
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolybasic nanogels for intracellular co-delivery of paclitaxel and carboplatin: a novel approach to ovarian cancer therapy†
Angela M.
Wagner
ab,
Olivia L.
Lanier
*bcdef,
Ani
Savk
ab and
Nicholas A.
Peppas
 *abcghi
*abcghi
aMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, TX, USA. E-mail: peppas@che.utexas.edu
bInstitute for Biomaterials, Drug Delivery, and Regenerative Medicine, The University of Texas at Austin, Austin, TX, USA
cDepartment of Biomedical Engineering, The University of Texas at Austin, Austin, TX, USA
dDepartment of Chemical and Biological Engineering, University of New Mexico, Albuquerque, NM, USA
eDepartment of Biomedical Engineering, University of New Mexico, Albuquerque, NM, USA
fCancer Therapeutics Program, University of New Mexico Comprehensive Cancer Center, Albuquerque, NM, USA
gDepartment of Surgery and Perioperative Care, Dell Medical School, University of Texas at Austin, Austin, TX, USA
hDepartment of Pediatrics, Dell Medical School, University of Texas at Austin, Austin, TX, USA
iDivision of Molecular Pharmaceutics and Drug Delivery, College of Pharmacy, The University of Texas at Austin, Austin, TX, USA
First published on 21st February 2025
Abstract
Ovarian cancer is one of the leading causes of cancer-related deaths in women, with limited progress in treatments despite decades of research. Common treatment protocols rely on surgical removal of tumors and chemotherapy drugs, such as paclitaxel and carboplatin, which are capable of reaching cancer cells throughout the body. However, the effectiveness of these drugs is often limited due to toxic reactions in patients, nonspecific drug distribution affecting healthy cells, and the development of treatment resistance. In this study, we introduce a polybasic nanogel system composed of poly(diethylaminoethyl methacrylate-co-cyclohexyl methacrylate)-g-poly(ethylene glycol) designed for the targeted co-delivery of paclitaxel and carboplatin directly to ovarian cancer cells. These nanogel systems can respond to the cellular microenvironment to achieve controlled, on-demand drug release, reducing off-target effects and enhancing therapeutic uptake. Additionally, we investigated nanoparticle degradation and controlled drug release as a function of various crosslinkers, including tetraethylene glycol dimethacrylate, bis(2-methacryloyl)oxyethyl disulfide, poly(lactic acid)-b-poly(ethylene glycol)-b-poly(lactic acid)dimethacrylate, and polycaprolactone dimethacrylate. Our results, using OVCAR-3 human ovarian cancer cells, demonstrated that this dual-delivery system outperformed free drugs in inducing cancer cell death, representing a promising advance in the field of nanoparticle-based therapies for ovarian cancer. By loading two chemotherapeutic agents into a single, environmentally responsive particle, this approach shows the potential to overcome common resistance mechanisms and achieve more effective tumor suppression. In summary, by delivering chemotherapy more precisely, it may be possible to enhance therapeutic outcomes while minimizing toxicity and nonspecific drug distribution, ultimately improving patient quality of life.
Introduction
Ovarian cancer is the most lethal type of gynecological cancer and ranks among the top five causes of cancer-related deaths in women.1,2 The International Agency for Research on Cancer (IARC) estimated that in 2022, there were approximately 9.7 million cancer-related deaths and 20 million new cancer cases around the world. By 2050, this number is predicted to increase to 35 million new cancer cases based on projected population growth.3 Despite having high mortality, no major public health or medical organizations recommend routine screening for ovarian cancer to the general public. This is due to the potential for several benign conditions, such as endometriosis, pregnancy, and liver disease, to cause elevated levels of biomarkers such as CA125, resulting in a high rate of false-positive results.4,5 Additionally, the challenge of detecting ovarian cancer at an early stage stems from its typically asymptomatic nature in the early phases, which means that most patients are diagnosed only after the disease has progressed to an advanced stage.6 This late detection, combined with the limitations of current screening methods, underscores the need for improved therapies.Current treatment plans for ovarian cancer include local and systemic therapies. Local treatment options include surgical removal of tumors, which is the cornerstone of treatment, and radiation therapy, which is less commonly used. Systemic treatments involve the use of drugs that can reach cancer cells throughout the body and include chemotherapy, which is typically administered after surgery, targeted drug therapy, hormone therapy, and immunotherapy. These treatments may be combined depending on the cancer's type and stage, as well as the patient's general health and specific conditions.7 Emerging targeted delivery approaches further include gene therapy, protein therapy, molecular therapy, dual-targeting therapy, and photodynamic therapy.8 However, chemotherapy protocols lack the ability to differentiate between healthy and cancerous cells, often causing severe side effects. Due to toxicity-related limitations in dosing, patients often face extended intervals between treatments. During this time, cancer cells that are affected but not eliminated by the drug may develop resistance to the treatment.9
Nanoparticles as delivery systems have demonstrated potential in improving the systemic administration of chemotherapeutic agents and addressing challenges associated with traditional methods of delivery.10 By precisely targeting and regulating drug release at the tumor site, these approaches can provide significant benefits compared to conventional chemotherapy, reducing toxicity to healthy cells and minimizing side effects for patients.6,11 Among these, nanoscale hydrogel systems, also called nano gels, represent a subclass of nanoparticles that are three-dimensional hydrophilic polymer matrices through physical and chemical cross-linking. Such structures can be customizable in size, bio-integration, and water affinity, allowing them to hold a wide range of drugs and respond to the surrounding environment such as pH, temperature, or biological agents. Some of the advantages of nanoscale hydrogel systems include high drug loading, biocompatibility, and the ability to deliver both hydrophilic and hydrophobic drugs. Their environmental responsiveness helps to minimize toxicity and improves therapeutic precision. However, these systems face disadvantages that include premature drug per, low drug-loading efficiency, and mechanical stability, which can limit their clinical application.12
Although substantial research has been conducted on nanoparticle systems, there is only one FDA-approved nanoparticle-based therapy for ovarian cancer: Doxil®. Approved in 1995, Doxil® was the first FDA-approved nanodrug, however, it is not recommended as the first-line treatment for ovarian cancer. Doxil® is a PEGylated liposomal formulation of doxorubicin, with a particle size of approximately 100 nm and a negative surface charge, engineered to leverage the enhanced permeability and retention (EPR) effect for passive drug targeting. This mechanism allows the drug to accumulate in tumor tissues, releasing its chemotherapeutic payload gradually to minimize off-target effects and improving tolerability.13,14 The PEGylation of Doxil® results in prolonged circulation time and the ability to extravasate into tumor sites, which collectively improve its safety profile by minimizing exposure to healthy tissues and reducing systemic toxicity.15–17 However, the therapeutic efficacy of Doxil® is still limited, providing only slight improvements over conventional treatments.18 While Doxil represents a significant advancement in nanoparticle-based therapy, there are still major challenges for nanoparticle systems, such as premature release, limited drug-loading capacity, insufficient cellular uptake, and nonspecific distribution.18–21
In the last decade, clinical treatment with multiple pharmacologically active agents has shown success in enhancing the treatment of many diseases. With these recent clinical successes, focus has now been shifting towards nanocarrier-mediated combination therapies. Combination therapies using nanoparticles provide multiple benefits, such as ability to signal various pathways within cancer cells, enhancing treatment efficacy against targeted areas, influencing distinct phases of the cell cycle, and bypassing resistance caused by efflux mechanisms of resistance.22 Further, it allows the pharmacokinetics and pharmacodynamics (PK/PD) to be controlled by how nanoparticles are distributed and absorbed in vivo, rather than the physicochemical characteristics of the free drugs, ensuring that each therapeutic agent reaches the cytosol in an optimal synergistic ratio.23,24 In ovarian cancer, there has been a strong history of clinical success in using two different pharmacologically active agents; most importantly, the combination of paclitaxel and carboplatin has become the first line of treatment.25 Paclitaxel is a highly lipophilic, water insoluble agent, and current FDA approved formulations rely on Cremophor EL for solubilization which leads to significant vehicle toxicities.21 Conversely, carboplatin is hydrophilic and insoluble in common organic solvents used to solubilize paclitaxel.
The polybasic nanoscale hydrogel (nanogel) system based upon poly(diethylaminoethyl methacrylate-cyclohexyl methacrylate)-g-poly(ethylene glycol) (P(DEAEMA-co-CHMA)-g-PEGMA) developed in this work has been tailored to exploit multiple environmental cues for the controlled, targeted, intracellular delivery of multiple low molecular weight chemotherapeutic agents. The nanogel molecular architecture is designed to simultaneously carry cargo with varying physicochemical properties, promote long circulation and increased cellular uptake, and release the cargo only in response to intracellular environmental cues. The objective of this study is to demonstrate the utility of this nanogel system in co-delivering paclitaxel and carboplatin for the treatment of ovarian cancer. To achieve this, multiple degradable crosslinkers—including tetraethylene glycol dimethacrylate (TEGDMA), bis(2-methacryloyl)oxyethyl disulfide (DisulfideMA), poly(lactic acid)-b-poly(ethylene glycol)-b-poly(lactic acid) dimethacrylate (PLA-b-PEGDMA), and polycaprolactone dimethacrylate (PCL-DMA)—were synthesized and investigated to improve long-term biocompatibility and enhance drug release through intracellular-triggered degradation. This novel nanogel system aims to overcome the limitations of conventional therapies and serve as a platform for the co-localized delivery of drug combinations in cancer therapy.
