
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and synthesis of amphiphilic statistical copolymers forming unimeric micelles with thermoresponsive behaviour in the physiological range†
Florian
Tondock
 a,
David
Nash
a,
Cathleen
Hudziak
b,
Kai
Ludwig
b and
Marie
Weinhart
*ab
a,
David
Nash
a,
Cathleen
Hudziak
b,
Kai
Ludwig
b and
Marie
Weinhart
*ab
aInstitute of Physical Chemistry and Electrochemistry, Leibniz Universität Hannover, Callinstr. 3A, 30167 Hannover, Germany
bInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: marie.weinhart@fu-berlin.de; marie.weinhart@pci.uni-hannover.de
First published on 7th April 2025
Abstract
A crucial aspect of drug development is designing carriers that efficiently solubilise therapeutic agents while ensuring stability, minimising cytotoxicity, and enabling targeted delivery. Multimolecular micelles are commonly used but often destabilise under physiological conditions. This study focuses on developing stable, unimolecular carriers with high loading capacity for hydrophobic cargos. The synthesis of amphiphilic copolymers based on oligoethylene glycol acrylate (OEGA) and butyl acrylate (BA) was optimised to achieve consistent statistical comonomer incorporation for molecular weights up to 600 kDa with low dispersity via activator regenerated by electron transfer atom transfer radical polymerisation (ARGET ATRP). The copolymers demonstrated reversible thermoresponsive behaviour in aqueous media, with adjustable lower critical solution temperature (LCST) between 25 and 70 °C based on the comonomer ratio. Below the LCST, unimolecular micelles formed with sizes tunable from 4 to 22 nm diameter through the copolymers’ molecular weight and hydrophobicity. Micellar stability was unaffected by dilution and physiological salt concentrations unless heated above the LCST, triggering aggregation into defined nanosized colloids, which holds potential for temperature-controlled accelerated drug release. The maximum loading capacity for pyrene as a small molecule proxy varied with molecular weight and copolymer composition and reached up to 36 molecules per unimolecular carrier, making these copolymers promising candidates for smart drug delivery systems.
Introduction
Targeted drug delivery using nanoscale polymer-based carriers is a cornerstone of modern therapies to enhance drug solubility and minimise side effects on healthy tissues and organs.1,2 Concurrently, drug stability is often increased by mitigating enzymatic degradation. Optimised nanocarrier dimensions, ranging from 6 to 200 nm, are crucial for evading renal clearance and immune cell recognition, thereby minimising premature opsonization.3 Micellar systems featuring hydrophobic cores formed through the self-assembly of amphiphiles in water are thermodynamically stable above their critical micelle concentration (CMC). However, their dynamic nature leads to instability under dilution or environmental changes such as temperature, pH, or ionic strength. As a result, most multimolecular micellar carriers become destabilised in physiological conditions,3 causing drug leakage and dissociation into their building blocks after injection into the bloodstream. This instability reduces therapeutic efficacy and increases toxicity risks,3 thereby limiting the biomedical application of many multimolecular micelles.1Amphiphilic copolymers in water with unique dendritic,4 hyperbranched, star-shaped, or other architectures like bottlebrushes1 can reorganize or intramolecularly self-assemble into defined unimolecular core–shell structured micelles driven by the hydrophobic effect. Such unimeric micelles form spontaneously and independently of the polymer concentration, exhibiting enhanced stability under physiological conditions compared to conventional micelles.5,6 For amphiphilic bottlebrush copolymers, a sufficiently high molecular weight with a statistical distribution of the pendant amphiphilicity-causing groups is a crucial structural requirement for self-folding and unimolecular micelle formation. Thus, below a certain threshold value for the polymer length, multimolecular micelles of similar size to the unimeric ones are obtained.7,8 Furthermore, the nature and architecture of the polymer backbone and the pendant groups determine the amphiphilic balance and govern its self-assembly.5,7 Hence, the self-assembly behaviour, including micellar size and aggregation number, as well as the drug loading capacity, is largely predefined by the molecular design and nature of the amphiphilic statistical copolymers.
For biomedical applications, the highly water-soluble outer shell of the unimeric micelles needs to exhibit intrinsic cytocompatibility, low fouling properties, and no systemic toxicity. Consequently, synthetic copolymers based on stealth building blocks such as poly (ethylene glycol) (PEG) derivatives and their lower molecular weight counterparts, oligoethylene glycol (OEG), are often employed. The use of PEG/OEG is further benefited by its approval for biomedical application via the U.S. Food and Drug Administration.3,9
Particularly, OEG-based (meth)acrylate monomers (OEGMA and OEGA) enable the direct statistical copolymerisation with hydrophobic alkyl (meth)acrylates to generate bottlebrush structures required for micelle formation in aqueous solution.10,11 According to literature,5,7,12 the molecularity and size of the micelles forming in aqueous media can be controlled by the proportion and type of the hydrophobic alkyl (meth)acrylate comonomer, the type and length of the used OEG-based monomer, and the overall molecular weight of the copolymer, as illustrated by color-coded arrows for unimeric micelles (green) and multimeric micelles (red) in Scheme 1.
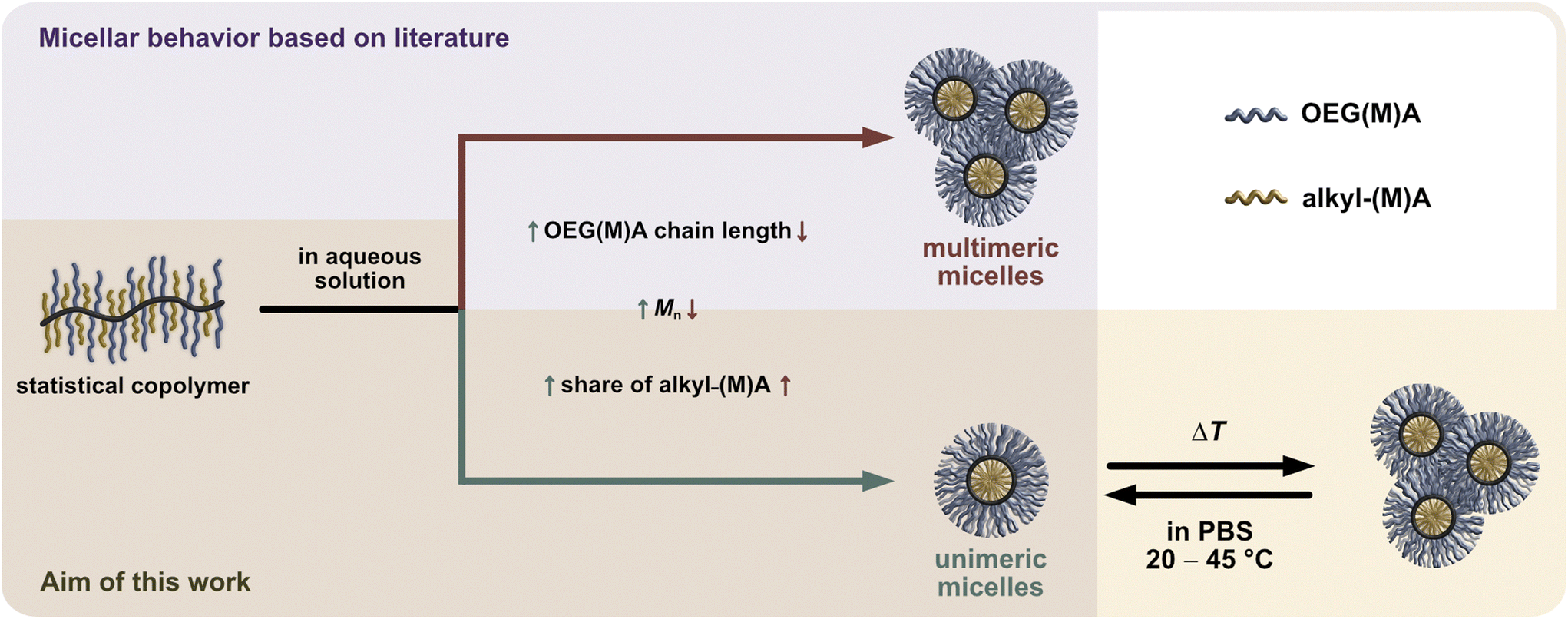 | ||
| Scheme 1 Structure of statistical amphiphilic bottlebrush copolymers based OEG(M)A (blue) and alkyl (meth)acrylate (yellow) comonomers and their dynamic self-assembly into polymeric micelles in aqueous solution.13 The annotated impact of the copolymers’ molecular features on the resulting molecularity and size of the forming micelles is based on literature (purple background).5,7,12 For thermoresponsive amphiphilic acrylate-based bottlebrushes, a change in temperature can induce a reversible aggregation of the unimers to mesoscopic aggregates of defined size, as shown in this work (ivory background). | ||
In addition to their spontaneous self-assembly behaviour, amphiphilic PEG- or OEG-based statistical copolymers can exhibit thermoresponsive behaviour with a lower critical solution temperature (LCST) and reversible phase separation in aqueous media as illustrated in the lower right side of Scheme 1. By carefully fine-tuning the spatial amphiphilic balance and architecture of the copolymer according to a rational polymer design, it is possible to generate versatile and predictable thermoresponsive behaviour.14,15 This has recurrently been demonstrated with short-chain OEGMA (300–475 g mol−1) monomers and various alkyl (meth)acrylate comonomers.7,12,13,16 Increasing the side chain length of the alkyl (meth)acrylate comonomer reduced the cloud point temperature (Tcp), as demonstrated with methyl methacrylate (MMA) and butyl methacrylate (BMA). Further increase of the alkyl chain length from butyl to octadecyl, conversely, increased the Tcp of the statistical copolymers. This behaviour was attributed to the strong interaction of the octadecyl side groups in the forming micellar core, diminishing their dehydrating and, thus, Tcp-lowering effect on the surrounding OEGMA units.13
Unimicellar systems based on thermoresponsive polymers hold great potential for the delivery and thermally induced drug release through micelle aggregation.15,17 Most thermoresponsive OEG-containing statistical copolymer systems forming unimeric micelles described in literature are based on OEGMA, often neglecting the corresponding acrylate. It is known that the nature of the polymer backbone can have a marked impact on the dehydration and phase transition mechanism of thermoresponsive polymers in solution.18 Furthermore, thermoresponsive OEGMA-based statistical copolymers, which form unimeric micelles, exhibit limited use for smart biomedical applications as they exhibit Tcp's far above the physiologically relevant regime.5,7,8,12,13 Promising P(OEGMA-co-BMA) candidates for biomedical applications with a Tcp of around 45 °C at 70 mol% BMA tend to assemble into multimeric micelles with increasing BMA content (50–70 mol%).7
We hypothesize that by substituting OEGMA with OEGA and increasing the proportion of hydrophobic comonomer in thermoresponsive statistical copolymers, we can reduce the hydrophobicity of the polymer backbone, shifting it to the side chains. Thereby, the cloud point temperature should be significantly lowered, enabling the development of thermoresponsive unimeric micelles exhibiting a Tcp within the physiologically relevant range.
