
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyglyoxylamide hydrogels for the traceless stimulus-mediated release of covalently-immobilized drugs†
Jue
Gong
a,
Burak
Tavsanli
a and
Elizabeth R.
Gillies
 *ab
*ab
aDepartment of Chemistry, The University of Western Ontario, 1151 Richmond St., London, Ontario, Canada N6A 5B7. E-mail: egillie@uwo.ca
bDepartment of Chemical and Biochemical Engineering, The University of Western Ontario, 1151 Richmond St., London, Ontario, Canada N6A 5B9
First published on 7th January 2025
Abstract
Hydrogels can be used in a wide range of applications from personal care products to drug delivery vehicles. Particularly for drug delivery, it is desirable to control the release of the loaded cargo as well as the hydrogel degradation time. Self-immolative hydrogels have been recently investigated to enable the stimulus-mediated breakdown of the hydrogel, which can also modulate to some extent the release of loaded drugs. However, when the drug was loaded into the hydrogel using non-covalent interactions, the background release rate of the drug in the absence of the stimulus was relatively rapid. Thus, we report here a new hydrogel system based on an acetal end-capped self-immolative polyglyoxylamide backbone with photo-responsive linkers as pendent groups to enable the covalent conjugation of amine-functionalized drugs. Using phenylalanine methyl ester as a model drug, we showed that hydrogels were successfully prepared with 96% equilibrium water content and a compressive modulus of 5.5 kPa. Light irradiation stimulated the rapid and traceless release of the model drug, while no detectable release was observed without irradiation. Furthermore, the PGAm backbone depolymerized selectively at mildly acidic pH. This system therefore provides a new hydrogel platform enabling a high level of control over both hydrogel breakdown and drug release.
Introduction
Hydrogels are cross-linked three-dimensional networks that can retain a high mass fraction of water. They can be composed of natural polymers including polysaccharides,1,2 nucleic acids,3 or proteins,4 or synthetic polymers such as poly(ethylene glycol) (PEG),5 polyesters,6 or poly(meth)acrylates.7,8 Cross-linking can be achieved by a variety of covalent bonding approaches9 or supramolecular interactions such as host–guest chemistry, ionic bonding, or hydrophobic interactions.10,11 As their high water content often imparts properties resembling those of biological tissues, hydrogels have been of substantial interest for biomedical applications such as tissue engineering,12 sensing,13 and drug delivery.14In the field of drug delivery, hydrogels can be used to encapsulate and release cargo ranging from small molecule drugs to therapeutic proteins. For example, the US Food and Drug Administration (FDA) approved Vantas®, a polymethacrylate-based hydrogel for the controlled release of Histrelin acetate in the treatment of prostate cancer.15 Hyaluronic acid (HA)-based hydrogels such as Belotero balance® and Revanesse® have been approved as fillers for the treatment of facial wrinkles, with loaded lidocaine providing pain relief upon injection.16 In addition, PEG hydrogels cross-linked by thiol-maleimide chemistry were developed for the intraocular delivery of bevacizumab, a monoclonal antibody for vascular endothelial growth factor for the treatment of neovascularization diseases.17 However, it is often challenging to control the release rates of therapeutics from hydrogels, as they are generally based on molecular diffusion and thus primarily depend on the dimensions of the pores within the material, as well as the hydrogel size and shape.18 Release can be tuned to some extent by modulating the mesh size of the hydrogel19 and through affinity-controlled release approaches,20,21 but control remains challenging.
To achieve increased control over drug release, stimuli-responsive hydrogels have garnered substantial interest in recent years.22–24 For example, hydrogels have been engineered to break down in response to intrinsic stimuli such as a mild pH reduction25,26 or elevated concentrations of reactive oxygen species27 associated with inflammation or cancer. Glucose-responsive hydrogels have been developed to release insulin as needed for the treatment of diabetes.28 Extrinsic stimuli such as light29 and ultrasound30 irradiation can also be used to stimulate drug release.
Our group and others have begun to explore self-immolative polymer hydrogels, incorporating backbones such as polyglyoxylates,31 polyglyoxylamides,32 poly(benzyl ether)s,33 polythioesters,34 and polydisulfides35 or cross-linkers containing self-immolative spacers.36 Self-immolative polymers can depolymerize fully from end-to-end upon the cleavage of their backbone or end-cap, thereby leading to breakdown of the hydrogel under specified conditions.37 However, very little research has been done on the incorporation of therapeutics into these hydrogels and when drugs were incorporated non-covalently, they exhibited relatively high levels of background release, even in the absence of the stimulus.31
Covalent conjugation can serve as an effective approach to slow the background release of bioactive molecules from hydrogels.38 For example, to extend their release time for intervertebral disc regeneration, bone morphogenic proteins were conjugated to fibrin-HA hydrogels by enzymatic cross-linking.39 When the linker between a drug and hydrogel contains a stimuli-responsive bond, the selective release of the drug in response to the stimulus can be achieved. For example, various aminoglycoside antibiotics were covalently cross-linked by imine linkages into hydrogels composed of oxidized polysaccharides with pendent aldehydes.40 While the hydrogels were stable at neutral pH, at pH 5–6 they degraded, releasing the antibiotic.
Herein, we describe the preparation of a polyglyoxylamide (PGAm) with pendent tri(ethylene glycol) (TEG) chains to provide hydrophilicity, azides for cross-linking by copper-catalysed azide–alkyne click chemistry (CuAAC), and activated o-nitrobenzyl moieties for the conjugation of amino-containing drugs. Conjugation of phenylalanine methyl ester (Phe methyl ester), as a model drug, followed by cross-linking with alkyne-terminated 4-arm-PEG provided hydrogels (Fig. 1). These hydrogels provided very low levels of background Phe methyl ester release in the dark, but rapid traceless release in the presence of light. The hydrogel network exhibited relatively high stability at pH 7.4, but underwent gradual degradation at pH 6, indicating that it was possible to modulate both drug release and hydrogel degradation in response to different stimuli.
