
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe livingness of poly(methyl acrylate) under visible light photoiniferter-RAFT polymerization mediated by trithiocarbonates†
Jungwook
Lee
 a,
Yonghwan
Kwon
a,
Changhoon
Yu
a,
Dominik
Konkolewicz
b and
Min Sang
Kwon
*a
a,
Yonghwan
Kwon
a,
Changhoon
Yu
a,
Dominik
Konkolewicz
b and
Min Sang
Kwon
*a
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: minsang@snu.ac.kr
bDepartment of Chemistry and Biochemistry, Miami University, Oxford, OH, USA
First published on 14th March 2025
Abstract
This study investigates the formation and evolution of dead chains during and after the photoiniferter-RAFT polymerization of poly(methyl acrylate). Thermal gradient high-performance liquid chromatography (TG-HPLC) was employed to quantify living and dead chains within the system. Through HPLC fractionation, dead chains were directly separated and further analyzed using 1H NMR spectroscopy and gel permeation chromatography. The results demonstrated a linear increase in dead chain formation over time, likely attributed to the continuous removal of thiocarbonylthio (TCT) end groups from living chains. The relationship between light intensity and dead chain formation was also investigated; however, the dead fraction exhibited a linear increase regardless of light intensity. These findings underscore the critical importance of managing these dynamics to preserve the “living” nature of photoiniferter-RAFT systems.
Introduction
Given the strong relationship between the properties of polymeric materials and their molecular structures, synthesizing polymers with precisely defined architectures is essential for polymer chemists.1 Reversible-deactivation radical polymerization (RDRP) methods, such as atom transfer radical polymerization (ATRP) and reversible addition-fragmentation chain transfer (RAFT), have been widely utilized for this purpose.2–8 These techniques enable the straightforward preparation of well-defined and structurally complex polymers from a diverse range of monomers under mild conditions.9Photoiniferter-RAFT polymerization, a type of RDRP, was first introduced by Otsu and colleagues in 1982.10,11 Renewed interest in this method arose after 2015 studies by Qiao and Boyer, which demonstrated the use of visible light, rather than high-energy UV light, as the excitation source.12–14 This approach employs a thiocarbonylthio (TCT)-based compound, which acts as an initiator, transfer agent, and reversible terminator. As shown in Scheme 1, light absorption by the TCT moiety excites it, leading to the homolysis of the C–S bond and the formation of an active initiating/propagating radical and a TCT radical (TCT˙).15,16 The initiating radical participates in the degenerative chain transfer mechanism by transferring to TCT-based molecules, while the TCT radical can reversibly deactivate growing radical species, ensuring controlled polymerization.
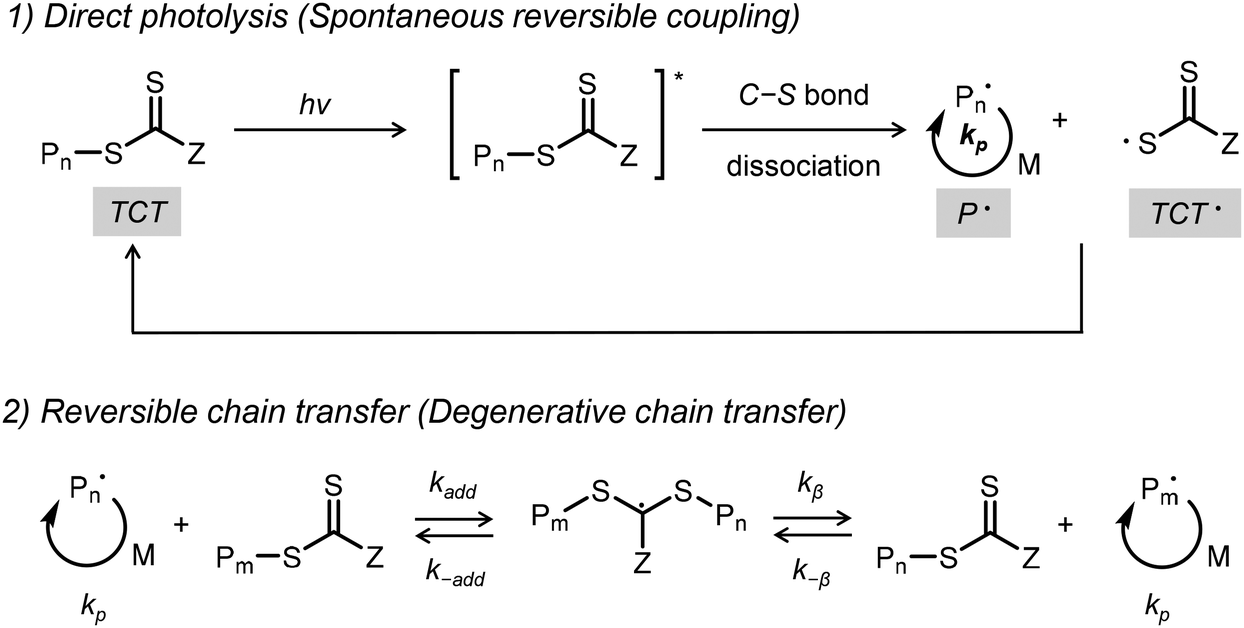 | ||
| Scheme 1 Scheme of the photoiniferter-RAFT process. | ||
Unlike ATRP or traditional RAFT, photoiniferter-RAFT polymerization proceeds without catalysts or conventional radical initiators.17–19 This eliminates challenges such as catalyst contamination, unwanted by-products, and initiation heterogeneity. Additionally, dual deactivation mechanisms reduce bimolecular radical termination and chain growth heterogeneity, providing advantages over conventional RDRP.20,21 This process enables the synthesis of acrylic polymers with ultra-high livingness,13 ultra-high molecular weight,22,23 new block copolymer sequences,24 and even single-unit monomer insertions.25–28 Its unique capabilities have led to a wide range of applications, including surface functionalization,29–32 novel thermoplastic polyurethane elastomers,33 crosslinking-free pressure sensitive adhesives,34,35 living additive manufacturing,36,37 and self-healing polymers.38
