
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpcycling polyethersulfones to luminescent materials by aminolysis†
Johan
Liotier
 a,
Leila
Issoufou Alfari
b,
Benoit
Mahler
b,
Thomas
Niehaus
b,
Christophe
Dujardin
bc,
Simon
Guelen
d,
Vincent
Schanen
d,
Véronique
Dufaud
*a,
Jean
Raynaud
*a and
Vincent
Monteil
*a
a,
Leila
Issoufou Alfari
b,
Benoit
Mahler
b,
Thomas
Niehaus
b,
Christophe
Dujardin
bc,
Simon
Guelen
d,
Vincent
Schanen
d,
Véronique
Dufaud
*a,
Jean
Raynaud
*a and
Vincent
Monteil
*a
aUniversite Claude Bernard Lyon 1, Catalyse Polymerization Processes Materials (CP2 M), UMR 5128, CNRS, 69616 Villeurbanne, France. E-mail: veronique.dufaud@univ-lyon1.fr; vincent.monteil@univ-lyon1.fr; jean.raynaud@cnrs.fr
bUniversite Claude Bernard Lyon 1, Institut Lumiere Matière (iLM), UMR 5306, CNRS, 69622 Villeurbanne, France
cInstitut universitaire de France (IUF), 1 rue Descartes 75231, Paris cedex 05, France
dSpecialty Operations France (member of SYENSQO group), Research and Innovation Center Lyon, 85 Avenue des Frères Perret, 69190 Saint-Fons, France
First published on 10th January 2025
Abstract
The upcycling of polyethersulfone (PES), a high-performance polymer based on an aromatic-rich aryl-ether-based backbone, can advantageously yield both the starting comonomer bisphenol S (BPS) and valuable OLED derivatives, providing a complete atom valorization strategy for PES waste. Deprotonated selected amines have proved particularly efficient at depolymerizing PES at moderate temperatures (∼120 °C). The recycled monomer yields validate the back-to-monomer chemical recycling method for industrial compliance. The OLED derivatives afforded by the same simple process can easily be isolated, promoting an innovative upcycling strategy that transforms polymer into valuable chemicals, a highly relevant approach for mitigating the ever-growing plastic waste accumulation.
1. Introduction
Polymer waste, often commonly called plastic waste, is a major problem in today's world. The disposal of polymers, including thermoplastics, using obsolete landfilling or incineration methods creates many pollution issues. Only a meagre 10% of worldwide plastic production is currently recycled.1 Recycling “plastics” is becoming a necessity to address the ever-growing waste and reduce the quantity released into the environment. One way of recycling polymers is through mechanical recycling. This involves reprocessing the polymers by melting them, using for instance optimized extrusion processes.2 This process is industry-compliant but usually yields downgraded materials.3 Indeed, thermoplastics are reprocessed in the melt at high temperatures and are thus prone to partial chain degradation (reduction in molar masses, functionalities and thus mechanical and chemical properties).4,5 To tackle this issue, the chemical transformation of this waste into useful intermediates or small molecules and even monomers, where possible, a.k.a chemical recycling, has attracted attention. Chemical cleavage of the polymer chain can be used in a circular strategy to return it to its monomer form and eventually recover the virgin polymer without any loss of intrinsic properties.6,7 Another strategy is to cleave the polymer to obtain functional molecules or intermediates that are useful in different industrial loops.8–10 If the properties of the new product are substantial, one can end up upcycling the polymer waste.11 It is the era of converting waste to chemicals rather than converting crude oil to chemicals.3,12,13 This strategy is also investigated for the valorisation of lignin to obtain functional aromatic rings from renewable bio-based polymer waste and abundant resources.14–16 For industrial polymers, this strategy could be used to recycle/upcycle polymer waste from the production chain or even post-consumer wastes at the end of their life cycle.1 This strategy is even more valuable and for the moment economically viable for high-value polymers, such as polyarylethers.17,18 In this vein, the aminolysis of polyethersulfone (PES) (Fig. 1) has been investigated with ethylenediamine.19 This aminolysis provided a waste-to-functionalised oligomers and/or polymers strategy, with amine- and phenol-functionalized products which might be useful for the synthesis of other polycondensates, albeit with a hampered control of functionality19 and more likely limited to downgraded platform industrial polymers or necessitating further complex separation steps to yield valuable products. Until now, aminolysis processes have been rather limited to the functionalization of existing polymer platforms rather than true chemical recycling.19,20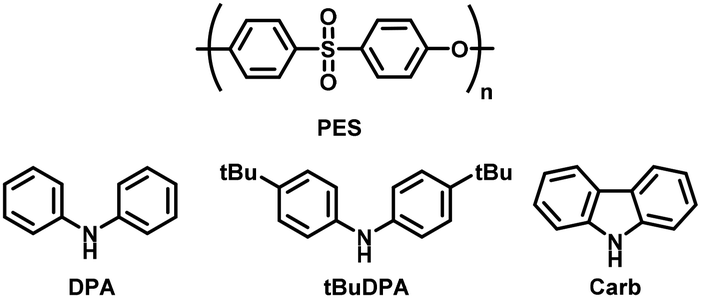 | ||