To ensure an efficient loading capacity of the desired unimers, high molecular weight copolymers were targeted by controlled radical polymerisation. The thermoresponsive properties of the resulting statistical copolymers were analysed in aqueous solution, and their micellar assembly and thermal aggregation behaviour under physiological conditions, as well as their loading capacity, were investigated.
Experimental section
Additional detailed information on chemicals, materials, and methods can be found in the ESI.†Cu(0)-mediated synthesis of P(OEGA-co-BA)
For the synthesis of copolymers with a comonomer-ratio of oligo(ethylene glycol) methyl ether acrylate (OEGA, 480 Da) and n-butyl acrylate (BA) = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (mol%) and a target molecular weight of 10 kDa, the following general procedure was applied. OEGA (7.24 mL, 16.4 mmol), BA (2.34 mL, 16.4 mmol), N,N-dimethylformamide (DMF, 9.9 mL), 1,2-bis(2′-bromisobutyryloxy)ethane (2f-BiB, 373.5 mg, 1.0 mmol) and a magnetic stir bar wrapped with a pre-activated copper wire (d = 0.15 mm, l = 30 cm) were mixed and degassed via argon bubbling for 20 min at 25 °C. In another reaction vessel, CuBr2 (6.5 mg, 29 μmol) and tris[2-(dimethylamino)ethyl]amine (Me6TREN, 27.9 μL, 0.1 mmol) were jointly dissolved in DMF (0.1 mL) and degassed with argon for 20 min. The polymerisation was started by adding the Cu(II)-containing solution to the thermostated (25 °C) monomer solution. Samples of the reaction mixture were taken periodically for conversion determination by 1H NMR analysis. The reaction was stopped by exposing the reaction mixture to air and removing the copper wire. The polymer was purified via dialysis (MWCO: 1 kDa P1, 3.5 kDa P2–P12) in water (6 × 2 L) followed by ethanol (3 × 1 L) to remove the reaction residues. The brush copolymer was dried under vacuum to a constant mass. According to Table 1, the 10 kDa copolymer with a 1:1 (mol%) ratio of OEGA to BA will be referred to as P1. Copolymerisations with a 1:1 (mol%) comonomer ratio targeting higher molecular weights (50 kDa (P2), 100 kDa (P3), 500 kDa (P4), 1000 kDa (P5)) as well as targeted 1000 kDa polymers with initial OEGA to BA ratios of 2:3 (mol%; P6), 3:7 (mol%; P7), 7:18 (mol%; P8), 13:37 (mol%; P9), 6:19 (mol%; P10), 11:39 (mol%; P11) and 1:4 (mol%; P12) were performed accordingly with adjusted proportions of reagents (Table S1†).
:1 (mol%) and a target molecular weight of 10 kDa, the following general procedure was applied. OEGA (7.24 mL, 16.4 mmol), BA (2.34 mL, 16.4 mmol), N,N-dimethylformamide (DMF, 9.9 mL), 1,2-bis(2′-bromisobutyryloxy)ethane (2f-BiB, 373.5 mg, 1.0 mmol) and a magnetic stir bar wrapped with a pre-activated copper wire (d = 0.15 mm, l = 30 cm) were mixed and degassed via argon bubbling for 20 min at 25 °C. In another reaction vessel, CuBr2 (6.5 mg, 29 μmol) and tris[2-(dimethylamino)ethyl]amine (Me6TREN, 27.9 μL, 0.1 mmol) were jointly dissolved in DMF (0.1 mL) and degassed with argon for 20 min. The polymerisation was started by adding the Cu(II)-containing solution to the thermostated (25 °C) monomer solution. Samples of the reaction mixture were taken periodically for conversion determination by 1H NMR analysis. The reaction was stopped by exposing the reaction mixture to air and removing the copper wire. The polymer was purified via dialysis (MWCO: 1 kDa P1, 3.5 kDa P2–P12) in water (6 × 2 L) followed by ethanol (3 × 1 L) to remove the reaction residues. The brush copolymer was dried under vacuum to a constant mass. According to Table 1, the 10 kDa copolymer with a 1:1 (mol%) ratio of OEGA to BA will be referred to as P1. Copolymerisations with a 1:1 (mol%) comonomer ratio targeting higher molecular weights (50 kDa (P2), 100 kDa (P3), 500 kDa (P4), 1000 kDa (P5)) as well as targeted 1000 kDa polymers with initial OEGA to BA ratios of 2:3 (mol%; P6), 3:7 (mol%; P7), 7:18 (mol%; P8), 13:37 (mol%; P9), 6:19 (mol%; P10), 11:39 (mol%; P11) and 1:4 (mol%; P12) were performed accordingly with adjusted proportions of reagents (Table S1†).
| Polymer | Feeda [OEGA:BA] |
M
nb [kDa] |
M
nc [kDa] |
M
nd [kDa] |
M
ne [kDa] |
Đ | Đ |
A
H2Of |
Compositiona,c [OEGA:BA] |
|---|---|---|---|---|---|---|---|---|---|
| a Molar ratio. b Targeted molecular weight. c Calculated from 1H NMR spectra acquired in CDCl3 at 25 °C. d Calculated from GPC traces obtained in THF with a MALS detector at 25 °C. e Calculated from GPC traces obtained in water with a MALS detector at 25 °C. f Calculated association number AH2O = Mn,H2O·Mn,THF−1 in H2O at 25 °C. g n.d. = not determined due to low Tcp in water of the polymers P11 and P12 or low scattering intensity of P1. | |||||||||
| P1 | 50:50 |
10 | 9.8 | n.d.g | n.d.g | n.d.g | n.d.g | n.d.g | 50:50 |
| P2 | 50:50 |
50 | 46.8 | 48.5 | 52.2 | 1.01 | 1.00 | 1.08 | 50:50 |
| P3 | 50:50 |
100 | 91.2 | 91.0 | 91.2 | 1.07 | 1.05 | 1.00 | 50:50 |
| P4 | 50:50 |
500 | 366.9 | 314.9 | 313.8 | 1.15 | 1.42 | 1.00 | 50:50 |
| P5 | 50:50 |
1000 | 615.2 | 477.3 | 477.9 | 1.14 | 1.62 | 1.00 | 49:51 |
| P6 | 40:60 |
1000 | 594.1 | 390.4 | 443.0 | 1.41 | 1.58 | 1.13 | 38:62 |
| P7 | 30:70 |
1000 | 626.9 | 460.1 | 505.5 | 1.41 | 1.51 | 1.10 | 29:71 |
| P8 | 28:72 |
1000 | 622.2 | 480.6 | 503.1 | 1.65 | 1.90 | 1.05 | 27:73 |
| P9 | 26:74 |
1000 | 633.4 | 559.4 | 581.3 | 1.67 | 2.06 | 1.04 | 26:74 |
| P10 | 24:76 |
1000 | 640.1 | 555.2 | 623.4 | 1.66 | 2.13 | 1.12 | 23:77 |
| P11 | 22:78 |
1000 | 650.6 | 565.8 | n.d.g | 1.82 | n.d.g | n.d.g | 22:78 |
| P12 | 20:80 |
1000 | 655.9 | 362.2 | n.d.g | 1.79 | n.d.g | n.d.g | 20:80 |
Nuclear magnetic resonance (NMR) spectroscopy
1H NMR spectra were recorded on a Bruker Ascend or Ultrashield (Bruker Corporation, Billerica, United States) at 400 MHz at ambient temperature and processed using MestReNova software Version 14. The samples were prepared by dissolving 15 mg of copolymer in 0.6 mL deuterated chloroform (CDCl3) or deuterium oxide (D2O). Chemical shifts were reported in parts per million (ppm) and referenced to the respective deuterated solvent peak. The corresponding spectrum for P1, confirming the chemical identity of the copolymer, is shown in Fig. S1.† The number-average molecular weights (Mn) of the synthesized copolymers were calculated from the proton ratios of the CH3-end group signals of OEGA and BA in relation to their vinyl signals in the 1H NMR spectra (Fig. S2†) as described in the ESI.†Gel permeation chromatography (GPC)
GPC measurements were performed with a PSS SECcurity2 GPC System (Polymer Standards Service GmbH, Mainz, Germany) in tetrahydrofuran (THF) at polymer concentrations of 8 mg mL−1 and a flow rate of 1 mL min−1 at 25 °C. One SDV pre-column with dimensions of 8 × 50 mm and a particle size of 5 μm and three SDV columns with dimensions of 8 × 300 mm, a particle size of 5 μm and a pore size of 1000, 10000, and 100000 Å, respectively, from PSS GmbH (Mainz, Germany) were used in line with a refractive index and a PSS SLD7x00/BI-MwA MALS detector.Aqueous GPC-MALS measurements were performed on the same PSS SECcurity2 GPC System in 0.05% NaNO3 solution at a polymer concentration of 1.5 mg mL−1 and a flow rate of 1 mL min−1 at 25 °C. The column set consisted of a PSS Suprema pre-column with dimensions of 8 × 50 mm and a particle size of 10 μm and three linear PSS Suprema columns with dimensions of 8 × 300 mm, a particle size of 10 μm and a pore size of 30 and 2 × 1000 Å, respectively.
The dn/dc values were determined by recording a calibration series with five concentrations using the MALS detector in the corresponding solvent (Table S2†). All molecular weight and dispersity values were calculated using the PSS WinGPC UniChrom® software Version 8.
