
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transparent diblock copolymer nanoparticle dispersions via efficient RAFT emulsion polymerisation in ionic liquid†
Anisha
Patel
 a,
Georgia L.
Maitland
a,
Evelina
Liarou
b,
Paul D.
Topham
a and
Matthew J.
Derry
*a
a,
Georgia L.
Maitland
a,
Evelina
Liarou
b,
Paul D.
Topham
a and
Matthew J.
Derry
*a
aAston Institute for Membrane Excellence, Aston University, Birmingham, B4 7ET, UK. E-mail: m.derry@aston.ac.uk
bDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
First published on 14th May 2025
Abstract
We report the first reversible addition–fragmentation chain transfer polymerisation-induced self-assembly (RAFT-PISA) in ionic liquid (IL) that proceeds under emulsion conditions. Moreover, this formulation exploits refractive index contrast matching to generate highly transparent nanoparticle dispersions. Specifically, 1-ethyl-3-methyl-imidazolium ethylsulfate, [EMIM][EtOSO3], was used as the solvent for the chain extension of poly(2-hydroxyethyl methacrylate) (PHEMA) macromolecular chain transfer agents (macro-CTAs) using n-butyl methacrylate (BuMA) via RAFT emulsion polymerisation. Two series of PHEMAx-b-PBuMAy diblock copolymers with target PBuMA degrees of polymerisation (DPs) varying from 50 to 1000 were synthesised using either a PHEMA21 or PHEMA77 macro-CTA. All resulting nanoparticle dispersions yielded highly transparent dispersions, even when nanoparticle diameters exceeded 100 nm, due to the closely matched refractive index values of the [EMIM][EtOSO3] solvent and PBuMA nanoparticle core. Detailed analysis using small-angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) confirmed the presence of spherical nanoparticles. Furthermore, the synthesis of PHEMA-b-PBuMA via this new PISA formulation was directly compared to equivalent block copolymer syntheses conducted in N,N-dimethylformamide (DMF) or ethanol/water mixtures. It was found that syntheses conducted in [EMIM][EtOSO3] resulted in the highest monomer conversions (up to >99%) and lowest dispersity (ĐM) values (as low as 1.16) in the shortest reaction times (2 hours) compared to the other solvent systems. This work demonstrates the use of ILs as a more sustainable and effective solvent for RAFT–PISA via the development of the first emulsion PISA formulation in IL.
Introduction
Ionic liquids (ILs) are salts that typically exhibit melting points below 100 °C.1,2 In contrast to conventional solvents, ILs have many unique and advantageous physicochemical properties including non-flammability, non-volatility, high ionic conductivity, recyclability and good thermal stability.3 This, combined with the vast array of possible anion and cation combinations, allows for numerous applications of ILs as solvents in catalysis and electrochemistry,4–6 as well as their ability to function as performance additives such as anti-static7,8 and dispersing agents.9,10 ILs serve as an environmentally friendly alternative to traditional organic solvents and have become increasingly popular,11,12 with their application also extending to the field of polymer chemistry.13–15 Previous work has reported that radical polymerisations in ILs occur faster and yield higher monomer conversions than those conducted in traditional organic solvents.16,17 This is attributed to factors such as increased viscosity and polarity of the system, which reduces the rate of termination and increases propagation rates.18,19 ILs also provide the potential for solvent reuse and recyclability, where monomer conversion in polymerisations conducted in recycled solvent was found to be unaffected.20 Additionally, it has been demonstrated that block copolymers can self-assemble into well-defined nanostructures in ILs.2,21Over the past 20 years, reversible addition–fragmentation chain transfer (RAFT) polymerisation has become a widely used reversible-deactivation radical polymerisation (RDRP) technique emplyed to synthesise well-defined block copolymers,22,23 which can be used in a broad range of applications such as inkjet printing,24 optoelectronics25,26 and drug delivery.27,28 Self-assembly of such block copolymers allows for the formation of different nanomorphologies in solution, however post-polymerisation processing such as solvent or pH switching and film rehydration is often required.29,30 Polymerisation-induced self-assembly (PISA) eliminates the requirement of these processing steps and enables syntheses to be conducted at higher concentrations (up to 50% w/w).31 The PISA process typically involves the chain extension of a solvophilic macromolecular chain transfer agent (macro-CTA) using a monomer that will form an insoluble polymer structure-directing block. This promotes the in situ formation of a range of sterically stabilised nano-objects, mainly spheres, worms or vesicles, due to the self-assembly of the AB diblock copolymer chains.32 PISA reactions can proceed via either dispersion30,33–40 or emulsion41–43 polymerisation, where the added monomer is miscible or immiscible with the solvent, respectively. Combining RAFT with PISA leads to a more versatile technique for the preparation of block copolymer nanoparticle dispersions, and RAFT-mediated PISA is frequently employed due to its high compatibility with monomers, solvents (polar solvents including water, or non-polar media44) and reaction conditions.45–47
There are many reports of RAFT-mediated PISA in the literature, however there are very few reports of PISA syntheses being conducted in ILs. This type of reaction was first reported by Zhang and Zhu39 whereby a trithiocarbonate-terminated poly(ethylene glycol) (PEG) macro-CTA was chain extended using styrene (St), n-butyl methacrylate (BuMA) or 2-hydroxyethyl methacrylate (HEMA) in the imidazolium-based IL, 1-butyl-3-methylimidazolium hexafluorophosphate, [BMIM][PF6]. This study demonstrated the use of IL as a solvent for RAFT dispersion PISA to form spherical nanoparticles (spheres and vesicles), as confirmed by transmission electron microscopy (TEM). Zhou et al.40 progressed this work by focusing on the effect of solvent on the synthesis of PEG-b-PSt diblock copolymers when employing [EMIM][PF6], methanol and methanol/water mixtures. Again, TEM successfully showed the formation of spherical nano-objects (spherical and vesicular morphologies). Additionally, it was shown that using IL as a solvent increased the rate of polymerisation, with 95% monomer conversion being achieved in 12 hours, whereas syntheses conducted in methanol or methanol/water mixtures only achieved 20% and 65%, respectively, for the same duration. Yamanaka et al.38 recently reported the first use of PISA to produce IL-based gels during the synthesis of ABA triblock copolymers in IL. In situ small-angle X-ray scattering (SAXS) and rheological studies demonstrated that after the initial induction period, the gelation process occurs alongside self-assembly due to the formation of interconnected spherical domains. Subsequently, Yamanaka et al.48 used functionalised PEG star polymers to polymerise St in [EMIM][PF6] using PISA to produce a gel. Hexagonally packed cylindrical (HEX) and hexagonal close-packed spherical (HCP) structures were also obtained from 2-arm PEG–PSt and 4/8-arm PEG–PSt, respectively.48 Maitland et al.37 recently established the synthesis of AB diblock copolymers via RAFT dispersion PISA in IL as a direct route to worm gels in IL, or worm ionogels. In this prior study, a PHEMA macro-CTA was successfully chain extended with benzyl methacrylate (BzMA) in 1-ethyl-3-methylimidazolium dicyanamide, [EMIM][DCA], to form worm-like nanoparticles that yield free-standing ionogels as confirmed using SAXS, TEM and rheology, while electrochemical studies demonstrated significant potential for use in energy storage applications. It should be noted that all reports of RAFT-mediated PISA conducted in ILs to date are examples of dispersion polymerisations.