Applications such as living additive manufacturing and self-healing polymer networks leverage the dynamic and reversible properties of the C–S bond in TCT moieties under light exposure. For instance, Matyjaszewski and colleagues demonstrated that TCT groups incorporated into polymer networks, when photoactivated in the absence of monomers, undergo C–S bond homolysis to generate active radicals. These radicals then participate in chain transfer reactions and reversible radical deactivation, reshuffling the matrix connections and enabling photoinduced self-healing (Fig. 1a).38 Similarly, Johnson and colleagues showed that embedding TCT moieties in a gel network enables network expansion under light (Fig. 1b).39 They used a symmetrical trithiocarbonate (TTC) with two tertiary R-groups, each containing a norbornene unit, to construct a network from polymeric precursors. These gels could be extended after crosslinking through additional photopolymerization steps, where the TTC units cleaved within the network, allowing monomer addition before deactivation. These applications necessitate the activation of TCT groups and the shuffling of polymer chains under monomer-free conditions. However, TCT groups are known to decompose under light irradiation, which endangers the reversibility of the system.40 Thus, for a more sophisticated design of systems utilizing the direct photolysis of TCT compounds, it is essential to deepen our understanding of the extent to which dead chains form during polymer chain exchange.
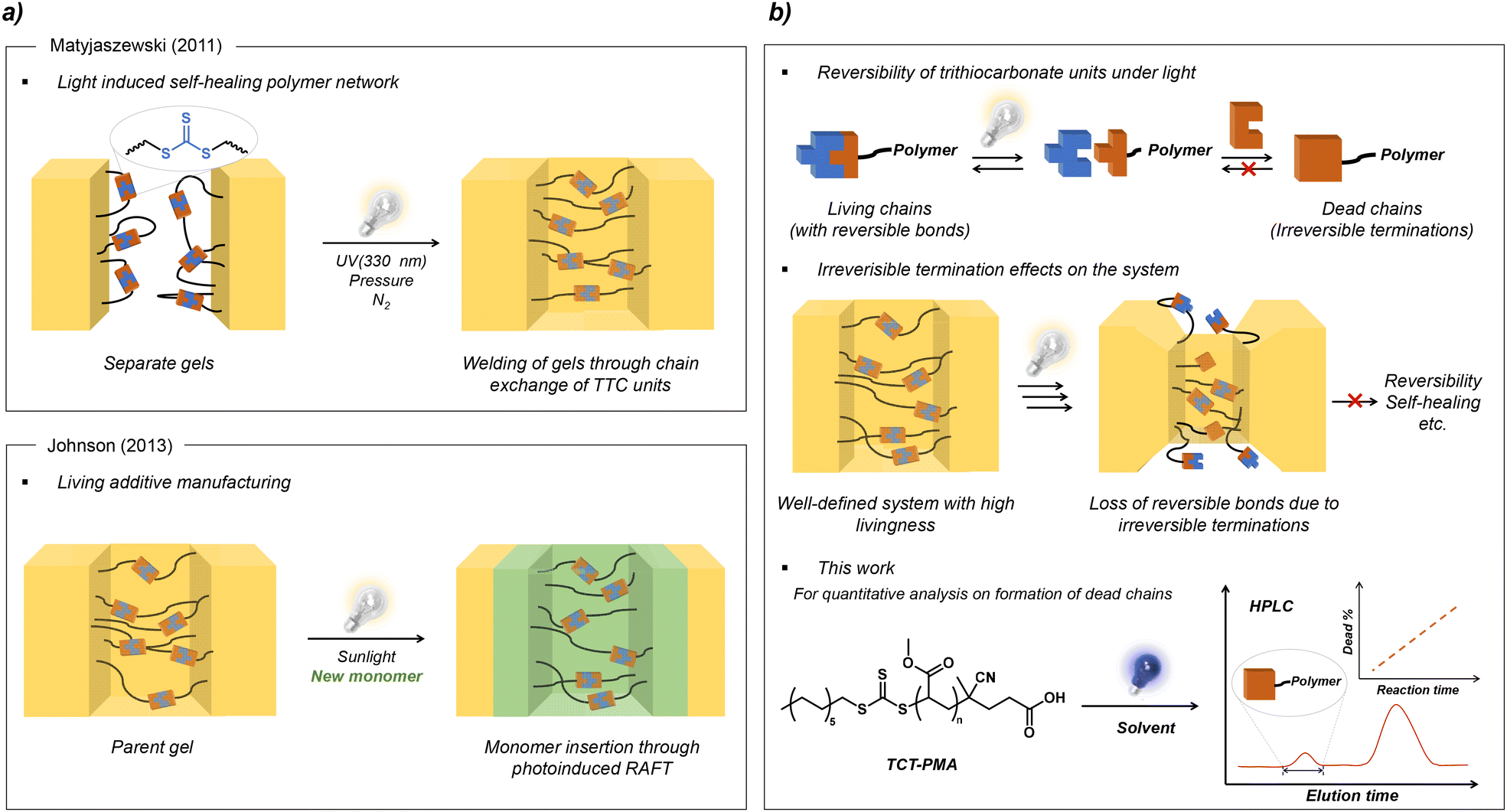 | ||
| Fig. 1 (a) Previous works involving trithiocarbonate units for light-induced self-healing polymer networks and living additive manufacturing. (b) Schematic overview of this work. | ||
To gain a clearer picture of dead chain formation during monomer-free chain reshuffling involving TCT units, it is essential to (i) identify the types of dead chains generated and (ii) quantify their formation over time. Yamago and Matyjaszewski separately investigated the termination of acrylates prepared via organotellurium-mediated radical polymerization and ATRP, respectively.41,42 However, these studies focused on different systems that did not incorporate TCT moieties and, therefore, could not provide a comprehensive understanding of TCT group involvement in the chain reshuffling process. Guymon and colleagues performed a chain-end analysis of poly(butyl acrylate) (PBA) prepared via photoiniferter-RAFT polymerization using gel permeation chromatography (GPC) and deconvolution of GPC chromatograms. Their study revealed a double molecular weight fraction at high conversions, resulting from the formation of dead chains via bimolecular coupling of growing radicals.43 However, due to the methodological limitations of GPC, this work could not include the amount of dead chains formed via disproportionation or similar pathways, which yield molecular weights comparable to those of living chains.