Experimental
Materials
Poly(ethylene glycol)methyl ether methacrylate (Mn 2080, 50 wt% in water), 2-(diethylamino)ethyl methacrylate (DEAEMA), tetraethylene glycol dimethacrylate (TEGDMA), and cyclohexyl methacrylate (CHMA) were acquired from Sigma-Aldrich (St Louis, MO), along with myristyl trimethylammonium bromide (MyTAB), chlorpromazine hydrochloride (98%), dynasore, and wortmannin (98%). Irgacure 2959 came from Ciba Inc. (Basel, Switzerland). Brij 30 and deuterium oxide (99.8% D) were obtained from Acros Organics. Additionally, acetone, tetrahydrofuran, 1× Dulbecco's phosphate-buffered saline (DPBS), hydrochloric acid, sodium hydroxide, and fetal bovine serum (Corning, Catalog 35010CV) were sourced from Thermo Fisher Scientific (Waltham, MA). The 5-N,N-dimethyl amiloride was supplied by Enzo Life Sciences (Farmingdale, NY), and Filipin III was purchased from Cayman Chemical (Ann Arbor, MI). OVCAR-3 cells (ATCC® HTB-161™) and RPMI-1640 medium (ATCC® 30-2001™) were obtained from ATCC (Manassas, VA). Insulin from bovine pancreas (10 mg mL−1 in 25 mM HEPES pH 8.2, Catalog I-0516) was provided by Sigma-Aldrich. Deionized water used in the experiments was obtained via a Milli-Q Plus Ultrapure Water System (Millipore), fitted with a 0.22 μm in-line filter, and all chemicals were utilized as received without further modification.Nanoparticle synthesis
Poly(2-diethylaminoethyl methacrylate-co-cyclohexyl methacrylate)-g-poly(ethylene glycol methyl methacrylate) (P(DEAEMA-co-CHMA)-g-PEGMA) nanoparticles were synthesized via UV-initiated, aqueous emulsion free radical polymerization.26 For nanoparticle synthesis, the pre-polymerization mixture was prepared by mixing DEAEMA (71.0 mol%), PEGMA (7.8 mol%), and CHMA (21.2 mol%) in deionized water using a round bottom flask. To stabilize the emulsion, Brij 30 (a non-ionic surfactant) and myristyl trimethylammonium bromide (MyTAB, a cationic surfactant) were added at concentrations of 4 mg mL−1 and 1.16 mg mL−1, respectively. The free radical initiator, Irgacure 2925, was added at 0.5 wt% relative to the total monomer content.Various crosslinkers were also investigated for the synthesis, including tetraethlyene glycol dimethacrylate (TEGDMA), bis(2-methacryloyl)oxyethyl disulfide (DisulfideMA), poly(lactic acid)-b-poly(ethylene glycol)-b-poly(lactic acid)dimethacrylate (PLA-b-PEG-DMA), and polycaprolactone dimethacrylate (PCL-DMA), as shown in Fig. 1. Disulfide-DMS contains a disulfide bond that is degradable by intracellular levels of glutathione (GSH) under reductive conditions, and is of a similar molecular weight and length to the non-responsive crosslinking agent used (TEGDMA). This degradation facilitates the rapid release of encapsulated drugs, ensuring effective intracellular delivery. We also synthesized (see below methods for crosslinker synthesis) and evaluated crosslinkers that degrade by carboxylesterase-triggered hydrolysis (PCL-DMA, PLA-PEG-DMA m = 2 and m = 5). Carboxylesterases are abundant in the intracellular environment of ovarian cancer cells, further enhancing the degradation of nanogels and promoting controlled drug release.
 | ||
| Fig. 1 Structures of degradable crosslinking agents investigated. (A) A disulfide crosslinking agent that can be reduced in the presence of intracellular glutathione, and (B–D) three ester-liable crosslinking agents that can be degraded by the presence of intracellular carboxylesterase. | ||
The reaction was typically carried out at a pH range of 9.5–10.0. The reagents were ultrasonicated for 20 minutes to create an oil-in-water emulsion, which was subsequently purged with nitrogen to remove any free radical scavengers. Finally, the emulsion was exposed to UV light (140 mW cm−2) for 2.5 hours with constant stirring, using a BlueWave 200 Spot Lamp System (Dymax Corporation, Torrington, CT).
The particles were purified by four repeated cycles of collapsing and resuspending the ionomers to remove any remaining surfactants and unreacted monomers. They were titrated to a pH of 1.0 using 6 N HCl and stirred for at least 5 minutes. Following this, the particles were diluted to 10% v/v using either acetone or tetrahydrofuran (THF). For purification using acetone, the suspension was centrifuged at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for about 5 minutes until a pellet was formed. When using THF, the suspension was centrifuged at 3000g for approximately 10 minutes to achieve pellet formation. After removing the supernatant, the nanoparticle pellets were resuspended in a 0.5 N HCl solution. Subsequently, the particles were dialyzed against distilled water for 7–10 days, with the water being replaced twice daily.
000g for about 5 minutes until a pellet was formed. When using THF, the suspension was centrifuged at 3000g for approximately 10 minutes to achieve pellet formation. After removing the supernatant, the nanoparticle pellets were resuspended in a 0.5 N HCl solution. Subsequently, the particles were dialyzed against distilled water for 7–10 days, with the water being replaced twice daily.
Synthesis of custom degradable crosslinking agents
To develop nanoparticles with hydrolytic degradability, ester groups susceptible to breakdown were added along a PEGDMA crosslinker chain. Methacrylate-poly(lactic acid)-b-poly(ethylene glycol)-b-poly(lactic acid)-methacrylate (PLA-b-PEG-DMA) crosslinkers were synthesized following a two-step method initially outlined by Hubbell et al.27 and further modified based on Diederich et al.28Ring opening polymerization of D,L-lactide on PEG
The poly(ethylene glycol)-co-poly(lactic acid) copolymers, with varying quantities of lactic acid units, were synthesized through ring-opening polymerization of D,L-lactide onto PEG chains. Diederich et al. demonstrated that approximately 70% of the lactic acid units were successfully integrated into the PEG backbone.28 For the incorporation of 4 and 10 lactic acid units per PEG chain, 5.6 and 11.2 units of lactic acid were used, respectively. To maintain a consistent crosslinker length, PEG with a lower molecular weight was chosen for chains with more lactic acid units. To achieve conjugation of 4 lactic acid units per PEG chain (where m = 2), 0.0125 mol of PEG (MW = 400, 5 g) and 0.035 mol of D,L-lactide (5.045 g) were introduced into a 50 mL round-bottom flask. The mixture was then purged with nitrogen for 20 minutes to remove oxygen or other oxidizing agents. The flask was placed in an oil bath and heated to 140 °C. Afterward, 0.681 mL of a nitrogen-purged SnOct2 solution in toluene (10 wt%) was added to the flask to achieve a molar ratio of 0.0012 mol SnOct2 per mol D,L-lactide. The reaction was allowed to proceed for 12 hours before being cooled to 30 °C, and the product was dissolved in 7 mL of dichloromethane. To purify the product, it was reprecipitated in 400 mL of cold diethyl ether (−80 °C) and allowed to sit for 6 hours. The product was redissolved in dichloromethane (7 mL), followed by a second precipitation. The intermediate was then dried under vacuum at 40 °C and 28 mmHg overnight before being stored at 4 °C. The purified product was analyzed by proton (1H) and carbon (13C) NMR in CDCl3, along with ATR-FTIR spectroscopy for further characterization.Dimethacrylation of PLA-b-PEG-b-PLA intermediate
After synthesizing and characterizing the PLA-b-PEG-b-PLA intermediate, the dimethacrylation process was initiated.27 The dried intermediate was mixed with a 10-fold molar excess of methacrylic anhydride in a 20 mL scintillation vial. The vial, with the cap loosely attached, was placed in a microwave set to full power for 5 minutes. Every 30 seconds, the vial was taken out, the cap tightened, and the contents were vortexed for 30 seconds before the cap was loosened again, and the vial was returned to the microwave. This cycle was repeated for the full duration. After microwaving, the vial was left to cool down to room temperature with the cap loosened. The methacrylated compound was then dissolved in 3 mL of dichloromethane, followed by precipitation in 400 mL of hexanes at room temperature for 6 hours. The precipitated product was collected using a vacuum filtration system with a Buchner funnel and then dissolved again in dichloromethane. After a second precipitation step, the product was vacuum-dried overnight and stored at 4 °C. To verify the methacrylation, proton (1H) and carbon (13C) NMR in CDCl3, as well as ATR-FTIR spectroscopy, were conducted on the resulting crosslinkers.Fluorescent polymer synthesis
Nanoparticles were fluorescently labeled by reacting their primary amine groups with 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD chloride), which forms fluorescent NBD-amine conjugates. Ethanol served as the reaction solvent. The fluorescent tag was covalently attached to the nanoparticles by including amino ethyl methacrylate (AEMA) during their synthesis. The presence of the primary amine group was confirmed using both 1H-NMR and a fluorescamine assay after the synthesis and purification processes. NBD chloride was added to the nanoparticle mixture at a 1:1 molar ratio to AEMA, and the reaction was carried out under gentle stirring in the dark for 6 hours. After the reaction was complete, excess unreacted dye was removed from the labeled nanoparticles through extensive dialysis in distilled water. The labeled nanogels were stored in a dark environment until further use.