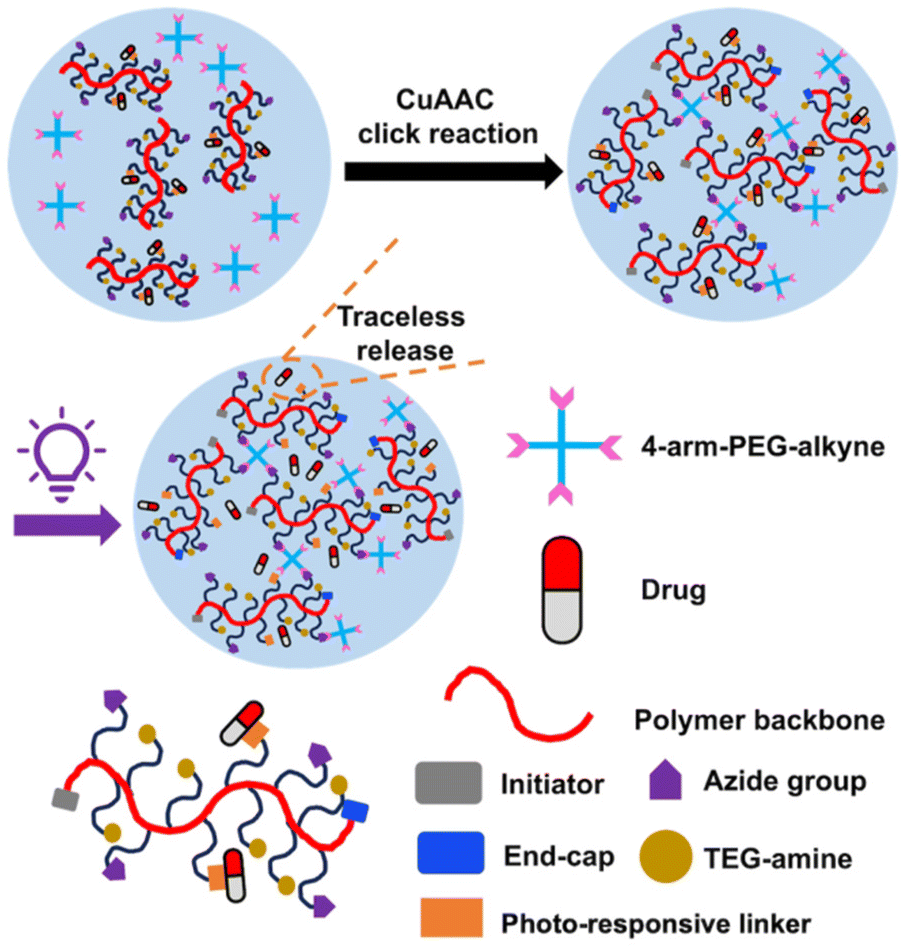 | ||
| Fig. 1 Schematic representation of a hydrogel containing a self-immolative polymer backbone and a traceless photo-responsive linker for conjugating an amine-containing drug. | ||
Experimental
General materials
2-Azidoethylamine,41 tri(ethylene glycol) methyl ether amine (TEG-amine),42 alkyne-functionalized o-nitrobenzyl alcohol (alkyne-NB-OH),43 and L-phenylalanine methyl ester (Phe methyl ester)44 were synthesized as previously reported. Alkyne-functionalized 5 kg mol−1 4-arm-PEG (4-arm-PEG-alkyne) was synthesized as previously reported.32 Ethyl glyoxylate (EtG) in toluene solution (50% w/w) and copper sulfate were obtained from Alfa Aesar (Ward Hill, MA, USA). n-Butanol, CH2Cl2, N,N-dimethylformamide (DMF), and trifluoroacetic acid (TFA) were purchased from Caledon Laboratory Chemicals (Georgetown, ON, Canada). Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS), Glutamax, and penicillin/streptomycin were obtained from VWR (Mississauga, ON, Canada). Tetrahydrofuran (THF), NaOH, ethyl acetate, (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT), and dialysis membranes (2 kDa molecular weight cut-off (MWCO) Spectra/Por® 6) were obtained from Thermo Fisher Scientific (Burlington, ON, Canada). Ethyl vinyl ether (EVE), sodium ascorbate, 1,1′-carbonyldiimidazole (CDI), fluorescamine, acetonitrile, CuSO4, NEt3, 1,4-dioxane, dimethyl sulfoxide-d6 (99.9 atom% D), chloroform-d (99.8 atom% D) and deuterium oxide (99.9 atom% D) were purchased from Millipore Sigma (Oakville, ON, Canada). CH2Cl2 and NEt3 were distilled over CaH2 under nitrogen at atmospheric pressure before use. n-Butanol was dried over 4 Å molecular sieves and distilled under nitrogen at atmospheric pressure onto 4 Å molecular sieves before use. EtG was purified by distillation over P2O5 as previously reported.45 1,4-Dioxane was obtained from a solvent purification system equipped with alumina columns. Phosphate buffered saline (PBS) contained 137 mM NaCl, 2.7 mM KCl, and 11.8 mM phosphate buffer (pH 7.4 or 6).General procedures
1H and 13C NMR spectra were obtained using a 400 MHz Bruker AvIII HD instrument. 1H NMR chemical shifts are reported in parts per million (ppm) and are referenced to the residual solvent signals of CDCl3 (7.26 ppm), D2O (4.79 ppm) or DMSO-d6 (2.50 ppm) while 13C NMR chemical shifts were referenced to the residual solvent signals of CDCl3 (77.2 ppm). FT-IR spectra were obtained using a PerkinElmer FT-IR Spectrum Two instrument in attenuated total reflectance mode. Size exclusion chromatography (SEC) in DMF was performed using an instrument equipped with a Waters 515 HPLC pump, Waters In-Line Degasser AF, two PLgel mixed D 5 μm (300 × 1.5 mm) columns attached to a corresponding PLgel guard column, and a Wyatt Optilab Rex RI detector. Samples were dissolved in DMF containing 10 mM LiBr and 1% (v/v) NEt3 at a concentration of ∼5 mg mL−1 and filtered through a 0.2 μm PTFE syringe filter prior to injection using a 50 μL loop. Samples were run at a flow rate of 1 mL min−1 for 30 min at 85 °C. Number average molar mass (Mn) and dispersity (Đ) were determined relative to poly(methyl methacrylate) (PMMA) standards.Synthesis of poly(ethyl glyoxylate) (PEtG)
Freshly distilled EtG45 (6.0 mL, 64 mmol, 300 equiv.) was added to a flame-dried Schlenk flask under nitrogen. To this flask, dry n-butanol (19 μL, 0.21 mmol, 1.0 equiv.) was added at room temperature and allowed to mix for 10 min and then 24 mL of dry CH2Cl2 was added at room temperature and the resulting solution was mixed for 30 min. The solution was subsequently cooled to −20 °C and stirred for 20 min. Then, dry NEt3 (0.18 mL, 1.3 mmol, 6.0 equiv.) was added to the polymerization flask and the reaction mixture was stirred for 20 min. Next, EVE (0.12 mL, 1.3 mmol, 6.0 equiv.) was added to the polymerization flask and the solution was stirred for 5 min at −20 °C, and then TFA (0.20 mL, 2.6 mmol, 12 equiv.) was added. The mixture was stirred for 1 h at −20 °C, and then placed in the freezer at −15 °C for 48 h. The reaction mixture was precipitated into 300 mL of methanol/water (4/1 v/v) while adding 0.1 M NaOH to maintain the pH basic (pH = ∼10). The flask was then sealed and transferred into a −20 °C freezer where it was kept for 5 h. After decanting off the liquid, the precipitate was dried under vacuum to yield PEtG as an off-white tacky solid. Yield: 30%. 1H NMR (CDCl3, 400 MHz): 5.75–5.50 (m, 335H), 4.25 (br s, 680H), 4.05 (br s, 2H), 3.79 (br s, 2H), 1.31 (br s, 1016H), 0.91 (br s, 3H). 13C{1H} NMR (CDCl3, 400 MHz): δ 165.5, 93.2, 62.1, 13.9. FT-IR: 2920, 1750 cm−1. SEC (DMF, PMMA): Mn = 33 kg mol−1, Mw = 55 kg mol−1, Đ = 1.67.Synthesis of TEG-N3-40-PGAm