The refractive index of a material can be defined as a dimensionless number related to the speed of light when it passes through a dielectric medium.49 Snell's law states that when light travels between two media with the same refractive index values, no refraction will occur,50 which results in optical transparency. This phenomenon has been utilised in the synthesis of highly transparent nanoparticle dispersions via PISA, and was first demonstrated by Semsarilar et al.51 by utilising the small difference in refractive index values of ethanol compared to the structure-directing poly(2,2,2-trifluoroethyl methacrylate) (PTFEMA). Other examples include work conducted by György et al.,33 who found that transparent nanoparticle dispersions could be generated only at temperatures for which the refractive indices of the n-alkane solvent and structure-directing polymer block were equivalent. In addition to this, Rymaruk et al.41 has also demonstrated isorefractivity in Pickering emulsions stabilised by anisotropic and spherical block copolymer nanoparticles. All of these examples made use of a fluorinated monomer to yield the polymer whose refractive index matches that of the solvent, whereas the focus of this work employs non-fluorinated monomers in an attempt to improve sustainability. The development of an emulsion-based polymerisation formulation that yields transparent refractive index matched dispersions using non-fluorinated monomers in IL would represent an advancement in the PISA field and may further demonstrate that ILs provide many advantages over traditional solvents (e.g. decreased reaction time, reduced flammability).
Herein, we report a RAFT–PISA emulsion formulation in IL for the first time to yield transparent diblock copolymer nanoparticle dispersions. Specifically, poly(2-hydroxyethyl methacrylate)x-b-poly(n-butyl methacrylate)y (PHEMAx-b-PBuMAy) diblock copolymer nanoparticles were synthesised via RAFT emulsion polymerisation in 1-ethyl-3-methylimidazolium ethyl sulfate, [EMIM][EtOSO3] (Scheme 1). Detailed characterisation was conducted, including 1H nuclear magnetic resonance (NMR) spectroscopy, gel permeation chromatography (GPC), dynamic light scattering (DLS), transmission electron microscopy (TEM), small-angle X-ray scattering (SAXS) and ultraviolet-visible light (UV-vis) spectroscopy. This new PISA formulation enables the formation of transparent dispersions containing spherical micelles up to ∼100 nm in diameter using non-fluorinated monomers without the requirement for organic co-solvents and post-polymerisation processing or purification.
 | ||
| Scheme 1 Synthesis of poly(2-hydroxyethyl methacrylate) (PHEMA) macro-CTA via RAFT solution polymerisation in methanol at 60 °C, followed by RAFT emulsion polymerisation of n-butyl methacrylate (BuMA) in 1-ethyl-3-methylimidazolium ethyl sulfate ([EMIM][EtOSO3]) at 70 °C to yield PHEMAx-b-PBuMAy diblock copolymers. | ||
Experimental
Materials
2-Hydroxyethyl methacrylate (HEMA) and n-butyl methacrylate (BuMA) were purchased from Sigma Aldrich and passed through a basic alumina column prior to use in order to remove the inhibitor. 2,2′-Azobisisobutyronitrile (AIBN) was purchased from Molekula and was recrystallised from methanol prior to use. 4-Cyano-4-(phenylcarbonothioylthio) pentanoic acid (CPTP) RAFT agent and 1-ethyl-3-methylimidazolium ethyl sulfate ([EMIM][EtOSO3]) were purchased from Sigma Aldrich and used as received. Reagent grade methanol, diethyl ether and N,N-dimethylformamide were purchased from Fisher Scientific. Dimethyl sulfoxide-d6 and chloroform-d for 1H NMR analysis were purchased from Cambridge Isotope Laboratories.Synthesis of poly(2-hydroxyethyl methacrylate) macromolecular chain transfer agent via RAFT solution polymerisation
The synthesis of the PHEMA21 macro-CTA at 50% w/w solids was conducted as follows. A 100 mL round-bottomed flask was charged with 2-hydroxyethyl methacrylate (HEMA; 10.0 g; 76.8 mmol), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPTP; 1.07 g; 3.84 mmol), 2,2′-azobisisobutyronitrile (AIBN; 126 mg; 768 μmol; CPTP/AIBN molar ratio = 5.0) and methanol (14.2 g). The sealed reaction flask was purged with nitrogen for 30 minutes prior to being placed in a preheated aluminium metal block at 60 °C and stirred for 6 hours. The resulting PHEMA (HEMA conversion = 67% (Fig. S1†), Mn = 3800 g mol−1, ĐM = Mw/Mn = 1.34) was purified by twice precipitating into a ten-fold excess of diethyl ether and dried on a rotary evaporator until all unreacted monomer was removed as judged by 1H NMR spectroscopy (see Fig. S2†). The resulting PHEMA macro-CTA was obtained as a pink solid. The mean degree of polymerisation (DP) of this macro-CTA was calculated to be 21 using 1H NMR spectroscopy by comparing the integrated signals corresponding to the five CPTP aromatic protons at 7.2–8.0 ppm relative to the peak at 4.0–4.1 ppm corresponding to the two oxymethylene protons of PHEMA (see Fig. S2†).Synthesis of poly(2-hydroxyethyl methacrylate)-block-poly(n-butyl methacrylate) (PHEMA-b-PBuMA) diblock copolymer via RAFT emulsion polymerisation in 1-ethyl-3-methylimidazolium ethyl sulfate
A typical RAFT emulsion polymerisation for the synthesis of PHEMA21-b-PBuMA96 at 10% w/w solids was conducted as follows. n-Butyl methacrylate (BuMA; 0.49 g; 3.45 mmol), 2,2′-azobisisobutyronitrile (AIBN; 2.8 mg; 24.3 μmol), PHEMA21 macro-CTA (0.10 g; 34.5 μmol; macro-CTA/initiator molar ratio = 2.0; PBuMA target DP = 100) and 1-ethyl-3-methylimidazolium ethyl sulfate ([EMIM][EtOSO3]; 5.38 g) were added to a 14 mL sample vial. The sealed reaction mixture was purged with nitrogen for 30 minutes prior to being placed in a preheated aluminium metal block at 70 °C whilst stirring for 2 hours (BuMA conversion = 96% (Fig. S5 and S6†); Mn = 7000 g mol−1, ĐM = 1.26).Synthesis of poly(2-hydroxyethyl methacrylate)-block-poly(n-butyl methacrylate) (PHEMA-b-PBuMA) diblock copolymer via RAFT solution polymerisation in N,N-dimethylformamide
A typical RAFT solution polymerisation for the synthesis of PHEMA21-b-PBuMA87 at 10% w/w solids was conducted as follows. n-Butyl methacrylate (BuMA; 0.47 g; 3.32 mmol), 2,2′-azobisisobutyronitrile (AIBN; 2.7 mg; 16.6 μmol), PHEMA21 macro-CTA (0.10 g; 33.2 μmol; macro-CTA/initiator molar ratio = 2.0; PBuMA target DP = 100) and N,N-dimethylformamide (DMF; 5.17 g) were added to a 14 mL sample vial. The sealed reaction mixture was purged with nitrogen for 30 minutes prior to being placed in a preheated aluminium metal block at 70 °C whilst stirring for 25 hours (BuMA conversion = 87%; Mn = 5900 g mol−1, ĐM = 1.59).Synthesis of poly(2-hydroxyethyl methacrylate)-block-poly(n-butyl methacrylate) (PHEMA-b-PBuMA) diblock copolymer via RAFT dispersion polymerisation in an ethanol–water mixture
A typical RAFT dispersion polymerisation for the synthesis of PHEMA21-b-PBuMA70 at 10% w/w solids was conducted as follows. n-Butyl methacrylate (BuMA; 0.57 g; 3.98 mmol), 2,2′-azobisisobutyronitrile (AIBN; 0.7 mg; 3.98 μmol), PHEMA21 macro-CTA (0.02 g; 7.97 μmol; macro-CTA/initiator molar ratio = 2.0; PBuMA target DP = 100), ethanol (EtOH; 4.26 g) and water (H2O; 1.06 g) were added to a 14 mL sample vial. The sealed reaction mixture was purged with nitrogen for 30 minutes prior to being placed in a preheated oil bath at 70 °C whilst stirring for 24 hours (BuMA conversion = 70%; Mn = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1, ĐM = 1.51).