Encapsulation and release profiles
Loading studies were completed with paclitaxel and carboplatin across a variety of conditions to optimize the mass loading and demonstrate the ability to achieve a ratiometric loading in a repeatable fashion. Briefly, nanoparticles were loaded by imbition at the specified conditions to allow the drugs to diffuse into the swollen polymer network, then collapsed by titration to pH 8.0. Non-loaded drug was separated by centrifugal filtration (twice washed with 1× DPBS pH 8.0) and quantified by UV absorbance and/or HPLC. Analysis was calculated against standard curves from known concentrations. The final products were stored in at 4 °C for up to 1 week until use. To obtain the release behavior and profile, the drug-loaded nanoparticles were incubated in 1× DPBS pH 7.4 (sink conditions) for 4 hours. The solution was stirred continuously throughout the release evaluation and samples were in closed containers to minimize evaporation. Samples were taken every 1 hour, and released drug was separated by centrifugal filtration (twice washed with 1× DPBS pH 8.0) and quantified by UV absorbance and/or HPLC. After the 4 hours time point, the pH was adjusted to 6.5 and diluted to low ionic strength through rapid addition of hydrochloric acid in water, and addition of the specified concentrations of enzyme for specified formulations only.Cell culture
OVCAR-3 human ovarian cancer cells (derived from 60yo patient of European ancestry) were grown in RPMI-1640 medium, which was supplemented with insulin from bovine pancreas (0.01 mg mL−1) and 20% fetal bovine serum (FBS). These cells, typically within passages 6 through 20, were washed using pre-warmed Dulbecco's phosphate-buffered saline (DPBS) lacking divalent cations to prepare them for passaging. After washing, the cells were treated with 0.25% trypsin–EDTA at 37 °C to detach them from the culture surface. The trypsinization process was stopped by adding fresh, pre-warmed medium, and the cells were pelleted by centrifugation. The pellet was resuspended in complete medium, and cell numbers were determined using a TC20™ Automated Cell Counter (Bio-Rad, Catalog 1450102) with trypan blue staining to assess viability. The suspension was diluted as needed and transferred to tissue-culture treated plates or flasks. OVCAR-3 cells were regularly split at a 1:3 ratio, with media changes every 2–3 days, and passaging was performed approximately every 7 days. RAW 264.7 murine macrophages (derived from adult male BALB/c) were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% FBS. These cells were typically used between passages 9 and 16. For passaging, the cells were first washed with pre-warmed DPBS, then the medium was replaced with fresh, complete medium. Cells were detached from the flask surface using a 25 cm cell scraper (BD Falcon, Franklin Lakes, NJ), and the detached cells were then counted. After counting, they were diluted and transferred to tissue-culture treated flasks or plates. RAW 264.7 cells were usually passaged every 4 days, with fresh media added every 2 days.
In vitro cytocompatibility and efficacy screening
A live cell assay to investigate nanogel in vitro cytocompatibility using two commercially available cytotoxicity assays (MTS and LDH) and in vitro efficacy using the MTS assay. Polymer solutions were prepared in 1× DPBS without divalent cations, titrated to pH 7.4. OVCAR-3 cells were cells were seeded in 96-well plates at a density of 30000 cells per well and incubated for 48 hours with 200 μL of complete medium. Media was aspirated and cells were washed twice with 1× DPBS. Polymer stock solutions at 10 times the final concentration were introduced to the cells for designated exposure periods. After 2 or 24 hours exposure time, the media and polymer were aspirated and fresh complete medium was added. For MTS assays, the CellTiter 96 AQueous one solution cell proliferation assay kit (Promega Corp., Madison, WI) was used, which contains the tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium]. This compound is reduced to a purple formazan product in the presence of viable cells absorbance was measured at 490 nm after a 4-hour incubation period. LDH assays were performed using the CytoTox-ONE™ homogeneous membrane integrity assay kit (Promega Corp., Madison, WI), which measures the release of lactate dehydrogenase (LDH) from cells with compromised membranes. For the LDH assay, cells were seeded in 96-well plates and exposed to polymer solutions under the same conditions as described. At specific time points, 50 μL of the medium was aspirated and mixed with 50 μL of LDH assay buffer in a black-walled 96-well plate. After a 10-minute room-temperature incubation, fluorescence was measured using 530 nm excitation and 590 nm emission wavelengths. To determine the concentration of drug required for 50% growth inhibition (IC50) Graphpad Prism 5 software was used. The fraction of cells affected (Fa) by each drug concentration was calculated, and for combination treatments, the combination index (CI) was calculated using CompuSyn software based on the Chou and Talalay method.29 The CI for binary drug combinations was computed using the following formula:| CI = D1/(Dx)1 + D2/(Dx)2 | (1) |
1H-nuclear magnetic resonance (NMR) spectroscopy analysis
The nanogel composition was assessed using 1H-nuclear magnetic resonance (NMR) spectroscopy. Spectral data were obtained at 25 °C using a 400 MHz NMR system, either the Varian Direct Drive 400 or the Agilent MR 400. For compositional analysis, dried linear polymers were dissolved in deuterium oxide at concentrations ranging from 10 to 15 mg mL−1. The resulting spectra were processed using MestReNova 10.0 software, with integration of specific peaks for DEAEMA (δ 1.22, 6H), PEGMA2k (δ 3.55 ppm, 176H), and hydrophobic monomer signals.Attenuated total reflectance fourier transform infrared (ATR-FTIR) spectroscopy analysis
Attenuated total reflectance fourier transform infrared (ATR-FTIR) spectroscopy was utilized to examine lyophilized crosslinked polymer nanoparticles. The analyses were carried out using a Thermo Scientific Nicolet iS10, equipped with a germanium crystal. Prior to analyzing each sample, background spectra were collected for subtraction purposes. The final spectra, representing each sample, were averaged over 64 scans, with a data spacing of 0.482 cm−1.Dynamic light scattering (DLS) and zeta potential
The hydrodynamic diameter of the nanoparticles was measured using dynamic light scattering (Zetasizer Nano, Malvern) with a particle concentration of 0.5 mg mL−1 in 1× DPBS. For each formulation (n = 3), the readings were averaged over at least 12 acquisitions, each lasting 10 seconds. Zeta potential measurements were taken to assess the surface charge, also using the Zetasizer Nano (Malvern). For this, a 0.5 mg mL−1 concentration of nanoparticles was prepared in 5 mM sodium phosphate buffer (n = 3 per formulation). Both the hydrodynamic diameter and the zeta potential were evaluated as a function of pH.Statistical analysis
The data are presented as an arithmetic mean ± standard uncertainty of the mean (n = 3), unless otherwise noted in a figure caption. Differences between groups were examined for statistical significance using two-tailed paired t-test or two-way ANOVA with Tukey test for multiple comparisons for a significance level α = 0.05 using GraphPad Prism.Results & discussion
Nanogel synthesis
The nanogel formulations were comprised of: (i) a hydrophilic, cationic monomer 2-(diethylamino)ethyl methacrylate (DEAEMA), which provides pH responsiveness through the ionization of amine pendant groups, (ii) a hydrophobic n-alkyl monomer cyclohexyl methacrylate that enables a tunable pH-responsive profile and improves drug–polymer interactions, (iii) crosslinker to improve drug retention and mechanical stability over self-assembled counterparts (see Fig. 1), and (iv) a poly(ethylene glycol)methacrylate (PEGMA) graft to enhance stability in solution and in serum.The poly(2-(diethylamino)ethyl methacrylate-co-cyclohexyl methacrylate)-g-poly(ethylene glycol methyl methacrylate) (P(DEAEMA-co-CHMA)-g-PEGMA) nanoparticles were synthesized using a previously developed a novel, robust method via aqueous, UV-initiated emulsion free radical polymerization.26,32–34 This polymerization method allows the hydrophobic cationic monomer to form the nanoparticle core, with PEGMA grafted predominantly on the surface, ensuring that the buffering properties of the polymer are not compromised.
