
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective polycondensation of levoglucosenone leading to water-degradable biopolymers†
Atsushi
Tahara
 *ab,
Shogo
Yashiro
b,
Toshio
Hokajo
c,
Shinji
Kudo
d,
Yuta
Yoshizaki
b,
Tomohiro
Konno
b and
Takayuki
Doi
b
*ab,
Shogo
Yashiro
b,
Toshio
Hokajo
c,
Shinji
Kudo
d,
Yuta
Yoshizaki
b,
Tomohiro
Konno
b and
Takayuki
Doi
b
aFrontier Research Institute for Interdisciplinary Sciences (FRIS), Tohoku University, Aoba 6-3, Aramaki, Aoba-ku, Sendai, Miyagi, 980-8578, Japan. E-mail: tahara.a.aa@tohoku.ac.jp
bGraduate School of Pharmaceutical Sciences, Tohoku University, Aoba 6-3, Aramaki, Aoba-ku, Sendai, Miyagi, 980-8578, Japan
cSeiko PMC corporation, Nihombashi 3-6, Honcho 3-chome, Chuo-Ku, Tokyo, 103-002, Japan
dInstitute for Materials Chemistry and Engineering (IMCE), Kyushu University, Kasugakoen 6-1, Kasuga, Fukuoka, 816-8580, Japan
First published on 24th December 2024
Abstract
Highly stereochemically controlled polymers were successfully synthesized from levoglucosenone (LGO), derived from cellulose. Since its discovery in the 1970s, the reactivity of LGO has been widely studied in organic chemistry, owing to its diverse functional groups that serve as linkages for polymer formation. However, most of the previous methods for synthesizing polymers from LGO lacked precise control over regio- and stereochemistry, making stereoselective polymerization from LGO a persistent challenge. Although the ketone moiety in LGO is typically reduced before polymerization, a new LGO polymer was designed, containing a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond obtained by condensation with dicarboxylic dihydrazide. NMR measurements revealed that condensation occurred with high stereoselectivity to produce the E-isomer. This selectivity extended from the model compound to polymer synthesis, achieving high E/Z selectivity. The resulting polymer exhibited optical rotation (up to +89), indicating its potential as a chiral polymer. In spite of these polymers showing high tolerance toward many solvents, they were degradable in water with a simple chemical treatment. The proposed approach facilitates the development of sustainable, high-performance materials that can address both environmental and industrial needs.
N bond obtained by condensation with dicarboxylic dihydrazide. NMR measurements revealed that condensation occurred with high stereoselectivity to produce the E-isomer. This selectivity extended from the model compound to polymer synthesis, achieving high E/Z selectivity. The resulting polymer exhibited optical rotation (up to +89), indicating its potential as a chiral polymer. In spite of these polymers showing high tolerance toward many solvents, they were degradable in water with a simple chemical treatment. The proposed approach facilitates the development of sustainable, high-performance materials that can address both environmental and industrial needs.
Introduction
Owing to the increasing global concern over environmental issues, the development of biomass polymers has become the focus of considerable research interest.1 Although biomass-derived polyethylene/polypropylene has been previously synthesized,2 polymers using lactic acid3 or 5-(hydroxymethyl)furfural (HMF)4 have also been reported with unique properties that are different from those of conventional polymers. Among them, biological or environmental degradability is crucial for facilitating sustainability.5Based on the aforementioned analysis, we focused on (1S,5R)-6,8-dioxabicyclo[3.2.1]oct-2-en-4-one, that is, levoglucosenone (LGO; 1), as a monomer.6 LGO is a biomass molecular compound composed of C6H6O3; it has an enone and a cyclic acetal moiety with a rigid dioxabicyclo[3.2.1]octane skeleton. Enantiomerically pure LGO is easily obtained from the acid-catalyzed pyrolysis of cellulose via the formation of an intramolecular 1,6-glycosidic bond to generate 1,6-anhydro-beta-D-glucose, that is, levoglucosan (LGA) as an intermediate, which is referred to as anhydrosugars.7
Since the discovery of LGO in the 1970s, its reactivity has been explored in terms of organic chemistry.8 For example, transformation reactions of the CC bond via 1,4-addition,9 hydrogenation,10 Diels–Alder reaction,11 other additions,12 α-substitution,13 and β-substitution14 have been reported. For the CO bond, 1,2-addition,15 hydride reduction,16 other additions,17 condensation with amines,18 aldol condensation,19 as well as the ring-opening reactions of the cyclic acetal20 have been investigated. Other dynamic skeletal rearrangements of LGO have also been developed to form HMF and21 γ-(hydroxymethyl)-α,β-butenolide (HBO),22 among others.23 Notably, the transformation of LGO proceeds with high regio- and stereoselectivities owing to its rigid skeleton; therefore, LGO chemistry has been applied to the total syntheses of natural products using chiral building blocks,24 pharmaceutical sciences,25 and carbohydrate chemistry.26
After the fundamental reactivity of LGO was established, the adoption of these approaches slowed owing to the challenges in obtaining a sufficient quantitative supply at the laboratory scale. However, Circa Group Ltd successfully industrialized the production of LGO and its hydrogenated product (Cyrene™), enabling the mass-utilization of LGO/Cyrene™ as a monomer of biomass polymers and the use of a greener polar solvent.27