Paik and colleagues successfully separated living and dead poly(styrene) (PS) chains synthesized via thermally initiated RAFT polymerization using thermal-gradient high-performance liquid chromatography (TG-HPLC), demonstrating effective separation based on end-group functionality.44 Similarly, Kwon and colleagues conducted a comparative analysis of dead chains in poly(methyl acrylate) (PMA) synthesized using photoiniferter-RAFT polymerization and thermally initiated RAFT polymerization.33 However, none of these studies chronologically tracked dead chain formation at a low monomer concentration. To the best of our knowledge, no prior work has quantitatively monitored the formation of dead chains during polymer chain reshuffling mediated by activation of TCT groups in the absence of monomers.
In this study, we observed that extended light exposure during photoiniferter-RAFT polymerization of methyl acrylate, including after monomer depletion (>95%), significantly reduces the proportion of living chains while increasing dead chains. GPC and TG-HPLC revealed a linear increase in dead chain formation, reaching approximately 17% after 168 hours. This behavior suggests that the stability of TCT˙ is indeed limited in a monomer-free environment, likely due to the inherent chemical instability of TCT and its radical form under constant light irradiation.
Results and discussion
Photoiniferter-RAFT polymerization of methyl acrylate
For this study, we utilized well-established photoiniferter-RAFT polymerization conditions from our previous work.33 The commercially available 4-cyano-4-(dodecylthiocarbonothioylthio)pentanoic acid (CDTPA) served as the iniferter, methyl acrylate (MA) as the monomer, and dimethyl sulfoxide (DMSO) as the solvent (Fig. 2a). The reaction was carried out under commercial blue light-emitting diodes (LEDs, 455 nm, 100 mW cm−2) under a nitrogen atmosphere at room temperature (Fig. S10†). To ensure consistency, the monomer to solvent volumetric ratio was fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) in all cases. The choice of CDTPA and MA under 455 nm light was based on two key considerations: (i) as shown in our prior study, this combination provides exceptional control over the polymerization process, achieving exceptionally narrow molecular weight distribution and high chain-end fidelity; and (ii) TG-HPLC analysis allows for complete separation of dead and living polymer chains by distinguishing the presence or absence of the long alkyl chain end derived from CDTPA. This capability facilitates the precise quantification of polymerization controllability.33,44,45 Photoiniferter-RAFT polymerization of MA was performed with a molar ratio of [MA]:[CDTPA] = 100:1 under the previously described conditions. The reaction spanned 24 hours, with samples collected hourly during the first 6 hours under inert conditions from the same batch, followed by additional collections at 18 hours and at the end of the reaction at 24 hours. Each sample was first analyzed by 1H-NMR to calculate monomer conversion. During the initial stages, an induction period of approximately one hour was observed (Fig. 2b and Fig. S1†). Following this, the reaction progressed rapidly, achieving approximately 90% monomer conversion within the first 6 hours. During this time, polymerization followed pseudo-first-order kinetics. Beyond the 6 hour mark, monomer conversion showed minimal further increase by the 24 hour endpoint. These observations align well with previously reported results for photoiniferter-RAFT polymerization,14–16,33 including our own prior findings.