800 g mol−1, ĐM = 1.51).
1H NMR spectroscopy
1H NMR spectra were obtained in either DMSO-d6 or CDCl3-d using a Bruker Avance Neo 300 MHz spectrometer. Typically, 16 scans were averaged per spectrum and all chemical shifts are expressed in ppm. 1,3,5-Trioxane (5 mol% with respect to BuMA monomer) was added to the reaction mixture for all block copolymer samples as an internal standard to facilitate the calculation of monomer conversion.Gel permeation chromatography
Molecular weight distributions were obtained by using an Agilent Infinity II gel permeation chromatography (GPC) instrument comprising a guard column and two PL gel mixed-C columns. The mobile phase contained 0.10% w/v LiBr in HPLC grade DMF and the flow rate was fixed at 1 mL min−1 at 80 °C. The GPC was calibrated using near-monodispersed poly(methyl methacrylate) standards (Mp range = 535–1591000 g mol−1).
Dynamic light scattering
Dynamic light scattering (DLS) studies were conducted using a Zetasizer Nano ZS instrument (Malvern Panalytical, UK) at a fixed scattering angle of 173°. The block copolymer dispersions were diluted in [EMIM][EtOSO3] (refractive index = 1.48 as determined by A. P. Fröba et al.,52 viscosity = 94.2 cP) to 0.10% w/w prior to light scattering studies at 25 °C. The polydispersity index (PDI) and average diameter (D) were calculated by cumulants analysis of the experimental correlation function using Dispersion Technology Software version 6.20, and data were averaged over three sets of approximately thirteen runs each of 30 seconds duration.Transmission electron microscopy
Bright field transmission electron microscopy (TEM) imaging was conducted using a JEOL2100 microscope operating at 200 kV. Prior to analysis, block copolymer dispersions were diluted with [EMIM][EtOSO3] to 0.10% w/w and 10 μL of this solution was deposited on lacey carbon coated copper grids, blotted using filter paper and allowed to dry for 4 days at ambient conditions. No staining agent was used. The images were analysed using ImageJ.Small-angle X-ray scattering
Small-angle X-ray scattering (SAXS) patterns were recorded for 1.0% w/w copolymer dispersions in [EMIM][EtOSO3] in 1.5 mm diameter polycarbonate capillaries at a synchrotron source (beamline B21,53 Diamond Light Source, UK) using monochromatic X-ray radiation (X-ray wavelength λ = 0.9464 Å, sample-to-detector distance of 3.685 m corresponding to scattering vector q ranging from 0.0045 to 0.34 Å−1) and an EigerX 4M detector (Dectris, Switzerland). Scattering data were reduced using standard protocols from the beamline and were further analysed using Irena SAS macros for Igor Pro. Background-subtracted SAXS data were fitted to a spherical micelle model54 (see ESI† for detailed information of models and fitting summaries).Ultraviolet–visible spectroscopy
Ultraviolet–visible (UV-Vis) spectra were recorded in absorbance mode between 800 and 400 nm, with a spectral resolution of 1 nm, for 10% w/w block copolymer dispersions/solutions synthesised in [EMIM][EtOSO3], DMF and EtOH/H2O mixtures, using an Implen NanoPhotometer® C40.Results and discussion
Synthesis of PHEMA macromolecular chain transfer agent
PHEMA was deemed a suitable macromolecular chain transfer agent (macro-CTA) due to its solubility in a wide range of solvents including DMF, EtOH/H2O mixtures and ILs, in particular [EMIM][EtOSO3]. Thus, it was considered a good candidate as the solvophilic stabiliser block in subsequent PISA syntheses. In preparation for these PISA reactions, the synthesis of a PHEMA macro-CTA was conducted via RAFT solution polymerisation in methanol at 60 °C using 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPTP) as a chain transfer agent (Scheme 1). The reaction was quenched after 6 hours, resulting in a final HEMA monomer conversion of 67% as judged by 1H NMR spectroscopy (Fig. S1†). This intermediate monomer conversion was deliberately targeted in order to preserve the end group functionality55 and enable use of this homopolymer as a macro-CTA. 1H NMR spectroscopy was used to determine the mean degree of polymerisation (DP) by end-group analysis, which was calculated to be 21 (Fig. S2†). The macro-CTA was analysed using GPC to obtain the number-average molecular weight (Mn) and dispersity (ĐM), which were found to be 3800 g mol−1 and 1.34, respectively. This procedure was repeated to synthesise an additional PHEMA macro-CTA, this time targeting a higher DP, resulting in a PHEMA77 as judged by 1H NMR spectroscopy (Fig. S3 and S4†). For this macro-CTA, an Mn of 9500 g mol−1 and a ĐM of 1.30 were obtained from GPC analysis, thus confirming good control over the RAFT polymerisation.Synthesis of PHEMA-b-PBuMA block copolymers via RAFT emulsion polymerisation
Initially, the kinetics for the chain extension of the PHEMA21 macro-CTA using BuMA were investigated to identify the optimum reaction duration to achieve ≥90% monomer conversion. This kinetic study was conducted for 2 hours at 70 °C using [EMIM][EtOSO3] as the solvent with a total solids concentration of 10% w/w with a target PBuMA DP of 100 (Fig. 1). Since BuMA is immiscible with [EMIM][EtOSO3] and PBuMA is insoluble in said IL, this reaction proceeds as an emulsion polymerisation.56 Throughout the kinetics study, samples were withdrawn every 5 minutes for the first 60 minutes, and every 10 minutes for the remaining duration of the reaction. An internal standard, 1,3,5-trioxane, was used to enable monitoring of the reduction in intensity of the BuMA vinyl proton signals in the 1H NMR spectrum and thus determine monomer conversion (Fig. S5–S8†). Due to difficulties arising from the two-phase nature of the emulsion system, early sample collection was found to be unreliable, and therefore kinetic data have been plotted beginning from 10 minutes (Fig. 1a). Analysis revealed that full monomer conversion was achieved after approximately 60 minutes. Furthermore, the time at which micellar nucleation occurs was observed to be approximately 36 minutes, which corresponds to a PBuMA DP of 58. This critical DP reflects the length of the PBuMA structure-directing block which triggers spontaneous self-assembly, which is observed by an approximate seven-fold increase in rate of reaction (Fig. 1a). In addition to this, both the linear increase of Mn with monomer conversion and the maintained low dispersity values demonstrate good RAFT control of the system (Fig. 1b). The success of the chain extension can be confirmed using GPC analysis, where a clear gradual shift of the distribution to the higher molecular weight is observed (Fig. 1c). The extension of the PHEMA77 macro-CTA, again targeting a PBuMA DP of 100, showed similar results (Fig. S9†). In the case of PHEMA77-b-PBuMAy in [EMIM][EtOSO3], the critical PBuMA DP for self-assembly was determined to be 40. Based on these kinetic analyses, the optimal polymerisation time for complete monomer conversion during the RAFT emulsion polymerisation of BuMA in [EMIM][EtOSO3] using the PHEMA macro-CTAs was identified as 2 hours. Subsequently, a series of PHEMAx-b-PBuMAy diblock copolymers with targeted PBuMA DPs varying from 50 to 1000 were synthesised using both the PHEMA21 and PHEMA77 macro-CTAs. | ||
