C were selected based on electronic differences in substituent structures at both ends of the thiourea framework and their distinct pKa values. The structural differences and varying pKa values significantly influence their ability to activate substrates and stabilize intermediates during polymerization. The superior performance of TU-B may be attributed to this, enabling optimal interaction with DBU during the catalytic process. This interaction will be discussed in detail in the subsequent section.
Furthermore, controlling stereoselectivity in PLA synthesis is vital because isotactic PLA, characterized by its regular stereochemical arrangement, exhibits significantly improved properties, including higher crystallinity, mechanical strength, thermal stability, and slower degradation rates, compared to atactic PLA.25,26 These superior characteristics make isotactic PLA ideal for advanced applications such as biomedical implants, durable packaging, and engineering materials, underscoring the importance of achieving precise stereoselectivity during synthesis.25 Although PLA was synthesized with the higher conversion at an equimolar ratio of DBU/TU-B, severe epimerization produced atactic PLA, as indicated by multiplets at 5.16 ppm in the methine region of the 1H NMR spectrum (Fig. S2†). To generate highly isotactic PLA, we increased the TU-B ratio to 1.5 and 3 relatives to DBU. At a 3:1 ratio, the same monomer conversion was achieved only in 2 h (entry 6 in Table 1). Notably, the 1H NMR spectrum of pure P(S-LA) showed a quartet in the methine region and a sharp single peak in the nuclear-decoupled 1H NMR, with a Pm (probability of meso dyad) value of 0.92, indicating the generation of highly isotactic PLA (Fig. 1b and c). Additionally, the structure of P(S-LA) was characterized using 13C and HSQC NMR (Fig. S3 and S4†). Similarly, the enantiomeric (R)-Me3DOX underwent ROP under identical conditions to yield isotactic P(R-LA), confirming that the stereoselectivity of the catalytic system is maintained regardless of monomer configuration (Table S1,† entries 7–9). These results highlight the importance of thiourea in the binary organocatalytic system, which demonstrates that increased its ratio relative to DBU significantly minimizes the epimerization of the chiral center, resulting in PLA transition from atactic to isotactic.
 | ||
| Fig. 1
1H NMR spectra of (a) (S)-Me3DOX monomer and (b) P(S-LA) (entry 6 in Table 1). (c) Homonuclear decoupled 1H NMR spectra of the methine region (5.16 ppm) (entries 3, 5, and 6 in Table 1, DBU:TU-B = 1:1, 1:1.5, 1:3) (400 MHz, CDCl3). | ||
To further improve reaction efficiency and obtain high-molecular-weight PLA with low dispersity, we attempted to extend the reaction time and elevate the reaction temperature. However, the results indicate that prolonged reaction time does not significantly increase monomer conversion. Instead, it leads to a decrease in polymer isotacticity and a slight increase in dispersity. For example, prolonging the reaction time from 2 h to 12 h resulted in an increase in dispersity from 1.44 to 1.55 (Table S1,† entries 1 and 2). Moreover, the polymer's stereoregularity was affected, with the changing from isotactic (Pm = 0.77) to atactic (Table S1,† entries 3 and 4). To better understand when epimerization occurs during the reaction, we performed time-resolved 1H NMR analyses at different stages of polymerization (Fig. S5†). The results show that the b′ peak gradually broadened into a multiplet over time, suggesting that epimerization becomes increasingly prominent at the late stage, when the monomer is nearly depleted. A similar trend is observed with an increase in reaction temperature (Table S1,† entries 5 and 6). The observation that monomer conversion does not improve with extended reaction time suggests that the polymerization reaches an equilibrium state, beyond which further conversion becomes thermodynamically or kinetically unfavorable. The slight broadening in dispersity over time primarily results from transesterification and slow chain transfer events, which become more pronounced at longer reaction times. Furthermore, the decrease in isotacticity suggests that prolonged exposure to reaction conditions affects stereoselectivity, possibly due to a loss of catalyst selectivity and increased chain-end mobility, which facilitates epimerization. It will be a subject of our future endeavor to further optimize the catalyst system to enhance the ROP efficiency of DOXs.
It is well-established that thiourea can be deprotonated by strong bases such as alkoxides, imidazolium, metal hydrides, and t-BuP4 (pKa 42.6 in MeCN) to generate an active reaction center of thiourea anion that follows an anionic mechanism.27,28 If the basicity is sufficiently high, anionic H-bonding based on thiourea anions (TU-H+-base) forms through complete thiourea deprotonation. However, for the relatively weak bases such as DBU, MTBD, or BEMP (pKa 24.2, 25.4, 27.5 in MeCN, respectively), the degree of deprotonation depends on the balance between the basicity of the base and the acidity of the thiourea.27,29 In the DBU/TU-B catalytic system, their similar pKa values (DBU: 13.9, TU-B: 13.2 in DMSO) creates a dynamic equilibrium between the anionic and synergistic mechanisms which collaboratively achieve the highest catalytic activity for the production of highly isotactic PLA.