| Fig. 1 Structure of PES and selected amines. | ||
Herein, we develop an upcycling strategy that goes beyond back-to-monomer chemical recycling, additionally providing useful luminescent moieties from PES waste and selected amines as depolymerizing reagents. Sulfone-based polymers already account for ∼80 kT of thermoplastics, and this is predicted to rise to reach ∼100 kT by 2029, which is very significant for high-performance and high-value polymers (∼representing almost a 2B$ business).21
Addressing the recycling of this ever-growing waste is becoming a necessity. This proof of concept validates the upcycling strategy from polymer chains to functional small molecules with added value, and valuable and quantifiable physical properties. Molecules allowing for the formulation of OLEDs hold a lot of potential within modern technologies.
Indeed, the repeating unit of PES is of interest for the synthesis of luminescent materials presenting Thermally Activated Delayed Fluorescence (TADF).22,23 This class of materials features valuable properties.24–26 These materials include aryl-amine motifs that could be harnessed through the use of parent anilines (or corresponding bases) for the aminolysis of the polymer. The three selected amines for the depolymerisation are diphenylamine (DPA), bis(4-(tert-butyl)phenyl)amine (tBuDPA) and carbazole (Carb). Their structures are presented in Fig. 1. The targeted class of luminescent materials is very useful for the fabrication of OLEDS with high External Quantum Efficiency (EQE).27–29 Interestingly, these luminescent molecules retain the bisphenol S (BPS) scaffold and could be reincorporated into PES-based formulations.30,31
Here, we report a new process for the complete depolymerisation of PES using aryl amines to cleave the aryl ether bonds to readily obtain high-value fluorescent compounds that can be easily isolated as well as the recovery of the BPS comonomer, thus validating the back-to-monomer chemical recycling and additionally the upcycling strategy for OLED production.
2. Materials and methods
All reactants and solvents were purchased from Sigma Aldrich. The polyethersulfone (PES) Veradel® product with Mn ≈ 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da was provided by SYENSQO. The absorption spectra were recorded using solutions in DCM (2 × 10−5 M) with an Evolution 220 UV-vis spectrometer from Thermo Scientific. The emission spectra and quantum yields (QY) of the luminescent materials were obtained using an FS5 spectrofluorometer equipped with an integration sphere for collecting the photons emitted in all directions (Edinburgh Instruments). Photoluminescence spectra were recorded with excitation at a wavelength corresponding to the maximum absorption, an integration time of 100 ms per data point, and a spectral resolution of 1 nm. QY measurements were conducted at a wavelength corresponding to maximum absorption, with an integration time of 100 ms per data point, and a spectral resolution of 0.4 nm. LC-MS was performed at the CCSM (Mass Spectrometry Center at Villeurbanne) using a U3000 UPLC from Thermo Fisher Scientific and an Impact II mass spectrometer from Bruker.
000 Da was provided by SYENSQO. The absorption spectra were recorded using solutions in DCM (2 × 10−5 M) with an Evolution 220 UV-vis spectrometer from Thermo Scientific. The emission spectra and quantum yields (QY) of the luminescent materials were obtained using an FS5 spectrofluorometer equipped with an integration sphere for collecting the photons emitted in all directions (Edinburgh Instruments). Photoluminescence spectra were recorded with excitation at a wavelength corresponding to the maximum absorption, an integration time of 100 ms per data point, and a spectral resolution of 1 nm. QY measurements were conducted at a wavelength corresponding to maximum absorption, with an integration time of 100 ms per data point, and a spectral resolution of 0.4 nm. LC-MS was performed at the CCSM (Mass Spectrometry Center at Villeurbanne) using a U3000 UPLC from Thermo Fisher Scientific and an Impact II mass spectrometer from Bruker.