Dynamic light scattering (DLS)
DLS measurements were performed with a Malvern Zetasizer μV (Malvern Panalytical GmbH, Kassel, Germany) at a 90° scattering angle or, for dilution experiments, with a Zetasizer nano at 173° scattering angle in glass and PS cuvettes. All copolymers were dissolved in ultra-pure water (c = 10 mg mL−1) and filtered using 0.45 μm CA syringe filters before use. Samples were equilibrated for 5 min at 20–25 °C or 15 min at elevated temperatures. Three technical replicate measurements were recorded (15 scans of 10 seconds, respectively) and analysed with the Malvern Zetasizer Software Version 7 using the refractive index of polystyrene latex particles (n = 1.59) and the general purpose model. In graphs and tables, size distributions by volume are plotted and their corresponding average hydrodynamic radius Rh ± standard deviation (SD) are stated. The optical measurement parameters were automatically optimized before each measurement, except for the measurements of concentration-dependent scattering intensity, where the attenuation was kept constant for all samples.Rheology
Intrinsic viscosities of the copolymers were determined at 25 °C in ultra-pure water via oscillatory rheology with an MCR 302 modular rheometer from Anton Paar Group AG (Graz, Austria) equipped with plate-plate geometry (40 mm diameter plate). The viscosity was determined from three independent measurements with a shear rate between 10 and 1000 Hz and analysed with OriginPro Version 10.Cryo-transmission electron microscopy (Cryo-TEM)
Microscopic 200-mesh grids covered with a perforated carbon film (batch R1/4 of Quantifoil, MicroTools GmbH, Jena, Germany) were cleaned with chloroform and hydrophilized by glow discharge at 8 W for 60 seconds in a BALTEC MED 020 instrument (Leica Microsystems, Wetzlar, Germany) before 4 μL aliquots of the polymer solution (1 mg mL−1 in ultra-pure water) were applied to the grids. Samples were vitrified by automated blotting and plunge freezing with a FEI Vitrobot Mark IV (Thermo Fisher Scientific Inc., Waltham, Massachusetts, United States) using liquid ethane as cryogen. The climate in the chamber was set to 20 °C and 100% humidity. The grids with the vitrified specimens were assembled into so-called autogrid cartridges and transferred to the autoloader of a FEI TALOS® ARCTICA electron microscope (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA). This microscope is equipped with a high-brightness field emission gun (X-FEG) operated at an accelerating voltage of 200 kV. Microscope images were acquired with a FEI Falcon 3EC direct electron detector (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA) using the microscope's low-dose protocol.UV-Vis spectroscopy
Turbidimetry measurements were performed on a V-750 UV-Vis spectrometer at λ = 500 nm with a 6-fold Peltier temperature programmer from JASCO Deutschland GmbH (Pfungstadt, Germany) in PS disposable cuvettes. Cloud points were determined in ultra-pure water and phosphate-buffered saline (PBS) solution employing heating/cooling rates of 0.5 °C min−1 with a data point recording every 0.5 °C. The temperature-dependent transmittance of the aqueous polymer solutions was measured for three up and down cycles, and the Tcp was defined as the temperature at the inflection point of the normalized transmittance versus temperature curves of all heating cycles. The obtained data were analysed using OriginPro Version 10.Pyrene loading
Polymers P1–P12 (20 mg) were dissolved in a saturated solution of pyrene in MeOH (2 mL) overnight. The solvent was then completely removed under high vacuum. Subsequently, the dry residue was dissolved in PBS or D2O (2 mL) at 5 °C overnight and filtered (CA, 0.45 μm) to remove the excess pyrene. The resulting pyrene-loaded micelles were characterised by DLS, 1H NMR, fluorescence and UV-Vis spectroscopy.Statistical section
Independent replicates N and technical replicates n of the experiments or measurements are stated in the figure captions. Data is presented as mean ± SD if not stated otherwise.Results
Controlled-radical polymerisations are the method of choice when aiming at complex structures with defined molecular weights and narrow dispersity (Đ). The activator regenerated by electron transfer-atom transfer radical polymerisation (ARGET ATRP)19 exhibits high monomer compatibility. Thus, it was employed to synthesise the desired high molecular weight statistical amphiphilic copolymers composed of OEGA (480 Da) and BA. In Fig. 1A, the copolymerisation using 2f-BiB as a bifunctional ATRP initiator is illustrated. Aiming at different molecular weight copolymers P1–P5, the ratios of catalytic metal salt (CuBr2) and chelating ligand (Me6TREN) have been adjusted and optimised based on monomer conversion after 6 h of polymerisation in DMF at 25 °C, while keeping the monomer concentration and comonomer ratio constant (Table S3†). At lower catalyst concentrations than the optimised ones stated in Fig. 1A, a decrease in conversion within 6 h, especially for P5 with the a priori lowest catalyst concentration, was observed. Conversely, at higher catalyst concentrations than the optimised ones, a slight trend of decreasing monomer conversion was observed, attributed to an increase in radical–radical termination reactions.20,21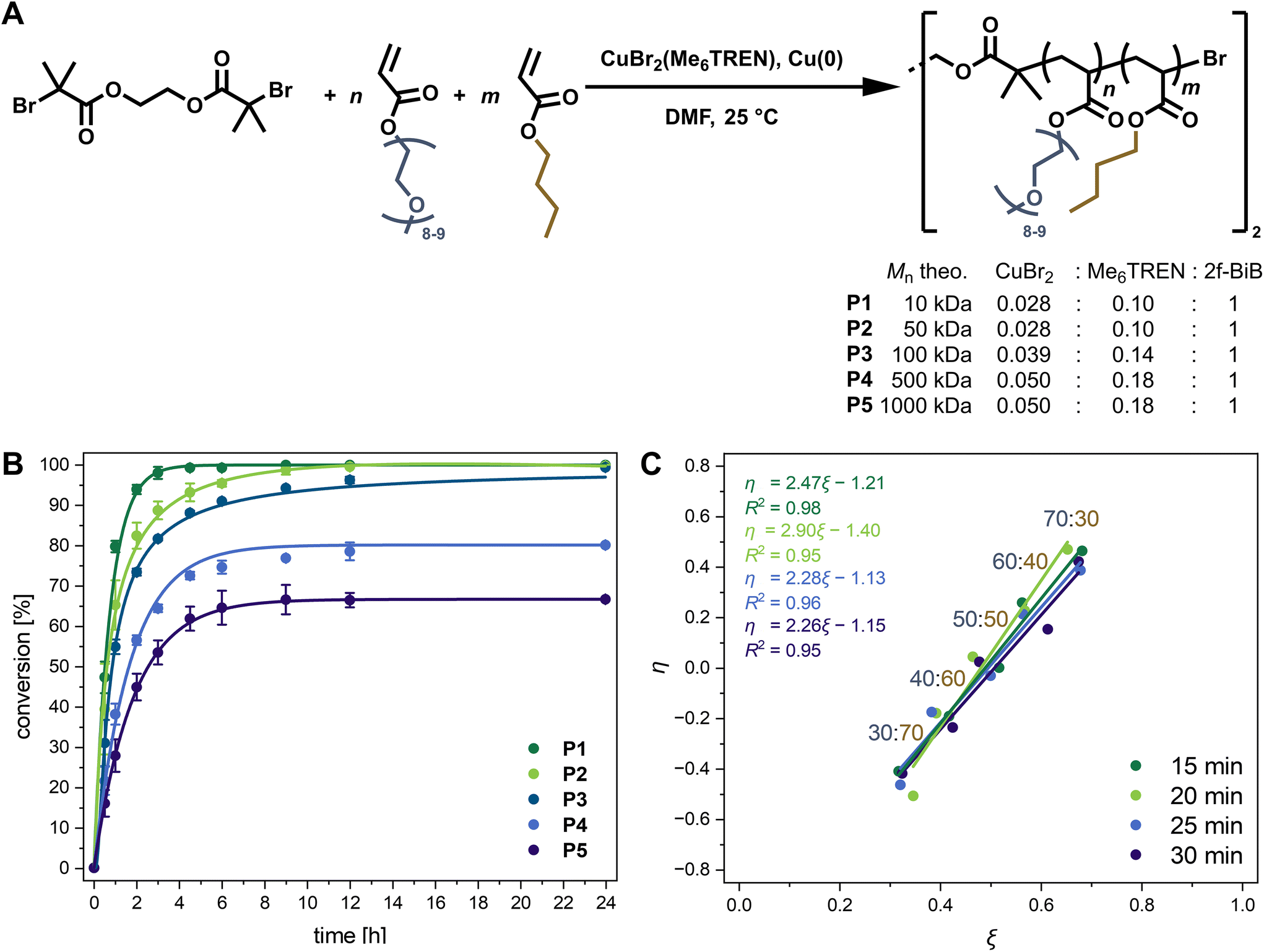 | ||
| Fig. 1 Synthesis of statistical P(OEGA-co-BA) copolymers via ARGET ATRP with 2f-BiB as initiator and [CuBr2(Me6TREN)] as catalyst in DMF at 25 °C. (A) Reaction scheme for the synthesis of copolymers P1–P5 with increasing targeted molecular weight applying an OEGA:BA = 1:1 ratio with optimised ratios of metal salt and ligand. (N ≥ 3). (B) Time-dependent monomer conversion. Curves are fitted by an exponential function. (N ≥ 2). (C) Extended Kelen–Tüdös plots at different time points for the copolymerisation with targeted molecular weights of 1000 kDa and feed ratios of OEGA:BA = 30:70–70:30 in increments of 10 mol% yielding the copolymerisation parameters rOEGA and rBA from the slope and the intercept with the y-axis of the linear fits. | ||
Time-dependent monomer conversions in the crude reaction mixtures were calculated according to eqn (S1)† from the 1H NMR signal integrals of the double bonds of both monomers at δ = 6.5–5.5 ppm, and the methyl groups of both monomers and the copolymer at δ = 0.91 and 3.35 ppm, respectively (Fig. S2†). Conversion versus time plots, as illustrated in Fig. 1B for copolymers P1–P5 synthesised under the optimised conditions, reveal maximum conversion after a reaction time of approximately 6 h. Generally, the kinetic plots of ln([M]0/[M]t) increased linearly with time (Fig. S3†), suggesting pseudo-first-order kinetics up to a reaction time of 4.5 h and indicating a constant concentration of propagating radicals during the polymerisation of P1–P5.21,22 Therefore, within the first few hours, the polymerisations fulfil the criteria of a controlled living radical polymerisation with a linearly increasing molecular weight Mn (Fig. 1B). While P1 and P2 showed conversions ≥95% after 4.5 h, a decrease in polymerisation rate was observed for P3–P5, resulting in a moderate drop in conversion from 90–60%.