:1 (v/v) in all cases. The choice of CDTPA and MA under 455 nm light was based on two key considerations: (i) as shown in our prior study, this combination provides exceptional control over the polymerization process, achieving exceptionally narrow molecular weight distribution and high chain-end fidelity; and (ii) TG-HPLC analysis allows for complete separation of dead and living polymer chains by distinguishing the presence or absence of the long alkyl chain end derived from CDTPA. This capability facilitates the precise quantification of polymerization controllability.33,44,45 Photoiniferter-RAFT polymerization of MA was performed with a molar ratio of [MA]:[CDTPA] = 100:1 under the previously described conditions. The reaction spanned 24 hours, with samples collected hourly during the first 6 hours under inert conditions from the same batch, followed by additional collections at 18 hours and at the end of the reaction at 24 hours. Each sample was first analyzed by 1H-NMR to calculate monomer conversion. During the initial stages, an induction period of approximately one hour was observed (Fig. 2b and Fig. S1†). Following this, the reaction progressed rapidly, achieving approximately 90% monomer conversion within the first 6 hours. During this time, polymerization followed pseudo-first-order kinetics. Beyond the 6 hour mark, monomer conversion showed minimal further increase by the 24 hour endpoint. These observations align well with previously reported results for photoiniferter-RAFT polymerization,14–16,33 including our own prior findings.
 | ||
| Fig. 2 (a) Scheme of photoiniferter-RAFT polymerization of poly(methyl acrylate) using CDTPA as an iniferter in DMSO under a blue LED (λmax = 455 nm, 100 mW cm−2). Adead (%) = area of fraction/(area of all polymer peaks) × 100. (b) Pseudo-first order kinetic plot (black) and respective monomer conversion plot (green). (c) GPC chromatogram of as-prepared PMA at 16 hours of reaction, RI (green): refractive index detector signal, R90 (red): multi-angle light scattering detector signal, A310 (blue): UV detector signal at 310 nm, and A235 (black): UV detector signal at 235 nm. (d) HPLC chromatogram of as-isolated PMA at 16 hours of reaction. (e) GPC chromatograms of fractioned “Fdead” and “Fliving”. | ||
GPC analysis of the prepared PMA
To characterize the synthesized polymers, GPC measurements were conducted to determine their molecular weights and dispersity (Đ) (Fig. 2a). All values were obtained using GPC coupled with a multi-angle light scattering (MALS) detector, as the conventional refractive index (RI) detector provided less reliable results due to band-broadening.46 Polymers sampled after 3 and 6 hours of reaction demonstrated a proportional increase in molecular weight with monomer conversion. The measured molecular weights closely agreed with theoretical predictions. Consistent with previous studies, the polymers exhibited exceptionally low dispersity values, indicative of excellent reaction controllability. Polymers collected after 12 hours showed a more pronounced increase in molecular weight relative to monomer conversion compared to the 6 hour sample. While the molecular weight distribution remained narrow, it was slightly broader than that of the 6 hour sample. Samples obtained at 16 and 24 hours exhibited trends similar to that of the 12 hour sample, with comparable molecular weight and dispersity profiles.A more detailed investigation was conducted on the polymer collected after 16 hours of reaction. GPC was performed using a setup equipped with ultraviolet (UV) detectors at two excitation wavelengths (235 and 310 nm), along with RI and MALS detectors. The chromatogram of the PMA sample showed distinct signals across all detectors (Fig. 2c). Notably, the MALS detector—more sensitive to higher molecular weight polymers than the other detectors—revealed a subtle high-molecular-weight “shoulder” at an elution time between 32 and 34 minutes. This feature is attributed to dead polymer chains formed via the bimolecular termination of growing polymer radicals.
TG-HPLC analysis of the prepared PMA
We further conducted a more detailed analysis of the resulting polymers using TG-HPLC to separate the PMA collected after 16 hours of reaction, employing four detectors. As shown in Fig. 2d, the polymers eluted in two distinct peaks based on the LS signal, RI detector, and UV detector at 235 nm. A clear distinction between the two peaks was observed in the UV detector at 310 nm, attributed to the absorption of the TCT group. The RI signals of each peak were integrated to determine the relative proportion of each fraction compared to the total number of chains. The major fraction, designated as “Fliving” and accounting for 97.7% of the sample, exhibited strong absorption at 310 nm. In contrast, the minor fraction, labeled as “Fdead” (2.3%), showed no absorption. The separation of these fractions is likely influenced by the TCT units in the living chains, which possess long alkyl groups. These groups significantly affect the interaction of the living chains with the column, enhancing their separation from the dead chains.45The two fractions were then collected and reanalyzed by GPC, as shown in Fig. 2e. The major fraction, “Fliving”, exhibited a slightly narrower peak compared to the as-prepared PMA (Đ = 1.006) after the removal of “Fdead”. This fraction is attributed to living chains, as they retain the TCT group. In contrast, the “Fdead” fraction displayed a broad bimodal peak (Đ = 1.140). Since this fraction lacks the TCT moiety, it is assigned to dead chains that were spontaneously terminated during the polymerization via coupling and disproportionation processes.41,42,47
The same TG-HPLC analysis was performed on polymer samples collected after 3, 6, 12, and 24 hours of reaction (Fig. 2a and Fig. S2†). The proportion of dead chains among the total polymer chains increased over time, rising from 0.3% at 3 hours to 1.9% at 12 hours. Interestingly, even in the 16 hour and 24 hour samples, where monomer conversion and molecular weight showed minimal increase, the proportion of dead chains continued to rise, reaching 2.3% and 2.5%, respectively. Unlike conventional RAFT processes, the remarkably low formation of dead chains in photoiniferter-RAFT seems to stem largely from the absence of an external initiator. Furthermore, as noted earlier, the presence of two distinct deactivation processes contributes to the low levels of dead chain formation and the narrow dispersity observed.