3. Results and discussion
3.1. Aminolysis of PES
In order to depolymerise the PES chains by aminolysis, we decided to use the deprotonated forms, amides, to increase the intrinsic reactivity of the amines. Selected aromatic amines were reacted with NaH in DMF as previously described.25 This solvent/base combination offers several advantages and results from optimisation. First, NaH has been chosen as a non-nucleophilic base. This is key, as the goal is to only activate the amine without having the base acting in the depolymerisation process yielding undesired side reactions. Other bases such as Li/Na/KHMDS or BuLi variations are not suitable for this transformation since they lead to unwanted competitive reactions, more notably with the amide/amine HMDS. Weaker bases such as Na, KOtBu were not efficient to fully deprotonate our selected amines. Solvents were also considered: DMF is more suitable than DMAc or DMSO for instance, as it does not bear a labile proton that would react with NaH, detrimental to the activation of the amines. HMPA was ruled out for toxicity reasons. The use of DMF had another interest since PES is soluble in this solvent. As a result, PES could be added directly to the amide solution once preformed. After the reaction time, the reaction mixture was poured into iced water and acidified using 2 M HCl. Upon acidification, some products precipitated and were filtered. This solid mixture contained luminescent materials alongside oligomer products. The filtrate was then extracted by ethyl acetate to obtain the pure monomer bisphenol S (BPS).Firstly, bis(4-(tert-butyl)phenyl)amine (tBuDPA) was used as the amine for the optimisation of the depolymerisation process. The temperature and the number of amine equivalents were optimised as presented in Table 1. The synthetic and purification protocols as well as a detailed characterization of the isolated products are provided in the ESI† for each entry in Table 1. The 1H NMR spectra of the filtered solids resulting from the optimisation are presented in Fig. 2 alongside the native PES and BPS spectra for comparison.
 | ||
| Fig. 2 1H NMR of the depolymerisation process obtained with tBuDPA at 100 °C and 120 °C with 2 and 3 equivalents of amine alongside the 1H NMR of the native PES and commercial BPS. The signals of the different products are labelled in the spectra of different crude products. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Amine | Amine equivalent | Temperature (°C) | BPS yield (%) | Mono-aminated product (%) | Di-aminated product (%) | Dimer yield (%) | Conversion (%) |
| 1 | tBuDPA | 2 | 100 | 0 | — | — | — | — |
| 2 | tBuDPA | 2 | 120 | 17 | — | — | — | — |
| 3 | tBuDPA | 3 | 120 | 52 | 25 | 0 | 17 | 94 |
| 4 | DPA | 3 | 120 | 34 | 13 | 21 | 31 | 100 |
| 5 | Carb | 3 | 120 | 50 | 12 | 21 | 17 | 100 |

Firstly, all the presented reactions have resulted in degradation or depolymerization of the native PES. Indeed, the signals corresponding to the starting polymer do not appear anymore in the filtered products, rather suggesting the formation of diverse products (see also the ESI,† with NMR spectroscopy and liquid chromatography corresponding analyses). With two equivalents of amine at 100 and 120 °C, a complex signal appears at 7.85 and 8.05 ppm corresponding to PES oligomers. These oligomers could not be characterised by size exclusion chromatography (SEC), as their molar masses precluded proper analysis with this method (outside the calibration window). However, we could perform Liquid Chromatography (LC) to qualitatively analyse the number of products obtained. The NMR signals diminish from 100 °C to 120 °C and this decrease corresponds to the diminution of the number of signals obtained by LC (see the ESI, Fig. S2 and S6†). With three equivalents at 120 °C, an excess of amine is present in large quantities but the signal corresponding to the oligomer product appears as better-resolved doublets likely belonging to a single product (see also the ESI, part 1†). The extracted BPS obtained from these reactions could thus be quantified since it matched our reference BPS monomer. At 100 °C, no BPS was obtained (Table 1, entry 1). However, at 120 °C, a 17% yield of BPS (calculated in comparison with the repeating unit) was obtained with 2 equivalents of amine and a 52% yield was obtained with 3 equivalents. More equivalents of amines were then needed to push the reaction to completion. This increase not only leads to the appearance of the aminated/nitrogen-containing compounds but also to increases in the formation of BPS. This surprising effect could be attributed to the strong basic behaviour of the amide tempering its nucleophilicity or more likely to the fact that these amines/amides are also excellent leaving groups. If the non-aromatic intermediates (Meisenheimer complexes) are stable enough, some –OH groups could be formed during the hydrolytic HCl quenching of the reaction. This effect could explain why the product distribution does not follow a statistical behaviour. This also suggests that the added arylamine moiety influences the addition of the second functionality. After purification of the solid product, we could show that under these conditions, the longest oligomer product obtained is a BPS-dimer, therefore validating nearly complete conversion of the native PES into small molecules.