PEtG (200 mg, 2.0 mmol of pendent ester, 1.0 equiv.) was placed into a flame-dried round-bottom flask and sealed with a rubber septum. The flask was evacuated-refilled three times. After the flask was refilled with nitrogen, 5 mL of dry 1,4-dioxane was added to dissolve the polymer. The polymer solution was then transferred into a flame-dried Schlenk flask under nitrogen. To this flask, 2-azidoethylamine (130 mg 1.5 mmol, 0.75 equiv.) was added at room temperature. The reaction was stirred under nitrogen at room temperature, and aliquots of the reaction mixture were removed periodically to check conversion by 1H NMR spectroscopy (Fig. S2†). The conversion of pendent ester groups to amides was determined by comparing the integral of the polymer backbone methine peak at 5.66 ppm with the integral of the –CH2 peak from the pendent ester groups at 4.25 ppm. When ∼40% of the pendent ester groups were converted to amides, the solvent and excess 2-azidoethylamine were removed in vacuo. After the flask was refilled with nitrogen, TEG-amine (960 mg, 5.9 mmol, 5.0 equiv. relative to remaining ester) was added. The reaction mixture was stirred at 50 °C for 24 h, and then dialyzed against deionized water using a 2 kDa MWCO dialysis membrane, and then lyophilized to yield TEG-N3-40-PGAm as a tacky solid. Yield: 90%. 1H NMR (CDCl3, 400 MHz): δ 8.87–7.92 (br s, 1.0H), 5.75 (br s, 1.0H), 3.74–3.50 (m, 7.1H), 3.45 (s, 1.7H), 3.38 (s, 2.0H). 13C{1H} NMR (CDCl3, 400 MHz): δ 167.2, 96.6, 71.9, 70.4, 69.1, 58.9, 39.3. FT-IR: 3647–3137, 2870, 2101, 1668, 1539 cm−1. SEC (DMF, PMMA): Mn = 63 kg mol−1, Mw = 106 kg mol−1, Đ = 1.68.Synthesis of oNB-TEG-N3-30-PGAm
TEG-N3-40-PGAm (100 mg, 0.21 mmol of azide, 1.0 equiv.) and alkyne-NB-OH43 (12.4 mg, 0.053 mmol, 0.25 equiv.) were dissolved in 4 mL of water/acetonitrile (1/1 v/v). Then, sodium ascorbate (10.5 mg, 0.053 mmol, 0.25 equiv.) was added and the reaction mixture was stirred for 5 min at room temperature. Copper sulfate (8.46 mg, 0.053 mmol, 0.25 equiv.) was then added. The reaction mixture was stirred for 2 h at room temperature, dialyzed against deionized water using a 2 kDa MWCO membrane, and then lyophilized to yield oNB-TEG-N3-30-PGAm as a yellow tacky solid. Yield: 83%. 1H NMR (DMSO-d6, 400 MHz): δ 8.90–7.76 (m, 1.5H), 5.59 (br s, 1.0H), 4.84 (br s, 0.2H), 4.47 (br s, 0.4H), 3.68–3.40 (m, 6.9H), 3.23 (s, 1.8H). FT-IR: 3516–3137, 2873, 2102, 1668, 1533 cm−1.Synthesis of Phe-oNB-TEG-N3-30-PGAm
oNB-TEG-N3-30-PGAm (130 mg, 0.061 mmol of oNB linker, 1.0 equiv.), CDI (99 mg, 0.61 mmol, 10 equiv.) and 10 mL of dry CH2Cl2 were combined in a flame-dried Schlenk flask under nitrogen. The solution was stirred overnight under nitrogen at room temperature. The reaction mixture was then precipitated in 100 mL diethyl ether to yield an off-white tacky solid (82 mg). The dried CDI-activated polymer (82 mg, 0.037 mmol of activated carbonate, 1.0 equiv.) and Phe methyl ester (20 mg, 0.11 mmol, 3.0 equiv.) were dissolved in 4 mL of dry THF. The reaction mixture was stirred overnight at room temperature. The reaction mixture was then dialyzed against deionized water using a 2 kDa MWCO membrane, and then lyophilized to yield an off-white tacky solid. Yield: 40% over the two steps. 1H NMR (DMSO-d6, 400 MHz): δ 9.14–8.10 (m, 1.6H), 7.22–7.00 (m, 0.4H), 5.60 (br s, 1H), 4.90 (br s, 0.2H), 4.60–4.24 (m, 0.5H), 3.68–2.82 (m, 28H).Hydrogel preparation
Hydrogels were prepared using a total of 15% w/v of 4-arm-PEG-alkyne and 30 kg mol−1 Phe-oNB-TEG-N3-PGAm in DMF/water (4/1 v/v), with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of azide:alkyne functional groups (Table S1†). The 4-arm-PEG-alkyne and Phe-oNB-TEG-N3-PGAm were dissolved in DMF and vortexed. Sodium ascorbate and CuSO4 were each dissolved separately in water and vortexed. The sodium ascorbate solution was then added to the DMF solution and it was vortexed, then the CuSO4 solution was added and the solution was briefly vortexed. After gelation overnight, the resulting gels were immersed in a 0.1 M EDTA solution (5 mL), changing the solution 3 times over 24 h to remove copper. The gels were then immersed in deionized water, changing the solution 3 times over 24 h and then lyophilized.
:1 molar ratio of azide:alkyne functional groups (Table S1†). The 4-arm-PEG-alkyne and Phe-oNB-TEG-N3-PGAm were dissolved in DMF and vortexed. Sodium ascorbate and CuSO4 were each dissolved separately in water and vortexed. The sodium ascorbate solution was then added to the DMF solution and it was vortexed, then the CuSO4 solution was added and the solution was briefly vortexed. After gelation overnight, the resulting gels were immersed in a 0.1 M EDTA solution (5 mL), changing the solution 3 times over 24 h to remove copper. The gels were then immersed in deionized water, changing the solution 3 times over 24 h and then lyophilized.
Measurement of gel content and equilibrium water content (EWC)
After gelation and washing of the Phe-oNB-TEG-N3-30-PGAm hydrogel as described above, the swollen mass (ms) of the hydrogel was recorded. The hydrogel was then lyophilized and its dry mass (md) was measured. The theoretical mass (mt) of polymer involved in cross-linking was calculated based on the total mass of polymer added to the formulation (Table S1†). The gel content and EWC were calculated using eqn (1) and (2), respectively.| Gel content = (md/mt) × 100% | (1) |
| EWC = [(ms − md)/ms] × 100% | (2) |
Gel content and EWC were measured for triplicate hydrogels and the results are reported as the mean ± standard deviation.