This decline in conversion can primarily be attributed to the lower concentration of initiating species used when targeting higher molar masses. Consequently, a substantially lower concentration of active propagating species prevails throughout the polymerization, rendering the system more susceptible to termination reactions and oxygen inhibition. Additionally, the increased viscosity during polymerization further slows the reaction rate, collectively leading to the observed plateau at lower conversions.23,24
Statistical copolymerisation can be a challenging task when aiming at high molecular weights and applying comonomers with very different reactivity ratios. In the latter case, a gradient polymer chain and, in extreme cases, copolymers that resemble more block than random copolymers are obtained.25 Kinetic studies were performed to ensure statistical incorporation of the comonomers into the copolymer without gradient formation under the optimised reaction conditions specified in Table S4† aiming at 1000 kDa. Therefore, samples were taken every five minutes between the first 15 and 30 minutes of the reaction to assess monomer conversion via1H NMR spectroscopy of the crude mixture and prepare extended Kelen–Tüdös plots with feed ratios of OEGA:BA from 30:70 to 70:30 (Fig. 1C).26,27 The average reactivity ratios rOEGA and rBA were determined from the linear fits according to eqn (S11)–(S23) in the ESI† to be in a similar range and close to 1, with 1.26 and 1.47, respectively. Thus, a statistically azeotropic copolymerisation prevails despite the slightly more reactive BA comonomer, as further evidenced during the course of the reaction by the matching copolymer composition of P5 with the initial feed ratio (Fig. S4†). Complementarily determined reactivity ratios via the Finemann-Ross approach according to eqn (S2)–(S10) in the ESI† yielded equivalent ratios of rOEGA = 1.23 and rBA = 1.42 (Fig. S5†).28 Moreover, the matching monomer feed with the final copolymer composition of P1–P12 (Table 1) underscores the statistical copolymerisation of OEGA and BA under the applied conditions. Similar results of the reaction kinetics for monomers with significantly different polarity based on OEG and alkyl methacrylates or acrylates prepared by living radical polymerisations have been reported.5,29 In analogous mixed acrylate and methacrylate copolymerisations, however, the methacrylate comonomers are typically more reactive and thus incorporate preferentially.30
GPC traces revealed a monomodal and narrow distribution of the copolymers P1–P12 both in THF and water (Table 1 and Fig. S6†). For molecular weight determination, a MALS detector coupled to the GPC was applied after measuring dn/dc values of the respective copolymers in the corresponding solvent (Table S2†). Molecular weights were further calculated from 1H NMR spectra of the crude reaction products in CDCl3 by using characteristic peaks of the copolymer and unreacted monomer according to eqn (S1).†
Molecular weights determined absolutely via MALS are generally considered more reliable than the ones obtained by 1H NMR spectroscopy, particularly for high molecular weight polymers. The number-average molecular weights Mn determined in THF and water by MALS matched well for all polymers P2–P10 despite their presumed structural differences in the two solvents. Interestingly, the MALS-derived molecular weights also strongly agreed with those calculated from 1H NMR spectra and aligned well with the theoretical molecular weights for polymers up to 100 kDa (Table 1). However, when targeting higher molecular weight copolymers, deviations of 30–50% from the theoretical values were observed, particularly for copolymers approaching 1000 kDa. This divergence is consistent with the reduced polymerisation kinetics and decreased monomer conversion at higher molecular weights. Additionally, a noticeable increase in polydispersity with increasing molecular weight P2–P5 was observed from GPC measurements both in THF (Đ = 1.01–1.15) and water (Đ = 1.00–1.62). Moreover, the dispersity also increased with a rising proportion of butyl acrylate from P5–P12 in THF (Đ = 1.14–1.82) and from P5–P10 in water (Đ = 1.62–2.13), indicating progressively reduced control at higher BA contents.
Hence, the employed optimised ARGET ATRP conditions provide reasonable control over the statistical P(OEGA-co-BA) copolymerisation with access to molecular weights up to 600 kDa and narrow distributions (Đ < 1.7) with up to 77% BA content. At the same time, the method overcomes the limitations of conventional ATRP by adding elemental copper as a reducing agent to significantly lower the amount of cytotoxic copper salt required for catalysis.
In this regard, we investigated several purification protocols for the crude polymerisation mixtures of P5 on a 10 g scale, compared their efficiency by 1H NMR, and quantified the remaining copper traces in the polymer by atom absorption spectroscopy (AAS) as listed in Table S5.† Filtration of the product mixture over alumina is among the most frequently reported methods for copper catalyst removal31 and reduced the copper content of the crude product by 75%. A similar efficiency of 67% reduction was detected for repeated liquid–liquid extraction with saturated aqueous ammonium chloride solution. Both methods require the removal of DMF from the crude reaction mixture prior to the purification. However, for both methods signals of residual unreacted monomer in the 1H NMR spectra were detected (Fig. S7†). Dialysis in various media with multiple cycles using regenerated cellulose tubings with a molecular weight cut-off of 3.5 kDa eliminates the need to remove DMF as the solvent from the crude reaction mixture and allows simultaneous removal of unreacted monomer. Among the different tested dialysis protocols listed in Table S5,† protocol D2 encompassing six dialysis cycles in 2 L water each, followed by three dialysis cycles in 1 L EtOH each, was identified as the most efficient way to remove the water-soluble catalyst, residual OEGA monomer, and DMF in water and residual BA monomer in ethanol. A reduction of the initial copper concentration in the crude product by >82% was achieved, resulting in a final copper concentration of 2.4 ± 0.1 ppm and no residual monomers according to the 1H NMR spectra (Fig. S7†). Besides the residual copper content, also the amphiphilic nature of the copolymers could induce cell toxicity, while PGE or OGE is generally considered highly bio- and cell-compatible.11,32 Representative for this type of copolymers, P5 purified according to the optimised protocol was evaluated for its concentration-dependent cytocompatibility by a metabolic assay using a human epithelial cell line (Fig. S8†). A non-significant drop in metabolic activity was detected after 24 and 48 h exposure up to a polymer concentration of 1 mg mL−1, demonstrating the necessary cytocompatibility for potential drug delivery applications. Of note, even at 10 mg mL−1, the metabolic activity of the epithelial cells was reduced only by around 30%.
Next, the assembly of the amphiphilic copolymers in aqueous solution due to their potential to form micellar structures was investigated. The first evidence for micelle formation in water was gained from 1H NMR spectra recorded in D2O and CDCl3. A general decrease in signal intensity and line broadening of signals attributed to the hydrophobic butyl side chains was observed in spectra of P5–P7 and P12 in D2O compared to CDCl3 (Fig. S9†). Furthermore, with an increasing fraction of the hydrophobic comonomer, the intensity of backbone and backbone-proximal protons of BA diminished by up to 49% and the BA methyl group by up to 44% in D2O compared to CDCl3 (Table S6†). This reduction in signal intensity is attributed to the restricted chain mobility within the micellar core. As this trend is not observed in CDCl3, micelle formation of the statistical copolymers in aqueous solution is strongly suggested.
Statistical amphiphilic copolymers tend to form monomeric micelles and single-chain nanoparticles due to dominating intramolecular interactions at high degrees of polymerisation.1,3,33 The defined, monomodal GPC distribution curves suggest the absence of undefined aggregates (Fig. S6†). The steric repulsion of hydrophilic OEGA side chains can isolate a single main chain of the copolymers in water and effectively shield the hydrophobic BA side chains, arranged in a micellar core, from intermolecular interactions preventing aggregation.13 A strong indication for unimolecular micelles was obtained from combined MALS data, yielding the association number AH2O of copolymers. AH2O is derived from the ratio of Mn determined from MALS in water and THF according to eqn (S24).†7,13 As can be seen in Table 1, the association number of all copolymers in water is approximately one and thus independent of the degree of polymerisation (P2–P5) and composition (P5–P10), indicating single-chain polymers that self-assemble into unimeric micelles at 25 °C. Notably, copolymer P1 was too small for detection via MALS. So far, in literature, single-chain micelle formation has only been reported for random oligo ethylene glycol-based amphiphilic copolymers with a methacrylate backbone and molecular weights up to 140 kDa.12 Comparative structural investigations have shown that stable unimeric self-assembly of these copolymers is primarily supported by the larger OEGMA monomer (475 vs. 300 Da), the less hydrophobic alkyl methacrylate comonomer (butyl vs. dodecyl), and a comparably low mol% of the hydrophobic units (≤30%). However, the limited hydrophobic fraction significantly restricts the achievable property matrix of these amphiphilic polymethacrylates, particularly in terms of tunable LCST.
Due to the shear stress experienced by polymers during GPC, which may cause disaggregation, additional DLS experiments were conducted to further validate the formation of well-defined unimeric micelles of P(OEGA-co-BA). The measurements were conducted in aqueous solution in a series of dilutions, both in the presence and absence of salts at 25 °C (Fig. 2A). The hydrodynamic radii Rh of the copolymers were identical within the range of error in the presence and absence of salts at all three concentrations as well as upon dilution of a distinct polymer solution from 10–0.1 mg mL−1. For the larger copolymers P3 and P5, a slight trend towards size increase upon dilution was observed, caused by the decrease in osmotic repulsion of the single-molecule micelles. This effect is more pronounced for P5 than P3 (Fig. 2A), as the osmotic repulsion of the single-molecule micelles increases with the molecular weight. Overall, the DLS data under dilution strongly support a unimodal size distribution of the copolymers P1, P3, and P5 in water and PBS with Rh at 10 mg mL−1 in the range of 3, 4, and 9 nm (Table 2).