The influence of other amines with varied steric hindrance and electronic properties on the depolymerisation extent and selectivity was then investigated under the optimized conditions (120 °C and 3 equivalents of amine) and the results are included in Table 1.
Once again, full depolymerisation of PES was achieved with 34% and 50% yields of BPS monomer for respectively diphenylamine (DPA) and carbazole (Carb).
This filtered product from the complete depolymerisation could be purified by silica gel column chromatography and three products were isolated: di-aminated bisphenyl S, mono-aminated phenylphenol S and BPS-dimer. These products were obtained in different ratios depending on the selected amine. Interestingly, the di-aminated product is not obtained with tBuDPA. Only the mono-aminated product (tBuDPA-OH) is obtained with a 25% yield. This could be explained by the steric hindrance of the tert-butyl unit and the low angle between the phenyl units of bisphenyl S (Fig. S24 to S26†).
In the case of depolymerisation with the DPA and Carb, it is interesting to note that the yields of the mono- and di-aminated products are very close. The mono-aminated product is obtained in 13% and 12% yields, respectively, and the di-aminated product is obtained in 21% in both cases.
For each experiment, BPS and BPS-dimer are obtained in large quantities. Interestingly, the yields of these products obtained with tBuDPA and Carb are very close (around 50% of BPS was obtained for each amine and 17% of BPS-dimer). In the case of the reaction with DPA, both products are obtained in similar yields (around 30%). These products hold great industrial potential because they can be used as starting materials to readily obtain recycled PES, since they represent one of the two complementary comonomers in the AA/BB type polycondensation reaction to produce PES.
3.2. Characterisation of the luminescent materials
Five luminescent products could be obtained from the aminolysis of PES and are referred to as DiDPA, DPA-OH, tBuDPA-OH, DiCarb and Carb-OH (Fig. 3). The absorption of the materials was recorded in DCM (2 × 10−5 M) and their luminescent properties were analysed with an excitation beam set at the maximum absorption of the analysed solution (solution diluted to obtain an optical density of 0.1). The optical characteristics are presented in Table 2 and the absorption and emission spectra are presented in Fig. 4a, b and ESI Section 2.† | ||
| Fig. 3 Structures of the five luminescent materials obtained by aminolysis of PES. | ||
 | ||
| Fig. 4 (a) Absorption spectra of DiDPA, DPA-OH, tBuDPA-OH, Carb-OH and DiCarb (2 × 10−5 M in DCM); (b) emission spectra of DiDPA, DPA-OH, tBuDPA-OH, Carb-OH and DiCarb (diluted to obtain an optical density of 0.1 in DCM, excitation at the λmax absorption); (c) xy colour coordinates obtained for the emission of each molecule placed on the 1931 chromaticity diagram (calculated with Python).33 | ||
| Product | λ maxabsorption (nm) | ε (L mol−1 cm−2) | Optical band gap (eV) | λ maxemission (nm) | Quantum yield QY (%) | Brightness B (L mol−1 cm−2) |
|---|---|---|---|---|---|---|
| DiDPA | 350 | 31800 |
3.21 | 435 | 85 | 26900 |
| DPA-OH | 330 | 20700 |
3.23 | 440 | 83 | 17300 |
| tBuDPA-OH | 335 | 21100 |
3.14 | 470 | 61 | 12800 |
| DiCarb | 340 | 27800 |
3.36 | 420 | 70 | 19500 |
| Carb-OH | 337 | 12400 |
3.42 | 420 | 49 | 6 100 |