| Fig. 1 Kinetic study for the RAFT emulsion polymerisation of BuMA, targeting a degree of polymerisation of 100, in [EMIM][EtOSO3] at 10% w/w solids using a PHEMA21 macro-CTA: (a) BuMA conversion vs. time (blue circles) and semi-log kinetic (red squares); (b) Mn (blue circles) and ĐM (red squares) vs. BuMA conversion; (c) DMF GPC chromatograms. GPC data were obtained against poly(methyl methacrylate) standards. | ||
In the RAFT emulsion polymerisations, high BuMA conversions were achieved using both PHEMA macro-CTAs when targeting PBuMA DPs up to 300, above which longer reaction times were required to achieve approximately 90% conversion (see Table S1†). For comparison, synthesis of PHEMA-b-PBuMA was also conducted in DMF, as a solution polymerisation, using each of the PHEMA21 and PHEMA77 macro-CTAs, targeting a PBuMA DP of 100 with a reaction time of 25 hours. In DMF, using the PHEMA21 macro-CTA resulted in a BuMA monomer conversion of 87%, whereas using PHEMA77 achieved a lower conversion of 69% (Table S2 and Fig. S10†). This data indicates that reactions conducted in DMF not only result in lower monomer conversion but also much slower reaction rates compared to those in IL, with the polymerisation rate in IL increasing ten-fold. The use of ILs as a more suitable solvent over traditional organic solvents is further demonstrated by good control throughout the polymerisation process, generating a series of amphiphilic block copolymer nanoparticle dispersions (Table S1† and Fig. 2).
 | ||
| Fig. 2 DMF GPC data obtained for PHEMAx-b-PBuMAy block copolymers synthesised via RAFT emulsion polymerisation of n-butyl methacrylate in [EMIM][EtOSO3] at 10% w/w solids, where PHEMA is denoted as H and PBuMA is denoted as B. GPC data were obtained against poly(methyl methacrylate) standards. Chromatograms obtained for a selection of (a) PHEMA21-b-PBuMAy and (c) PHEMA77-b-PBuMAy block copolymers, with the corresponding plots of Mnvs. PBuMA DP (blue circles) and ĐM (red squares) vs. PBuMA DP for (b) PHEMA21-b-PBuMAy and (d) PHEMA77-b-PBuMAy. | ||
Analysis from GPC indicated lower dispersity values ranging between 1.15 and 1.32 for PHEMAx-b-PBuMAy synthesised in IL (Fig. 2), compared to those of 1.27–2.09 for equivalent diblock copolymers synthesised in DMF (see Table S2 and Fig. S11†). This is presumably due to the fact that longer polymerisation times are required to reach higher monomer conversions in DMF, meaning that more radicals are generated during the reaction and thus termination events are more prevalent.57 Successful chain extension using both PHEMA macro-CTAs was confirmed due to the notable shift in molecular weight to higher values when targeting increasing PBuMA DPs. It can be noted that the PHEMA21 macro-CTA (Fig. 2a) demonstrated more efficient chain extension than the PHEMA77 macro-CTA, which is evident from the low molecular weight shoulder in GPC traces when targeting PBuMA DPs ≥ 250, which is characteristic of unreacted macro-CTA (Fig. 2b). Despite this, the dispersity values for both series of block copolymers in IL remained low (≤1.32) when targeting PBuMA DPs up to 500 using PHEMA21 and 300 using PHEMA77, both sets of data exhibiting a linear evolution of Mn with increasing PBuMA DP. For higher targeted PBuMA DPs, it was observed that there was a deviation from the expected trend, indicating a loss of control in the synthesis, this has been previously observed in several PISA formulations.45
Transparent dispersions of PHEMA-b-PBuMA block copolymer nanoparticles in [EMIM][EtOSO3]
Most PISA formulations result in the formation of turbid dispersions due to the light scattering events caused by the presence of (sufficiently large) nanoparticles of different refractive index to that of the solvent. Optically transparent dispersions have received some attention as model colloidal systems to explore structure and dynamics,58 and such formulations have use in the paint and coatings industry.59,60 However, it was observed that all the resulting PHEMA-b-PBuMA dispersions in [EMIM][EtOSO3] presented in this work were optically transparent (see Fig. 3a and b), despite clear evidence for polymerisation-induced self-assembly, and thus the formation of block copolymer nanoparticles, from the kinetics studies presented earlier. This observation is particularly interesting given that initial reaction mixtures were turbid prior to syntheses being conducted, as expected for an emulsion polymerisation where the monomer is immiscible with the solvent.42 The optical transparency of final dispersions was quantified using UV-vis spectroscopy, where the transmittance values were measured across the visible range (see Fig. S12 and S13†) with wavelength-independent transmittance values being observed between 600 and 800 nm, where the absorbance from the RAFT CTA chromophore does not impact the spectrum. Thus, the transmittance at 700 nm was noted and plotted against the PBuMA DP value for each dispersion (Fig. 3a and b). Transmittance values for all dispersions were found to be ≥95%, indicating the formation of near-isorefractive dispersions where the refractive index of the solvent and nanoparticle core are very similar. Indeed, this aligns with the refractive index values reported in the literature for [EMIM][EtOSO3] and PBuMA, which are both 1.48,52,61,62 explaining the solution transparency. For comparison, PHEMA-b-PBuMA block copolymers were prepared via RAFT dispersion PISA in an EtOH/H2O solvent mixture (4:1 w:w) to demonstrate the appearance of a turbid dispersion for this formulation (Fig. 3c and d). The refractive index value for the EtOH/H2O solvent mixture has been reported to be 1.36,63 therefore as expected these dispersions do not provide high solution transmittance at 700 nm.