LGO dimerizes at the C3C4 bond under basic conditions;28 hence, forming LGO homopolymers via a direct connection between the C3- and C4-positions in the LGO ring is ideal. However, further treatment of the LGO homodimer with LGO results in the formation of a cyclic trimer.28 Therefore, in most cases, LGO was reportedly incorporated into biomass polymers after chemical modification. For example, Banwell,29 Fadlallah, and Allais30 independently reported the ring-opening metathesis polymerization (ROMP) of an LGO derivative prepared by the Diels–Alder reaction of LGO with cyclopentadiene, where LGO was incorporated as a side chain in the polymers obtained. For biopolymers containing an LGO skeleton in the main chain, the C2- and C4-diol derivatives of LGO were utilized as components of the copolymers. Mouterde, Miller, and Allais reported the synthesis of bio-based polyesters from the diol derivatives of LGO (HO-LGOL).31 The same approach was applied to the polymerization of LGA,32 or skeletally transformed compounds from LGO such as hydrated HBO (HO-HBO)33 and dimerized HBO (2H-HBO-HBO).34 There are several examples in which the main chain of the polymers was constructed only with LGO. Schlaad reported the ROMP of levoglucosenol and O-alkyl levoglucosenyl ether to form a series of polyacetals.35 They also reported the cationic ring-opening polymerization (CROP) of the same monomers via the formation of the intermolecular 1–6 glycosidic bonds forming other polyacetals.36
Unlike other biomass compounds, LGO contains diverse reactive functional groups that can be utilized as linkages to form polymers. However, previously reported methods for polymer synthesis using LGO do not precisely control the regio- and stereochemistry. Notably, Niu achieved the formation of precisely aligned polyacetals via CROP of the cyclic acetals of LGA derivatives.37 Sato also reported the CROP of 6,8-dioxabicyclo[3.2.1]octane (6,8-DBO) derivatives prepared by the Wolff–Kishner reduction of LGO derivatives, where the formation of stereoselective intermolecular 1–6 glycosidic bonds was performed, providing enantiomerically enriched polyacetals.38 Compared with these studies that focused on C–O cleavage, there are no studies regarding chiral polymers including the enone moiety in the main chain. In addition, the carbonyl moiety tends to be removed, increasing the reactivity of the other CC or acetal moieties. Pollard recently reported that using DBU as an initiator provided the LGO homopolymer, which demonstrated high thermal stability.39 Although this was an impactful achievement, the 1H NMR spectrum of the homopolymer obtained appeared complicated. Therefore, the stereoselective synthesis of polymers from LGO remains challenging.
Based on the aforementioned background, we designed a new concept for the precise polymerization of LGO using the 1,4-addition of dithiol to the enone moiety, and the condensation of dihydrazide at the C2-carbonyl group, both of which have been regarded as effective polymerization methods using multicomponent monomers (Fig. 1).40 Whether the condensation occurred stereoselectively, leading to bishydrazone, was analyzed. In addition, unlike previous studies, the resulting CN bonds were easily degradable under weakly acidic conditions compared to the ester bonds in polyesters. Herein, we report the synthesis of tricomponent polymers containing LGO via the stereoselective C–S and CN bond formation with the enone moiety.
 | ||
| Fig. 1 Designs of LGO-containing polymers. | ||
Results and discussion
Condensation using model compounds
The thia-Michael addition of thiophenol to LGO (1) is known to proceed under basic conditions with exclusive exo-selectivity to form compound 2 (eqn (1)).9 Although the stereochemistry with the addition reaction of various types of nucleophiles to the enone moiety in 1 has been reported,8 that for the condensation reaction of the carbonyl moiety in 1 with acylhydrazine has not been studied. Poplausky reported the reaction of 1 with thiosemicarbazide to form the corresponding semicarbazone compound, for which the stereochemistry was not determined; however, the 3D-model structure with a Z-form stereochemistry was determined.18 To study the reactivity of the condensation with acylhydrazide and the stereoselectivity of the hydrazone produced, the condensation reaction of 2 with acetohydrazide was initially investigated (eqn (1)). | (1) |
The treatment of exo-adduct 2 with 1.0 equiv. of acetohydrazide in the presence of solid acid (montmorillonite K10; MK10) and molecular sieves (MS3A) resulted in the formation of the corresponding hydrazone compound (3) with a 79% isolated yield. The 1H NMR spectrum of 3 measured in DMSO-d6 at room temperature indicated a mixture of two isomers (ratio of 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38), which may be due to several possibilities; one was a mixture of E/Z isomers (Fig. 2a), and the other was a mixture of rotamers (Fig. 2b). An irradiation experiment was performed to clarify this (Fig. 2c–e). When the 1H NMR spectrum of compound 3 in DMSO-d6 was measured under irradiation at δ 2.09 ppm, which was derived from the acetyl methyl protons of the major isomer, the integration of the signal observed at δ 1.91 ppm, which was derived from the acetyl methyl protons of the minor isomer, decreased by 82% (Fig. 2d). When the same measurement was performed under irradiation at δ 1.91 ppm, the signal observed at δ 2.09 ppm also decreased by 87% (Fig. 2e). These results indicate that a spin-saturation transfer occurred between the two isomers, and that their isomerization proceeded rapidly. The 1H NMR total correlation spectroscopy (TOCSY) of 3 was also performed to exclude the possibility of simultaneous irradiation owing to the close chemical shifts. Because the ratio of the two isomers gradually varied with the mixing time, they were found to be interchangeable on the NMR time scale (see the ESI†). Finally, the 1H NMR nuclear Overhauser effect spectroscopy (NOESY) of 3 demonstrated that both isomers had strong correlations between the NH protons (δ 10.49 and 10.32 ppm) and H3exo (δ 3.05 and 2.93 ppm), indicating that both were assigned E-forms. Thus, we concluded that the two isomers are rotamers of the amide bonds, as shown in Fig. 2b. The NMR analyses revealed that the condensation reaction on LGO proceeded stereoselectively, leading to the formation of E-isomers. Notably, the NMR spectrum of 3 measured in CDCl3 showed a single diastereomer (see the ESI†). However, as the polymers we prepared were not dissolved in CDCl3, DMSO-d6 was used as a solvent for measuring the NMR spectra of the polymers discussed below.