 | ||
| Fig. 2 (A) Representative DLS curves of P1, P3, and P5 at varying polymer concentrations in water and PBS at 25 °C. (B) Representative Mark–Houwink plot showing the intrinsic viscosity vs. molar mass on a log–log scale for P(OEGA-co-BA) P1–P5 in water at 25 °C with Mn in g mol−1. (D) Cryo-transmission electron micrograph of P5 embedded in amorphous ice with an enlargement of the indicated area and arrows indicating the nearly spherical unimers. | ||
| Polymer | Compositiona [OEGA:BA] |
M
nb [kDa] |
c [°C] |
T
PBScpc [°C] |
|
R PBSh (25 °C) [nm] |
|
R PBSh (37 °C) [nm] |
|---|---|---|---|---|---|---|---|---|
| a Molar ratio. b Calculated from 1H NMR spectra acquired in CDCl3 at 25 °C. c Determined from the inflection point of turbidity curves obtained during heating cycles. d Deviations from values stated in Fig. 2A are due to the use of different devices. e Determined by DLS at 20 °C. f Large aggregates outside the detection limit of the device. g Bi- or multimodal distribution in DLS. | ||||||||
| P1 | 50:50 |
9.8 | 68.6 ± 0.3 | 67.6 ± 0.5 | 2.5 ± 0.7d | 2.6 ± 0.8d | 2.2 ± 0.4 | 2.0 ± 0.3 |
| P2 | 50:50 |
46.8 | 73.5 ± 0.1 | 68.4 ± 0.3 | 3.1 ± 0.9 | 3.2 ± 1.0 | 3.0 ± 0.7 | 3.0 ± 0.7 |
| P3 | 50:50 |
91.2 | 71.1 ± 0.5 | 66.6 ± 0.3 | 4.3 ± 1.3d | 4.3 ± 1.3d | 3.2 ± 0.7 | 4.3 ± 1.3 |
| P4 | 50:50 |
366.9 | 70.5 ± 0.1 | 66.5 ± 0.5 | 7.6 ± 3.0 | 7.7 ± 3.0 | 7.9 ± 2.2 | 7.7 ± 2.9 |
| P5 | 49:51 |
615.2 | 69.1 ± 0.2 | 64.0 ± 0.5 | 8.3 ± 4.1d | 8.9 ± 4.2d | 8.6 ± 3.9 | 9.3 ± 4.1 |
| P6 | 38:62 |
594.1 | 64.0 ± 0.1 | 58.4 ± 0.3 | 7.7 ± 3.6 | 8.2 ± 3.5 | 8.0 ± 3.3 | 8.1 ± 3.3 |
| P7 | 29:71 |
626.9 | 48.4 ± 0.4 | 45.4 ± 0.1 | 7.2 ± 3.1 | 7.4 ± 3.3 | 7.6 ± 2.8 | 8.3 ± 3.0 |
| P8 | 27:73 |
622.2 | 45.3 ± 0.1 | 44.7 ± 0.4 | 7.9 ± 3.6 | 8.2 ± 3.6 | 9.5 ± 4.2 | 11.0 ± 4.9 |
| P9 | 26:74 |
633.4 | 41.3 ± 0.3 | 38.9 ± 0.3 | 8.8 ± 4.2 | 9.5 ± 4.3 | 13.9 ± 6.2 | 19.8 ± 10.3 |
| P10 | 23:77 |
640.1 | 38.8 ± 0.6 | 34.1 ± 0.0 | 10.6 ± 4.5 | 11.7 ± 6.1 | 27.7 ± 21.0 | 16.3 ± 4.2 |
| 1565.9 ± 687.5g | 530.5 ± 203.6g | |||||||
| P11 | 22:78 |
650.6 | 33.2 ± 0.2 | 30.4 ± 0.3 | 14.7 ± 6.9 | 19.2 ± 9.0 | >1000f | >1000f |
| P12 | 20:80 |
655.9 | 26.4 ± 0.2 | 24.1 ± 0.3 | 20.7 ± 9.4e | 24.6 ± 10.0e | 59.4 ± 24.6 | 66.0 ± 29.3 |
| 297.5 ± 137.8g | 577.4 ± 433.4g | |||||||
| 2614.8 ± 405.2 | 2537.0 ± 434.1 | |||||||
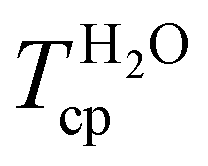


In addition, the scattering intensities of copolymer P5 were examined at constant incident intensity in water, PBS, THF, and ethanol during dilution. In all solvents, a linear decrease in scattering intensity with decreasing polymer concentration was observed (Fig. S10†). The significantly lower slope in THF compared to the other solvents can be attributed to the polymer's low refractive index contrast in THF. Furthermore, copolymer P5 exhibits a substantially higher degree of swelling in organic solvents upon dilution than in aqueous systems (Fig. S11 and Table S7†). This observation substantiates the hypothesis that the polymers form unimeric micelles in aqueous solution rather than existing as solvated polymer coils, as seen in ethanol and particularly in THF. The deviation from this trend at the lowest concentration in THF is attributed to the very low scattering intensities, which result in significant measurement errors.
The structure of the unimers of copolymers P1–P5 in aqueous solution was further investigated via a Mark–Houwink plot (Fig. 2B). Therefore, the intrinsic viscosities [η] of the aqueous solutions were measured on a plate-plate rheometer and plotted against the copolymers’ molecular weight Mn. The molecular weights determined by GPC-MALS were used except for P1, whose molecular weight was derived from 1H NMR spectroscopy. The corresponding Mark–Houwink-Sakurada coefficients α and K were calculated to be α = 0.23 and K = 5.90 × 10−1 mL g−1 after fitting the experimental data using linear regression (Fig. 2B and Fig. S12†). A coefficient α < 0.5 is typical for branched polymers of high density and spherical structure. Hence, the measured coefficient α of the copolymers in water agrees well with a highly compact micellar structure.34,35 These structural results were qualitatively confirmed by imaging analysis via cryo-TEM of copolymer P5, which clearly indicates dark spheroids of nanometer size without any signs of larger aggregate formation (Fig. 2C).
Polymers based on OEGA typically exhibit LCST behaviour in aqueous solutions, with their transition temperature adjustable through amphiphilic balance, similar to their methacrylate-based counterparts.36 The thermoresponsiveness of the copolymers P1–P12 at a concentration of 10 mg mL−1 was investigated via turbidity measurements in water and PBS as a biological fluid. While the OEGA homopolymers exhibit an LCST of 92 °C in water,36 statistical copolymerisation with BA shifted the location of the cloud points with increasing BA content towards the physiologically relevant regime. In Fig. 3A, the normalised turbidity curves from heating cycles for copolymers P5–P12 with a molecular weight of approximately 600 kDa in water are shown. Additional heating and cooling cycles in water and PBS are presented in Fig. S13† for P(OEGA-co-BA) copolymers grouped according to increasing molecular weight at a fixed 1:1 comonomer ratio and increasing BA content at a fixed molecular weight around 600 kDa. Generally, all copolymers exhibited a sharp and reversible, thermally induced phase separation in water and PBS with little to no hysteresis. Turbidity curves of copolymers P7–11 are particularly interesting as the phase transition occurs in the temperature range relevant to biological settings.
 | ||
| Fig. 3 (A) Representative normalised turbidity curves of P5–P12 in water determined at a heating rate of 0.5 °C min−1 and a concentration of 10 mg mL−1 with photographs of the aqueous solution of P12 below and above the phase transition temperature. (n = 3). (B) Plots of the measured cloud point temperature Tcp in water and PBS as a function of the average mole fraction of BA units per chain for the statistical P(OEGA-co-BA) copolymers. | ||
The temperature at the inflection point of the turbidity curves in water and PBS was set as the Tcp, summarised for copolymers P1–P12 in Table 2. Equally composed copolymers P1–P5 of varying molar mass exhibited a comparable Tcp, indicating that the phase transition is independent of the copolymers’ molecular weight. This is consistent with the observations for other thermoresponsive copolymers for which the molecular weight dependency of the cloud point is only pronounced at a low degree of polymerisation.12,25,37,38 Furthermore, for copolymers P5–P12, a steady decrease of the Tcp from 69 to 26 °C with increasing BA content between 50–80 mol% was observed in aqueous solution. The cloud point was more sensitive to variations in the copolymer composition when the content of hydrophobic BA in the polymer chain increased above 60 mol% (Fig. 3B). In the range of 60–80 mol% BA, a linear decrease in Tcp of about 1.9 °C per mol% BA was observed in aqueous solution. For similar OEGMA-based copolymers with di(ethylene glycol) methyl ether methacrylate (MEO2MA) as the hydrophobic comonomer, the Tcp decreased by 1.04 °C per mol% of MEO2MA in the copolymer in the range of 0–30 mol% OEGMA475 Da.39 The observed larger incremental decrease of the Tcp for P(OEGA-co-BA) compared to P(OEGMA-co-MEO2MA) can be attributed to the higher hydrophobicity of BA compared to the MEO2MA comonomer. Notably, when compared to the analogous methacrylate-based copolymers (OEGMA475 Da:BMA) with 30–50 mol% OEGMA and cloud points between 45 and 63 °C,7 the cloud points of the polyacrylates reported here were 3–7 °C higher. In contrast to the methacrylate-based system that likely becomes insoluble at BMA contents >70%, as it has not been reported in the literature so far, a further increase in the BA content up to 80% is possible for the acrylate-based system to lower and adjust the Tcp within the physiologically-relevant regime.
For LCST-type polymers, the phase transition temperature is often shifted in the presence of ions, which are omnipresent in biological settings.16,40,41 Kosmotropic anions like Cl− and HPO42− present in PBS reduce hydrogen bonding interactions between thermoresponsive copolymers and water, favouring polymer–polymer interactions and leading to polymer aggregation and phase separation. For the P(OEGA-co-BA) copolymers, a 2–6 °C decrease of the Tcp in PBS compared to ultra-pure water was observed (Table 2). This so-called salting-out effect became slightly more pronounced with increasing molecular weight of the copolymers (P1–P5). A similar decrease in PBS by 3–7 °C was observed for P(HEMA-co-OEGMA300 Da) copolymers.16 While in water no hysteresis between heating and cooling curves was observed, the presence of salts, in some cases, induced a slight hysteresis due to partial, barely detectable macroscopic phase separation and retarded dissolution of the precipitated polymer fraction (Fig. S13D–F†). In any case, no complete macroscopic phase separation was observed upon heating, as represented by photographs of aqueous P12 solutions above and below the cloud point in Fig. 3A. Similar observations were made with P(HEMA-co-OEGMA300 Da) copolymers in the presence of Na2HPO4 and NaCl salts when the Tcp was exceeded.16 Overall, we were able to show that P(OEGA-co-BA) copolymers are completely soluble below their LCST and show fully reversible phase transitions in water and PBS, irrespective of the molecular weight up to 600 kDa and composition up to 80 mol% BA. Importantly, the Tcp could be adjusted to the physiological regime in water and PBS by adjusting the comonomer ratio.
Unimeric micelle-forming polymers in aqueous media hold great potential for drug delivery applications, as they remain stable even under extreme dilution, unlike conventional micelles, making them superior drug carriers.1,3,33 Therefore, the stability of the unimeric micelles under physiological conditions at 37 and 25 °C in the presence of salts (PBS buffer) was investigated via DLS measurements, and the resulting hydrodynamic radii Rh were compared to the ones in water (Table 2). Generally, with increasing molecular weight of the copolymers at a fixed comonomer ratio, the hydrodynamic radii increased from around 3 to 9 nm in water and PBS for P1–P5 (Table 2 and Fig. S14, S15†). For copolymers with a Tcp far above 37 °C, physiological temperature and salt conditions did not markedly alter the hydrated size of the unimers. The micellar size of the polyacrylate P3 in water (4.3 ± 1.3 nm) is similar to the corresponding polymethacrylate P(OEGMA475 Da-co-BMA) reported in literature (4.9 nm) at comparable molecular weight and comonomer ratio.7 In contrast to our acrylate-based copolymers, the methacrylate analogues showed an increase in the association number with increasing BMA content (50–70 mol%).7 This phenomenon can be attributed to the larger hydrophobic core of the P(OEGMA475 Da-co-BMA) micelles, which results in a smaller hydrophilic shell compared to the corresponding micelles of the acrylate-based copolymers. Thus, reduced shielding properties of the shell of polymethacrylate unimers may lead to their instability, favouring intermolecular micellisation. To date, none of the reported OEGMA-based copolymers has been shown to form unimeric micelles with dimensions exceeding the renal clearance cut-off or to exhibit thermoresponsive properties in the physiologically relevant regime,5,7,8,12,13 limiting their potential as smart drug nanocarriers. For the P(OEGA-co-BA) copolymers, a molecular weight ≥90 kDa (P3) ensures unimer formation with a hydrodynamic radius Rh > 4 nm in PBS at 37 °C, exceeding the lower renal clearance threshold. The discrepancy in the estimated radii of P5 by cryo-TEM (∼3 nm) (Fig. 2C) and DLS (∼8 nm) arises from the fact that cryo-TEM visualises primarily the micellar core, leading to a smaller size due to contrast limitations, compared to the hydrodynamic radius determined by DLS, which includes the solvated corona, resulting in larger values.