The comparison between the molecules DiDPA and DiCarb with their mono-aminated homologues DPA-OH and Carb-OH shows that the presence of two donating units implies a higher molar extinction coefficient ε than their mono-aminated counterparts going from 31800 L mol−1 cm−1 to 20700 L mol−1 cm−1 for the DPA products and from 27800 L mol−1 cm−1 to 12400 L mol−1 cm−1 for the Carb products. In addition, we observe a small hypsochromic shift between the absorption maximum of the di-aminated and mono-aminated products going from 350 nm to 330 nm for the DPA products and from 340 nm to 337 nm for the Carb. The fact that the absorption of the compounds bearing Carb is lower than that of the ones bearing DPA could be explained by DFT (Fig. S24 and S25†). In fact, the HOMO–LUMO overlap is lower in the case of Carb compared to DPA. This could facilitate the π–π* transition in the case of DPA explaining the lower absorption intensity of the Carb family.32 In the case of the tBuDPA-OH, the absorption maximum is located at 335 nm, which corresponds to a 5 nm bathochromic shift of the absorption band compared to DPA-OH. This is due to the addition of the tert-butyl units making the DPA unit slightly more donating. The ε of tBuDPA-OH is 21100 L mol−1 cm−1, which is very close to that of DPA-OH since the conjugated part is not modified.
For the emission, DiCarb and Carb-OH both have a maximum emission at 420 nm with similar full width at half maximum (FWHMem) values of 75 and 76 nm. The quantum yields (QY) of the two molecules differ and are equal to 75% for DiCarb and 49% for Carb-OH. As a result, the brightness of DiCarb (19500 L mol−1 cm−1) is higher than that of Carb-OH (6100 L mol−1 cm−1). DiDPA and DPA-OH have maximum emissions at 435 and 440 nm, respectively, with FWHMem values of 77 and 79 nm. The QY and brightness of the DPA family are 85% and 26900 L mol−1 cm−1 and 83% and 17300 L mol−1 cm−1 respectively. In the case of tBuDPA-OH, the maximum emission is at 470 nm with a FWHMem of 91 nm. Its QY is 61% and its brightness is 12800 L mol−1 cm−1. We can see an interesting shift of the emission band with the increase in the donating strength of the amine. We observe that we go from 420 nm for Carb, the least donating unit, to 470 nm for tBuDPA, the most donating unit. The FWMHem of the compounds DiDPA, DPA-OH, DiCarb and Carb-OH are around 75 nm but the FWMHem of tBuDPA-OH is 91 nm. This is visible to the naked eye as the emitted light of this compound is whiter. The xy coordinates could be calculated from the emission spectra and are presented in Fig. 4c. Only tBuDPA-OH stands out from the group by being closer to the neutral white colour.
The photoluminescence (PL) decay was measured for DiDPA as it has the most intense luminescence. The results are presented in the ESI (Fig. S27†). As already described in the literature,25 the PL decay follows a bi-exponential behaviour as follows:
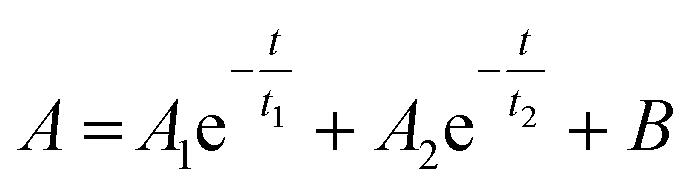 | (1) |
The first exponential component corresponds to normal fluorescence behaviour with a decay time t1 of 5.1 ns. The second exponential corresponds to thermally activated delayed fluorescence with a decay time of 79.2 ns. These properties correspond to the literature,25 which confirms that our methods of upcycling can yield the same product with similar properties to those obtained through classical synthesis.
4. Conclusions
We provide a new strategy to recycle/upcycle PES in a single synthetic step. We developed an aminolysis process achieving complete conversion of PES into useful separable small molecules. On the one hand, this reaction allows the recovery of ∼50% of BPS monomer and some dimer which could be used as starting materials for the repolymerisation of PES – with Cl- or F-based comonomers – by providing the necessary OH-containing comonomer for polycondensation. On the other hand, by carefully selecting the amine structural features, we obtained five luminescent materials, which could be fully characterised. To push the formation of these materials, one could work on the quenching methodology to avoid the formation of the –OH substituted compounds.Harnessing PES depolymerization, we could for instance derive the straightforward synthesis of the DiDPA compound yielding an 85% quantum yield with a FWMHem of 77 nm and similar PL properties from industrial waste material. We will continue developing the waste-to-chemicals strategy to valorise efficiently other high-value polymer wastes, in particular within the polyarylether family.