:38), which may be due to several possibilities; one was a mixture of E/Z isomers (Fig. 2a), and the other was a mixture of rotamers (Fig. 2b). An irradiation experiment was performed to clarify this (Fig. 2c–e). When the 1H NMR spectrum of compound 3 in DMSO-d6 was measured under irradiation at δ 2.09 ppm, which was derived from the acetyl methyl protons of the major isomer, the integration of the signal observed at δ 1.91 ppm, which was derived from the acetyl methyl protons of the minor isomer, decreased by 82% (Fig. 2d). When the same measurement was performed under irradiation at δ 1.91 ppm, the signal observed at δ 2.09 ppm also decreased by 87% (Fig. 2e). These results indicate that a spin-saturation transfer occurred between the two isomers, and that their isomerization proceeded rapidly. The 1H NMR total correlation spectroscopy (TOCSY) of 3 was also performed to exclude the possibility of simultaneous irradiation owing to the close chemical shifts. Because the ratio of the two isomers gradually varied with the mixing time, they were found to be interchangeable on the NMR time scale (see the ESI†). Finally, the 1H NMR nuclear Overhauser effect spectroscopy (NOESY) of 3 demonstrated that both isomers had strong correlations between the NH protons (δ 10.49 and 10.32 ppm) and H3exo (δ 3.05 and 2.93 ppm), indicating that both were assigned E-forms. Thus, we concluded that the two isomers are rotamers of the amide bonds, as shown in Fig. 2b. The NMR analyses revealed that the condensation reaction on LGO proceeded stereoselectively, leading to the formation of E-isomers. Notably, the NMR spectrum of 3 measured in CDCl3 showed a single diastereomer (see the ESI†). However, as the polymers we prepared were not dissolved in CDCl3, DMSO-d6 was used as a solvent for measuring the NMR spectra of the polymers discussed below.
 | ||
| Fig. 2 (a) Possibility of a mixture of E/Z isomers. (b) Possibility of a mixture of rotamers. (c) Irradiation experiments of 3. (d) 1H NMR chart of 3 without irradiation. (e) Irradiation at δ 2.09 ppm. (f) Irradiation at δ 1.91 ppm. | ||
Next, 2 equiv. of exo-adduct 2 were treated with adipic dihydrazide in the presence of acetic acid (AcOH) to provide the bishydrazone compound 4a with an 81% yield (eqn (2)). In this reaction, 4a was obtained as a precipitate in EtOH; therefore, acetic acid was used instead of solid MK10, and the product was easily separated via centrifugation.
Similar to 4a, sebacic and succinic dihydrazides were condensed with compound 2 to obtain solids 4b and 4c, respectively, with good yields. In the synthesis of 4d from carbohydrazide, MK10 was used because 4d is soluble in EtOH. The former procedure was applied to aromatic dicarboxylic dihydrazides such as terephthalic dihydrazide to obtain compound 4e with an 87% yield.
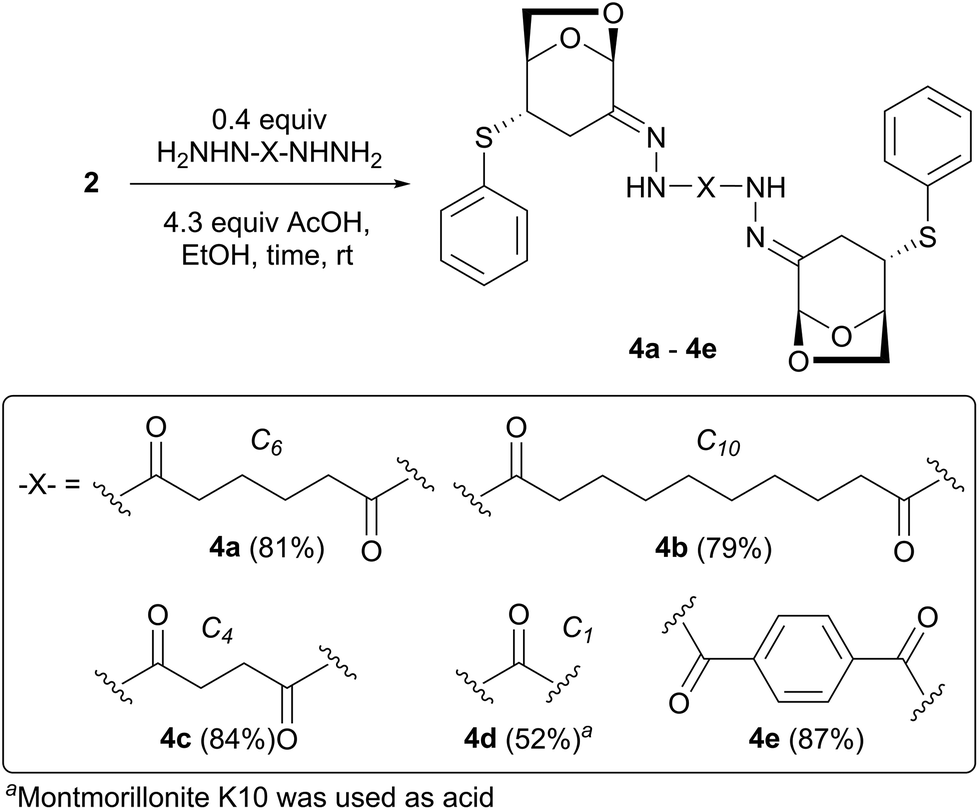 | (2) |