Formation of dead chains under continuous light irradiation
The results from the 16 hour and 24 hour samples prompted us to further investigate how continuous light exposure influences the formation of dead chains. To explore this, we extended the photoiniferter-RAFT polymerization to a week (a total of 168 hours). Samples were collected at 24 hour intervals and analyzed using GPC and TG-HPLC (Fig. 3a and Fig. S2, S3†). | ||
| Fig. 3 (a) GPC chromatograms of samples under prolonged irradiation. (b) HPLC chromatograms of isolated samples under prolonged irradiation. The peak between “Fdead” and “Fliving” represents residual solvents as well as small molecule impurities. (c) GPC chromatograms of separated “Fdead”. (d) Plot of dead chain fraction vs. irradiation time. Portion of combination (purple) and disproportionation (orange) calculated by deconvolution of fractioned “Fdead”. | ||
Fig. 3b shows the plots obtained by HPLC with an RI detector for samples collected from 24 hours to 168 hours. As observed earlier, the peak corresponding to the dead chains appears at a shorter elution time, while the broad peak at a longer elution time of 4.8 minutes corresponds to the living chains. This was further confirmed by separating each fraction using HPLC, collecting the fractions, and reanalyzing the fraction suspected to be dead chains by GPC (Fig. 3c). As observed previously, a bimodal peak was detected. The peak at the shorter elution time can be attributed to recombination, while the peak at the longer elution time is assigned to disproportionation.
The structures of the dead and living chains were further confirmed through 1H NMR analysis. To facilitate detailed 1H NMR characterization, photoiniferter-RAFT polymerization of MA was performed with a molar ratio of [MA]:[CDTPA] = 25:1, while keeping all other conditions unchanged. Samples irradiated for one week were fractionated using the same TG-HPLC procedure and the resulting chains were characterized by 1H NMR (Fig. S4†). The 1H NMR spectra of the living fraction displayed signals corresponding to hydrogens adjacent to TCT units (δ 3.38 ppm, 2H; δ 4.9 ppm, 1H), confirming that the fractionated living chains were indeed “living” (Fig. S4b†). In contrast, the dead fraction lacked these signals, indicating the absence of TCT units, consistent with the lack of 310 nm absorption in the HPLC spectra. Additionally, the 1H NMR spectra of the dead fraction revealed the presence of vinylic and alkyl hydrogens, suggesting a mixture of termination products formed through various pathways. However, a detailed structural analysis of the dead chains was not possible due to the limitations of the fractionation method used in this study, which could not distinguish between the different termination products (Fig. S4c†).
Based on the HPLC results, we quantified the fraction of dead chains over time and calculated the proportions of disproportionation and recombination in the dead chains using GPC (Fig. S5† and Table 1). Importantly, the fraction of dead chains increased linearly with time (Fig. 3d), reaching a total of ∼17% after 168 h. Meanwhile, the ratio of disproportionation to recombination remained consistent at approximately 8:2, showing no clear directional trend. As demonstrated in the recent work by Perrier and coworkers, TCT˙ can also initiate polymerization, suggesting that it is not completely persistent and can participate in other reactions beyond reversible coupling with transient radicals.48–50 From the experimental observation as well as the work done by Perrier and coworkers, we could hypothesize that the observation made above could stem from the instability of TCT˙ (see the ESI†).51–53 However, we were unable to experimentally observe the evidence of TCT˙ instability, such as CS2 generation or isolation of thiols resulting from degradation of TCTs.