Preparation of drug-loaded nanogels
Loading studies were completed with paclitaxel and carboplatin across a variety of conditions to understand the key process parameters, maximize the achievable drug loading and demonstrate the ability to achieve a ratio metric loading in a repeatable fashion. Studies were performed by investigating several factors, including: total ionic strength of the loading solution, incubation time, incubation temperature, incubation pH, the ratio of drug to polymer, and the ratio of organic solvent (Table 1). For the organic solvent, ethanol was used as carboplatin more readily degrades in DMSO and paclitaxel more readily degrades in methanol.| Inputs | Input range tested | Outputs |
|---|---|---|
| Incubation time | 24, 48, 72 hours | Mass loading (μg API to mg polymer) |
| Incubation temperature | 22 °C, 30 °C, 37 °C | Loading efficiency (%) |
| Incubation pH | pH 3.5, 4.5, 6.0 | |
| Total ionic strength | 15 mM, 150 mM, 300 mM | |
| Ratio of total API: polymer | 10:100, 50:100, 100:100 |
|
| Organic solvent ratio | 2.5 v/v%, 13 v/v%, 25 v/v% |
To explore each of these variables one by one, with three levels (low, medium, and high) – also known as a full factorial design – it would require more than 700 experiments. To analyze this effect for both single agent loading and competitive loading, it would require more than 2100 experiments. Instead, this study was approached using a statistical design of experiments (DoE). With a central composite design (CCD), it is possible to create a map of the response surface and explore any interactions between variables, while also minimizing the number of experiments. With this design, only 63 experiments were required, which is a much more manageable number and can be easily accomplished. The method was further streamlined through the use of plate-based filtration.
To aid in the DoE design and analysis, both the JMP and SigmaPlot software were used. Briefly, the JMP software was used to design the experimental arms and compute a power analysis. The data from the experiment was analyzed it in JMP, which provides an effect summary (not shown). This effect summary is a good visual cue to see which variables are impacting the output the most, but it is not as useful for an in-depth analysis. From here, a predictive model was established to fit the experimental data of mass API loaded per mass of nanoparticle. The predicted versus actual values of the mass loading of API per nanoparticle resulted in an R-square value of 0.98 (p < 0.0001) (Fig. 2A). This fit then enabled the generation of a prediction profiler (Fig. 2B) and 3D response surface maps (not shown).
 | ||
| Fig. 2 Summary of conditions to maximize the loading of paclitaxel and carboplatin into P(DEAEMA-co-CHMA)-g-PEGMA nanogels. Data was analyzed in JMP software. (A) Actual versus predicated values for mass of API loaded per mass of nanoparticle with an R-square value of 0.98 (p < 0.0001). (B) Resulting predication profiler showing maximal loading conditions. | ||
The data in Fig. 2 is from the competitive agent co-loading studies, illustrating the conditions that yield the maximum output. Overall, with both therapeutic agents, the most prominent changes were seen with the factors that increase the particle swelling and mesh size, and with the factors that create a less favorable environment in the bulk solution or promote a larger equilibrium concentration. By using this approach, we were able to improve the achievable loading by over 2-fold over what was observed initially.
Synthesis of nanoparticles with glutathione degradable crosslinkers (disulfide-DMA)
Polymers that respond to internal biological signals, such as degradation triggered by the environment of specific body regions or disease conditions (like the intracellular endosome), are highly valuable for the development of advanced drug delivery systems.34 The most effective approach is to design biomaterials that are sensitive to changes in the physiological or pathological environment, aligning with native biological characteristics.Oxidation–reduction responsive polymers react to environmental changes by altering the oxidation state of redox-sensitive groups.35 These materials often share design strategies with pH-responsive polymers, which undergo acid-sensitive cleavage or degradation.36 On a molecular level, these polymers are often engineered with functional groups that have multiple oxidation states (such as iron, selenium, and sulfur), or with linkages like disulfide, diselenide, and ditellurium.20,35,37,38
One effective strategy for degradation is the use of disulfide linkers, which are cleaved by intracellular glutathione (GSH) at concentrations ranging from 1 to 11 mM.39 By incorporating disulfide-based crosslinkers or conjugates, degradation points can be introduced into the polymer structure while maintaining the material's mechanical strength and desired macroscopic properties. To this extent, the crosslinking agent bis(2-methacryloyl)oxyethyl disulfide (disulfide-DMA, Fig. 1) was investigated for use in the P(DEAEMA-co-CHMA)-g-PEGMA formulation.
Nanoparticles were successfully synthesized with the biodegradable disulfide-crosslinking agent without necessary changes to the UV-initiated emulsion polymerization method. The synthesis was conducted in identical fashion to the non-degradable nanoparticles. Further, replacing the non-degradable linker TEGDMA with disulfide-DMA had little to no effect on the resulting nanoparticle physicochemical properties (Fig. 3). There was a minor effect to the pH-dependent swelling profile. There was no identifiable change to the effective surface ζ-potential. Composition was confirmed via1H-NMR and ATR-FTIR analysis (ESI Fig. S.1†), and demonstrated no significant changes until exposure to 10 mM glutathione.
 | ||
| Fig. 3 Nanoparticle formulations synthesized with variations in crosslinking agent, disulfide-DMA and TEGDMA. (A) Hydrodynamic diameter and polydispersity index in 1× phosphate buffered saline determined by DLS, and (B) nanogel effective surface ζ-potential as a function of pH as determined by zeta potential. Data represent mean ± SEM (n = 3). | ||
The degradation kinetics were analyzed by dynamic light scattering, and is demonstrated by the change in relative count rate, nanoparticle diameter, and polydispersity index over time when maintained at conditions mimicking that of the intracellular environment (pH 6.5, 37 °C, 10 mM glutathione, ESI Fig. S.2†). Nanoparticles fabricated with the disulfide-DMA crosslinkers exhibit degradation behavior as compared to those made with the non-responsive TEGDMA crosslinking agent. It is important to note that degradation was not observed that the levels of extracellular glutathione (1 mM).
During the first 2 hours, a notable decrease in the count rate is observed, with the rate dropping by 50% within 90 minutes for particles made using the disulfide-DMA crosslinking agent. Moreover, the particle formulations exhibited a continued decline in count rate over time, eventually stabilizing at a plateau after approximately 20 to 21 hours. Similarly, the nanoparticle diameter and polydispersity index increased dramatically over similar time periods.
The cytocompatibility of the intact particles was investigated with an ovarian cancer cell line (OVCAR-3). The data in ESI Fig. S.3† shows that there was minimal change to the cytocompatibility when compared to the non-degradable particles over the concentrations tested. As with the prior section, cumulative drug release was analyzed and compared with the non-degradable particles.
While the current study provides cytocompatibility data for OVCAR-3 ovarian cancer cells, we acknowledge the importance of evaluating the toxicity of the polybasic nanogels in non-cancerous cells, such as ovarian epithelial cells or fibroblasts, to ensure their safety and selectivity. We have previously tested related polybasic nanogels for toxicity in Caco-2 cells40,41 and L929 mouse fibroblasts42 and seen favorable cytocompatibility. Future work should include a comprehensive toxicity profile using a broader range of non-cancerous cell types to validate the safety of these nanogels.
Synthesis of custom crosslinking agents
Two custom poly(lactic acid)-b-poly(ethylene glycol)-b-poly(lactic acid) dimethacrylate crosslinking agents were synthesized. After each synthesis step, the intermediate and final products were characterized using 1H-NMR and ATR-FTIR (Fig. S.4 and S.5†). This process was conducted to ensure that the PLA units were successfully incorporated and that the chain end groups were methacrylated.In the control PEG and PEGDMA crosslinker samples, a prominent peak at 3.6 ppm was observed, which is characteristic of PEG units.28 The spectrum also displayed three distinct peaks at 1.8, 5.7, and 6.2 ppm, corresponding to the methacrylate ends. Additionally, a peak at 4.25 ppm, indicating a carbon bond to the PEG repeating units, was detected. Notably, this peak was absent in the non-methacrylated PEG spectrum, suggesting its presence is due to the connection between the repeating units and the methacrylate groups. When analyzing the m = 2 and m = 5 custom degradable crosslinkers, these same five peaks were observed, along with additional peaks at 1.5 ppm and 5.2 ppm, which are indicative of lactic acid units.
From the NMR data, it was noted that as the number of degradable units on the crosslinker increased, the intensity of the PEG peak decreased. This change was attributed to the use of PEG with molecular weights of 400 for m = 2 and 200 for m = 5, ensuring that the overall crosslinker length remained constant. Additionally, the areas under peaks c and g grew larger as the crosslinker transitioned from m = 2 to m = 5, due to an increased number of lactic acid units.
Finally, the peaks corresponding to the methacrylate end groups of the polymer chains were similar for all the custom crosslinkers when compared to the PEGDMA control sample. This similarity occurs because each crosslinker, regardless of the number of lactic acid units or the length of the PEG chain, contains two methacrylate groups, one at each end. Quantitative analysis of the NMR spectra confirmed that each chain contains two methacrylate groups, with an average of 4.2 lactic acid units per chain for m = 2 and 10.5 lactic acid units per chain for m = 5.
Nanoparticle degradation and release by carboxylesterases
Carboxylesterase-triggered hydrolysis of the nanoparticle crosslinking agent was explored as a function of three ester containing crosslinkers (PCL-DMA, PLA-PEG-DMA m = 2 and m = 5) (Fig. 1).Carboxylesterases (CES) are major esterases that metabolize a wide variety of compounds (including esters, thioesters, carbamates, and amides).43–54 They are important in phase I metabolism, significantly contribute to first-pass metabolic hydrolysis, and are widely known to degrade prodrugs for activation. Further, they mediate the detoxification of many xenobiotic compounds.