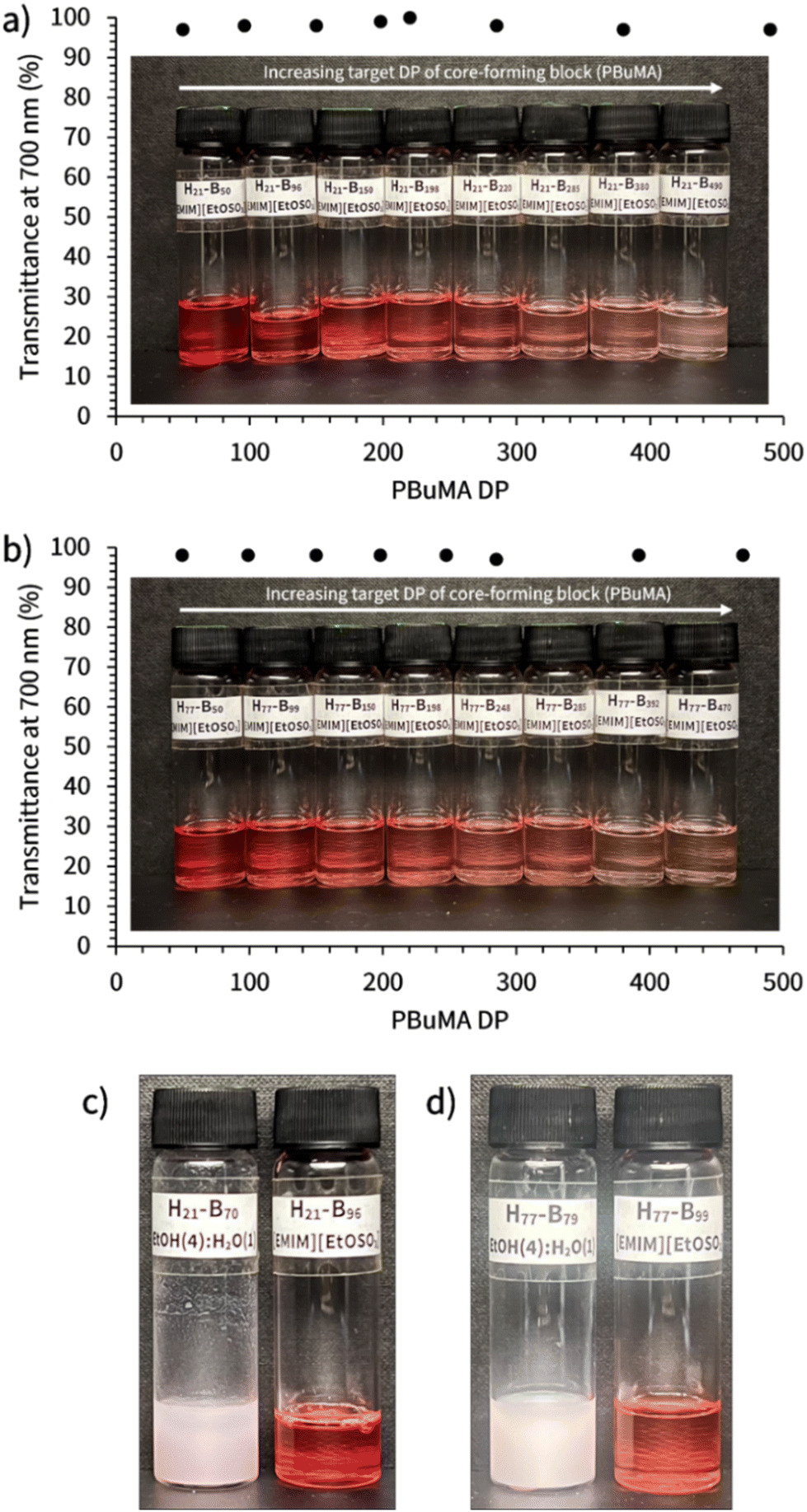 | ||
| Fig. 3 Images showing the physical appearance of the series of (a) PHEMA21-b-PBuMAy, and (b) PHEMA77-b-PBuMAy block copolymer dispersions in [EMIM][EtOSO3] at 10% w/w solids alongside corresponding transmittance values obtained from UV-vis spectroscopy. Labels on sample vials denote the actual PHEMAx-b-PBuMAy composition, denoted as Hx–By, as determined using 1H NMR spectroscopy. Images comparing the PHEMA21-b-PBuMAy (c) and PHEMA21-b-PBuMAy (d) prepared in an EtOH/H2O solvent mixture (left) and [EMIM][EtOSO3] (right). | ||
It can also be observed from that the extension of both PHEMA macro-CTAs in EtOH/H2O via dispersion polymerisation resulted in block copolymers with high dispersity values of 1.51 and 1.61 (Fig. S10†), implying that the polymerisation in this solvent system was less well controlled than when [EMIM][EtOSO3] was used as the solvent system. Again, this is attributed to the longer reaction times and thus increased change of termination events compared to the shorter polymerisations conducted in [EMIM][EtOSO3].