| Fraction | 24 h | 48 h | 72 h | 96 h | 168 h |
|---|---|---|---|---|---|
| a A dead or living (%) = area of fraction/(area of all polymer peaks) × 100. b A comb or disp (%) = area of deconvoluted peak/(total area of all peaks) × Adead; the numbers inside parentheses represent the percentage of Acomb or disp with respect to Adead. | |||||
|
A
deada (%) |
2.5 | 5.3 | 8.7 | 10 | 16.5 |
|
A
combb (%) |
0.35 (14) | 1.39 (26) | 1.66 (19) | 2.81 (28) | 4.74 (28) |
| A disp (%) | 2.15 (86) | 3.91 (74) | 7.04 (81) | 7.19 (72) | 11.96 (72) |
| A living (%) | 97.5 | 94.7 | 91.3 | 90 | 83.3 |
Effect of light intensity on dead chain formation
To track the effect of light intensity on dead chain formation after full monomer conversion, we conducted photoiniferter-RAFT polymerization at 45 mW cm−2, 80 mW cm−2, and 200 mW cm−2. Fig. 4a presents the plots of monomer conversion over time under different light intensities, demonstrating pseudo-first-order kinetics under all conditions. As the light intensity increased, the apparent rate of propagation also increased. Since all other reaction conditions were identical, the enhanced reaction rate with higher light intensity can be attributed to an increase in radical concentration.39,43Fig. 4b illustrates the amount of dead chains formed over time. Polymer samples were observed for an additional 72 hours after approximately 3% of dead chains formed. Such an arbitrary number was chosen because a dead chain fraction below 2% makes it challenging to distinguish the small RI signal from the baseline (Fig. S6–S8†). Higher light intensities led to faster formation of dead chains, which is reasonable, as higher radical concentration increases the likelihood of radical cross-termination. However, interestingly, the formation of dead chains increased linearly with time in the observed timeframe, regardless of intensity. | ||
| Fig. 4 (a) Pseudo-first order kinetic plot of photoiniferter-RAFT polymerization conducted under different light intensities. (b) Plot of the dead chain fraction of samples irradiated under different light intensities. | ||
Conclusion
In summary, we investigated the formation of dead chains during and after the photoiniferter-RAFT polymerization of poly(methyl acrylate). Using GPC, TG-HPLC, and 1H NMR, we successfully demonstrated the synthesis of poly(methyl acrylate) with minimal dead chain formation (<2%) with exceptionally low dispersity. We quantified dead chain formation after monomer depletion and observed a previously unreported phenomenon: the fraction of dead chains increased linearly over time. HPLC fractionation of the dead chains, followed by GPC reanalysis, revealed termination products with molecular weights both similar to and higher than those of the living chains. Furthermore, we found that dead chain formation was influenced by light intensity, with higher-intensity irradiation accelerating the process at low monomer concentrations. This study quantitatively demonstrated the significance of identifying the optimal reaction conditions, such as monomer conversion, light intensity and irradiation time, to minimize dead chain formation.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI.† All data may be obtained from the corresponding author upon request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Samsung Research Funding & Incubation Center of Samsung Electronics under project number SRFC-MA2101-05 and the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (RS-2024-00354184). The authors are thankful to Dr Yungyeong Lee (Department of Materials Science and Engineering, Seoul National University) for her constructive suggestions and comments on this research work.References
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, From precision polymers to complex materials and systems, Nat. Rev. Mater., 2016, 1(5), 16024 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, Controlled/“living” radical polymerization. atom transfer radical polymerization in the presence of transition-metal complexes, J. Am. Chem. Soc., 1995, 117(20), 5614–5615 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, et al., Living Free-Radical Polymerization by Reversible Addition−Fragmentation Chain Transfer: The RAFT Process, Macromolecules, 1998, 31 (16), 5559–5562 CrossRef.
- Y. Lee, C. Boyer and M. S. Kwon, Photocontrolled RAFT polymerization: past, present, and future, Chem. Soc. Rev., 2023, 52(9), 3035–3097 RSC.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, A comparison of RAFT and ATRP methods for controlled radical polymerization, Nat. Rev. Chem, 2021, 5(12), 859–869 CrossRef CAS PubMed.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Reversible-deactivation radical polymerization (Controlled/living radical polymerization): From discovery to materials design and applications, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- Y. Song, Y. Kim, Y. Noh, V. K. Singh, S. K. Behera, A. Abudulimu, K. Chung, R. Wannemacher, J. Gierschner and L. Lüer, et al., Organic Photocatalyst for ppm-Level Visible-Light-Driven Reversible Addition–Fragmentation Chain-Transfer (RAFT), Polymerization with Excellent Oxygen Tolerance, Macromolecules, 2019, 52(15), 5538–5545 CrossRef CAS.
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel and L. S. Park, et al., Highly efficient organic photocatalysts discovered via a computer-aided-design strategy for visible-light-driven atom transfer radical polymerization, Nat. Catal., 2018, 1 (10), 794–804 CrossRef.
- M. Chen, M. Zhong and J. A. Johnson, Light-Controlled Radical Polymerization: Mechanisms, Methods, and Applications, Chem. Rev., 2016, 116(17), 10167–10211 CrossRef CAS PubMed.
- T. Otsu, M. Yoshida and T. Tazaki, A model for living radical polymerization, Die Makromol. Chem., Rapid Commun., 1982, 3(2), 133–140 CrossRef CAS.
- T. Otsu and A. Kuriyama, Polymer Design by Iniferter Technique in Radical Polymerization: Synthesis of AB and ABA Block Copolymers Containing Random and Alternating Copolymer Sequences, Polym. J., 1985, 17(1), 97–104 CrossRef CAS.
- Q. Fu, T. G. McKenzie, S. Tan, E. Nam and G. G. Qiao, Tertiary amine catalyzed photo-induced controlled radical polymerization of methacrylates, Polym. Chem., 2015, 6(30), 5362–5368 RSC.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Visible Light Mediated Controlled Radical Polymerization in the Absence of Exogenous Radical Sources or Catalysts, Macromolecules, 2015, 48(12), 3864–3872 CrossRef CAS.
- J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, Catalyst-Free Visible Light-Induced RAFT Photopolymerization, in Controlled Radical Polymerization: Mechanisms, ACS Symposium Series, American Chemical Society, 2015, vol. 1187, pp. 247–267 Search PubMed.
- M. Hartlieb, Photo-Iniferter RAFT Polymerization, Macromol. Rapid Commun., 2022, 43(1), e2100514 CrossRef PubMed.