1H NMR analysis confirmed the close interaction between DBU and TU-B. Fig. 2b shows that the a peak of thiourea in TU-B shifted slightly upfield from 6.43 to 6.29 ppm and became broader upon addition of the TU-B to a BnOH and DBU mixture (BnOH/DBU/TU-B = 1:1:3) (full spectrum in Fig. S6†). Similarly, the b peak associated with the other thiourea in TU-B shifted upfield to 4.54 ppm. These chemical shifts suggest deprotonation of TU-B by DBU, leading to the formation of an anionic thioimidate species. In addition, the deshielding of benzyl protons (c peaks) of BnOH in the BnOH/DBU/TU-B mixture, compared to free BnOH, indicates hydrogen bond formation with DBU or TU-B. Furthermore, we hypothesize that excess thiourea stabilizes the thioimidate, possibly through intramolecular hydrogen bonding, thereby enhancing its catalytic activity in the ROP of the (S)-Me3DOX.30,31
 | ||
| Fig. 2 (a) Proposed mechanisms for the DBU/TU-B mediated ROP of (S)-Me3DOX. (b) 1H NMR spectra from BnOH/DBU/TU-B (1:1:3), TU-B, and BnOH (400 MHz, toluene-d8). | ||
In traditional ROP of DOXs, particularly under basic or Lewis base catalysis, the chiral α-carbon is highly susceptible to protonation/deprotonation, causing epimerization. In this study, thiourea stabilizes (S)-Me3DOX through hydrogen bonding, effectively reducing α-carbon deprotonation and minimizing chiral inversion, which enhances stereoselectivity and facilitates the formation of highly isotactic PLA through a combination of anionic and synergistic mechanisms.
After identifying DBU/TU-B as the optimal catalyst system for (S)-Me3DOX, we explored its ROP in detail. The polymerization of (S)-Me3DOX by the binary organocatalyst DBU/TU-B exhibited characteristics of controlled polymerization, including a distinct first-order kinetic relationship between ln([M]0/[M]t) and polymerization time, with an apparent propagation rate constant (kp,app) of 9.03 × 10−5 ± 0.49 × 10−5 s−1 (Fig. 3a). It is of note that our system achieved a significantly higher polymerization rate than Salen-based catalysts (Table S2†), attributed to efficient monomer activation by the binary organocatalyst and the absence of solvent effects, which enhance monomer accessibility and catalytic efficiency in bulk polymerization. Furthermore, the solvent-free nature of this approach provides a more sustainable and scalable alternative for polyester synthesis.
 | ||
| Fig. 3 (a) Kinetic plots for ROP of (S)-Me3DOX (entry 7 in Table 1), (b) plots of Mn (black) and Đ (red) versus conversion (entry 7 in Table 1), and (c) GPC traces of the P(S-LA) obtained (entries 6–8 in Table 1). (d) Representative MALDI-ToF spectrum of P(S-LA) (entry 7 in Table 1). (e) Analyzed distribution histogram of the respective initiating groups in the synthesized P(S-LA) polymers and their corresponding chemical structures (P1–P3) with varying initiating groups. | ||
Additionally, effective control of polymerization in this catalytic system is evidenced by the observation of a linear correlation between Mn and monomer conversion in the ROP of (S)-Me3DOX (Fig. 3b). We then investigated whether the catalytic systems could produce PLA with high molecular weights predictable from the monomer-to-initiator feed ratios (Fig. 3c). When the ratio of (S)-Me3DOX to BnOH was initially set to 50, the ROP reactions proceeded smoothly. Increasing the feed ratio to 100 yielded a polymer with an Mn value of 2630 g mol−1, Đ value of 1.51, and high stereoregularity (Pm = 0.93) (entry 7 in Table 1). However, at a ratio of 150, the ROP deteriorated, as evidenced by a Đ value of 1.51 and reduced stereoregularity (Pm = 0.85) (entry 8 in Table 1). This decline in performance likely results from higher monomer concentrations promoting side reactions such as reversible transesterification, leading to broader molecular weight distribution. Additionally, slower monomer consumption at elevated ratios increases epimerization, reducing stereoregularity. In bulk polymerization, rising viscosity limits monomer and catalyst diffusion, creating heterogeneous polymerization conditions that broaden Đ and lower Pm. These factors collectively make maintaining control at high monomer-to-initiator ratios challenging.