Scanning electron microscopy
The hydrogels based on Phe-oNB-TEG-N3-30-PGAm were prepared as described above, flash frozen in liquid nitrogen, and then lyophilized. The dried hydrogels were mounted on stubs covered in carbon tape and coated with osmium using a SPI Supplies, OC-60A plasma coater. SEM was performed using a Zeiss LEO 1530 instrument, operating at 2.0 kV and a working distance of 7 mm.Measurement of compressive moduli under unconfined compression
Cylindrical samples of Phe-oNB-TEG-N3-30-PGAm hydrogels with diameters of 4 mm and heights of ∼6 mm were prepared as described above but in 1 mL syringes and equilibrated in PBS for 3 days. The compressive moduli of the hydrogels were then determined using a UniVert system (CellScale, Waterloo, ON, Canada) equipped with a 10 N load cell. During the measurement, the samples were immersed in a 37 °C PBS bath, preloaded at 0.01 N and compressed to a total strain of 40% at a rate of 0.5% s−1. The compressive moduli were calculated from the slope of the linear region of the stress–strain curve between 5 and 15% strain. The hydrogels were prepared and measured in triplicate. The results are reported as the mean ± standard deviation.Phe methyl ester release
The Phe-oNB-TEG-N3-30-PGAm hydrogels were prepared as described above in 1 mL syringes. Cylindrical samples with diameters of 4 mm and heights of ∼6 mm were washed and then lyophilized to provide ∼10 mg of lyophilized hydrogel, accurately weighed. The samples were then immersed in 2 mL of PBS (pH = 7.4) at 37 °C. The amine release was monitored from hydrogels stored in the dark (control) or subjected to cycles of irradiation with a mercury lamp (12 mW cm−2 of UVA, 10 mW cm−2 of UVB, 2.7 mW cm−2 of UVC, 9.6 mW cm−2 of UVV) followed by storage in the dark (1 h light, 1 h dark). Before and after each irradiation, a 0.1 mL aliquot of each sample was diluted to 1 mL with PBS. 75 μL of the diluted sample in PBS was then mixed with 25 μL of 3 mg mL−1 fluorescamine in DMSO and the mixture was incubated in the dark at room temperature for 15 min. The amine content was determined relative to a calibration curve for Phe methyl ester (Fig. S21†) using a plate reader (Tecan M1000-Pro) by measuring the fluorescence intensity at 470 nm. Following each time point, the release media was replaced with fresh PBS. The amine release experiments were performed in triplicate and the results are presented as the mean ± standard deviation.Hydrogel degradation
13 mg of lyophilized hydrogel prepared from Phe-oNB-TEG-N3-30-PGAm and 4-arm-PEG-alkyne were immersed in 1.0 mL of deuterated PBS (pH = 6 or 7.4) containing 10 μL of acetonitrile in an NMR tube. An initial 1H NMR spectrum was obtained. The samples were stored 37 °C. Degradation was monitored by acquiring 1H NMR spectra of the sample at specific time points and the percentage degradation was determined by comparing the integral of the –CH3 peak from tri(ethylene glycol) methyl ether pendent groups at 3.32 ppm to that of the acetonitrile standard at 2.10 ppm as the soluble depolymerization products were released from the hydrogel.Cytotoxicity assay
Mouse myoblast (C2C12) cells were cultured in DMEM that contained 1% Glutamax (100×), antibiotics (Streptomycin and Penicillin at 100 units per mL), and 10% FBS. The cells were then seeded at a density of 10000 cells per well (in 100 μL of culture medium per well) in a 96-well plate. They were then incubated for 24 h at 37 °C and 5% CO2. Meanwhile, Phe-oNB-TEG-N3-30-PGAm hydrogel was prepared as described above in a 1 mL syringe, and then washed and lyophilized. A 3 mg sample of dry hydrogel was immersed in 3 mL of cell culture media. The hydrogel in the media was irradiated with UV light using the mercury lamp described above for 1 h and then incubated for 24 h at 37 °C, allowing potentially toxic species to diffuse out of the hydrogel. After removal of the hydrogel, the leachate media and serial 2-fold dilutions of this leachate in culture media were then added to the cells in the 96-well plate (3 replicates per concentration). As positive controls, 0.05–0.2 mg mL−1 sodium dodecyl sulfate (SDS) was added to cells, while fresh culture medium served as a negative control. Wells containing only the culture media served as the background. The cells were incubated for 24 h at 37 °C, and then the media was aspirated and fresh media (110 μL) containing 0.5 mg mL−1 of MTT was added to each well. After incubation for 4 h at 37 °C, the solution was aspirated and the purple crystals were dissolved in 50 μL per well of dimethyl sulfoxide (DMSO). After mixing, the absorbance at 540 nm was read for each well using a Tecan M1000-Pro plate reader. The absorbance was compared to the control cells exposed to media alone to calculate the % metabolic activity. The data are presented as the mean ± standard deviation.
Results and discussion
Polymer synthesis
A PGAm backbone was selected for the preparation of the hydrogel. PGAms can be readily prepared from PEtG via post-polymerization amidation, allowing different pendent functional groups to be incorporated to tune the structure, solubility, and function of the polymer.46 An acetal end-cap was selected to enable gradual degradation and depolymerization of the PGAm backbone under mildly acidic conditions.47 This degradation could potentially allow the hydrogels to break down in vivo. To synthesize the PEtG, EtG monomer was purified by distillation over P2O5 twice, and then polymerization was initiated with n-butanol in the presence of NEt3 at −20 °C (Scheme 1). The polymer was end-capped using ethyl vinyl ether (EVE) in the presence of TFA.48 End-group analysis by 1H NMR spectroscopy suggested the degree of polymerization (DPn) was about 335, corresponding to an Mn of 33 kg mol−1, in reasonable agreement with the targeted DPn of 300 (Fig. S1†). 13C NMR and FT-IR spectroscopic data further supported the successful synthesis of the polymer (Fig. S10 and S13†). SEC analysis in DMF relative to poly(methyl methacrylate) (PMMA) standards indicated an Mn of 33 kg mol−1 and Đ was 1.67 (Fig. 2).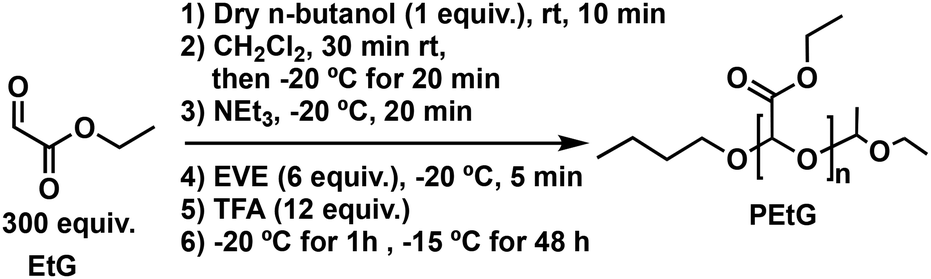 | ||
| Scheme 1 Synthesis of PEtG. | ||
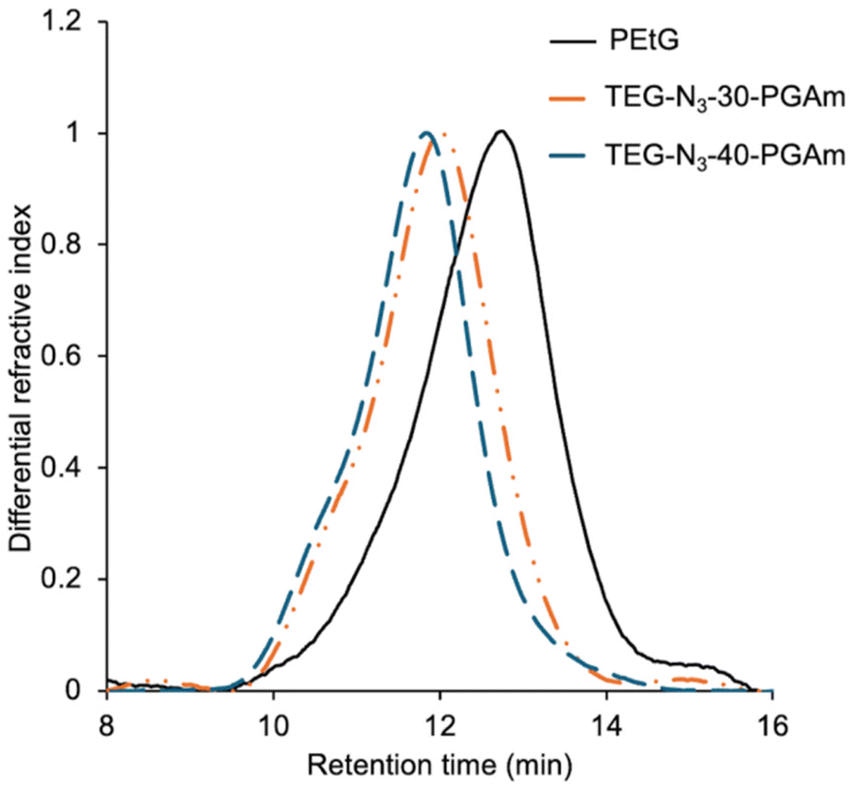 | ||
| Fig. 2 DMF SEC traces for PEtG, TEG-N3-30-PGAm, and TEG-N3-40-PGAm. | ||
Next, the PEtG was converted to PGAms. Our recent work on PGAm hydrogels indicated that having about 30% pendent azides available for cross-linking was ideal.32 Higher percentages of pendent azides (i.e., 40%) led to lower gel content, possibly due to the reduced reactivity of remaining azides after the initial cross-linking of a portion of the pendent azides. Therefore, we aimed for 30 and 40% pendent azides, with the objective of leaving 20 and 30% azides respectively after conversion of a portion of these azides to photo-cleavable drug linkages. To introduce the azides, PEtG was reacted with 2-azidoethylamine in 1,4-dioxane (Scheme 2). The use of 0.3 and 0.4 stoichiometric equivalents of the amine per pendant ester were insufficient, but 0.56 and 0.75 equivalents of 2-azidoethylamine were used to achieve 30 and 40% amidation respectively. The reactions were monitored by 1H NMR spectroscopy and when the targeted conversion was achieved after ∼72 h, the excess 2-azidoethylamine was removed in vacuo (Fig. S2 and S3†). Then, the polymers were treated with excess TEG-amine at 50 °C for 24 h to generate the corresponding copolymers TEG-N3-30-PGAm and TEG-N3-40-PGAm with 30% and 40% pendent azides, respectively, after purification by dialysis. SEC analysis indicated that TEG-N3-30-PGAm had a Mn of 56 kg mol−1 and Đ of 1.61 and TEG-N3-40-PGAm had a Mn of 63 kg mol−1 and Đ of 1.68 (Fig. 2). 1H NMR spectroscopic analyses confirmed the disappearance of –CH2 peak from ester at 4.25 ppm and appearance of a broad –NH peak in a range of ∼7.8–8.9 ppm as well as peaks corresponding to the pendent TEG and azidoethyl moieties from 3.3–3.7 ppm, indicating the complete conversion of ester to amide was achieved (Fig. S4 and S5†). Moreover, in addition to a new peak at 2100 cm−1 corresponding to the azide stretch, FT-IR spectroscopy revealed two peaks at ∼1670 and 1540 cm−1 corresponding to the new amides and disappearance of the initial ester carbonyl stretch at 1750 cm−1 (Fig. S14 and S15†). 13C NMR spectroscopic data further supported the successful synthesis of TEG-N3-30-PGAm and TEG-N3-40-PGAm (Fig. S11 and S12†).