Given that two rotamers were observed for compound 3 in DMSO-d6 at room temperature, 4a–4c could exist as three rotamers: 4-1, 4-2, and 4-3 (Fig. 3a). 4-1 and 4-3 provide one magnetically equivalent NH proton signal (2H) owing to C2-symmetry, whereas 4-2 provides two magnetically inequivalent NH signals (1H each). Thus, up to four signals may appear despite compound 4 being a single diastereomer. In fact, the 1H NMR spectrum of 4c measured in DMSO-d6 at 25 °C demonstrated four singlets at δ 10.39, 10.41, 10.52, and 10.53 ppm, corresponding to the NH protons (Fig. 3b). The valuable temperature (VT) 1H NMR of 4c from 25 °C to 100 °C showed that these signals gradually coalesced into a single broad singlet at 80 °C, followed by a sharp signal at 100 °C. Along with the NH protons, all the other signals derived from the three rotamers coalesced into one (see the ESI†). These results clearly show that compounds 4a–4c are single diastereomers with an E-configuration in the hydrazone moieties determined by NOESY measurements. While four NH proton signals were observed in the 1H NMR spectrum of 4c, they decreased to three (δ 10.30, 10.46, and 10.47 ppm) for 4a and two (δ 10.28 and 10.44 ppm) for 4b. As the distance between two intramolecular LGO fragments was increased by changing the carbon lengths of the dihydrazides, their interactions weakened, and the magnetic inequivalency between the rotamers decreased. Consequently, the two NH signals of 4b-2 overlapped completely with those of 4b-1 and 4b-3 (Fig. 3b).
 | ||
| Fig. 3 (a) Isomerization of rotamers 4. (b) Comparison of the 1H NMR spectra of 4 (NH region). | ||
Conversely, the 1H NMR spectra of 4d and 4e were obtained as single diastereomers with slight broadening. This indicated that the amide rotations in 4d and 4e were faster than those in 4a–4c. In summary, the condensation of LGO with hydrazides proceeded with a high stereoselectivity to form E-isomers, regardless of the difference between monohydrazides and dihydrazides.
Polymer syntheses
The polymer synthesis was performed based on these model experiments. As a linker, 1,4-benzenedithiol was selected instead of thiophenol with the expectation of simple 1H NMR observation derived from magnetically equivalent four aromatic protons. In the presence of MgO as a base, treatment of 1,4-benzenedithiol with a small excess of LGO at 0 °C resulted in the formation of compound 5. After filtration and washing with Et2O to remove the LGO, the pure compound 5 was isolated with a 91% yield. The 1H NMR spectrum of 5 suggested a C2-symmetric structure, based on the observation of one sharp singlet (δ 7.41 ppm) as an aromatic ring.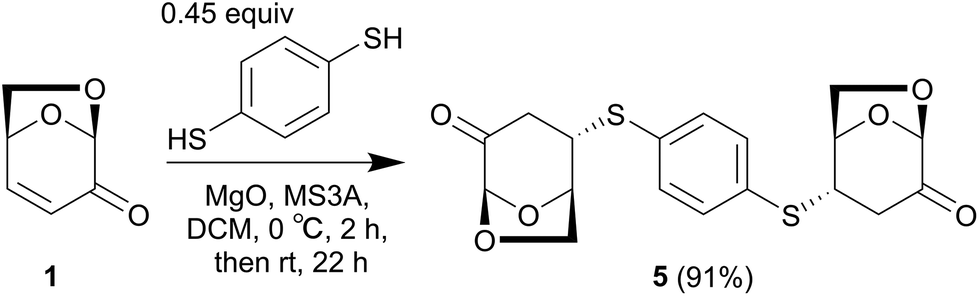 | (3) |
By using the resulting compound 5 as a monomer, polymerization was subsequently performed. After the initial optimization shown in the ESI,† the effect of the solvents was investigated using acetic acid (4.3 equiv.), a solution concentration of 250 mM, a 1:1 ratio of 5 and adipic dihydrazide, and purification procedures. Table 1 presents the results of the study. In EtOH, precipitation was observed (entry 1). Compared with THF and DCM (entries 2 and 3), polar DMF and HFIP showed high Mn and Mw values (entries 4 and 5). Notably, the selection of the combination of solvents was important for the elongation of 6a. Although the polymerization did not sufficiently proceed in a mixed solvent of DCM/DMF (entry 6) or DMF/HFIP (entry 7), in a 1:2 mixture of DCM and HFIP, the molecular weight of polymer 6a dramatically increased (Mn = 5400, Mw = 17400, entry 8). Increasing the reaction time from 2 to 24 h did not affect Mn (entry 9). The ratio of HFIP to DCM (1:2 to 1:1) could be maintained without decreasing Mn (entry 10). Finally, after washing the product with THF, the Mn and Mw values of 6a reached 6400 and 15600, respectively (entry 11).