Mammalian carboxylesterases are primarily intracellular proteins that are predominantly found in the cytoplasm and within the microsomal fraction associated with the endoplasmic reticulum. In humans, there are three membrane-bound CES isozymes—CES1, CES2, and CES3—that demonstrate differences in their tissue distribution and substrate preferences. CES1 is expressed abundantly in organs such as the liver, kidneys, lungs, and brain, as well as in macrophages, but its expression is significantly lower in the gastrointestinal tract. CES2 is mainly located in the intestines, kidneys, and liver, while CES3 is found in the trachea, intestines, and placenta.
CES2, in particular, is overexpressed in various cancers and cancer cell lines, such as ovarian cancer, multiple myeloma, thyroid papillary carcinoma, esophageal squamous carcinoma, and kidney adenocarcinoma.48,54 CES2 expression in tumor tissues and cancer cell lines is strongly linked to the bioactivation of several cancer prodrugs, including irinotecan and gemcitabine. These findings indicate that developing CES2-bioactivated nanoparticles could offer a promising approach for targeted cancer treatment.
To this extent, carboxylesterase-triggered hydrolysis of the nanoparticle crosslinking agent was explored as a function of three ester containing crosslinkers (PCL-DMA, PLA-PEG-DMA m = 2 and m = 5). As with the disulfide-DMA crosslinker, nanoparticles were successfully synthesized with all three of the biodegradable crosslinking agents without necessary changes to the UV-initiated emulsion polymerization method. The synthesis was conducted in identical fashion to the non-degradable nanoparticles. Composition was confirmed via1H-NMR and ATR-FTIR analysis (Fig. S.6†), and demonstrated no significant changes until exposure to relevant levels of CES2 (10 U mL−1).
Again, the degradation kinetics were analyzed by dynamic light scattering, and was demonstrated by the change in relative count rate, nanoparticle diameter, and polydispersity index over time when maintained at conditions mimicking that of the intracellular environment (pH 6.5, 37 °C, 10 U mL−1 CES2, Fig. S.7†). Again, nanoparticles fabricated with the three ester-containing crosslinkers exhibited degradation behavior as compared to those made with the non-responsive TEGDMA crosslinking agent.
Further, it is again important to note that degradation was not observed that the levels of extracellular CES2 (estimated maximal concentration of 2.5 U mL−1, Fig. S.8†). Fig. 4 demonstrated the degradation of one nanoparticle (PLA-PEG-DMA m = 5) with varying concentrations of CES2. Conversely, exposure to CES1 showed no significantly degradation.
 | ||
| Fig. 4 Comparison of the degradation time-scale for all nanoparticles at intracellular relevant conditions on a single graph, (A) showing the full time scale and (B) showing up to 120 minutes. Nanoparticles were synthesized with the non-responsive crosslinker (TEGDMA), the glutathione responsive crosslinker (disulfide-DMA), and the three crosslinkers capable of ester-mediated hydrolysis (PCL-DMA, PLA-PEG-DMA m = 2, PLA-PEG-DMA m = 5). Degradation occurred in the presence of either glutathione or human carboxylesterase 2 (CES2) at a relevant intracellular conditions. Dynamic light scattering was measured sequentially (approximately every 3 minutes) for nanogels at 0.5 mg mL−1 in 5 mM sodium phosphate pH 6.5, and the count rate normalized to initial. | ||
Compared to the disulfide-DMA crosslinking agent, all three ester-containing nanoparticles degraded at significantly faster time scales. This is ideal for ensuring maximal release of the loaded cargo upon cell uptake and intracellular trafficking. In the first 10 minutes, a significant reduction in the count rate is observed, with the count rate reduced by half within 11, 7, and 4.5 minutes for particles fabricated with the PCL-DMA, m = 2, and m = 5 crosslinking agents, respectively (Fig. 4). In addition, the particle formulations continued to see a decrease in count rate over time and reached a plateau after around 115, 32, and 25 minutes, respectively. Similarly, the nanoparticle diameters and polydispersity indexes increased dramatically over similar time periods.
The cytocompatibility of the intact particles was investigated with an ovarian cancer cell line (OVCAR-3). As with the prior section, cumulative drug release was analyzed and compared the non-degradable particles.
Release studies
Cumulative release of paclitaxel and carboplatin was demonstrated from dual-loaded nanogels made with the various crosslinkers. To obtain the release behavior and profile, the drug-loaded nanoparticles were incubated in 1× DPBS pH 7.4 (sink conditions) for 4 hours. Samples were taken periodically, and released drug was separated by centrifugal filtration (twice washed with 1× DPBS pH 8.0). After the 4-hour mark, the pH was brought down to 6.5, and the solution was diluted to a low ionic strength—mimicking the intracellular environment of the early endosome—by quickly adding hydrochloric acid diluted in water.As shown in Fig. 5, the release of carboplatin and paclitaxel generated distinctly different profiles as a function of time. For both therapeutic agents, there was minimal release (less than 5%) at the pH of the bloodstream (pH 7.4). When the pH was shifted to that of the early endosome after 4 hours, the particle swelled in response to the acidic condition and resulted in the controlled diffusion out of both therapeutic agents.
 | ||
| Fig. 5 Cumulative release of paclitaxel and carboplatin from dual-loaded nanogel. To obtain the release behavior and profile, the drug-loaded nanoparticles were incubated in 1× DPBS pH 7.4 (sink conditions) for 4 hours. Samples were taken periodically, and released drug was separated by centrifugal filtration (twice washed with 1× DPBS pH 8.0). After the 4 hours time point, the pH was adjusted to 6.5 and low ionic strength (to mimic the conditions of the intracellular early endosome) through rapid addition of hydrochloric acid in water. Data represent mean ± SEM (n = 3). (A) TEGDMA NPs; (B) disulfide-DMA NPs, at 4 hours time point, the pH was adjusted to 6.5 and low ionic strength through rapid addition of hydrochloric acid and 10 mM glutathione (GSH) in water. (C) PCL-DMA; (D) PLA-PEG-DMA; and (E) PLA-PEG-DMA, for (C–E) after the 4 hours time point, the pH was adjusted to 6.5 through rapid addition of hydrochloric acid and 10 U mL−1 CES2. | ||
Carboplatin released at a faster rate than was observed for paclitaxel. This makes sense as carboplatin is much more hydrophilic than paclitaxel. The release of carboplatin proceeded at a relatively steady rate for 10 hours, and reached a plateau around 65% in cumulative release after 18 hours. Paclitaxel release was much slower, and did not demonstrate a plateau in the release after 18 hours. However, only approximately 39% of the paclitaxel loaded was released at 24 hours. Overall, the release of both components was slower than expected. Ideally, both agents would release in the desired ratiometric level across all time points. To achieve this, incorporation of intracellularly-targeted degradable crosslinking agents was investigated. This step helps ensure complete drug release within the cell to maximize its therapeutic effect while also minimizing long-term nanogel toxicity and promoting its clearance from the body after the cargo is delivered.
As shown in Fig. 5B, the release of carboplatin and paclitaxel again generated distinctly different profiles as a function of time. Still for both therapeutic agents, there was minimal release (less than 5%) at the pH of the bloodstream (pH 7.4). When the pH and ionic strength were shifted to that of the early endosome after 4 hours, the particle swelled in response to the acidic condition and resulted in the controlled diffusion out of both therapeutic agents. Conversely to the non-degradable nanoparticle, paclitaxel released at a much faster rate than carboplatin. This observation can be explained by the reactivity of the free drug. Carboplatin is stable at high API and salt concentrations due to dimer formation. However, it becomes much more reactive at dilute conditions and low ionic strength. Hydrolysis to activate carboplatin occurs in low salt solutions (inside cell), where water replaces chloride leaving groups.
This hydrolysis is what enables carboplatin to be cytotoxic and form DNA-adducts. However, once hydrolyzed, both carboplatin and cisplatin will react with nucleophilic molecules, such as free thiols and thio-esters, to form adducts. Additionally, it has been noted that cells develop resistance to both carboplatin and cisplatin through intracellular mechanisms, including enhanced drug detoxification by thiol groups present in enzymes like glutathione.55,56 Due to the weaker interaction between platin-thiol adducts and DNA, the formation of these complexes reduces the amount of drug available for DNA binding. By binding to thiol groups, the cell is able to repair itself and increase its tolerance to nuclear damage, which in turn leads to a reduction in apoptosis and lower intracellular accumulation of carboplatin. To address this limitation, degradable crosslinking agents activated by carboxylesterase were explored.
As shown in Fig. 5C–E, the release of carboplatin and paclitaxel again generated relatively similar profiles as a function of time, as compared to the TEGDMA and disulfide-DMA nanoparticles. Additionally, for both therapeutic agents, there was minimal release (less than 5%) at the pH of the bloodstream (pH 7.4). When the pH and ionic strength were shifted to that of the early endosome after 4 hours, the particles swelled in response to the acidic condition and resulted in the controlled diffusion out of both therapeutic agents. Overall, the release of both components was faster than expected, and resulted in maximal release between 85 and 93% cumulative release. This is ideal for intracellular-targeted delivery.