Author contributions
Conceptualization: JL, VD, JR, VM; methodology: JL, VD, VM, JR; resources: SG, VS; OLED characterization: JL, LIA, BM, TN, CD; project administration, supervision: SG, VS, VD, JR, VM; writing – original draft, review & editing: JL, VD, VM and JR with participation from all authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
Dr B. Guerdener and S. Bouveyron are thanked for the preliminary experiments realized on PES aminolysis. The CCSM lab, and particularly Dr E. Fromentin, are thanked for the LC and LC-MS measurements. Dr J. Liotier also thanks the CNRS/Solvay-Syensqo research fund for his fellowship. The authors also acknowledge fruitful discussions with Drs F. Batt, P. Marion, K. Jouvin & S. Mastroianni from SYENSQO.References
- Plastics – the Facts 2022·Plastics Europe. Plastics Europe. https://plasticseurope.org/knowledge-hub/plastics-the-facts-2022/.
- G. W. Coates and Y. D. Y. L. Getzler, Chemical Recycling to Monomer for an Ideal, Circular Polymer Economy, Nat. Rev. Mater., 2020, 5(7), 501–516, DOI:10.1038/s41578-020-0190-4.
- A. Rahimi and J. M. García, Chemical Recycling of Waste Plastics for New Materials Production, Nat. Rev. Chem., 2017, 1(6), 0046, DOI:10.1038/s41570-017-0046.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. Van Harmelen, P. De Wild, G. P. Van Der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Beyond Mechanical Recycling: Giving New Life to Plastic Waste, Angew. Chem., Int. Ed., 2020, 59(36), 15402–15423, DOI:10.1002/anie.201915651.
- Z. O. G. Schyns and M. P. Shaver, Mechanical Recycling of Packaging Plastics: A Review, Macromol. Rapid Commun., 2021, 42(3), 2000415, DOI:10.1002/marc.202000415.
- N. Wang, G. Strong, V. DaSilva, L. Gao, R. Huacuja, I. Konstantinov, M. Rosen, A. Nett, S. Ewart, R. Geyer, S. Scott and D. Guironnet, Chemical Recycling of Polyethylene by Tandem Catalytic Monomerization to Propylene, J. Am. Chem. Soc., 2022, 144(40), 18526–18531, DOI:10.1021/jacs.2c07781.
- R. J. Conk, S. Hanna, J. X. Shi, J. Yang, N. R. Ciccia, L. Qi, B. J. Bloomer, S. Heuvel, T. Wills, J. Su, A. T. Bell and J. F. Hartwig, Catalytic Deconstruction of Waste Polyethylene with Ethylene to Form Propylene, Science, 2022, 377(6614), 1561–1566, DOI:10.1126/science.add1088.
- A. Ahrens, A. Bonde, H. Sun, N. K. Wittig, H. C. D. Hammershøj, G. M. F. Batista, A. Sommerfeldt, S. Frølich, H. Birkedal and T. Skrydstrup, Catalytic Disconnection of C–O Bonds in Epoxy Resins and Composites, Nature, 2023, 617(7962), 730–737, DOI:10.1038/s41586-023-05944-6.
- Z. Xu, N. E. Munyaneza, Q. Zhang, M. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. Liu, Chemical Upcycling of Polyethylene, Polypropylene, and Mixtures to High-value Surfactants, Science, 2023, 381(6658), 666–671, DOI:10.1126/science.adh0993.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Mixed Plastics Waste Valorization through Tandem Chemical Oxidation and Biological Funneling, Science, 2022, 378(6616), 207–211, DOI:10.1126/science.abo4626.