 | ||
| Scheme 2 Synthesis of the functionalized PGAms and their conversion into hydrogels using 4-arm-PEG-alkyne. Linker cleavage to release Phe methyl ester in a traceless manner is also illustrated. | ||
To incorporate a traceless linker, the UV-responsive o-nitrobenzyl linker was selected as a model system, as it is easy to synthesize and light can be readily applied as a stimulus in the laboratory, leading to the release of an amine-functionalized molecule in a traceless manner (Scheme 2). To achieve a suitable drug loading, while retaining sufficient azides for cross-linking, alkyne-NB-OH43 was conjugated to 10% of the pendent groups (1/3 of the pendent azides for TEG-N3-30-PGAm and 1/4 of the azides for TEG-N3-40-PGAm) using CuAAC to provide oNB-TEG-N3-20-PGAm and oNB-TEG-N3-30-PGAm (Scheme 2). 1H NMR spectroscopic analyses confirmed the incorporation of the linker at the targeted loading based on integrals of new peaks corresponding to the linker, compared to those of the polymer backbone (Fig. S6 and S7†).
To investigate the potential covalent attachment of drugs onto the PGAms, Phe methyl ester was selected as a model compound. The hydroxyl groups on the light-responsive linker were activated with CDI and then reacted with Phe methyl ester (Scheme 2). The resulting polymers were purified by dialysis to give Phe-oNB-TEG-N3-20-PGAm and Phe-oNB-TEG-N3-30-PGAm. 1H NMR spectroscopic analysis of the polymers showed broad peaks from 7.0–7.4 ppm corresponding to the aromatic protons from Phe methyl ester, indicating the drug was conjugated to the polymer (Fig. S8 and S9†). Overall, assuming full functionalization of the linker hydroxyls, the Phe methyl ester model drug was calculated to comprise 7.4 and 7.7% of the mass of Phe-oNB-TEG-N3-20-PGAm and Phe-oNB-TEG-N3-30-PGAm respectively, typical loadings for polymer–drug conjugates.26
Hydrogel preparation and characterization
In our previous research using a PGAm backbone with water-solubilizing hydroxyl and cross-linkable azide pendent groups, 10–20% polymer in the hydrogel formulation was suitable for gelation with alkyne-functionalized PEGs.32 Therefore, we focused here on 15% w/v using Phe-oNB-TEG-N3-20-PGAm and Phe-oNB-TEG-N3-30-PGAm to compare the effect of the percentage of pendent azide groups (20 and 30% respectively). The cross-linking reaction with 4-arm-PEG-alkyne by CuAAC was initially performed in pure water. However, due to the poor solubility of the PGAms in water, hydrogels were not successfully formed. It is likely that conjugation of the relatively hydrophobic Phe methyl ester impaired the water-solubility of the polymers. Therefore, the gelation was then performed in DMF/H2O (4/1) (Scheme 2). The resulting materials were purified by washing with 0.1 M EDTA, followed by deionized water. We have previously demonstrated this method to be highly effective for removal of >99% of the Cu.31,49 Phe-oNB-TEG-N3-20-PGAm formed hydrogels with 4-arm-PEG-alkyne. However, the hydrogels collapsed after washing, making them unsuitable for further study. This behavior can be attributed to the lack of sufficient azide groups to form a well cross-linked network. On the other hand, the cross-linking of Phe-oNB-TEG-N3-30-PGAm with 4-arm-PEG-alkyne resulted a hydrogel that could be washed and transferred to water (Fig. S19†).The resulting hydrogel was then characterized by measurement of its gel content, equilibrium water content (EWC), FT-IR spectroscopy, SEM, and mechanical testing. The gel content was 72 ± 5%. A high EWC (96.3 ± 0.2%) was obtained, which is likely because both PGAm and PEG domains swell in water. With these properties, it is estimated that 1 mg of Phe methyl ester, a typical dose of a peptide therapeutic,50 could be delivered in about 300 μL of hydrogel. Given that Phe methyl ester has a much lower molar mass than an oligopeptide, this 1 mg in 300 μL represents a lower limit. The lyophilized hydrogel was analyzed by FT-IR spectroscopy. In contrast to the Phe-oNB-TEG-N3-30-PGAm which showed an intense peak corresponding to the azide stretch at ∼2100 cm−1, the spectrum of the resulting hydrogel had no observable peak in this region, indicating the absence of any significant levels of residual azide groups (Fig. S18†). SEM was also performed on the lyophilized hydrogel, revealing different sizes of pores on the order of a few micrometers and tens of micrometers in diameter (Fig. 3). In addition, compression testing performed on the hydrogel immersed in PBS at 37 °C indicated a compressive modulus of 5.5 ± 0.5 kPa (Fig. S20†).