At a fixed molecular weight of around 600 kDa, an increasing BA content from 51–71% – accompanied by the decrease in Tcp from 69 to 48 °C in water and 64 to 45 °C in PBS – progressively reduced the hydration of the OEGA-based shell of P5–P7 unimers, both in the presence and absence of salts, leading to a 1–2 nm reduction in hydrodynamic radius (Fig. S14 and S15†). However, further increase in the BA content from 71–77% for P7–P10 – decreasing the Tcp from 48 to 38 °C in water and 45 to 34 °C in PBS – caused a progressive increase of Rh at 25 °C from around 7 to 11 nm in water and PBS attributed to spatial limitations in the hydrophobic core of the unimolecular micelles. The monomodal distribution and compact size of the copolymers in water detected by DLS support the GPC results of single hydrated polymer chains below the Tcp without aggregation. The same trend is observed for P7–P9 below their respective Tcp at 37 °C, resulting in progressive Rh increases from around 8 to 14 nm in water and up to 20 nm in PBS. For P9 in PBS at 37 °C, the higher Rh value (20 nm) is already influenced by the proximity to its Tcp (39 °C), at least partially contributing to the observed micellar size increase, likely due to aggregation. Clear signs of thermally induced micelle aggregation, according to the illustration shown in Scheme 1, were observed for P10–P12 at temperatures close to or above the copolymers’ Tcp, which is located between 38–26 °C in water and 34–24 °C in PBS, resulting in Rh > 20 nm (Table 2) and occasional bi- or multimodal size distribution in DLS, particularly at 37 °C (Fig. S15†).
To further investigate the thermally induced aggregation behaviour of the amphiphilic copolymers P1–P12, DLS curves were acquired at RT and 10 °C above the respective Tcp in water and PBS (Fig. S14, S16 and Table S8†). Except for P11 and P12, all investigated copolymer micelles reproducibly formed stable and defined colloidal aggregates above the cloud point at T = Tcp + 10 °C in water and PBS, as can be seen for the 600 kDa copolymers in Fig. 4 from their narrow size distribution curves and the small SD. In water, all copolymers – except P12 – form thermally triggered aggregates with Rh < 200 nm. In PBS, a trend towards slightly larger aggregates is observed with Rh up to 300 nm for the smallest copolymer P1, but the Rh of the majority (P2–P9) of copolymer aggregates remains below 200 nm (Table S8†).
 | ||
| Fig. 4 Representative DLS curves of P(OEGA-co-BA) copolymers P5–P10 (10 mg ml−1) and their corresponding Rh below and above the respective Tcp (Tcp + 10 °C) in water and PBS. (n = 3). | ||
Interestingly, the specific amphiphilic ratios in P11 and P12 do not support colloidal stabilisation at T > Tcp, resulting in large aggregates (>1 μm) or multimodal distributions after 15 min equilibration. The comparative DLS results of copolymer P10 at 25, 37, and 44 °C (Tcp + 10 °C) further highlight the dynamics of the thermally induced aggregation process (Table 2 and Table S8†). While monomodal size distributions well below and above the Tcp at 25 and 44 °C were detected, bimodal size distributions at 37 °C close to the Tcp indicate the metastable transitional state of the system. Hence, the size of thermally induced aggregates in the transitional stage will depend not only on the comonomer composition but, most likely, also on the rate of temperature change.42
Based on the combined DLS data, the statistical copolymers’ BA content should not exceed 74%, particularly at high molecular weights (≥600 kDa), to prevent thermally induced aggregation under physiological conditions. For renal clearance and evasion of immune cell recognition,3 ideal nanoparticle diameters are between 6 and 200 nm, making copolymers P3–P9 formally suitable candidates in physiological salt solution at 37 °C (Table 2).
In literature, a thermally induced, faster drug release from polymeric, thermoresponsive micelles has been demonstrated at temperatures above their Tcp,17,43 underscoring their potential for drug delivery applications. To determine the loading capacity of the thermoresponsive P(OEGA-co-BA)-based micelles and study their hydrated size and colloidal stability under physiological conditions, we loaded them with the hydrophobic, UV-active pyrene dye as a hydrophobic drug proxy. Water-insoluble pyrene dissolved in methanol was added to the copolymers, the solvent was removed, and the residue was redispersed in PBS or D2O to generate the core-loaded polymeric micelles. Excessive and water-insoluble pyrene was removed from the aqueous solution by filtration. The pyrene-loading capacity of the micelles as the number of pyrene molecules per polymer (Npyr/Npol) (Table 3) was independently determined by UV-Vis (Fig. S17†) and 1H NMR (Fig. S18†) spectroscopic measurements, as described in detail in the ESI.†
| Polymer | Compositiona [OEGA:BA] |
M
nb [kDa] |
T
PBScpc [°C] |
T
pyrcpc [°C] |
R PBSh (37 °C) [nm] | R pyrh (37 °C) [nm] |
N
pyr/Npole |
N
pyr/Npolf |
|---|---|---|---|---|---|---|---|---|
| a Molar ratio. b Calculated from 1H NMR spectra acquired in CDCl3 at 25 °C. c Determined from the inflection point of turbidity curves obtained during heating cycles. d Large aggregates outside the detection limit of the DLS device. e Determined by UV-Vis in PBS at 25 °C. f Calculated from 1H NMR spectra acquired in D2O at 25 °C. g Bi- or multimodal distribution in DLS. | ||||||||
| P1 | 50:50 |
9.8 | 67.6 ± 0.5 | 70.7 ± 1.2 | 2.0 ± 0.3 | 2.5 ± 0.3 | 0.2 ± 0.1 | 0.2 ± 0.1 |
| P2 | 50:50 |
46.8 | 68.6 ± 0.3 | 70.7 ± 0.3 | 3.0 ± 0.7 | 3.3 ± 0.6 | 0.8 ± 0.1 | 1.0 ± 0.3 |
| P3 | 50:50 |
91.2 | 66.6 ± 0.3 | 67.2 ± 0.3 | 4.3 ± 1.3 | 4.4 ± 0.8 | 1.6 ± 0.2 | 1.5 ± 0.6 |
| P4 | 50:50 |
366.9 | 66.5 ± 0.5 | 69.0 ± 0.1 | 7.7 ± 2.9 | 7.6 ± 2.6 | 5.7 ± 0.7 | 5.9 ± 0.9 |
| P5 | 49:51 |
615.2 | 64.0 ± 0.5 | 63.9 ± 0.6 | 9.3 ± 4.1 | 9.2 ± 4.0 | 9.7 ± 0.4 | 12.8 ± 1.4 |
| P6 | 38:62 |
594.1 | 58.4 ± 0.3 | 58.5 ± 0.5 | 8.1 ± 3.3 | 7.9 ± 3.3 | 17.4 ± 0.4 | 19.2 ± 0.2 |
| P7 | 29:71 |
626.9 | 45.4 ± 0.1 | 46.2 ± 0.3 | 8.3 ± 3.0 | 8.9 ± 3.1 | 29.7 ± 0.6 | 28.8 ± 0.4 |
| P8 | 27:73 |
622.2 | 44.7 ± 0.4 | 42.3 ± 0.3 | 11.0 ± 4.9 | 19.5 ± 8.6 | 34.1 ± 1.5 | 35.1 ± 0.7 |
| P9 | 26:74 |
633.4 | 38.9 ± 0.3 | 37.9 ± 0.3 | 19.8 ± 10.3 | 13.3 ± 6.5 | 37.7 ± 1.0 | 36.2 ± 0.2 |
| P10 | 23:77 |
640.1 | 34.1 ± 0.1 | 34.0 ± 0.1 | 16.3 ± 4.2 | >1000d | 38.3 ± 0.2 | 40.9 ± 2.8 |
| 530.5 ± 203.6g | ||||||||
| P11 | 22:78 |
650.6 | 30.4 ± 0.3 | 30.4 ± 0.3 | >1000d | 58.9 ± 22.0 | 46.2 ± 1.3 | 46.2 ± 1.2 |
| 1088.0 ± 606.5g | ||||||||
| P12 | 20:80 |
655.9 | 24.1 ± 0.3 | 27.7 ± 0.6 | 66.0 ± 29.3 | >1000d | 49.3 ± 0.3 | 50.0 ± 2.8 |
| 577.4 ± 433.4g | ||||||||
| 2537.0 ± 434.1 | ||||||||
As expected, the amount of encapsulated pyrene increased with the molecular weight and micellar size of the copolymers P1–P5 at a fixed 1:1 comonomer ratio. While the 10–90 kDa copolymers P1–P3 did not encapsulate significant amounts of pyrene, copolymers of Mn = 365 kDa (P4) and Mn = 615 kDa (P5) could load 6 and 12 molecules per micelle. Interestingly, at a persistent high molecular weight of around 600 kDa, the loading capacity for pyrene could be drastically enhanced from 12 to 50 molecules via a 30% increase in the BA content of the copolymers P5–P12. Notably, under physiological conditions, only polymers P1–P9 assemble into unimolecular micelles. The high loading capacity of these P(OEGA-co-BA) copolymer-based micelles is outstanding compared to reported capacities of similar unimolecular host systems based on PEG, polyglycerol, or sodium maleate-based bottlebrushes or dendrimers in the literature.44–48 Due to the comparably low molecular weight (<50 kDa), these systems encapsulate only minor quantities of pyrene (0.2–6.2 molecules) per carrier.