a
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Dihydrazide | Polymer | Solvent | Time (h) | Conv. (%) | M n | M w | M w/Mn | DP |
| a 5 (0.25 mmol), dicarboxylic dihydrazide (1.00 equiv.), AcOH (4.3 equiv.), solvent (1 mL total), room temperature, 2 h, and precipitation with Et2O. b Rinsed with THF. c 30 °C, rinsed with DCM. d 0.75 mmol of 5 and carbohydrazide were used (750 mM), 40 °C, and rinsed with DCM (10 times). e Not determined owing to the monitoring time course. | |||||||||
| 1 | Adipic dihydrazide | 6a | Ethanol (EtOH) | 2 | 43 | 171 | 1711 | 9.99 | — |
| 2 | Adipic dihydrazide | 6a | Tetrahydrofuran (THF) | 2 | 73 | 1000 | 1300 | 1.29 | 1.9 |
| 3 | Adipic dihydrazide | 6a | Dichloromethane (DCM) | 2 | 85 | 2400 | 5000 | 2.05 | 4.5 |
| 4 | Adipic dihydrazide | 6a | N,N-Dimethylformamide (DMF) | 2 | 89 | 4100 | 10100 |
2.48 | 7.7 |
| 5 | Adipic dihydrazide | 6a | 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP) | 2 | 76 | 4800 | 16200 |
3.40 | 9.0 |
| 6 | Adipic dihydrazide | 6a | DCM:DMF = 1:1 |
2 | 86 | 1800 | 2900 | 1.59 | 3.4 |
| 7 | Adipic dihydrazide | 6a | DMF:HFIP = 1:1 |
2 | 77 | 1300 | 1900 | 1.53 | 2.4 |
| 8 | Adipic dihydrazide | 6a | DCM:HFIP = 1:2 |
2 | —e | 5400 | 17400 |
3.25 | 10.1 |
| 9 | Adipic dihydrazide | 6a | DCM:HFIP = 1:2 |
24 | —e | 5400 | 20400 |
3.74 | 10.1 |
| 10 | Adipic dihydrazide | 6a | DCM:HFIP = 1:1 |
2 | 91 | 5300 | 15400 |
2.92 | 10.0 |
| 11b | Adipic dihydrazide | 6a | DCM:HFIP = 1:1 |
2 | 80 | 6400 | 15600 |
2.42 | 12.2 |
| 12c | Sebacic dihydrazide | 6b | DCM:HFIP = 1:1 |
2 | 70 | 10000 |
21600 |
2.15 | 17.2 |
| 13c | Succinic dihydrazide | 6c | DCM:HFIP = 1:1 |
8 | 48 | 4900 | 14200 |
2.93 | 9.7 |
| 14d | Carbohydrazide (eqn (4)) | 6d | DCM:HFIP = 1:1 |
24 | 50 | 6200 | 14400 |
2.31 | 13.8 |
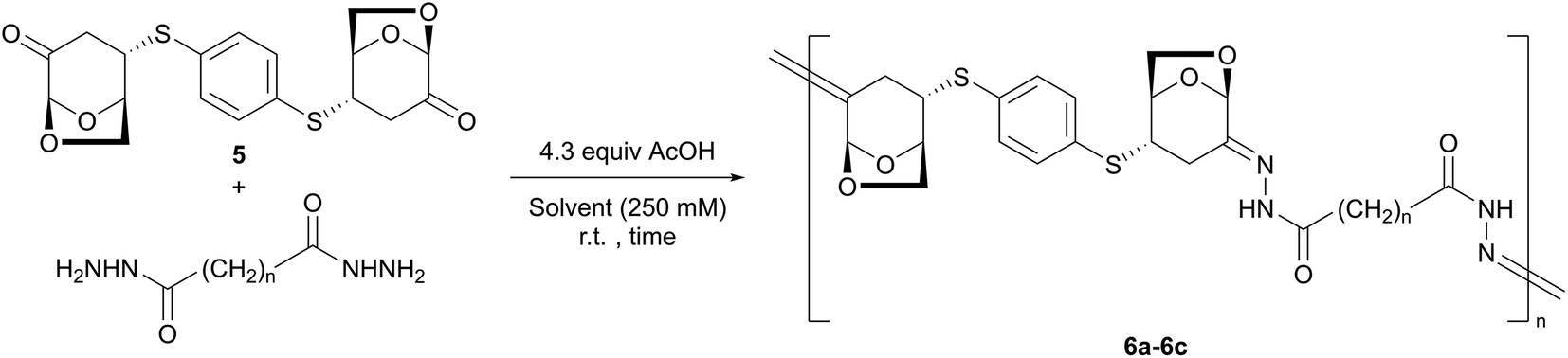
Fig. 4 shows the 1H NMR spectrum of 6a in DMSO-d6 accompanied by those of model compounds 2, 3, and 4a. Regarding the LGO fragment, two magnetically inequivalent H3 (H3exo/H3endo) appear at approximately δ 2.7–3.0 ppm as a doublet and doublet of doublet with large geminal coupling (JH–H ∼ 16 Hz). H6exo and H6endo independently appear at δ 3.7–4.0 ppm overlapped with H4. A methine proton H5 was observed as a multiplet at ca. δ 4.7 ppm. The remaining acetal proton H1 exists at a lower magnetic field at δ 5.5 ppm. Regarding the carbohydrazone moieties, NH protons were observed in two areas: at approximately δ 10.3 and δ 10.5 ppm, owing to the rotamers as indicated above. These observations in the 1H NMR spectrum of 6a are in good agreement with those of model compounds 2, 3, and 4a, which clearly indicates that polymer 6a has a high stereoselectivity. The NOESY correlations of 6a showed that the stereochemistry of the CN bond was the E-form, similar to 4a.