- R. W. Hughes, M. E. Lott, J. I. Bowman and B. S. Sumerlin, Excitation Dependence in Photoiniferter Polymerization, ACS Macro Lett., 2023, 12(1), 14–19 CrossRef CAS PubMed.
- D. A. Corbin and G. M. Miyake, Photoinduced Organocatalyzed Atom Transfer Radical Polymerization (O-ATRP): Precision Polymer Synthesis Using Organic Photoredox Catalysis, Chem. Rev., 2022, 122(2), 1830–1874 CrossRef CAS PubMed.
- R. W. Hughes, M. E. Lott, R. A. Olson S and B. S. Sumerlin, Photoiniferter polymerization: Illuminating the history, ascendency, and renaissance, Prog. Polym. Sci., 2024, 156, 101871 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, RAFTing down under: Tales of missing radicals, fancy architectures, and mysterious holes, J. Polym. Sci., Part A: Polym. Chem., 2003, 41(3), 365–375 CrossRef CAS.
- Q. Ma, G. G. Qiao and Z. An, Visible Light Photoiniferter Polymerization for Dispersity Control in High Molecular Weight Polymers, Angew. Chem., Int. Ed., 2023, 62(48), e202314729 CrossRef CAS PubMed.
- A.-C. Lehnen, J. A. M. Kurki and M. Hartlieb, The difference between photo-iniferter and conventional RAFT polymerization: high livingness enables the straightforward synthesis of multiblock copolymers, Polym. Chem., 2022, 13(11), 1537–1546 RSC.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Ultra-High Molecular Weights via Aqueous Reversible-Deactivation Radical Polymerization, Chem, 2017, 2(1), 93–101 CAS.
- M. E. Lott, L. Trachsel, E. Schué, C. L. G. Davidson IV, R. A. Olson S, D. I. Pedro, F. Chang, Y. Hong, W. G. Sawyer and B. S. Sumerlin, Ultrahigh-Molecular-Weight Triblock Copolymers via Inverse Miniemulsion Photoiniferter Polymerization, Macromolecules, 2024, 57(9), 4007–4015 CrossRef CAS.
- C. P. Easterling, Y. Xia, J. Zhao, G. E. Fanucci and B. S. Sumerlin, Block Copolymer Sequence Inversion through Photoiniferter Polymerization, ACS Macro Lett., 2019, 8(11), 1461–1466 CrossRef CAS PubMed.
- T. Gruendling, M. Kaupp, J. P. Blinco and C. Barner-Kowollik, Photoinduced Conjugation of Dithioester- and Trithiocarbonate-Functional RAFT Polymers with Alkenes, Macromolecules, 2011, 44(1), 166–174 CrossRef CAS.
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Synthesis of Discrete Oligomers by Sequential PET–RAFT Single–Unit Monomer Insertion, Angew. Chem., Int. Ed., 2017, 56(29), 8376–8383 CrossRef CAS PubMed.
- C. Fu, Z. Huang, C. J. Hawker, G. Moad, J. Xu and C. Boyer, RAFT-mediated, visible light-initiated single unit monomer insertion and its application in the synthesis of sequence-defined polymers, Polym. Chem., 2017, 8(32), 4637–4643 RSC.
- W. He, W. Tao, Z. Wei, G. Tong, X. Liu, J. Tan, S. Yang, J. Hu, G. Liu and R. Yang, Controlled switching thiocarbonylthio end-groups enables interconvertible radical and cationic single-unit monomer insertions and RAFT polymerizations, Nat. Commun., 2024, 15(1), 5071 CrossRef CAS PubMed.
- K. Kiani, D. J. T. Hill, F. Rasoul, M. Whittaker and L. Rintoul, Raft mediated surface grafting of t–butyl acrylate onto an ethylene–propylene copolymer initiated by gamma radiation, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(6), 1074–1083 CrossRef CAS.
- M. Barsbay and O. Güven, RAFT mediated grafting of poly(acrylic acid) (PAA) from polyethylene/polypropylene (PE/PP) nonwoven fabric via preirradiation, Polymer, 2013, 54(18), 4838–4848 CrossRef CAS.
- J. H. Wu, Z. Lan, J. M. Lin, M. L. Huang, S. C. Hao, T. Sato and S. Yin, A Novel Thermosetting Gel Electrolyte for Stable Quasi–Solid–State Dye–Sensitized Solar Cells, Adv. Mater., 2007, 19(22), 4006–4011 CrossRef CAS.
- A. Bagheri, H. Arandiyan, N. N. M. Adnan, C. Boyer and M. Lim, Controlled Direct Growth of Polymer Shell on Upconversion Nanoparticle Surface via Visible Light Regulated Polymerization, Macromolecules, 2017, 50(18), 7137–7147 CrossRef CAS.
- C. Yu, J. Choi, J. Lee, S. Lim, Y. Park, S. M. Jo, J. Ahn, S. Y. Kim, T. Chang, C. Boyer and M. S. Kwon, Functional thermoplastic polyurethane elastomers with α, ω-hydroxyl end-functionalized polyacrylates, Adv. Mater., 2024, 36(40), 2403048 CrossRef CAS PubMed.