 | ||
| Fig. 3 SEM image of the Phe-oNB-TEG-N3-30-PGAm hydrogel after lyophilization. | ||
Stimulus-mediated release
To evaluate the hydrogel's potential to release a payload in a stimuli-responsive manner, hydrogel samples were immersed in PBS at 37 °C and release of the Phe methyl ester in response to UV light irradiation was studied. A mercury lamp was used, which emits a wide range of wavelengths, but the relevant range for photochemical cleavage of o-nitrobenzyl moieties is generally from 300–350 nm.51 Released Phe methyl ester was quantified using the fluorescamine assay. Fluorescamine is nonfluorescent, but its reaction with primary amines at room temperature generates a highly fluorescent product that can be detected even in the picomolar range.52 In an alternating sequence, the hydrogels were irradiated for 1 h and then stored in the dark for 1 h, while the control non-irradiated hydrogels were stored in the dark over the same time period. During the first 1 h irradiation, ∼28% of phenylalanine methyl ester was released (Fig. 4). While stored in the dark for 1 h, no additional release was detected. The next 1 h irradiation period increased the level of release to ∼50%. Meanwhile, still no further release was detected while the hydrogel was stored in the dark. After a third irradiation period, ∼70% of phenylalanine methyl ester had been released from the irradiated hydrogels. In contrast, no release was detected from the non-irradiated hydrogel over the same time period, indicating that the Phe methyl ester remained covalently attached to the o-nitrobenzyl carbamate linker. Unlike our previous hydrogel system, where the drug was loaded into the PEtG domain during the hydrogel preparation and a background level of drug release was seen when the hydrogels were non-irradiated,31 these results indicate that the covalently conjugated drug can be released highly selectively from the hydrogel only in the presence of the stimulus.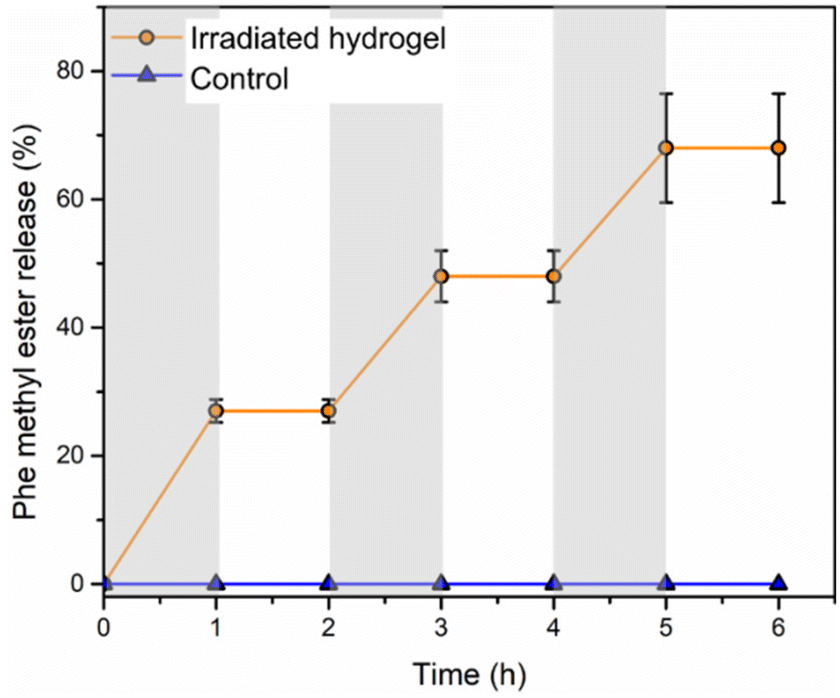 | ||
| Fig. 4 Release of Phe methyl ester from irradiated Phe-oNB-TEG-N3-30-PGAm hydrogel immersed in PBS at 37 °C. Periods of irradiation with light are indicated by grey shading. Data are presented as the mean ± standard deviation (n = 3), except for the control where no error bars are included as no amine was detected. | ||
Hydrogel degradation
To evaluate the degradability of the hydrogel, accurately measured quantities of hydrogel were immersed in deuterated PBS at pH 7.4 or pH 6, containing acetonitrile as the internal standard for quantification. The percent hydrogel degradation was assessed by 1H NMR spectroscopy based on the integral of peak 4 corresponding to the methoxy protons from the TEG pendent group of the monomer hydrate depolymerization product compared to that of the CH3CN internal standard (Fig. 5a). At pH 7.4, there were no detectable monomer hydrate peaks even at day 22, indicating that the acetal end-cap is quite stable at neutral pH. At pH 6, initially no peaks corresponding to degradation products were observed, but over the first few days, new monomer hydrate peaks appeared at 5.22, 3.57, 3.45, 3.38, 3.32 ppm. The methine proton peak at 5.22 ppm took longer to emerge than other peaks due to its overlap with the large peak corresponding to HOD. The hydrogel underwent ∼50% degradation over 22 days (Fig. 5b). These results indicate that the hydrogel can potentially undergo degradation in response to physiological conditions such as inflammation, which may be beneficial to accelerate the delivery of certain therapeutics.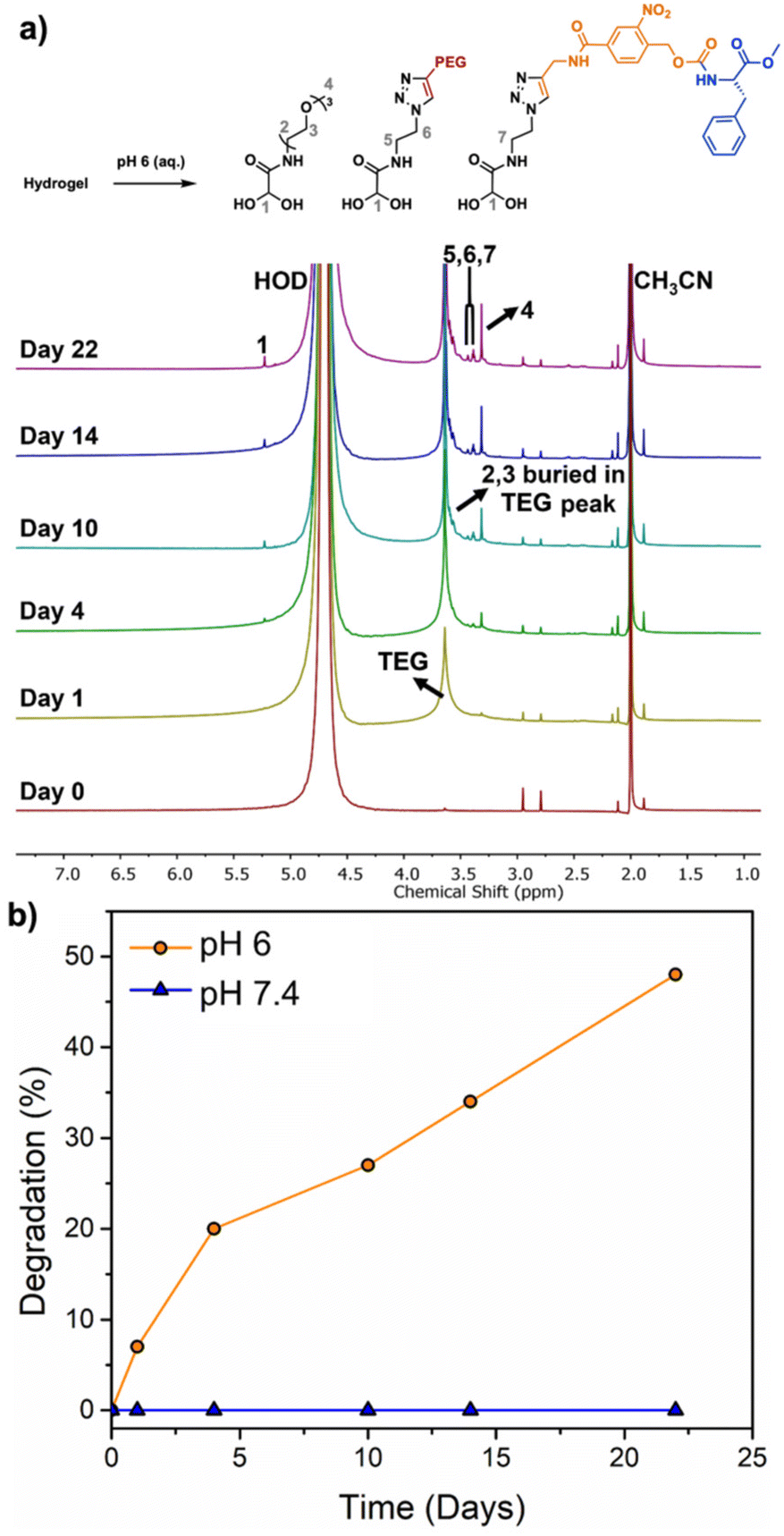 | ||
| Fig. 5 (a) 1H NMR spectra of the Phe-oNB-TEG-N3-30-PGAm hydrogel in deuterated PBS (pH 6) containing 1% CH3CN; (b) percent hydrogel degradation versus time at pH 6 and 7.4 as determined based on the integral of peak 4 compared to the CH3CN internal standard peak. | ||
Cytotoxicity
Finally, the potential for the hydrogel to leach toxic species into its surrounding environment was evaluated. Phe-oNB-TEG-N3-30-PGAm hydrogel was immersed in cell culture media, irradiated for 1 h, and then incubated in the culture media for 24 h. This culture media was then added to C2C12 mouse myoblast cells at varying dilutions. Even when incubated in 100% of this media, the metabolic activities of the cell were greater than 80% those of control cells (Fig. 6). This result indicated that the hydrogel didn't leach toxic species.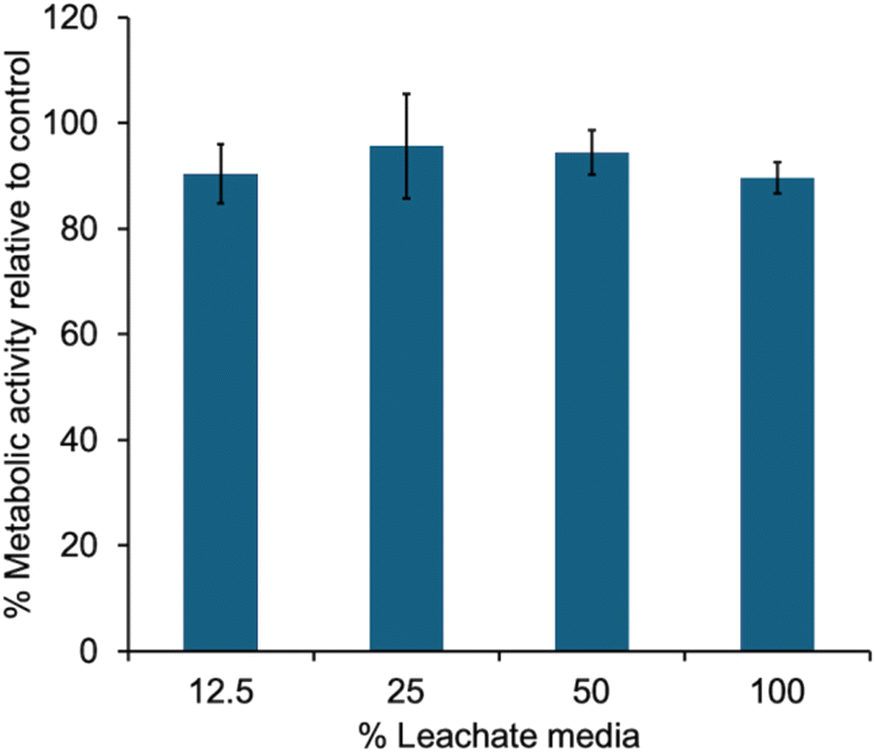 | ||