 | ||
| Fig. 4 Comparison of the 1H NMR spectra of compounds (a) 2, (b) 3, and (c) 4a; and (d) polymer 6a (LGO ring region). | ||
Using the optimized polycondensation conditions, the scope of the dicarboxylic dihydrazides was investigated. The polymerization of 5 with sebacic dihydrazide proceeded to form polymer 6b (Mw = 21600, entry 12). When succinic dihydrazide was used, a longer reaction time was required to obtain a sufficient length of the corresponding polymer 6c (Mw = 14200, entry 13). For carbohydrazide, a higher concentration (750 mM) was necessary to consume 5, and polymer 6d was obtained with a moderate yield (50%, Mw = 14400, entry 14) after washing the precipitate thoroughly with DCM (10 times). Notably, when the filtrate was concentrated in vacuo, the cyclic dimer 7 was obtained. In the 1H NMR spectrum of 7, a pair of similar signals derived from the LGO fragment were observed at a ratio of 1:1. Conversely, two doublet signals at δ 7.42 and 7.73 ppm, corresponding to the p-phenylene protons, were observed to be magnetically inequivalent. Although one of the NH protons appeared at δ 9.05 ppm, the other was downfield-shifted to δ 10.90 ppm, suggesting intramolecular hydrogen bonding. Both hydrazone moieties were determined as the E-form based on the NOESY correlations. Note that in the 13C NMR spectrum of 7, only one carbonyl signal was observed.
 | (4) |
The molecular structure of 7 was determined via X-ray diffraction.41Fig. 5 clearly shows that 7 is a cyclic dimer consisting of four LGO fragments, two 1,4-benzenedithiol, and two carbazone moieties. The crystal structure of the compound exhibits C2-symmetry, which is consistent with the symmetry inferred from the NMR data in the solution. All stereochemistries of the four hydrazone moieties were determined to be the E-form, and all C–S bonds were connected from the exo-faces of the LGO moiety. Both cis- and trans-amide bonds were observed in the carbazone. The NH of the latter amide and the oxygen atom of the other carbonyl group are within close proximity with a distance of 1.934 Å, indicating the existence of intramolecular hydrogen bonding.
 | ||
| Fig. 5 Molecular structure of the cyclic dimer 7.41 C: grey, H: white, N: blue, O: red, and S: yellow. | ||
The attempt for the polymerization of monomer 5 with terephthalic dihydrazide failed due to low solubility of the oligomers produced (see the ESI†).
Properties of polymers 6a–6d
The properties of the synthesized polymers, 6a–6d, were evaluated. They showed high tolerance (insolubility) toward many solvents such as Et2O, THF, DCM, EtOH and acetone, and were only soluble in DMF or DMSO. As shown in Fig. 6a, a thermogravimetric analysis (TGA) revealed that the 10% weight-loss temperature (Td,10%) increased as the number of carbon chains increased as follows: C1 (233.0 °C for 6d), C4 (248.4 °C for 6c), C6 (265.7 °C for 6a), and C10 (268.5 °C for 6b). Conversely, the melting points (m.p.) of 6a–6c decreased as follows: C4 (218.4 °C for 6c), C6 (205.7 °C for 6a), and C10 (155.2 °C for 6b), owing to the increasing flexibility of the main chains. All four polymers demonstrated no glass transition temperature (Tg) in the differential scanning calorimetry (DSC) analysis. Notably, polymers 6a–6d showed positive optical rotation values. These results clearly demonstrate that the obtained polymers have the potential for application as chiral polymers (Table 2). Unexpectedly, the shortest chain length of the carbazone polymer 6d demonstrated a relatively low viscosity. When the Hawk–Marking–Sakurada formula was applied, the slope was approximately 0.5, suggesting that the polymer was flexible in the solution (Fig. 6b). | ||
| Fig. 6 (a) TGA of polymers 6a–6d. (b) Viscosity measurement of polymers 6a–6d. | ||
Degradation experiments and modification attempts
An advantage of the polymer described herein is the presence of relatively weak CN bonds, which can be cleaved via hydrolysis or interchanged with other amino/hydrazide groups. To elucidate the reactivity of the CN bonds, polymer 6a was treated with excess water in the presence of AcOH. Hydrolysis of 6a resulted in an increase in the retention time, as demonstrated by the SEC measurements (Fig. 7a). However, the desired compound 5 was not obtained cleanly.
 | ||
| Fig. 7 (a) Degradation of polymer 6a with water in an organic solvent. (b) Degradation of polymers 6a–6d with the “capping reagent”. (c) Degradation of polymer 6a with H2O2 to full dissolution in water. | ||
Subsequently, 6a was treated with acetohydrazide as the “capping reagent”. Chemical degradation rapidly occurred even at room temperature to form compound 8 (73%), an analogue of compound 5 capped with an acetohydrazone moiety. Similarly, polymers 6b–6d were depolymerized into compound 8 in good yields along with the corresponding dicarboxylic dihydrazides (Fig. 7b).
Based on the literature regarding late-stage modifications of polymers,39 we attempted the Baeyer–Villiger oxidation of 6a. Notably, when 6a was treated with H2O2 in an aqueous medium at room temperature for 12 h, the white suspension gradually transitioned into a colorless clear solution. The viscous products obtained by reprecipitation in THF were a mixture of degradation products. The main product was identified as compound 9, an analogue of HBO linked to the 1,4-benzenedisulfonyl group (see the ESI†). Compound 9 is thought to be obtained from the Baeyer–Villiger oxidation of the LGO skeleton, followed by hydrolysis and γ-lactone formation, as well as the oxidation of sulfur atoms and hydrolysis of the CN bonds.
Conclusions
The synthesis and properties of a new polymer using LGO, a biomass compound obtained from cellulose, are reported herein. The sequential treatment of LGO with 1,4-benzenedithiol and mono- or dicarboxylic dihydrazides resulted in the formation of tricomponent biomass copolymers. In all the polymers and model compounds, both C–S and CN bonds formed stereoselectively as exo-adducts and E-isomers, respectively.