In vitro efficacy of nanoparticle-mediated dual-delivery
Currently the combination of paclitaxel and carboplatin has become the first-line chemotherapy for ovarian cancer in addition to many other types of cancer.57–61 It has been shown that paclitaxel acts in the cell cytoplasm to stabilize microtubules, preventing replication and causing cell death by stopping mitosis in the M-Phase. Carboplatin acts in the nucleus, where it is activated to form reactive platinum complexes that cause intra- and inter-strand DNA crosslinking. This inhibits DNA synthesis in all phases of the cell cycle. Paclitaxel has been shown to significantly increase the amount of carboplatin-DNA adduct formation by preventing repair through natural cell mechanisms.59The in vitro efficacy of free drug was then compared to five nanoparticle formulations using OVCAR-3 cells as determined using the MTS assay. Analysis was carried out for paclitaxel alone (all formulations), carboplatin alone (all formulations), varying ratios of paclitaxel and carboplatin combination (free drugs only), and finally with a constant ratio of paclitaxel and carboplatin combined (all formulations).
Data compares the free drug; the original nanoparticle composition, the optimized P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with varying crosslinking agents of TEGDMA and PLA-PEG-DMA m = 8; and finally the optimized P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with TEGDMA crosslinking agent and either non-CathepsinB responsively linked PEG grafts (PEG-Glycine-Glycine-Glycine-Glycine (GGGG)) and responsive PEG grafts (PEG-Glycine-Phenylalanine-Leucine-Glycine (GFLG)).
As shown in Fig. S.8–S.11,† three nanoparticle formulations out-performed the free drug for both paclitaxel delivery alone and carboplatin delivery alone. These three formulations were (in order of lowest IC50 value): P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with CathepsinB responsively linked PEG grafts (PEG-GFLG), P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with PLA-PEG-DMA m = 8, and P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with TEGDMA. The original nanoparticle (without CHMA) performed worse than the free drug alone in both cases. The non-CathepsinB responsively linked PEG grafts (PEG-GGGG) nanoparticle also performed significantly worse than all formulations and free drugs.
Fig. 6 shows the combination index (CI) versus fraction of cells affected (Fa) for in vitro efficacy of free drug paclitaxel and carboplatin combination at 1:1 (black), 1:2.5 (orange), 1:5 (red), 5:1 (green), and 2.5:1 (blue) ratios of the respective EC50 concentrations using OVCAR-3 cells as determined using the MTS assay. CI analysis based on the Chou and Talalay method was done using CompuSyn software.29 Generally, between Fa = 0.1 and Fa = 0.9 is considered valid. CI values below 0.9 or above 1.1 suggest drug synergy and antagonism, respectively, values between 0.9 and 1.1 are generally regarded as additive, and values below 0.3 are considered strongly synergistic.30,31 The 1:2.5 and 1:5 ratios exhibited strong synergy across a wide Fa values. The 1:5 ratio was then used to test the efficacy of the nanoparticle formulations.
 | ||
| Fig. 6 Combination index (CI) versus fraction of cells affected (Fa) for in vitro efficacy of free drug paclitaxel and carboplatin combination at 1:1 (black), 1:2.5 (orange), 1:5 (red), 5:1 (green), and 2.5:1 (blue) ratios of the respective EC50 concentrations using OVCAR-3 cells as determined using the MTS assay. CI analysis based on the Chou and Talalay method was done using CompuSyn software. Generally, the CI values between Fa = 0.1 and Fa = 0.9 are considered valid. Additionally, CI values below 0.9 or above 1.1 suggest drug synergy and antagonism, respectively. Values between 0.9 and 1.1 are generally regarded as additive, and values below 0.3 are considered strongly synergistic. | ||
As shown in Fig. 7 and 8, three nanoparticle formulations again out-performed the free drug for paclitaxel and carboplatin dual-delivery. Again, these three formulations were (in order of lowest IC50 value): P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with CathepsinB responsively linked PEG grafts (PEG-GFLG), P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with PLA-PEG-DMA m = 8, and P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with TEGDMA. The original nanoparticle (without CHMA) performed worse than the free drug until 10 μM, and the non-CathepsinB responsively linked PEG grafts (PEG-GGGG) nanoparticle also performed significantly worse than all formulations and free drugs at concentrations >2 μM.
 | ||
| Fig. 7
In vitro efficacy of free drug compared to 5 nanoparticle formulations with paclitaxel and carboplatin loaded at the 5:1 ratio using OVCAR-3 cells as determined using the MTS assay. Data compares the free paclitaxel (black); the original nanoparticle composition (green); the optimized P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with varying crosslinking agents of TEGDMA (purple) and PLA-PEG-DMA m = 8 (orange); and finally the optimized P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with TEGDMA crosslinking agent and either non-CathepsinB responsively linked PEG grafts (PEG-GGGG, blue) and responsive PEG grafts (PEG-GFLG, red). Two-way ANOVA was used to test significance at each concentration of various nanoparticles compared to free drug. Significance represents ** < 0.025, **** < 0.0001. | ||
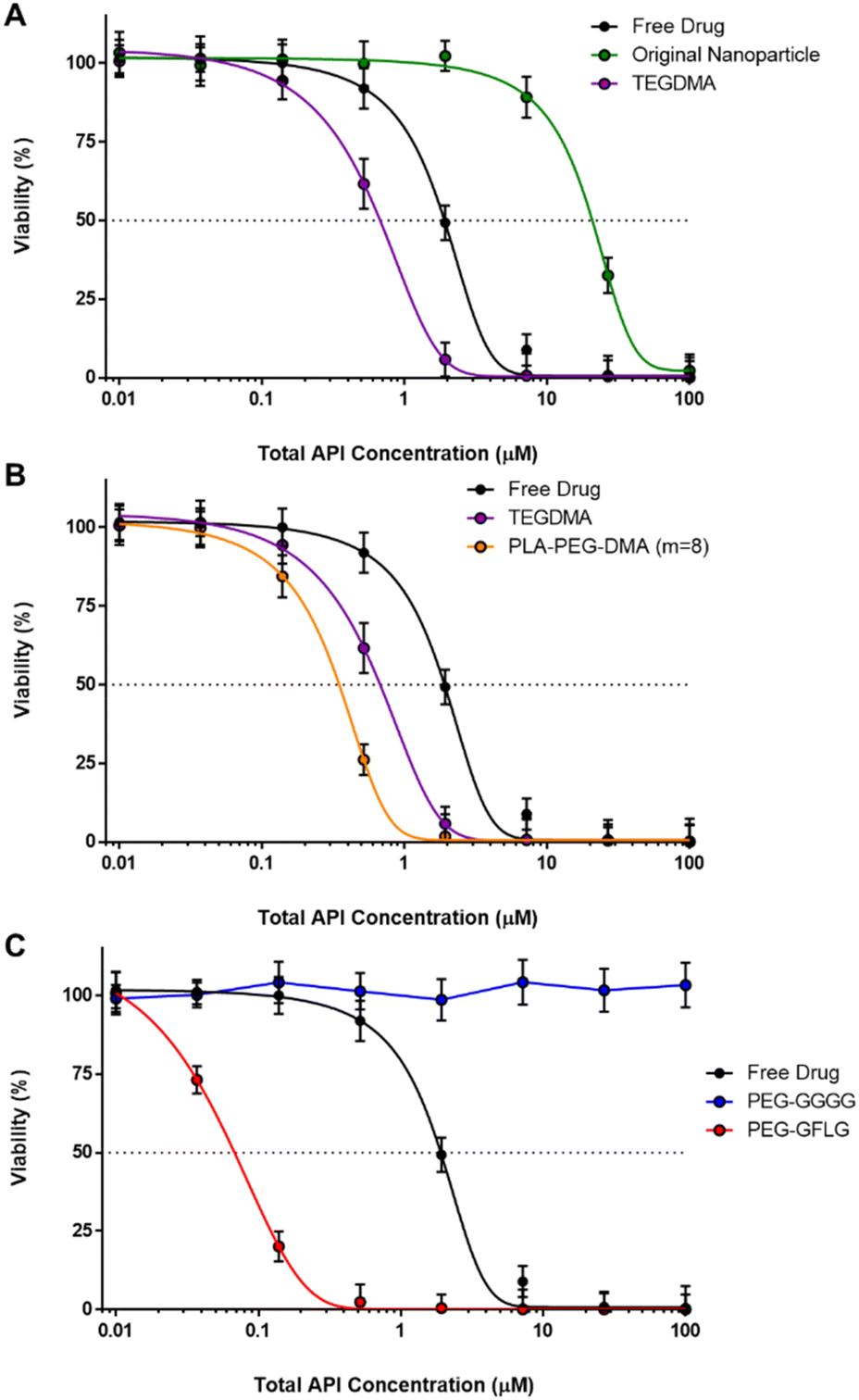 | ||
| Fig. 8
In vitro efficacy of free drug compared to 5 nanoparticle formulations paclitaxel and carboplatin loaded at the 5:1 ratio alone using OVCAR-3 cells as determined using the MTS assay. (A) Data compares the free paclitaxel (black) with the original nanoparticle composition (green) and the optimized P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with TEGDMA crosslinking agent. (B) Data compares the free paclitaxel (black) with the optimized P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with varying crosslinking agents of TEGDMA (purple) and PLA-PEG-DMA m = 8 (orange). (C) Data compares the free paclitaxel (black) with the optimized P(DEAEMA-co-CHMA)-g-PEGMA nanoparticles synthesized with TEGDMA crosslinking agent and either non-CathepsinB responsively linked PEG grafts (PEG-GGGG, blue) and responsive PEG grafts (PEG-GFLG, red). | ||
Finally, it is important to note that in vivo studies with nanoparticles containing amphipathic esters should be conducted with caution in rodent models. It has been demonstrated that their esterase-rich plasma will alter biodistribution.62 However, this limitation may be overcome to an extent through the generation or use of a carboxylesterase-deficient mouse model.