Generally, the presence of encapsulated pyrene did not adversely affect or substantially shift the copolymers’ Tcp according to turbidity experiments with the loaded and unloaded micelles (Table 3 and Fig. S19†). Surprisingly, however, the pyrene loading numbers determined by UV-Vis and 1H NMR showed remarkable agreement. Given its hydrophobic and water-insoluble nature, pyrene is expected to preferentially reside in the micellar core, where it is shielded from the surrounding environment, leading to a decrease in 1H NMR signal intensity. Consequently, pyrene concentrations determined by NMR spectroscopy would typically be underestimated compared to those obtained from UV-Vis spectroscopy, leading to discrepancies in the determined values.
To estimate the localisation of pyrene in the copolymer micelles, its solvatochromic fluorescence properties were utilised by determining the ratio of the first and third emission band I1 (∼373 nm)/I3 (∼384 nm) in the fluorescence emission spectra of the pyrene-loaded micelles (Fig. S20 and Table S9†).49 While the first vibronic band corresponds to a relatively polar environment, the third vibronic band indicates a more unpolar surrounding for pyrene. At a polymer concentration of 10 mg mL−1in PBS at 25 °C, the I1/I3 values of pyrene-loaded P1–P5 (1.54–1.45) indicate that, given the very low proportion of BA, pyrene seems to be distributed throughout the entire unimeric micelles. An increase in the hydrophobic BA content in P5–P7 and P12 leads, as expected, to a progressive reduction of the I1/I3 ratio from 1.55 to 0.83 for the pyrene-loaded micelles. Upon diluting the loaded micelles to 0.01 mg mL−1, the I1/I3 ratio increases for all systems to 1.61–1.69 (Table S9†). This effect can be attributed to a reduced osmotic repulsion between micelles upon dilution, allowing them to expand (see Fig. 2A). The increased water penetration into the expanding micelles generates a more hydrophilic environment for pyrene, thereby raising the I1/I3 ratio. The observed ratios remain lower than that of surfactants below their CMC in water (I1/I3 > 1.8)49 and fall within the typical range of surfactants like Triton X-10049 or polyethylene oxide-polypropylene oxide-polyethylene oxide block copolymers,50 which exhibit values of I1/I3 = 1.4–1.72 above their CMC. Based on these results, it can be concluded that pyrene is not exclusively located in the core but is also partially localised within the OEGA-based shell of the micelle. This finding fully agrees with the matching numbers of encapsulated pyrene determined by NMR and UV-Vis spectroscopy.
A comparison of the hydrodynamic radii of the loaded (Fig. S21†) and corresponding unloaded micelles in PBS at 25 °C revealed no significant size alteration of the unimeric micelles of copolymers P1–P3 upon pyrene loading, likely because each micelle contains less than two pyrene molecules (Table S10†). In contrast, an increasing number of encapsulated pyrene molecules (P4–P12) tends to result in more compact micellar structures (Rh = 8–13 nm), particularly with the more hydrophobic copolymers (≥77% BA), compared to the unloaded micelles (Rh = 9–20 nm). For P1–P7 with Tcp's > 37 °C, no effect of pyrene loading on the size of the unimeric micelles was observed (Table 3). For more hydrophobic copolymers with a Tcp near 37 °C (P8–P9), pyrene loading induced an earlier entry into the metastable transitional state of the micelles at 37 °C. The pyrene loading of copolymers with a Tcp below 37 °C (P10–P12) results in the formation of aggregates larger than 1000 nm at 37 °C. Overall, pyrene-loading leaves the micellar size and stability unaffected at temperatures well below the polymers’ Tcp.
To demonstrate the reversibility of the aggregation of pyrene-loaded micelles, we subjected unimolecular micelles of P9, loaded with around 36 pyrene molecules, to a stepwise heating and cooling process (Fig. S22†). Initially, the temperature was increased from 25 to 37 °C and subsequently equilibrated above the systems Tcp (38 °C) at 40 °C, mimicking inflammation conditions in the human body. A gradual increase in the hydrodynamic micelle radius from 8.5 to 13.3 nm and, finally, 18.3 nm due to thermally triggered aggregation was observed. Upon cooling to 37 and 25 °C, the micelle size decreased from 18.3 to 13.2 nm and, finally, 8.3 nm, indicating a fully reversible aggregation process of the loaded unimolecular micelles.
In summary, the comprehensive data on P(OEGA-co-BA) copolymers in this study yield molecular design guidelines for unimeric micelle-forming amphiphilic copolymers with high loading capacity for prospective applications as smart drug delivery vehicles (Fig. 5).
 | ||
| Fig. 5 Illustration of unimer size depending on amphiphilic balance and molecular weight in physiological salt solution at 37 °C with corresponding loading capacity for pyrene as drug proxy and thermally triggered aggregation into defined nanosized colloids due to their thermoresponsive properties. The most promising candidates for drug delivery applications P7–P9 are highlighted in grey. | ||
The schematically visualised OEGA- and BA-based copolymers P1–P9 form defined unimolecular micelles in both water and physiological salt solutions at 37 °C. The size of these micelles increases with the molecular weight of the copolymers at a constant monomer ratio (P1–P5) but decreases as the BA content rises from 50 to 70% (P5–P7). The size decrease at constant molecular weight can be attributed to a shift in the balance between the hydrophobic core and the hydrophilic corona. However, with further increase in BA content up to 74% (P7–P9), the micelle size increases again, likely due to spatial limitations in the hydrophobic core. At temperatures close to the Tcp of a micelle-forming copolymer, the unimeric micelles enter a metastable transitional state driven by weakened polymer chain–water interactions. When the Tcp is exceeded, the micelles reversibly form defined nanosized aggregates, also when loaded with pyrene. We infer that the de/rehydration of the hydrophilic ether oxygen groups of the OEGA-based shell primarily drives the reversible aggregation process. Shell dehydration results in its collapse, drawing it closer to the hydrophobic core, reducing osmotic repulsion, and ultimately leading to aggregation with other dehydrated unimeric micelles.51 Given the statistical distribution of comonomers within the P(OEGA-co-BA) polymers, microphase separation of the unimeric micelles prior to their aggregation is thermodynamically unlikely.52 Therefore, we propose that the copolymers undergo reversible phase separation via the self-assembly of dehydrated unimeric micelles into larger aggregates, as illustrated in Fig. 5. The rapid and fully reversible phase transition during heating and cooling further supports this interpretation.
Based on the size of the unimolecular micelles and their thermal aggregates in PBS, polymers P3–P9 fall in the relevant size regime for drug delivery applications.3 Significant loading capacity is achieved only for high molecular weight (∼600 kDa) copolymers (P5–P12). Considering also the micelles’ LCST as a potential trigger for drug release, copolymers P7–P9 with a Tcp close to physiological temperature are most interesting. Hence, with a Tcp of 38.9 °C in PBS, corresponding to typical inflammation temperature, polymer P9 emerges as an excellent candidate for drug delivery applications.
Conclusion
ARGET ATRP provided good control over the polymerisation of OEGA and BA, achieving high molecular weights of up to 600 kDa and statistical monomer incorporation essential for the formation of unimeric micelles with high loading capacities. The reduced amount of catalytic copper needed is essential to guarantee biocompatibility of the resulting polymers. In vitro cytocompatibility assays revealed no significant impact of residual copper or the copolymer on mammalian cell metabolic activity at polymer concentrations up to 1 mg mL−1. Importantly, copolymers with up to 74% BA content formed unimeric micelles below their LCST and well-defined aggregates above. The size and loading capacity of the resulting micelles could be finely tuned by precisely adjusting molecular weight and the amphiphilic balance. Remarkable loading capacities for pyrene exceeding 30 molecules per nanocarrier with a BA content >71% were achieved. With a phase transition temperature of approximately 39 °C in PBS, matching that of inflamed tissue, copolymer P9 shows great promise for thermally triggered drug delivery applications. Future work will focus on drug uptake and release kinetics to fully explore the capabilities of these smart PEG-based unimeric micelles. Potential limitations due to emerging PEG-antibody formation53in vivo can be addressed by replacing the OEGA macromonomers with glycidyl ether-based macromonomers.54,55Author contributions
Florian Tondock: conceptualization, investigation, methodology, writing – original draft; David Nash: investigation, methodology, writing – review & editing; Cathleen Hudziak: investigation, methodology, Kai Ludwig: investigation methodology, writing – review & editing; Marie Weinhart: conceptualization, methodology, funding acquisition, supervision, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† The raw data required to reproduce these findings are available at Zenodo at https://doi.org/10.5281/zenodo.15019507.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. T. gratefully acknowledges the support of Antje Walter in cell culture experiments. Parts of this work were funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project ID 431232613 – SFB 1449 (M. W.; K. L.). The authors acknowledge the assistance of the Core Facility BioSupraMol of Freie Universität Berlin supported by the DFG.References
- G. Chen, Y. Wang, R. Xie and S. Gong, A review on core-shell structured unimolecular nanoparticles for biomedical applications, Adv. Drug Delivery Rev., 2018, 130, 58–72 CrossRef CAS PubMed.
- D. Braatz, M. Cherri, M. Tully, M. Dimde, G. Ma, E. Mohammadifar, F. Reisbeck, V. Ahmadi, M. Schirner and R. Haag, Chemical Approaches to Synthetic Drug Delivery Systems for Systemic Applications, Angew. Chem., Int. Ed., 2022, 61, e202203942 CAS.
- M. Müllner, Molecular polymer bottlebrushes in nanomedicine: therapeutic and diagnostic applications, Chem. Commun., 2022, 58, 5683–5716 Search PubMed.
- B. N. S. Thota, L. H. Urner and R. Haag, Supramolecular Architectures of Dendritic Amphiphiles in Water, Chem. Rev., 2016, 116, 2079–2102 CAS.
- G. Hattori, Y. Hirai, M. Sawamoto and T. Terashima, Self-assembly of PEG/dodecyl-graft amphiphilic copolymers in water: consequences of the monomer sequence and chain flexibility on uniform micelles, Polym. Chem., 2017, 8, 7248–7259 CAS.
- R. Kanno, M. Ouchi and T. Terashima, Self-assembly and salt-induced thermoresponsive properties of amphiphilic PEG/cation random terpolymers in water, Polym. Chem., 2023, 14, 1718–1726 CAS.
- S. Imai, Y. Hirai, C. Nagao, M. Sawamoto and T. Terashima, Programmed Self-Assembly Systems of Amphiphilic Random Copolymers into Size-Controlled and Thermoresponsive Micelles in Water, Macromolecules, 2018, 51, 398–409 CAS.
- Y. Hirai, T. Terashima, M. Takenaka and M. Sawamoto, Precision Self-Assembly of Amphiphilic Random Copolymers into Uniform and Self-Sorting Nanocompartments in Water, Macromolecules, 2016, 49, 5084–5091 CAS.