All the obtained polymers showed positive optical rotations, indicating their potential as chiral monomers. Their thermal properties can be tuned by changing the number of carbon chains in the dihydrazides. To the best of our knowledge, this is the first study on an LGO-containing polymer connected by CN bonds which demonstrated water-degradability owing to its hydrolysis property. As shown in the Introduction, the general strategies for LGO polymerization are limited to the CROP of cyclic acetals, ROMP of alkenes, or polyester formation after the removal of the carbonyl group. These results clearly demonstrate a novel option for connecting LGO via a carbonyl group without reduction.
In this study, 1,4-benzenedithiol was selected as the bis-nucleophile, whose skeleton appeared to be as hard as the LGO moiety. To increase flexibility, the modification of aromatic dithiols into aliphatic compounds is under development. The use of diols instead of dithiols is also being investigated.
Author contributions
Conceptualization by A. T.; data curation by A. T., S. Y., and T. H.; formal analysis by A. T., S. Y., T. H., and Y. Y.; funding acquisition by A. T., T. H., and S. K.; investigation by A. T., S. Y., and T. H.; methodology by A. T., S. Y., T. H., and Y. Y.; project administration by A. T.; resources by A. T., T. K., and T. D.; supervision by A. T.; validation by A. T., T. H., and S. K.; visualization by A. T., S. Y., and T. H.; writing (original draft) by A. T., T. H., and T. D.; and writing (review & editing) by A. T. All authors discussed the results.Data availability
Experimental details and characterization data of the compounds can be found in the ESI.†The molecular structure of compound 7 is openly available in CCDC 2322689 at https://doi.org/10.5517/ccdc.csd.cc2hyygw.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a project (JPNP16002) subsidized by the New Energy and Industrial Technology Development Organization (NEDO) [A. T., T. H. and S. K.], JSPS KAKENHI Grant Numbers JP22H02089 [A. T.], JP22H05554 [A. T.], and JP24H01304 [A. T.], JSPS LEADER Grant Number JPMXS0320200558 [A. T.] and JST SPRING Grant Number JPMJSP2114 [S. Y.]. X-ray diffraction study for compound 7 was operated by Dr Hiroyasu Sato (RIGAKU). We also would like to thank Ms Honoka Ishikawa for her experimental support.References
- C. Tang and C. Y. Ryu, Sustainable Polymers from Biomass, Wiley-VCH, Weinheim, 2017 Search PubMed.
- V. Siracusa and I. Blanco, Polymers, 2020, 12, 1641 CrossRef CAS and references cited therein.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y.-X. Chen, Chem. Rev., 2024, 124, 4393 CrossRef CAS and references cited therein.
- (a) R. A. Sheldon, Green Chem., 2014, 16, 950 RSC; (b) A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2015, 116, 1637 CrossRef PubMed; (c) C. F. Araujo, M. M. Nolasco, P. J. A. Ribeiro-Claro, S. Rudić, A. J. D. Silvestre, P. D. Vaz and A. F. Sousa, Macromolecules, 2018, 51, 3515 CrossRef CAS.
- (a) G. W. M. Vandermeulen, A. Boarino and H.-A. Klok, J. Polym. Sci., 2022, 60, 1797 CrossRef CAS and references cited therein. (b) V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612 CrossRef CAS and references cited therein.
- S. Kudo, X. Huang, S. Asano and J.-I. Hayashi, Energy Fuels, 2021, 35, 9809 CrossRef CAS and references cited therein.
- S. Kudo, N. Goto, J. Sperry, K. Norinaga and J.-I. Hayashi, ACS Sustainable Chem. Eng., 2017, 5, 1132 CrossRef CAS.
- (a) J. E. Camp and B. W. Greatrex, Front. Chem., 2022, 10, 902239 CrossRef CAS; (b) J. Kühlborn, J. Groß and T. Opatz, Nat. Prod. Rep., 2020, 37, 380 RSC.
- (a) F. Shafizadeh, R. H. Furaneaux and T. T. Stevenson, Carbohydr. Res., 1979, 71, 169 CrossRef CAS; (b) S. Kim, E. T. Ledingham, S. Kudo, B. W. Greatrex and J. Sperry, Eur. J. Org. Chem., 2018, 2028 CrossRef CAS; (c) Y. Tsai, C. M. B. Etichetti, S. Cicetti, J. E. Girardini, R. A. Spanevello, A. G. Suárez and A. M. Sarotti, Bioorg. Med. Chem. Lett., 2020, 30, 12747 CrossRef.
- J. Sherwood, M. De bruyn, A. Constatinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650 RSC.
- (a) M. B. Comba, Y. Tsai, A. M. Sarotti, M. I. Mangione, A. G. Suárez and R. A. Spanevello, Eur. J. Org. Chem., 2018, 590 CrossRef CAS; (b) F. A. Valeev, I. N. Gaisina and M. S. Miftakhov, Russ. Chem. Bull., 1996, 45, 2454 Search PubMed.
- (a) M. B. Comba, A. G. Suárez, A. M. Sarotti, M. I. Mangione, R. A. Spanevello and E. D. V. Giordano, Org. Lett., 2016, 18, 1748 CrossRef CAS; (b) C. Lefebvre, T. V. Gysel, C. Michelin, E. Rousset, D. Djiré, F. Allais and N. Hoffmann, Eur. J. Org. Chem., 2022, e202101298 CrossRef CAS.
- K. P. Stockton, C. J. Merritt, C. J. Sumby and B. W. Greatrex, Eur. J. Org. Chem., 2015, 6999 CrossRef CAS.