Characterisation of PHEMA-b-PBuMA block copolymer nanoparticles in [EMIM][EtOSO3]
A range of analytical techniques, specifically dynamic light scattering (DLS), transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS), were employed to characterise the nanoparticles present in the PHEMA21-b-PBuMAy and PHEMA77-b-PBuMAy block copolymer dispersions in [EMIM][EtOSO3] (Table 1). Firstly, DLS was employed to demonstrate the presence of particles within the system. Although these results display a trend up to a certain point which aligns with other data, they must be interpreted with caution. In the case of PHEMA-b-PBuMA nanoparticles in [EMIM][EtOSO3], the refractive index (RI) values of the nanoparticle cores (PBuMA, RI = 1.48362) and the solvent ([EMIM][EtOSO3], RI = 1.48052,61) are extremely similar, meaning that the particles would be virtually undetectable by DLS and hence result in unreliable data (Fig. S14†). TEM was used to characterise the synthesised nanoparticles, however the imaging proved challenging due to the inevitable presence of residual ionic liquid in the sample (Fig. 4). This is due to the extremely low volatility of ionic liquids, which is explained by the strong ionic bonding between molecules within the fluid, resulting in extremely low vapour pressure values.64,65 This resulted in challenges arising in the analysis of many of the samples as the presence of residual ILs produced unclear imaging in the majority of cases (Fig. S15†), despite allowing the sample to dry on the TEM grid for 4 days. | ||
| Fig. 4 TEM images obtained for 0.10% w/w emulsions of PHEMA-b-PBuMA block copolymers prepared in [EMIM][EtOSO3]. | ||
| Actual composition | DLS | SAXS | |
|---|---|---|---|
| Diameter (nm) | PDI | Core diameter (nm) | |
| a Estimated from the position of the peak minumum as indicated in Fig. S16 and S17† (using core diameter = 2 × core radius = (2 × 4.49)/q). Fitting to the spherical micelle model for all data sets was not possible due to: (1) the q range not being sufficient, and (2) some patterns showing the presence of multiple populations (in this case, the particle with the largest diameter was recorded). | |||
| H21 | — | — | — |
| H21–B50 | 431 | 0.28 | 14.4a |
| H21–B96 | 44 | 0.14 | 21.5 |
| H21–B150 | 57 | 0.22 | 35.1 |
| H21–B198 | 59 | 0.17 | 41.0 |
| H21–B220 | 70 | 0.16 | 35.3 |
| H21–B285 | 102 | 0.07 | 61.7a |
| H21–B380 | 91 | 0.22 | 83.7a |
| H21–B490 | 86 | 0.14 | 113a |
| H77 | — | — | — |
| H77–B50 | 34 | 0.37 | 10.5a |
| H77–B99 | 44 | 0.14 | 20.4 |
| H77–B150 | 62 | 0.10 | 27.0 |
| H77–B198 | 65 | 0.24 | 33.4 |
| H77–B248 | 66 | 0.14 | 38.2 |
| H77–B285 | 66 | 0.04 | 43.4 |
| H77–B392 | 74 | 0.22 | 49.5 |
| H77–B470 | 103 | 0.14 | 49.6 |
Thus, SAXS was used as a more reliable method to determine not only the presence of particles, but also their morphology (Table 1, Fig. 5, Fig. S16 and S17†).
 | ||
| Fig. 5 Background-subtracted SAXS patterns recorded at 1.0% w/w for PHEMA21-b-PBuMAy spheres prepared via RAFT emulsion polymerisation, where dashed lines represent fits to the spherical micelle model. For clarity, data are offset on the y-axis by a factor of 10. | ||
The SAXS data indicate the presence of only spherical morphologies. This was determined from the plateauing of the gradient at lower q values, as a gradient of zero is indicative of spherical morphologies.66 This is mostly clear from block copolymers H21–B96 to H21–B220 and H77–B99 to H77–B470 (Fig. 5 and S17†). The q range accessible meant that this plateauing was not visible for larger particles, however from the trend in increasing core diameter values in Table 1 it is implied that all morphologies present were spherical. The lowest PBuMA DP block copolymers, H21–B50 and H77–B50, are not expected to have this trend as only loose aggregates would be present. From this, a spherical micelle model54 was used to fit each background-subtracted data set, which can be observed from the black dashed line. It should be noted that although the presence of spheres for all samples was confirmed, it was not possible to fit all data sets, especially those obtained when targeting PBuMA DPs ≥ 500. Despite seeing clear evidence for the formation of nanoparticles in the SAXS patterns obtained for PHEMA21-b-PBuMA50 or PHEMA77-b-PBuMA50 dispersions, the spherical micelle model could not be used to satisfactorily fit these data. This is most likely due to the presence of a high proportion of freely dissolved polymer chains and/or the formation of loose, less well-defined aggregates. This is unsurprising since, as previously discussed, the DP at which micellization occurs was determined to be 58 when extending the PHEMA21 macro-CTA and 40 when extending the PHEMA77 macro-CTA. Nevertheless, the spherical micelle model was successfully applied to SAXS data obtained for PHEMA21-b-PBuMAy nanoparticles with PBuMA DP values ≤220 and for PHEMA77-b-PBuMAy nanoparticles with PBuMA DP values ≤470. A summary of the fitting parameters obtained in each of these fits can be found in Table S3.†
The relationship between actual PBuMA DP and the sphere core diameter (D) was plotted (Fig. 6). The equation D ∼ k·DPα, where k represents a constant, DP represents degree of polymerisation of PBuMA and α represents a scaling exponent (the alpha parameter), which can then be used to analyse the data further. A value of 0.5 represents unperturbed chains within the micelle cores, meaning that the polymer chains behave as if under theta conditions. This behaviour results in random (Gaussian) coils which are ideal – meaning that there is an equal balance between the interactions of the polymer chains with either the solvent or other polymer chains within proximity.67,68 Conversely, an alpha value of 1 indicates that the polymer chain is completely stretched inside of the micelle and there is a linear relationship between core diameter and DP. In this case, it can be observed that the series for the PHEMA21-b-PBuMAy spherical nanoparticles exhibit a steeper gradient with an alpha value of 0.90, whereas the PHEMA77-b-PBuMAy series exhibits a shallower gradient with a lower alpha value of 0.71. From the literature, it has been suggested that the alpha parameter for block copolymer micelles in theory should be around 0.67,68,69 which is closer to the blue data for the PHEMA77-b-PBuMAy nanoparticles, whereas the data set for the PHEMA21-b-PBuMAy nanoparticles is closer to 1. Such a difference in alpha values between series of spherical micelles prepared via PISA using macro-CTAs of varying length has also been reported in the literature by both Derry et al.70 for a dispersion PISA formulation and Akpinar et al.71 for an emulsion PISA formulation. Both of these examples demonstrate that when preparing different block copolymer series using macro-CTAs with different DPs, the extension of longest DP macro-CTA results in a decreased alpha (α) parameter. This is also in agreement with this research, which has shown that the alpha value obtained for the extension of the PHEMA77 macro-CTA was lower (0.71) than the extension of the shorter PHEMA21 macro-CTA (0.90). The reasoning behind this phenomenon is due to the diblock copolymers with longer stabiliser blocks exhibiting weaker segregation, and therefore adapting a more coiled conformation, rather than stretched which is common for shorter stabiliser block copolymers.68,70Table 1 shows how the PHEMA77 polymer nanoparticles have a lower diameter compared to those of the corresponding nanoparticles yielded from the PHEMA21 polymers, which is also in agreement with this theory and the literature.68,70
 | ||
| Fig. 6 Relationship between core radius and target DP of the PBuMA block (y) for series of PHEMA21-b-PBuMAy (red circles) and PHEMA77-b-PBuMAy (blue squares) diblock copolymer spheres prepared via RAFT emulsion polymerisation of BuMA in [EMIM][EtOSO3] at 70 °C. The error bars represent the standard deviation of the diameter and α is the scaling factor. | ||
The combination of DLS, SAXS and TEM as a suite of nanoparticle characterisation techniques provides confidence that well-defined spherical nanoparticles are formed during the RAFT emulsion polymerisation of BuMA in [EMIM][EtOSO3] using PHEMA macro-CTAs as steric stabilisers. From the available images, it is clear that spherical morphologies were formed. These spherical nanoparticles increase in size with increasing target PBuMA DP, as suggested by DLS and SAXS analysis.