In addition, MALDI-ToF analysis was performed to elucidate the structure of the polymer. Fig. 3d shows three main distributions: (1) the major peaks correspond to PLA chains initiated by BnOH, (2) minor peaks indicate the presence of a hydroxyl group (OH–) at the terminus, likely due to trace water or BnOH degradation during measurement, and (3) the leftmost peak represents the proton adduct of the OH-terminal product with ketal bridge retention. Taken together, the initiation efficiency of BnOH exceeded 70%. Notably, the Tishchenko reaction was significantly suppressed, minimizing ketal bridge formation within the polymer backbone, a common issue in metal-catalyzed DOX polymerizations. To further verify this, 1H NMR analysis of the crude polymer confirmed acetone release, further supporting this conclusion (Fig. S8†).
Differential scanning calorimetry (DSC) was used to assess the thermal properties of isotactic PLA. Previously, PLA produced from DOXs exhibited a low melting transition temperature (Tm = 125.2 °C), limiting their use in commodity and biomedical applications. Physical blending of enantiomeric chiral polymers in a stoichiometric ratio enables the formation of crystalline stereocomplexes with enhanced properties. In this context, isotactic polymers P(S-LA) and P(R-LA), synthesized by ROP of respective enantiopure (S)-Me3DOX and (R)-Me3DOX, exhibited melting peaks (Tm: 143.6 °C and 135.1 °C, respectively) in the first heating scan, illustrating the influence of tacticity on polymer properties. In the second heating scan, the enantiomeric polymers displayed glass transition temperatures (Tg) of 22.9 °C and 33.9 °C. However, the absence of melting peaks in the second heating cycle suggests a slow crystallization rate, consistent with previous literature.17,32
Stereocomplexation studies were conducted to further improve the crystallization behavior of isotactic PLA. An equimolar blend of P(S-LA) and P(R-LA), prepared by solution mixing and solvent removal, formed stereocomplexed PLA (sc-PLA) between P(S-LA) and P(R-LA) (Fig. 4). The sc-PLA displayed a Tm of 195.1 °C, approximately 55 °C higher than that of the component enantiomeric PLA. Unlike enantiomeric parent polymers, which exhibited a Tm only during the first heating scan, sc-P(LA) retained a distinct Tm of 169.1 °C in the second heating scan. This indicates that recrystallization occurred during the first cooling process, demonstrating that stereocomplexation accelerates polymer crystallization, which is crucial for the practical processing of polymeric materials. The crystallization behavior observed during the second heating scan is attributed to the highly isotactic structures and stereocomplexation between the enantiomeric polymers, consistent with previous studies.26,33
 | ||
| Fig. 4 DSC curves for enantiomeric P(S-LA) and P(R-LA) and their stereocomplexed PLA (sc-PLA). | ||
Conclusions
In summary, we present the first example of the ROP of DOX derivatives using a binary organocatalyst system under mild conditions. In this approach, the binary organocatalysts activate both the monomer and initiator through hydrogen bonding, enabling the efficient synthesis of highly isotactic P(S-LA) with excellent stereoregularity (Pm = 0.92). The isotactic P(S-LA) exhibits high crystallinity and forms a stereocomplex with a melting temperature of 195.1 °C when mixed with enantiomeric PLA chains. Future studies will expand the monomer scope to produce functional polyesters with tailored properties. This sustainable and scalable method advances innovative polymer materials for biodegradable plastics and other high-performance applications.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. M. W.: experimentation, analysis, and writing; S. L. and H. L. analysis; B.-S. K.: conceptualization, supervision, and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (RS-2025-00558644, and RS-2024-00413183) and the China Scholarship Council (202306450065).References
- G. W. Coates and Y. D. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- A. Salimi, S. Ahmadi, M. Faramarzi and J. Faghihi, Macromol. Res., 2023, 31, 873–881 CrossRef CAS.
- S. Wang, J. Li, X. Li and Y. Tu, Fundam. Res., 2024 DOI:10.1016/j.fmre.2024.05.015.
- A. J. Lasprilla, G. A. Martinez, B. H. Lunelli, A. L. Jardini and R. M. Filho, Biotechnol. Adv., 2012, 30, 321–328 CrossRef CAS PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- A. Basterretxea, E. Gabirondo, C. Jehanno, H. Zhu, O. Coulembier, D. Mecerreyes and H. Sardon, Macromolecules, 2021, 54, 6214–6225 CrossRef CAS PubMed.