- E. T. Ledingham, C. J. Merritt, C. J. Sumby, M. K. Taylor and B. W. Greatrex, Synthesis, 2017, 2652 CAS.
- V. K. Brel, A. V. Samet, L. D. Konyushkin, A. I. Stash, V. K. Belsky and V. V. Semenov, Mendeleev Commun., 2015, 25, 44 CrossRef CAS.
- M. E. Jung and M. Kiankarimi, J. Org. Chem., 1998, 63, 8133 CrossRef CAS.
- E. T. Ledingham and B. W. Greatrex, Tetrahedron, 2018, 74, 6107 CrossRef CAS.
- A. Czubatka-Bieńkowska, J. Sarnik, A. Macieja, G. Galtia, Z. J. Witczak and T. Poplawski, Bioorg. Med. Chem. Lett., 2017, 27, 2713 CrossRef.
- L. Hughes, C. R. McElroy, A. C. Whitwood and A. J. Hunt, Green Chem., 2018, 20, 4423 RSC.
- A. R. Tagirov, I. M. Biktagirov, Yu. S. Galimova, L. Kh. Faizullina, Sh. M. Salikhov and F. A. Valeev, Russ. J. Org. Chem., 2015, 51, 569 CrossRef CAS.
- X. Huang, S. Kudo, J. Sperry and J. Hayashi, Green ACS Sustainable Chem. Eng., 2019, 7, 5892 CrossRef CAS.
- G. Bonneau, A. A. M. Peru, A. L. Flourat and F. Allais, Green Chem., 2018, 20, 2455 RSC.
- K. Matsumoto, T. Ebata and H. Matsushita, Carbohydr. Res., 1995, 267, 187 CrossRef CAS.
- T. Nishikawa and M. Isobe, Chem. Rec., 2013, 13, 286 CrossRef CAS PubMed.
- (a) X. Liu, P. Carr, M. G. Gardiner, M. G. Banwell, A. H. Elbanna, Z. G. Khalil and R. J. Capon, ACS Omega, 2020, 5, 13926 CrossRef CAS PubMed; (b) C. Müller, M. A. G. Frau, D. Ballinari, S. Colombo, A. Bitto, E. Martegani, C. Airoldi, A. S. Neuren, M. Stein, J. Weiser, C. Battistini and F. Peri, ChemMedChem, 2009, 4, 524 CrossRef PubMed.
- Z. J. Witczak, J. Sarnik, A. Czubatka, E. Forma and T. Poplawski, Bioorg. Med. Chem. Lett., 2014, 24, 5606 CrossRef CAS PubMed.
- https://circa-group.com/products/lgo-derivatives/ (24th Septemper 2024).
- F. Shafizadeh, R. H. Furaneaux, D. Pang and T. T. Stevenson, Carbohydr. Res., 1982, 100, 303 CrossRef CAS.
- M. G. Banwell, X. Liu, L. A. Connal and M. G. Gardiner, Macromolecules, 2020, 53, 5308 CrossRef CAS.
- S. Fadlallah, A. A. M. Peru, A. L. Flourat and F. Allais, Eur. Polym. J., 2020, 138, 109980 CrossRef CAS.
- F. Diot-Néant, L. M. M. Mouterde, C. Veith, J. Couvreur, S. A. Miller and F. Allais, ACS Sustainable Chem. Eng., 2022, 10, 10132 CrossRef.
- J. Bassut, K. Nasr, G. Stoclet, M. Bria, J.-M. Raquez, A. Favrelle-Huret, R. Wojcieszak, P. Zinck and I. Itabaiana, ACS Sustainable Chem. Eng., 2022, 10, 16845 CrossRef CAS.
- F. Diot-Néant, L. M. M. Mouterde, J. Couvreur, F. Brunois, S. A. Miller and F. Allais, Eur. Polym. J., 2021, 159, 110745 CrossRef.
- S. Fadlallah, A. L. Flourat, L. M. M. Mouterde, M. Annatelli, A. A. M. Peru, A. Gallos, F. Aricò and F. Allais, Macromol. Rapid Commun., 2021, 42, 2100284 CrossRef CAS.
- T. Debsharma, F. N. Behrendt, A. Laschewsky and H. Schlaad, Angew. Chem., Int. Ed., 2019, 58, 6718 CrossRef CAS.
- (a) T. Debsharma, Y. Yagci and H. Schlaad, Angew. Chem., 2019, 131, 18663 CrossRef; (b) T. Debsharma, B. Schimdt, A. Laschewsky and H. Schlaad, Macromolecules, 2021, 54, 2720 CrossRef CAS.
- L. Wu, Z. Zhou, D. Sathe, J. Zhou, S. Dym, Z. Zhao, J. Wang and J. Niu, Nat. Chem., 2023, 15, 1276 CrossRef CAS.
- Y. Mizukami, Y. Kakehi, F. Li, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, ACS Macro Lett., 2024, 13, 252 CrossRef CAS.
- B. Pollard, M. G. Gardiner, M. G. Banwell and L. A. Connal, ChemSusChem, 2024, 17, e202301165 CrossRef CAS.
- S. Luleburgaz, E. Cakmakci, H. Durmaz and U. Tunca, Eur. Polym. J., 2024, 209, 112897 CrossRef CAS.
- A. Tahara, S. Yashiro, T. Hokajo and T. Doi, CSD Commun., 2023 DOI:10.5517/ccdc.csd.cc2hyygw.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, characterization of compounds, and spectroscopic data. See DOI: https://doi.org/10.1039/d4py01094a |
| This journal is © The Royal Society of Chemistry 2025 |