- J. X. Shi, N. R. Ciccia, S. Pal, D. D. Kim, J. N. Brunn, C. Lizandara-Pueyo, M. Ernst, A. M. Haydl, P. B. Messersmith, B. A. Helms and J. F. Hartwig, Chemical Modification of Oxidized Polyethylene Enables Access to Functional Polyethylenes with Greater Reuse, J. Am. Chem. Soc., 2023, 145(39), 21527–21537, DOI:10.1021/jacs.3c07186.
- H. Li, J. Wu, Z. Jiang, J. Ma, V. M. Zavala, C. R. Landis, M. Mavrikakis and G. W. Huber, Hydroformylation of Pyrolysis Oils to Aldehydes and Alcohols from Polyolefin Waste, Science, 2023, 381(6658), 660–666, DOI:10.1126/science.adh1853.
- W. Zhang, S. Kim, L. Wahl, R. Khare, L. Hale, J. Hu, D. M. Camaioni, O. Y. Gutiérrez, Y. Liu and J. A. Lercher, Low-temperature Upcycling of Polyolefins into Liquid Alkanes via Tandem Cracking-alkylation, Science, 2023, 379(6634), 807–811, DOI:10.1126/science.ade7485.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis, Angew. Chem., Int. Ed., 2016, 55(29), 8164–8215, DOI:10.1002/anie.201510351.
- P. de Wild, R. Van der Laan, A. Kloekhorst and E. Heeres, Lignin Valorisation for Chemicals and (Transportation) Fuels via (Catalytic) Pyrolysis and Hydrodeoxygenation, Environ. Prog. Sustainable Energy, 2009, 28(3), 461–469, DOI:10.1002/ep.10391.
- R. R. Singhania, A. K. Patel, T. Raj, C.-W. Chen, V. K. Ponnusamy, N. Tahir, S.-H. Kim and C.-D. Dong, Lignin Valorisation via Enzymes: A Sustainable Approach, Fuel, 2022, 311, 122608, DOI:10.1016/j.fuel.2021.122608.
- Y. Minami, N. Matsuyama, Y. Takeichi, R. Watanabe, S. Mathew and Y. Nakajima, Depolymerization of Robust Polyetheretherketone to Regenerate Monomer Units Using Sulfur Reagents, Commun. Chem., 2023, 6(1), 14, DOI:10.1038/s42004-023-00814-8.
- Y. Minami, Y. Inagaki, T. Tsuyuki, K. Sato and Y. Nakajima, Hydroxylation-depolymerization of Oxyphenylene-based Super Engineering Plastics To Regenerate Arenols, JACS Au, 2023, 3(8), 2323–2332, DOI:10.1021/jacsau.3c00357.
- L. Wang, Y. Cui, N. Wang, H. Zhang, B. Zhu, L. Zhu and Y. Xu, Aminolytic Depolymerization of Polyarylsulfones, Polym. Degrad. Stab., 2014, 103, 69–74, DOI:10.1016/j.polymdegradstab.2014.03.013.
- N. R. Ciccia, J. X. Shi, S. Pal, M. Hua, K. G. Malollari, C. Lizandara-Pueyo, E. Risto, M. Ernst, B. A. Helms, P. B. Messersmith and J. F. Hartwig, Diverse Functional Polyethylenes by Catalytic Amination, Science, 2023, 381(6665), 1433–1440, DOI:10.1126/science.adg6093.
- Global Sulfone Polymers (PSU, PPSU & PESU) Industry Overview 2024: Owing to Unmatched Product Properties, Sulfone Polymers Demand to Reach 92 K Metric Tons by 2029. GlobeNewswire News Room. https://www.globenewswire.com/en/news-release/2024/04/10/2860553/28124/en/Global-Sulfone-Polymers-PSU-PPSU-PESU-Industry-Overview-2024-Owing-to-Unmatched-Product-Properties-Sulfone-Polymers-Demand-to-Reach-92K-Metric-Tons-by-2029.html.
- J.-M. Teng, Y.-F. Wang and C.-F. Chen, Recent Progress of Narrowband TADF Emitters and Their Applications in OLEDs, J. Mater. Chem. C, 2020, 8(33), 11340–11353, 10.1039/D0TC02682D.
- B. Wex and B. R. Kaafarani, Perspective on Carbazole-based Organic Compounds as Emitters and Hosts in TADF Applications, J. Mater. Chem. C, 2017, 5(34), 8622–8653, 10.1039/C7TC02156A.