Conclusions
The polybasic nanoscale hydrogel (nanogel) system based upon poly(diethylaminoethyl methacrylate-cyclohexyl methacrylate)-g-poly(ethylene glycol) (P(DEAEMA-co-CHMA)-g-PEGMA) developed in this dissertation has been tailored to exploit multiple environmental cues for the controlled, targeted, intracellular delivery of multiple low molecular weight chemotherapeutic agents.The utility and application of this nanogel system was successfully demonstrated to co-deliver paclitaxel and carboplatin for the treatment of ovarian cancer. To the best of our knowledge, this study will represent the first time that paclitaxel and carboplatin are formulated and delivered together in a polybasic polymeric nanoparticle for the treatment of ovarian cancer. Further, multiple degradable crosslinking strategies were carefully investigated and developed to both improve long term biocompatibility and improve drug release through intracellular-triggered degradation.
Ultimately, in vitro studies revealed that the co-delivery of paclitaxel and carboplatin via P(DEAEMA-co-CHMA)-g-PEGMA nanogels could induce a potent anticancer effect and out-perform the free drug (single or in combination) and single drug-loaded nanogels. These results further demonstrate the potential of the engineered polybasic nanogels as a novel drug delivery system for the co-localized delivery of drug combinations in cancer therapy. Future studies should expand on these findings by performing comprehensive safety evaluations in non-cancerous cells, conducting detailed imaging studies, evaluating long-term stability under various storage conditions, and exploring in vivo models to further validate the clinical potential of this system.
Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported in part by a grant from the National Institutes of Health (EB012726, EB022025) and the Cockrell Family Regents Chair in Engineering (UT Austin). A. M. W. was supported by the National Science Foundation Graduate Research Fellowship (DGE-1610403) and the Philanthropic Educational Organization Scholar Award. A. M. W. was also supported by the S.E.S.H.A. Endowed Graduate Fellowship in Engineering (UT Austin). OLL's work was supported by the Provost Early Career Fellowship at the University of Texas at Austin and the University of New Mexico's Chemical and Biological Engineering Department.References
- Ovarian Cancer Studies Aim to Reduce Racial Disparities. National Cancer Institute [Internet]. Available from: https://www.cancer.gov/news-events/cancer-currents-blog/2020/ovarian-cancer-racial-disparities-studies.
- Ovarian Cancer Research Alliance [Internet]. [cited 2024 Sep 5]. Ovarian Cancer Statistics. Available from: https://ocrahope.org/for-patients/gynecologic-cancers/ovarian-cancer/ovarian-cancer-statistics/.
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel and I. Soerjomataram, et al., Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2024, 74(3), 229–263 CrossRef.
- Final Recommendation Statement: Ovarian Cancer: Screening | United States Preventive Services Taskforce [Internet]. [cited 2024 Sep 5]. Available from: https://www.uspreventiveservicestaskforce.org/uspstf/document/RecommendationStatementFinal/ovarian-cancer-screening#citation1.
- A. Hasenburg, D. Eichkorn, F. Vosshagen, E. Obermayr, A. Geroldinger and R. Zeillinger, et al., Biomarker-based early detection of epithelial ovarian cancer based on a five-protein signature in patient's plasma – a prospective trial, BMC Cancer, 2021, 21(1), 1037 CrossRef CAS.
- S. Lheureux, C. Gourley, I. Vergote and A. M. Oza, Epithelial ovarian cancer, Lancet, 2019, 393(10177), 1240–1253 CrossRef.
- Ovarian Cancer Treatment | How to Treat Ovarian Cancer | American Cancer Society [Internet]. [cited 2024 Sep 5]. Available from: https://www.cancer.org/cancer/types/ovarian-cancer/treating.html.
- Z. Wang, F. Meng and Z. Zhong, Emerging targeted drug delivery strategies toward ovarian cancer, Adv. Drug Delivery Rev., 2021, 178, 113969 CrossRef CAS.
- N. Vasan, J. Baselga and D. M. Hyman, A view on drug resistance in cancer, Nature, 2019, 575(7782), 299–309 CrossRef CAS.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Engineering precision nanoparticles for drug delivery, Nat. Rev. Drug Discovery, 2021, 20(2), 101–124 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Nanoparticles in the clinic: An update, Bioeng. Transl. Med., 2019, 4(3), e10143 CrossRef.
- S. F, M. S. Umashankar and D. Narayanasamy, A Comprehensive Review of Nanogel-Based Drug Delivery Systems, Cureus, 2024, 16(9), e68633, DOI:10.7759/cureus.68633.
- Y. (Chezy) Barenholz, Doxil®—The first FDA-approved nano-drug: Lessons learned, J. Controlled Release, 2012, 160(2), 117–134 CrossRef PubMed.
- M. J. Nirmala, U. Kizhuveetil, A. Johnson, G. Balaji, R. Nagarajan and V. Muthuvijayan, Cancer nanomedicine: a review of nano-therapeutics and challenges ahead, RSC Adv., 2023, 13(13), 8606–8629 RSC.
- C. M. Dawidczyk, C. Kim, J. H. Park, L. M. Russell, K. H. Lee and M. G. Pomper, et al., State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines, J. Controlled Release, 2014, 187, 133–144 CrossRef CAS PubMed.
- O. S. Fenton, K. N. Olafson, P. S. Pillai, M. J. Mitchell and R. Langer, Advances in Biomaterials for Drug Delivery, Adv. Mater., 2018, 30(29), 1705328 CrossRef.
- R. Deshmukh, P. Sethi, B. Singh, J. Shiekmydeen, S. Salave and R. J. Patel, et al. Recent Review on Biological Barriers and Host–Material Interfaces in Precision Drug Delivery: Advancement in Biomaterial Engineering for Better Treatment Therapies, Pharmaceutics, 2024, 16(8), 1076 CrossRef CAS.
- K. Park, Facing the Truth about Nanotechnology in Drug Delivery, ACS Nano, 2013, 7(9), 7442–7447 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol., 2015, 33(9), 941–951 CrossRef CAS PubMed.
- A. M. Wagner, D. S. Spencer and N. A. Peppas, Advanced architectures in the design of responsive polymers for cancer nanomedicine, J. Appl. Polym. Sci., 2018, 135(24), 46154 CrossRef.
- H. Gelderblom, J. Verweij, K. Nooter, A. Sparreboom and E. L. Cremophor, Eur. J. Cancer, 2001, 37(13), 1590–1598 CrossRef CAS.
- N. Kolishetti, S. Dhar, P. M. Valencia, L. Q. Lin, R. Karnik and S. J. Lippard, et al., Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(42), 17939–17944 CrossRef CAS.
- P. M. Glassman and V. R. Muzykantov, Pharmacokinetic and Pharmacodynamic Properties of Drug Delivery Systems, J. Pharmacol. Exp. Ther., 2019, 370(3), 570–580 CrossRef CAS.
- P. Pandey, J. Patel, S. Kumar and Y. Pathak, Pharmacokinetics and Pharmacodynamics of Liposomal Nanoparticles, in Pharmacokinetics and Pharmacodynamics of Nanoparticulate Drug Delivery Systems, ed. J. K. Patel and Y. V. Pathak, Springer International Publishing, Cham, 2022, pp. 143–158 [cited 2024 Sep 5]. Available from: https://link.springer.com/10.1007/978-3-030-83395-4_8 Search PubMed.
- J. A. Ledermann and R. S. Kristeleit, Optimal treatment for relapsing ovarian cancer, Ann. Oncol., 2010, 21, vii218–vii222 CrossRef.
- O. Z. Fisher, T. Kim, S. R. Dietz and N. A. Peppas, Enhanced Core Hydrophobicity, Functionalization and Cell Penetration of Polybasic Nanomatrices, Pharm. Res., 2009, 26(1), 51–60 CrossRef CAS PubMed.
- A. S. Sawhney, C. P. Pathak and J. A. Hubbell, Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(.alpha.-hydroxy acid) diacrylate macromers, Macromolecules, 1993, 26(4), 581–587 CrossRef CAS.
- V. E. G. Diederich, T. Villiger, G. Storti and M. Lattuada, Modeling of the Degradation of Poly(ethylene glycol)-co-(lactic acid)-dimethacrylate Hydrogels, Macromolecules, 2017, 50(14), 5527–5538 CrossRef CAS.