Conclusions
RAFT emulsion polymerisation in ionic liquid was employed for the first time to prepare series of PHEMA21-b-PBuMAy and PHEMA77-b-PBuMAy block copolymer nanoparticles in [EMIM][EtOSO3] at 70 °C and at 10% w/w solids. The resulting nanoparticle dispersions were highly transparent owing to the matched refractive index values of the ionic liquid solvent and the insoluble poly(n-butyl methacrylate) structure-directing block, even when particle diameters exceeded 100 nm. The formation of spherical nanoparticles was confirmed using a combination of DLS, TEM and SAXS analyses. When investigating three different solvent systems to facilitate the synthesis of PHEMA-b-PBuMA block copolymers, it was demonstrated that higher monomer conversions were obtained at a much shorter polymerisation time (2 hours) for syntheses conducted in [EMIM][EtOSO3] (via emulsion polymerisation) compared to those in DMF (via solution polymerisation) and an EtOH/H2O mixture (via dispersion polymerisation). In addition to this, lower molar mass dispersity values were obtained for reactions performed in IL. This work demonstrates the advantages and ability of ILs to replace traditional solvents for block copolymer synthesis, as well as showcasing the use of RAFT emulsion PISA in IL for the first time. Moreover, this formulation produces transparent nanoparticle dispersions even when particle sizes exceed 100 nm owing to the closely matched refractive indices of the IL and the insoluble PBuMA structure-directing block.Author contributions
The authors contributed to this work in the following ways: Conceptualisation (M. J. D.); Formal analysis (G. L. M., M. J. D); Funding acquisition (M. J. D.); Investigation (A. P., G. L. M., E. L.); Methodology (A. P., M. J. D.); Project administration (M. J. D.); Supervision (P. D. T., M. J. D.); Validation (A. P.); Visualisation (A. P., M. J. D.); Writing – original draft (A. P., M. J. D.); Writing – review & editing (A. P., G. L. M., E. L., P. D. T., M. J. D.).Data availability
Additional data to support this article, including polymer and nanoparticle characterisation data, UV-vis measurements, small-angle X-ray scattering data and models and transmission electron microscopy images, have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. J. D. wishes to thank EPSRC for funding (grant number EP/Y005309/1) and for providing a DTP studentship for A. P. (EPSRC DTP 2020–2021, Aston University, Grant Ref: EP/T518128/1). The Aston Institute for Membrane Excellence (AIME) is funded by UKRI's Research England as part of their Expanding Excellence in England (E3) fund. E. L. thanks the Leverhulme Trust for an Early Career Fellowship (ECF-2023-602) and the Electron Microscopy RTP (University of Warwick) for access to the microscopes. Small-angle X-ray scattering (SAXS) experiments were conducted remotely with the support of Diamond Light Source, instrument B21 (experiment sm39167), and we would like to specifically acknowledge the support received by Dr Nathan Cowieson, Katsuaki Inoue, and Jodie Lavender during our experiment.References
- Z. Lei, B. Chen, Y.-M. Koo and D. R. MacFarlane, Chem. Rev., 2017, 117, 6633–6635 CrossRef PubMed.
- R. Tamate, K. Hashimoto, T. Ueki and M. Watanabe, Phys. Chem. Chem. Phys., 2018, 20, 25123–25139 Search PubMed.
- P. Sharma, S. Sharma and H. Kumar, J. Mol. Liq., 2024, 393, 123447 Search PubMed.
- A. J. Greer, J. Jacquemin and C. Hardacre, Molecules, 2020, 25, 5207 CrossRef CAS PubMed.
- G. A. O. Tiago, I. A. S. Matias, A. P. C. Ribeiro and L. M. D. R. S. Martins, Molecules, 2020, 25, 5812 CrossRef CAS PubMed.
- K. Sood, Y. Saini and K. K. Thakur, Mater. Today: Proc., 2023, 81, 739–744 CAS.
- A. Tsurumaki, T. Iwata, M. Tokuda, H. Minami, M. A. Navarra and H. Ohno, Electrochim. Acta, 2019, 308, 115–120 CrossRef CAS.
- Y. Ding, H. Tang, X. Zhang, S. Wu and R. Xiong, Eur. Polym. J., 2008, 44, 1247–1251 CrossRef CAS.
- C. M. Caldas, B. G. Soares, T. Indrusiak and G. M. O. Barra, J. Appl. Polym. Sci., 2021, 138, 49814 Search PubMed.
- V. Bugatti, G. Viscusi, A. Di Bartolomeo, L. Iemmo, D. C. Zampino, V. Vittoria and G. Gorrasi, Polymers, 2020, 12, 495 CrossRef CAS PubMed.
- J. R. Lim, L. S. Chua and A. A. Mustaffa, Process Biochem., 2022, 122, 292–306 CrossRef CAS.
- Z. Yang and W. Pan, Enzyme Microb. Technol., 2005, 37, 19–28 CrossRef CAS.
- G. Durga, P. Kalra, V. Kumar Verma, K. Wangdi and A. Mishra, J. Mol. Liq., 2021, 335, 116540 CrossRef CAS.
- P. Kubisa, Prog. Polym. Sci., 2009, 34, 1333–1347 Search PubMed.
- N. Winterton, J. Mater. Chem., 2006, 16, 4281–4293 RSC.
- P. Kubisa, Eur. Polym. J., 2020, 133, 109778 Search PubMed.
- N. K. Singha, K. Hong and J. W. Mays, in Polymerized Ionic Liquids, ed. A. Eftekhari, The Royal Society of Chemistry, 2017 Search PubMed.
- S. Harrisson, S. R. Mackenzie and D. M. Haddleton, Chem. Commun., 2002, 2850–2851 RSC.