- J. Yu, S. Qiu, K. Zhang, T. Zhou, X. Ban, Y. Duan, D. Jia, Q. Zhu and T. Zhang, A Novel Thermally-activated Delayed Fluorescent Probe Based on Hydroxyl as Identify Group for Detection of Iron Ions, J. Mol. Struct., 2022, 1251, 132074, DOI:10.1016/j.molstruc.2021.132074.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, Design of Efficient Thermally Activated Delayed Fluorescence Materials for Pure Blue Organic Light Emitting Diodes, J. Am. Chem. Soc., 2012, 134(36), 14706–14709, DOI:10.1021/ja306538w.
- X. Zhou, M. Huang, X. Zeng, C. Zhong, G. Xie, S. Gong, X. Cao and C. Yang, Sky-blue Thermally Activated Delayed Fluorescence Polymers with π-Interrupted Polymer Mainchain via Friedel-Crafts Polycondensation, Polymer, 2020, 204, 122722, DOI:10.1016/j.polymer.2020.122722.
- J.-X. Chen, W.-W. Tao, W.-C. Chen, Y.-F. Xiao, K. Wang, C. Cao, J. Yu, S. Li, F.-X. Geng, C. Adachi, C.-S. Lee and X.-H. Zhang, Red/Near-Infrared Thermally Activated Delayed Fluorescence OLEDs with Near 100% Internal Quantum Efficiency, Angew. Chem., Int. Ed., 2019, 58(41), 14660–14665, DOI:10.1002/anie.201906575.
- T. Huang, W. Jiang and L. Duan, Recent Progress in Solution Processable TADF Materials for Organic Light-emitting Diodes, J. Mater. Chem. C, 2018, 6(21), 5577–5596, 10.1039/C8TC01139G.
- J.-H. Lee, C.-H. Chen, P.-H. Lee, H.-Y. Lin, M. Leung, T.-L. Chiu and C.-F. Lin, Blue Organic Light-emitting Diodes: Current Status, Challenges, and Future Outlook, J. Mater. Chem. C, 2019, 7(20), 5874–5888, 10.1039/C9TC00204A.
- C. S. Anggraini, D. Wahyuningrum and A. Alni, The Synthesis of Polyethersulfone (PES) and its Derivatives as Polymer Light Emitting Diode (PLED) Material, Key Eng. Mater., 2019, 811, 126–132, DOI:10.4028/www.scientific.net/KEM.811.126.
- S. J. Lee, E. J. Lee, I.-K. Kang, S.-Y. Park, K.-B. Yoon, G. Kwak and L. S. Park, Fabrication and Performance of Flexible OLEDs with AGZO/Ag/AGZO Multilayer Anode on Polyethersulfone Film, Mol. Cryst. Liq. Cryst., 2011, 550(1), 172–182, DOI:10.1080/15421406.2011.599712.
- R. Venkatraman, S. V. K. Panneer, E. Varathan and V. Subramanian, Aromaticity–Photovoltaic Property Relationship of Triphenylamine-based D-π-A Dyes: Leads from DFT Calculations, J. Phys. Chem. A, 2020, 124(17), 3374–3385, DOI:10.1021/acs.jpca.9b10245.
- T. Mansencal, M. Mauderer, M. Parsons, N. Shaw, K. Wheatley, S. Cooper, J. D. Vandenberg, L. Canavan, K. Crowson, O. Lev, K. Leinweber, S. Sharma, T. J. Sobotka, D. Moritz, M. Pppp, C. Rane, P. Eswaramoorthy, J. Mertic, B. Pearlstine, M. Leonhardt, O. Niemitalo, M. Szymanski, M. Schambach, S. Huang, M. Wei, N. Joywardhan, O. Wagih, P. Redman, J. Goldstone, S. Hill, J. Smith, F. Savoir, G. Saxena, S. Chopra, I. Sibiryakov, T. Gates, G. Pal, N. Tessore, A. Pierre, F.-X. Thomas, S. Srinivasan and T. Downs, Colour 0.4.2, 2022. DOI:10.5281/zenodo.7367239.
Footnote |
| † Electronic supplementary information (ESI) available: General procedure for aminolysis; absorption and emission spectra; DFT modelling; photoluminescence decay. See DOI: https://doi.org/10.1039/d4py01250j |
| This journal is © The Royal Society of Chemistry 2025 |