- C. Hwang, S. Shin, D. Ahn, H.-J. Paik, W. Lee and Y. Yu, Realizing Cross-linking-free Acrylic Pressure-Sensitive Adhesives with Intensive Chain Entanglement through Visible-Light-Mediated Photoiniferter-Reversible Addition–Fragmentation Chain-Transfer Polymerization, ACS Appl. Mater. Interfaces, 2023, 15(50), 58905–58916 CrossRef CAS PubMed.
- L. E. Diodati, A. J. Wong, M. E. Lott, A. G. Carter and B. S. Sumerlin, Unraveling the Properties of Ultrahigh Molecular Weight Polyacrylates, ACS Appl. Polym. Mater., 2023, 5(12), 9714–9720 CrossRef CAS.
- M. Chen, Y. Gu, A. Singh, M. Zhong, A. M. Jordan, S. Biswas, L. T. Korley, A. C. Balazs and J. A. Johnson, Living Additive Manufacturing: Transformation of Parent Gels into Diversely Functionalized Daughter Gels Made Possible by Visible Light Photoredox Catalysis, ACS Cent. Sci., 2017, 3(2), 124–134 CrossRef CAS PubMed.
- H. Zhou and J. A. Johnson, Photo-controlled growth of telechelic polymers and end-linked polymer gels, Angew. Chem., Int. Ed., 2013, 52(8), 2235–2238 CrossRef CAS PubMed.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Repeatable photoinduced self-healing of covalently cross-linked polymers through reshuffling of trithiocarbonate units, Angew. Chem., Int. Ed., 2011, 50(7), 1660–1663 CrossRef CAS PubMed.
- H. Zhou and J. A. Johnson, Photo–controlled Growth of Telechelic Polymers and End–linked Polymer Gels, Angew. Chem., 2013, 125(8), 2291–2294 CrossRef.
- T. G. McKenzie, L. P. D. M. Costa, Q. Fu, D. E. Dunstan and G. G. Qiao, Investigation into the photolytic stability of RAFT agents and the implications for photopolymerization reactions, Polym. Chem., 2016, 7(25), 4246–4253 RSC.
- Y. Nakamura, R. Lee, M. L. Coote and S. Yamago, Termination Mechanism of the Radical Polymerization of Acrylates, Macromol. Rapid Commun., 2016, 37(6), 506–513 CrossRef CAS PubMed.
- T. G. Ribelli, K. F. Augustine, M. Fantin, P. Krys, R. Poli and K. Matyjaszewski, Disproportionation or Combination? The Termination of Acrylate Radicals in ATRP, Macromolecules, 2017, 50(20), 7920–7929 CrossRef CAS.
- T. L. Grover and C. A. Guymon, Regulating copolymer architecture using photoiniferter polymerization to direct composite morphology, Polymer, 2023, 280, 126036 CrossRef CAS.
- K. Kim, J. Ahn, M. Park, H. Lee, Y. J. Kim, T. Chang, H. B. Jeon and H.-J. Paik, Molecular-Weight Distribution of Living Chains in Polystyrene Prepared by Reversible Addition–Fragmentation Chain-Transfer Polymerization, Macromolecules, 2019, 52(19), 7448–7455 CrossRef CAS.
- T. Chang, Polymer characterization by interaction chromatography, J. Polym. Sci., Part B: Polym. Phys., 2005, 43(13), 1591–1607 CrossRef CAS.
- W. Lee, H. C. Lee, T. Chang and S. B. Kim, Characterization of Poly(methyl methacrylate) by Temperature Gradient Interaction Chromatography with On-Line Light Scattering Detection, Macromolecules, 1998, 31(2), 344–348 CrossRef CAS.
- N. Ballard, S. Hamzehlou, F. Ruipérez and J. M. Asua, On the Termination Mechanism in the Radical Polymerization of Acrylates, Macromol. Rapid Commun., 2016, 37(16), 1364–1368 CrossRef CAS PubMed.
- H. Fischer, The Persistent Radical Effect In “Living”, Radical Polymerization, Macromolecules, 1997, 30(19), 5666–5672 CrossRef CAS.
- M. A. Beres, J. Y. Rho, A. Kerr, T. Smith and S. Perrier, Photoiniferter-RAFT polymerization mediated by bis(trithiocarbonate) disulfides, Polym. Chem., 2024, 15(6), 522–533 RSC.
- D. Leifert and A. Studer, The Persistent Radical Effect in Organic Synthesis, Angew. Chem., Int. Ed., 2020, 59(1), 74–108 CrossRef CAS PubMed.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Reversible Addition−Fragmentation Chain Transfer Polymerization Initiated with Ultraviolet Radiation, Macromolecules, 2002, 35(20), 7620–7627 CrossRef CAS.
- A. Theis, A. Feldermann, N. Charton, M. H. Stenzel, T. P. Davis and C. Barner-Kowollik, Access to Chain Length Dependent Termination Rate Coefficients of Methyl Acrylate via Reversible Addition−Fragmentation Chain Transfer Polymerization, Macromolecules, 2005, 38(7), 2595–2605 CrossRef CAS.
- J. Chiefari, J. Jeffery, R. T. A. Mayadunne, G. Moad, E. Rizzardo and S. H. Thang, Chain Transfer to Polymer: A Convenient Route to Macromonomers, Macromolecules, 1999, 32(22), 7700–7702 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00151j |
| This journal is © The Royal Society of Chemistry 2025 |