- X. Fan, Z. Li and X. J. Loh, Recent development of unimolecular micelles as functional materials and applications, Polym. Chem., 2016, 7, 5898–5919 CAS.
- N. Badi, Non-linear PEG-based thermoresponsive polymer systems, Prog. Polym. Sci., 2017, 66, 54–79 CAS.
- J.-F. Lutz, Polymerization of oligo(ethylene glycol) (meth)acrylates: Toward new generations of smart biocompatible materials, J. Polym. Sci., Part A:Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- M. Shibata, M. Matsumoto, Y. Hirai, M. Takenaka, M. Sawamoto and T. Terashima, Intramolecular Folding or Intermolecular Self-Assembly of Amphiphilic Random Copolymers: On-Demand Control by Pendant Design, Macromolecules, 2018, 51, 3738–3745 CrossRef CAS.
- T. Terashima, T. Sugita, K. Fukae and M. Sawamoto, Synthesis and Single-Chain Folding of Amphiphilic Random Copolymers in Water, Macromolecules, 2014, 47, 589–600 CrossRef CAS.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Thermoresponsive polymers with lower critical solution temperature: from fundamental aspects and measuring techniques to recommended turbidimetry conditions, Mater. Horiz., 2017, 4, 109–116 RSC.
- R. Liu, M. Fraylich and B. R. Saunders, Thermoresponsive copolymers: from fundamental studies to applications, Colloid Polym. Sci., 2009, 287, 627–643 CrossRef CAS.
- M. Kasprów, J. Machnik, Ł. Otulakowski, A. Dworak and B. Trzebicka, Thermoresponsive P(HEMA-co-OEGMA) copolymers: synthesis, characteristics and solution behavior, RSC Adv., 2019, 9, 40966–40974 RSC.
- C. Chang, H. Wei, D.-Q. Wu, B. Yang, N. Chen, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Thermo-responsive shell cross-linked PMMA-b-P(NIPAAm-co-NAS) micelles for drug delivery, Int. J. Pharm., 2011, 420, 333–340 CrossRef CAS PubMed.
- A. Schweigerdt, D. D. Stöbener, A. Schäfer, S. Kara and M. Weinhart, Impact of Amphiphilicity Balance in Hydroxy-Functional, Isomeric, Thermoresponsive Poly(meth)acrylates, Macromolecules, 2023, 56, 8602–8613 CrossRef CAS.
- W. A. Braunecker, N. V. Tsarevsky, A. Gennaro and K. Matyjaszewski, Thermodynamic Components of the Atom Transfer Radical Polymerization Equilibrium: Quantifying Solvent Effects, Macromolecules, 2009, 42, 6348–6360 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Controlled/living radical polymerization: Features, developments, and perspectives, Prog. Polym. Sci., 2007, 32, 93–146 CAS.
- K. Matyjaszewski, Controlled Radical Polymerization, American Chemical Society, Washington, D.C., 1998 Search PubMed.
- A. Simula, G. Nurumbetov, A. Anastasaki, P. Wilson and D. M. Haddleton, Synthesis and reactivity of α,ω-homotelechelic polymers by Cu(0)-mediated living radical polymerization, Eur. Polym. J., 2015, 62, 294–303 CAS.
- K. Matyjaszewski, N. V. Tsarevsky, W. A. Braunecker, H. Dong, J. Huang, W. Jakubowski, Y. Kwak, R. Nicolay, W. Tang and J. A. Yoon, Role of Cu 0 in Controlled/“Living” Radical Polymerization, Macromolecules, 2007, 40, 7795–7806 CrossRef CAS.
- P. Krys and K. Matyjaszewski, Kinetics of Atom Transfer Radical Polymerization, Eur. Polym. J., 2017, 89, 482–523 CAS.
- S. Heinen, S. Rackow, A. Schäfer and M. Weinhart, A Perfect Match: Fast and Truly Random Copolymerization of Glycidyl Ether Monomers to Thermoresponsive Copolymers, Macromolecules, 2017, 50, 44–53 CAS.
- F. Tüdos, T. Kelen, T. Földes-berezsnich and B. Turcsányi, Analysis of Linear Methods for Determining Copolymerization Reactivity Ratios. III. Linear Graphic Method for Evaluating Data Obtained at High Conversion Levels, J. Macromol. Sci., Part A, 1976, 10, 1513–1540 Search PubMed.
- T. Kelen and F. Tüdos, Analysis of the Linear Methods for Determining Copolymerization Reactivity Ratios. I. A New Improved Linear Graphic Method, J. Macromol. Sci. Chem. A, 1975, 9, 1–27 Search PubMed.
- M. Fineman and S. D. Ross, Linear method for determining monomer reactivity ratios in copolymerization, J. Polym. Sci., 1950, 5, 259–265 CAS.
- T. Ikami, Y. Kimura, M. Takenaka, M. Ouchi and T. Terashima, Design guide of amphiphilic crystalline random copolymers for sub-10 nm microphase separation, Polym. Chem., 2021, 12, 501–510 CAS.
- D. Neugebauer, M. Theis, T. Pakula, G. Wegner and K. Matyjaszewski, Densely Heterografted Brush Macromolecules with Crystallizable Grafts. Synthesis and Bulk Properties, Macromolecules, 2006, 39, 584–593 CAS.
- I. Ydens, S. Moins, F. Botteman, P. Degée and P. Dubois, Removal of copper-based catalyst in atom transfer radical polymerization using different extraction techniques, e-Polym., 2004, 4, 39–45 Search PubMed.
- H. Le Pham Khanh, D. Nemes, Á. Rusznyák, Z. Ujhelyi, P. Fehér, F. Fenyvesi, J. Váradi, M. Vecsernyés and I. Bácskay, Comparative Investigation of Cellular Effects of Polyethylene Glycol (PEG) Derivatives, Polymers, 2022, 14, 279–293 Search PubMed.
- S. Ordanini and F. Cellesi, Complex Polymeric Architectures Self-Assembling in Unimolecular Micelles: Preparation, Characterization and Drug Nanoencapsulation, Pharmaceutics, 2018, 10, 209–227 CAS.
- M. Bohdanecky and M. Netopilik, The Mark-Houwink-Kuhn-Sakurada exponent of polymers with long side groups: is a,0 = 1/2 a reliable criterion of the theta state?, Polymer, 1995, 36, 3377–3384 CAS.
- M. Sadao and G. B. Howard, Size Exclusion Chromatography, Springer Berlin Heidelberg, Berlin, 2010 Search PubMed.
- G. Vancoillie, D. Frank and R. Hoogenboom, Thermoresponsive poly(oligo ethylene glycol acrylates), Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- J.-F. Lutz, O. Akdemir and A. Hoth, Point by point comparison of two thermosensitive polymers exhibiting a similar LCST: is the age of poly(NIPAM) over?, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- Z.-Y. Qiao, F.-S. Du, R. Zhang, D.-H. Liang and Z.-C. Li, Biocompatible Thermoresponsive Polymers with Pendent Oligo(ethylene glycol) Chains and Cyclic Ortho Ester Groups, Macromolecules, 2010, 43, 6485–6494 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Preparation of Ideal PEG Analogues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- P. Jungwirth and P. S. Cremer, Beyond Hofmeister, Nat. Chem., 2014, 6, 261–263 CrossRef CAS PubMed.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, Effects of Hofmeister Anions on the LCST of PNIPAM as a Function of Molecular Weight, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS PubMed.
- Ł. Otulakowski, M. Kasprów, A. Strzelecka, A. Dworak and B. Trzebicka, Thermal Behaviour of Common Thermoresponsive Polymers in Phosphate Buffer and in Its Salt Solutions, Polymers, 2020, 13, 90–103 CrossRef PubMed.
- H. Wei, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Thermo-sensitive polymeric micelles based on poly(N-isopropylacrylamide) as drug carriers, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- A. Ueda, A. Hashidzume and T. Sato, acs_MA_ma-2010-02635y 1.8, Macromolecules, 2011, 44, 2970–2977 CrossRef.
- M. Liu, K. Kono and J. M. J. Fréchet, Water-soluble dendritic unimolecular micelles - Their potential as drug delivery agents, J. Controlled Release, 2000, 65, 121–131 CrossRef CAS PubMed.
- R. K. Kainthan, C. Mugabe, H. M. Burt and D. E. Brooks, Unimolecular micelles based on hydrophobically derivatized hyperbranched polyglycerols: ligand binding properties, Biomacromolecules, 2008, 9, 886–895 CrossRef CAS PubMed.
- C. Xia, X. Ding, Y. Sun, H. Liu and Y. Li, Hyperbranched–upon–dendritic macromolecules as unimolecular hosts for controlled release, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4013–4019 CrossRef CAS.
- B. Xu, G. Gu, C. Feng, X. Jiang, J. Hu, G. Lu, S. Zhang and X. Huang, (PAA-g-PS)-co-PPEGMEMA asymmetric polymer brushes: synthesis, self-assembly, and encapsulating capacity for both hydrophobic and hydrophilic agents, Polym. Chem., 2016, 7, 613–624 RSC.
- J. Aguiar, P. Carpena, J. A. Molina-Bolívar and C. Carnero Ruiz, On the determination of the critical micelle concentration by the pyrene 1:3 ratio method, J. Colloid Interface Sci., 2003, 258, 116–122 CrossRef CAS.
- J. M. Taylor, K. Scale, S. Arrowsmith, A. Sharp, S. Flynn, S. Rannard and T. O. McDonald, Using pyrene to probe the effects of poloxamer stabilisers on internal lipid microenvironments in solid lipid nanoparticles, Nanoscale Adv., 2020, 2, 5572–5577 RSC.
- B. Zhang, H. Tang and P. Wu, In Depth Analysis on the Unusual Multistep Aggregation Process of Oligo(ethylene glycol) Methacrylate-Based Polymers in Water, Macromolecules, 2014, 47, 4728–4737 CrossRef CAS.
- M. Qi and Y. Zhou, Multimicelle aggregate mechanism for spherical multimolecular micelles: from theories, characteristics and properties to applications, Mater. Chem. Front., 2019, 3, 1994–2009 RSC.
- J. Fu, E. Wu, G. Li, B. Wang and C. Zhan, Anti-PEG antibodies: Current situation and countermeasures, Nano Today, 2024, 55, 102163 CrossRef CAS.
- H. Frey, R. Matthes and P. Dreier, US Pat., 2024317934A1, 2022 Search PubMed.
- D. D. Stöbener, D. Donath and M. Weinhart, Fast and solvent–free microwave–assisted synthesis of thermoresponsive oligo(glycidyl ether)s, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2496–2504 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01450b |
| This journal is © The Royal Society of Chemistry 2025 |