- S. Harrisson, S. R. Mackenzie and D. M. Haddleton, Macromolecules, 2003, 36, 5072–5075 CrossRef CAS.
- A. R. S. Santha Kumar, M. Roy and N. K. Singha, Eur. Polym. J., 2018, 107, 294–302 CrossRef CAS.
- Y. He, Z. Li, P. Simone and T. P. Lodge, J. Am. Chem. Soc., 2006, 128, 2745–2750 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. E. Parkes, H. J. Hutchins-Crawford, C. Bourdin, S. Reynolds, L. J. Leslie, M. J. Derry, J. L. Harries and P. D. Topham, Polym. Chem., 2020, 11, 2869–2882 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Chem. – Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- X. Tian, J. Ding, B. Zhang, F. Qiu, X. Zhuang and Y. Chen, Polymers, 2018, 10, 318 CrossRef PubMed.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 Search PubMed.
- B. D. Fairbanks, P. A. Gunatillake and L. Meagher, Adv. Drug Delivery Rev., 2015, 91, 141–152 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- P. J. Docherty, M. J. Derry and S. P. Armes, Polym. Chem., 2019, 10, 603–611 RSC.
- C. György, M. J. Derry, E. J. Cornel and S. P. Armes, Macromolecules, 2021, 54, 1159–1169 CrossRef PubMed.
- D. Ikkene, J.-L. Six and K. Ferji, Eur. Polym. J., 2023, 188, 111848 CrossRef CAS.
- E. R. Jones, O. O. Mykhaylyk, M. Semsarilar, M. Boerakker, P. Wyman and S. P. Armes, Macromolecules, 2016, 49, 172–181 CrossRef CAS PubMed.
- B. R. Parker, M. J. Derry, Y. Ning and S. P. Armes, Langmuir, 2020, 36, 3730–3736 Search PubMed.
- G. L. Maitland, M. Liu, T. J. Neal, J. Hammerton, Y. Han, S. D. Worrall, P. D. Topham and M. J. Derry, Chem. Sci., 2024, 15, 4416–4426 RSC.
- R. Yamanaka, A. Sugawara-Narutaki and R. Takahashi, Macromolecules, 2023, 56, 4354–4361 Search PubMed.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755–758 CrossRef CAS PubMed.
- H. Zhou, C. Liu, C. Gao, Y. Qu, K. Shi and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1517–1525 Search PubMed.
- M. J. Rymaruk, K. L. Thompson, M. J. Derry, N. J. Warren, L. P. D. Ratcliffe, C. N. Williams, S. L. Brown and S. P. Armes, Nanoscale, 2016, 8, 14497–14506 Search PubMed.
- A. Takashima, Y. Maeda and S. Sugihara, ACS Omega, 2022, 7, 26894–26904 Search PubMed.
- M. Zeng, X. Li, Y. Zhang, X. Chen, X. Sui and J. Yuan, Polymer, 2020, 206, 122853 CrossRef CAS.
- C. György and S. P. Armes, Angew. Chem., Int. Ed., 2023, 62, e202308372 Search PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- F. d'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 Search PubMed.
- J. Wan, B. Fan and S. H. Thang, Chem. Sci., 2022, 13, 4192–4224 Search PubMed.
- R. Yamanaka, A. Sugawara-Narutaki and R. Takahashi, ACS Macro Lett., 2024, 13, 1050–1055 Search PubMed.
- U. Rivera-Ortega, C. R. Hernández-Gómez, G. Vega-Torres and M. E. Lopez-Medina, Measurement, 2019, 134, 658–661 CrossRef.
- J. Ziming Sun, M. C. Erickson and J. W. Parr, Int. J. Cosmet. Sci., 2005, 27, 355–356 CrossRef.
- M. Semsarilar, E. R. Jones and S. P. Armes, Polym. Chem., 2014, 5, 195–203 RSC.
- A. P. Fröba, H. Kremer and A. Leipertz, J. Phys. Chem. B, 2008, 112, 12420–12430 CrossRef PubMed.
- N. P. Cowieson, C. J. Edwards-Gayle, K. Inoue, N. S. Khunti, J. Doutch, E. Williams, S. Daniels, G. Preece, N. A. Krumpa and J. P. Sutter, Synchrotron Radiat., 2020, 27, 1438–1446 CrossRef CAS PubMed.
- J. S. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- A. Czajka and S. P. Armes, J. Am. Chem. Soc., 2021, 143, 1474–1484 CrossRef CAS PubMed.
- C. H. J. Johnson, T. H. Spurling and G. Moad, Polymers, 2022, 14, 5013 CrossRef CAS PubMed.
- G. N. Smith, M. J. Derry, J. E. Hallett, J. R. Lovett, O. O. Mykhaylyk, T. J. Neal, S. Prévost and S. P. Armes, Proc. R. Soc. A, 2019, 475, 20180763 CrossRef PubMed.
- S. Panush and M. F. Hills, Transparent Pigment Dispersion Process, U.S. PatentUS4017448A, 1975 Search PubMed.
- X.-W. Han, X.-F. Zeng, J. Zhang, H. Huan, J.-X. Wang, N. R. Foster and J.-F. Chen, Chem. Eng. J., 2016, 296, 182–190 CrossRef CAS.
- A. N. Soriano, B. T. Doma and M.-H. Li, J. Taiwan Inst. Chem. Eng., 2010, 41, 115–121 CrossRef CAS.
- J. Stevens, D. Jackson and J. Champion, Mol. Phys., 1975, 29, 1893–1905 CrossRef CAS.
- T. A. Scott Jr, J. Phys. Chem., 1946, 50, 406–412 Search PubMed.
- M. Bier and S. Dietrich, Mol. Phys., 2010, 108, 211–214 CrossRef CAS.
- L. A. DiCecco, A. D'Elia, C. Miller, K. N. Sask, L. Soleymani and K. Grandfield, ChemBioChem, 2021, 22, 2488–2506 CrossRef CAS PubMed.
- I. W. Hamley, Small-Angle Scattering: Theory, Instrumentation, Data, and Applications, Wiley, 2021 Search PubMed.
- K. S. Kontturi, L. Solhi, E. Kontturi and T. Tammelin, Langmuir, 2024, 40, 568–579 CrossRef CAS PubMed.
- G. Battaglia and A. J. Ryan, J. Am. Chem. Soc., 2005, 127, 8757–8764 CrossRef CAS PubMed.
- S. Förster, M. Zisenis, E. Wenz and M. Antonietti, J. Chem. Phys., 1996, 104, 9956–9970 CrossRef.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- B. Akpinar, L. A. Fielding, V. J. Cunningham, Y. Ning, O. O. Mykhaylyk, P. W. Fowler and S. P. Armes, Macromolecules, 2016, 49, 5160–5171 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00076a |
| This journal is © The Royal Society of Chemistry 2025 |