| Fig. 6 Metabolic activity, as measured by an MTT assay, for cells incubated for 24 h with varying percentages of culture media that was exposed to irradiated hydrogel to leach potentially toxic species. Error bars correspond to the standard deviations on triplicate samples. | ||
Conclusions
The covalent immobilization of an amino acid model drug onto a 30 kg mol−1 acetal end-capped PGAm copolymer by a light-sensitive linker was demonstrated. Two different (20% and 30%) pendent azide functionalizations were explored for hydrogel preparation with a 4-arm-PEG-alkyne cross-linker. We found that 20% azide groups gave hydrogels with insufficient properties for further examination, while 30% azide groups provided hydrogels with 72% gel content, 96% EWC, and a compressive modulus of 5.5 kPa. Irradiation with light led to cleavage of the linker and rapid release of Phe methyl ester from the hydrogel. In contrast, no release was observed during the storage in the dark, indicating no significant background release. This behavior contrasts with that of our previously reported self-immolative hydrogels with non-covalently incorporated drugs,31 where a relatively rapid background release occurred even in the absence of stimulus. However, a drawback of the current system is that the linker in the current study is sensitive to UV light, which would not be compatible with various biological applications. This limitation could be addressed by replacing the o-nitrobenzyl motif with a different stimuli-responsive linker, such as boronate linker (H2O2) and disulfide bond (reduction).53 Moreover, we investigated the degradation of the hydrogel at both neutral and acidic pH, demonstrating the hydrogel was degraded slowly at pH 6. This feature could be of interest for the delivery of drugs to treat inflammatory conditions. Finally, we confirmed that the hydrogel didn't leach toxic species, even upon UV light irradiation. In future work, it would be desirable to develop an injectable hydrogel by tuning the pendent groups of the polyglyoxylamide, making the polymer water-soluble, and then cross-linking such a system using strained alkyne click chemistry rather than the copper-catalysed reaction. It is anticipated that the water-solubility of the PGAm can be readily enhanced by changing from TEG to tetra or hexa(ethylene glycol) pendent groups. This approach could also potentially enable the fraction of water-solubilizing groups to be reduced, allowing a higher fraction of pendent groups to be dedicated to drug loading.Author contributions
JG: investigation, methodology, writing – original draft preparation; BT: investigation, writing – review & editing; ERG: conceptualization, funding acquisition, supervision, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Natural Sciences and Engineering Research Council of Canada (RGPIN-2021-03950) and the Canada Research Chair program (E.R.G. CRC-2020-00101) are acknowledged for funding this work. We thank Xueli Mei for SEM imaging, Aneta Borecki for performing the cytotoxicity assay, and the Ragogna laboratory for use of their photochemical reactor.References
- Q. Yang, J. Peng, H. Xiao, X. Xu and Z. Qian, Carbohydr. Polym., 2022, 278, 118952 CrossRef CAS PubMed.
- Z. Li and Z. Lin, Aggregate, 2021, 2, e21 CrossRef CAS.
- Z. Wang, R. Chen, S. Yang, S. Li and Z. Gao, Mater. Today Bio, 2022, 16, 100430 CrossRef CAS PubMed.
- Q. Zhang, Y. Liu, G. Yang, H. Kong, L. Guo and G. Wei, Chem. Eng. J., 2023, 451, 138494 CrossRef CAS.
- Z. Wang, Q. Ye, S. Yu and B. Akhavan, Adv. Healthcare Mater., 2023, 12, 2300105 CrossRef CAS PubMed.
- Y. S. Fomina, A. Semkina, Y. D. Zagoskin, M. Aleksanyan, S. Chvalun and T. Grigoriev, Colloid J., 2023, 85, 795 CrossRef CAS.
- R. H. Galib, Y. Tian, Y. Lei, S. Dang, X. Li, A. Yudhanto, G. Lubineau and Q. Gan, Nat. Commun., 2023, 14, 6707 CrossRef CAS PubMed.
- A. Rosenfeld, T. Göckler, M. Kuzina, M. Reischl, U. Schepers and P. A. Levkin, Adv. Healthcare Mater., 2021, 10, 2100632 CrossRef CAS PubMed.
- Y. Gao, K. Peng and S. Mitragotri, Adv. Mater., 2021, 33, 2006362 CrossRef CAS PubMed.
- G. Sinawang, M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Chem. Commun., 2020, 56, 4381 RSC.