- R. M. Michell, V. Ladelta, E. Da Silva, A. J. Müller and N. Hadjichristidis, Prog. Polym. Sci., 2023, 101742 CrossRef CAS.
- G. L. Fiore, F. Jing, V. G. Young Jr, C. J. Cramer and M. A. Hillmyer, Polym. Chem., 2010, 1, 870–877 RSC.
- J. M. Becker, R. J. Pounder and A. P. Dove, Macromol. Rapid Commun., 2010, 31, 1923–1937 CrossRef CAS.
- T. Fuoco, D. Pappalardo and A. Finne-Wistrand, Macromolecules, 2017, 50, 7052–7061 CrossRef CAS.
- B. Martin-Vaca and D. Bourissou, ACS Macro Lett., 2015, 4, 792–798 CrossRef CAS.
- Z. Huo, X. Xie and R. Tong, Sustainable Green Chemistry in Polymer Research. Volume 2. Sustainable Polymers and Applications, 2023, pp. 99–123 Search PubMed.
- A. Buchard, D. R. Carbery, M. G. Davidson, P. K. Ivanova, B. J. Jeffery, G. I. Kociok-Köhn and J. P. Lowe, Angew. Chem., Int. Ed., 2014, 53, 13858–13861 CrossRef CAS PubMed.
- J. Wang and Y. Tao, Macromol. Rapid Commun., 2021, 42, 2000535 CrossRef CAS PubMed.
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768–1773 RSC.
- S. A. Cairns, A. Schultheiss and M. P. Shaver, Polym. Chem., 2017, 8, 2990–2996 RSC.
- Y. Xu, M. R. Perry, S. A. Cairns and M. P. Shaver, Polym. Chem., 2019, 10, 3048–3054 RSC.
- K. Sakai, Y. Yagi and N. Nomura, Synlett, 2023, 2520–2524 CrossRef CAS.
- S. Gazzotti, M. A. Ortenzi, H. Farina and A. Silvani, Polymers, 2020, 12, 2396 CrossRef CAS PubMed.
- J. Liang, W. Meng and J. Yang, Macromolecules, 2023, 56, 7043–7054 CrossRef CAS.
- Y. Liu, X. Wang, Z. Li, F. Wei, H. Zhu, H. Dong, S. Chen, H. Sun, K. Yang and K. Guo, Polym. Chem., 2018, 9, 154–159 RSC.
- Q. Song, C. Pascouau, J. Zhao, G. Zhang, F. Peruch and S. Carlotti, Prog. Polym. Sci., 2020, 110, 101309 CrossRef CAS.
- H. Li, S. M. Guillaume and J. F. Carpentier, Chem. – Asian J., 2022, 17, e202200641 CrossRef CAS PubMed.
- Y. Yu, M. Kim, G. S. Lee, H. W. Lee, J. G. Kim and B.-S. Kim, Macromolecules, 2021, 54, 10903–10913 CrossRef CAS.
- M. J.-L. Tschan, R. M. Gauvin and C. M. Thomas, Chem. Soc. Rev., 2021, 50, 13587–13608 RSC.
- M. Li, Y. Tao, J. Tang, Y. Wang, X. Zhang, Y. Tao and X. Wang, J. Am. Chem. Soc., 2018, 141, 281–289 CrossRef PubMed.
- J. Xu, X. Wang, J. Liu, X. Feng, Y. Gnanou and N. Hadjichristidis, Prog. Polym. Sci., 2022, 125, 101484 CrossRef CAS.
- M. S. Zaky, G. Guichard and D. Taton, Macromolecules, 2023, 56, 3607–3616 CrossRef CAS.
- B. Lin and R. M. Waymouth, Macromolecules, 2018, 51, 2932–2938 CrossRef CAS.
- R. S. Hewawasam, U. I. Kalana, N. U. Dharmaratne, T. J. Wright, T. J. Bannin, E. T. Kiesewetter and M. K. Kiesewetter, Macromolecules, 2019, 52, 9232–9237 CrossRef CAS.
- I. Jain and P. Malik, Eur. Polym. J., 2020, 133, 109791 CrossRef CAS.
- H. Z. Fan, X. Yang, J. H. Chen, Y. M. Tu, Z. Cai and J. B. Zhu, Angew. Chem., 2022, 134, e202117639 CrossRef.
- Y. Popowski, Y. Lu, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2022, 144, 8362–8370 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00205b |
| This journal is © The Royal Society of Chemistry 2025 |