- T. C. Chou and P. Talalay, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors, Adv. Enzyme Regul., 1984, 22, 27–55 CrossRef CAS PubMed.
- I. V. Bijnsdorp, E. Giovannetti and G. J. Peters, Analysis of Drug Interactions, in Cancer Cell Culture, ed. I. A. Cree, Humana Press, Totowa, NJ, 2011, pp. 421–434, [cited 2024 Sep 5]. (Methods in Molecular Biology; vol. 731). Available from: https://link.springer.com/10.1007/978-1-61779-080-5_34 Search PubMed.
- Y. Han, Z. He, A. Schulz, T. K. Bronich, R. Jordan and R. Luxenhofer, et al., Synergistic Combinations of Multiple Chemotherapeutic Agents in High Capacity Poly(2-oxazoline) Micelles, Mol. Pharmaceutics, 2012, 9(8), 2302–2313 CrossRef CAS PubMed.
- O. Z. Fisher and N. A. Peppas, Polybasic Nanomatrices Prepared by UV-Initiated Photopolymerization, Macromolecules, 2009, 42(9), 3391–3398 CrossRef CAS.
- W. B. Liechty, R. L. Scheuerle and N. A. Peppas, Tunable, responsive nanogels containing t-butyl methacrylate and 2-(t-butylamino)ethyl methacrylate, Polymer, 2013, 54(15), 3784–3795 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Stimuli-responsive nanocarriers for drug delivery, Nat. Mater., 2013, 12(11), 991–1003 CrossRef CAS PubMed.
- W. Tao and Z. He, ROS-responsive drug delivery systems for biomedical applications, Asian J. Pharm. Sci., 2018, 13(2), 101–112 Search PubMed.
- E. G. Kelley, J. N. L. Albert, M. O. Sullivan and T. H. Epps, Stimuli-responsive copolymer solution and surface assemblies for biomedical applications, Chem. Soc. Rev., 2013, 42(17), 7057–7071 RSC.
- J. Wang, Y. Zhang, E. Archibong, F. S. Ligler and Z. Gu, Leveraging H2O2 Levels for Biomedical Applications, Adv. Biosyst., 2017, 1(9), 1700084 CrossRef PubMed.
- S. H. Lee, M. K. Gupta, J. B. Bang, H. Bae and H. J. Sung, Current progress in Reactive Oxygen Species (ROS)-Responsive materials for biomedical applications, Adv. Healthcare Mater., 2013, 2(6), 908–915 CrossRef CAS.
- P. Baudouin-Cornu, G. Lagniel, C. Kumar, M. E. Huang and J. Labarre, Glutathione Degradation Is a Key Determinant of Glutathione Homeostasis, J. Biol. Chem., 2012, 287(7), 4552–4561 CrossRef CAS PubMed.
- A. Shodeinde, O. L. Lanier, S. Nallaparaju, M. Lam, A. Savk and N. A. Peppas, Tuning the pH-Responsive Properties of (2-Diisopropylamino) Ethyl Methacrylate-Based Cationic Nanoparticles: Toward Enhanced Nanoparticle Stability and Cytocompatibility, Biomed. Mater. Devices, 2024, 2(1), 529–540 CrossRef CAS.
- W. B. Liechty, R. L. Scheuerle, J. E. Vela Ramirez and N. A. Peppas, Uptake and function of membrane-destabilizing cationic nanogels for intracellular drug delivery, Bioeng. Transl. Med., 2019, 4(1), 17–29 CrossRef CAS.
- M. K. Notabi, E. C. Arnspang, N. A. Peppas and M. Ø. Andersen, siRNA delivery mediated by pH and redox responsive p(DEAEMA-co-HEMA-g-PEGMA) nanogels, J. Drug Delivery Sci. Technol., 2023, 86, 104510 CrossRef CAS.
- S. Casey Laizure, V. Herring, Z. Hu, K. Witbrodt and R. B. Parker, The Role of Human Carboxylesterases in Drug Metabolism: Have We Overlooked Their Importance?, Pharmacotherapy, 2013, 33(2), 210–222 CrossRef PubMed.
- M. Rooseboom, J. N. M. Commandeur and N. P. E. Vermeulen, Enzyme-Catalyzed Activation of Anticancer Prodrugs, Pharmacol. Rev., 2004, 56(1), 53–102 CrossRef CAS PubMed.
- B. Li, M. Sedlacek, I. Manoharan, R. Boopathy, E. G. Duysen and P. Masson, et al., Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma, Biochem. Pharmacol., 2005, 70(11), 1673–1684 CrossRef CAS PubMed.
- T. Satoh, P. Taylor, W. F. Bosron, S. P. Sanghani, M. Hosokawa and B. N. L. Du, Current Progress on Esterases: From Molecular Structure to Function, Drug Metab. Dispos., 2002, 30(5), 488–493 CrossRef CAS.
- T. Imai, Human Carboxylesterase Isozymes: Catalytic Properties and Rational Drug Design, Drug Metab. Pharmacokinet., 2006, 21(3), 173–185 CrossRef CAS.
- B. M. Liederer and R. T. Borchardt, Enzymes involved in the bioconversion of ester-based prodrugs, J. Pharm. Sci., 2006, 95(6), 1177–1195 CrossRef CAS.
- G. Xu, W. Zhang, M. K. Ma and H. L. McLeod, Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan, Clin. Cancer Res., 2002, 8(8), 2605–2611 CAS.
- M. Hosokawa, Structure and Catalytic Properties of Carboxylesterase Isozymes Involved in Metabolic Activation of Prodrugs, Molecules, 2008, 13(2), 412–431 CrossRef CAS PubMed.
- D. M. Maxwell, The specificity of carboxylesterase protection against the toxicity of organophosphorus compounds, Toxicol. Appl. Pharmacol., 1992, 114(2), 306–312 CrossRef CAS.
- D. Shi, J. Yang, D. Yang, E. L. LeCluyse, C. Black and L. You, et al., Anti-Influenza Prodrug Oseltamivir Is Activated by Carboxylesterase Human Carboxylesterase 1, and the Activation Is Inhibited by Antiplatelet Agent Clopidogrel, J. Pharmacol. Exp. Ther., 2006, 319(3), 1477–1484 CrossRef CAS PubMed.
- T. Satoh, M. Hosokawa, R. Atsumi, W. Suzuki, H. Hakusui and E. Nagai, Metabolic Activation of CPT-11, 7-Ethyl-10-(4-(1-piperidino)-1-piperidino)carbonyloxycamptothecin, a Novel Antitumor Agent, by Carboxylesterase, Biol. Pharm. Bull., 1994, 17(5), 662–664 CrossRef CAS PubMed.
- M. Aghi, F. Hochberg and X. O. Breakefield, Prodrug activation enzymes in cancer gene therapy, J. Gene Med., 2000, 2(3), 148–164 CrossRef CAS PubMed.
- D. J. Stewart, Mechanisms of resistance to cisplatin and carboplatin, Crit. Rev. Oncol. Hematol., 2007, 63(1), 12–31 CrossRef PubMed.
- D. Wang and S. J. Lippard, Cellular processing of platinum anticancer drugs, Nat. Rev. Drug Discovery, 2005, 4(4), 307–320 CrossRef CAS.
- D. K. Shah, J. Veith, R. J. Bernacki and J. P. Balthasar, Evaluation of combined bevacizumab and intraperitoneal carboplatin or paclitaxel therapy in a mouse model of ovarian cancer, Cancer Chemother. Pharmacol., 2011, 68(4), 951–958 CrossRef CAS PubMed.
- M. Munk Jensen, K. D. Erichsen, F. Björkling, J. Madsen, P. B. Jensen and M. Sehested, Imaging of Treatment Response to the Combination of Carboplatin and Paclitaxel in Human Ovarian Cancer Xenograft Tumors in Mice Using FDG and FLT PET, PLoS One, 2013, 8(12), e85126 CrossRef PubMed , G. E. Woloschak, editor.
- S. Jiang, A. W. Pan, T.-y. Lin, H. Zhang, M. Malfatti and K. Turteltaub, et al., Paclitaxel Enhances Carboplatin-DNA Adduct Formation and Cytotoxicity, Chem. Res. Toxicol., 2015, 28(12), 2250–2252 Search PubMed.
- J. A. Smith, H. Ngo, M. C. Martin and J. K. Wolf, An evaluation of cytotoxicity of the taxane and platinum agents combination treatment in a panel of human ovarian carcinoma cell lines, Gynecol. Oncol., 2005, 98(1), 141–145 CrossRef CAS.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs, Chem. Rev., 2016, 116(5), 3436–3486 CrossRef CAS PubMed.
- X. Lu, M. D. Howard, D. R. Talbert, J. J. Rinehart, P. M. Potter and M. Jay, et al., Nanoparticles Containing Anti-inflammatory Agents as Chemotherapy Adjuvants II: Role of Plasma Esterases in Drug Release, AAPS J., 2009, 11(1), 120–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00330f |
| This journal is © The Royal Society of Chemistry 2025 |