- C. G. Wang, N. E. B. Surat'man, J. J. Chang, Z. L. Ong, B. Li, X. Fan, X. J. Loh and Z. Li, Chem. – Asian J., 2022, 17, e202200604 CrossRef CAS.
- K. Zöller, D. To and A. Bernkop-Schnürch, Biomaterials, 2025, 312, 122718 CrossRef PubMed.
- M. Song, J. Zhang, K. Shen, Y. Hu, W. Shen, S. Tang and H. K. Lee, Biosens. Bioelectron., 2025, 267, 116803 CrossRef CAS PubMed.
- Y. Yu, Y. Cheng, J. Tong, L. Zhang, Y. Wei and M. Tian, J. Mater. Chem. B, 2021, 9, 2979 RSC.
- N. Shore, Eur. Urol., Suppl., 2010, 9, 701 CrossRef CAS.
- A. Mandal, J. R. Clegg, A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2020, 5, e10158 CrossRef CAS PubMed.
- J. Yu, X. Xu, F. Yao, Z. Luo, L. Jin, B. Xie, S. Shi, H. Ma, X. Li and H. Chen, Int. J. Pharm., 2014, 470, 151 CrossRef CAS PubMed.
- S. Simovic, K. R. Diener, A. Bachhuka, K. Kant, D. Losic, J. D. Hayball, M. P. Brownc and K. Vasilev, Mater. Lett., 2014, 130, 210 CrossRef CAS.
- M. S. Rehmann, K. M. Skeens, P. M. Kharkar, E. M. Ford, E. Maverakis, K. H. Lee and A. M. Kloxin, Biomacromolecules, 2017, 18, 3131 CrossRef CAS PubMed.
- M. M. Pakulska, S. Miersch and M. S. Shoichet, Science, 2016, 351, aac4750 CrossRef PubMed.
- V. Huynh and R. G. Wylie, Angew. Chem., Int. Ed., 2018, 57, 3406 CrossRef CAS PubMed.
- A. C. Marques, P. J. Costa, S. Velho and M. H. Amaral, Drug Discovery Today, 2021, 26, 2397 CrossRef CAS PubMed.
- Z. Li, Y. Zhou, T. Li, J. Zhang and H. Tian, View, 2022, 3, 20200112 CrossRef CAS.
- X. Ma, K. P. Sekhar, P. Zhang and J. Cui, Biomater. Sci., 2024, 12, 5468 RSC.
- T. Thambi, J. M. Jung and D. S. Lee, Biomater. Sci., 2023, 11, 1948 RSC.
- E. R. Gillies, Chem. Mater., 2024, 36, 9139 CrossRef CAS.
- R. Kilic Boz, D. Aydin, S. Kocak, B. Golba, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2022, 33, 839 CrossRef CAS PubMed.
- J. Wang, Z. Wang, J. Yu, A. R. Kahkoska, J. B. Buse and Z. Gu, Adv. Mater., 2020, 32, 1902004 CrossRef CAS.
- Y. Xing, B. Zeng and W. Yang, Front. Bioeng. Biotechnol., 2022, 10, 1075670 CrossRef PubMed.
- J. H. Arrizabalaga, M. Smallcomb, M. Abu-Laban, Y. Liu, T. J. Yeingst, A. Dhawan, J. C. Simon and D. J. Hayes, ACS Appl. Bio Mater., 2022, 5, 3212 CrossRef CAS PubMed.
- J. Gong, A. Borecki and E. R. Gillies, Biomacromolecules, 2023, 24, 3629 CrossRef CAS PubMed.
- J. D. Pardy, B. Tavsanli, Q. E. Sirianni and E. R. Gillies, Chem. – Eur. J., 2024, e202401324 CrossRef CAS.
- D. Jung, K. M. Lee, T. Tojo, Y. Oh, H. Yoon and H. Kim, Chem. Mater., 2019, 31, 6249 CrossRef CAS.
- S. M. Soars, B. E. Kirkpatrick, B. D. Fairbanks, J. T. Kamps, K. S. Anseth and C. N. Bowman, Polym. Int., 2022, 71, 906 CrossRef CAS.
- A. H. Agergaard, A. Sommerfeldt, S. U. Pedersen, H. Birkedal and K. Daasbjerg, Angew. Chem., Int. Ed., 2021, 60, 21543 CrossRef CAS PubMed.
- D. Jung, K. M. Lee, J. Y. Chang, M. Yun, H.-J. Choi, Y. A. Kim, H. Yoon and H. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 42985 CrossRef CAS.
- J. Gong, B. Tavsanli and E. R. Gillies, Annu. Rev. Mater. Res., 2024, 54, 47 CrossRef CAS.
- H. Thakar, S. M. Sebastian, S. Mandal, A. Pople, G. Agarwal and A. Srivastava, ACS Biomater. Sci. Eng., 2019, 5, 6320 CrossRef CAS PubMed.
- M. Peeters, S. E. Detiger, L. S. Karfeld-Sulzer, T. H. Smit, A. Yayon, F. E. Weber and M. N. Helder, BioRes. Open Access, 2015, 4, 398 CrossRef CAS.
- J. Hu, Y. Quan, Y. Lai, Z. Zheng, Z. Hu, X. Wang, T. Dai, Q. Zhang and Y. Cheng, J. Controlled Release, 2017, 247, 145 CrossRef CAS PubMed.
- J. Hannant, J. H. Hedley, J. Pate, A. Walli, S. A. F. Al-Said, M. A. Galindo, B. A. Connolly, B. R. Horrocks, A. Houlton and A. R. Pike, Chem. Commun., 2010, 46, 5870 RSC.
- D. Komáromy, M. C. Stuart, G. Monreal Santiago, M. Tezcan, V. V. Krasnikov and S. Otto, J. Am. Chem. Soc., 2017, 139, 6234 CrossRef.
- B. Fan, J. F. Trant, A. D. Wong and E. R. Gillies, J. Am. Chem. Soc., 2014, 136, 10116 CrossRef CAS.
- H. Adams, R. A. Bawa and S. Jones, Org. Biomol. Chem., 2006, 4, 4206 RSC.
- A. Rabiee Kenaree and E. R. Gillies, Macromolecules, 2018, 51, 5501 CrossRef CAS.
- Q. E. A. Sirianni, A. R. Kenaree and E. R. Gillies, Macromolecules, 2019, 52, 262 CrossRef CAS.
- D. R. Dryoff, J. Lynch and V. D. Papanu, US Pat, 4302564, 1981 Search PubMed.
- D. R. Dryoff and V. D. Papanu, US Pat, 4226959, 1980 Search PubMed.
- K. Gill, X. Mei and E. R. Gillies, Chem. Commun., 2021, 57, 11072 RSC.
- A. I. d'Aquino, C. L. Maikawa, L. T. Nguyen, K. Lu, I. A. Hall, C. K. Jons, C. M. Kasse, J. Yan, A. N. Prossnitz, E. Chang, S. W. Baker, L. Hovgaard, D. B. Steensgaard, H. B. Andersen, L. Simonsen and E. A. Appel, Cell Rep. Med., 2023, 4, 101292 CrossRef PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef PubMed.
- S. Udenfriend, S. Stein, P. Boehlen, W. Dairman, W. Leimgruber and M. Weigele, Science, 1972, 178, 871 CrossRef CAS PubMed.
- B. Fan and E. R. Gillies, Mol. Pharmaceutics, 2017, 14, 2548 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures, hydrogel formulations, NMR and IR spectra, SEC traces, photos of hydrogels, stress-strain curves, calibration curve for Phe methyl ester. See DOI: https://doi.org/10.1039/d4py01214c |
| This journal is © The Royal Society of Chemistry 2025 |