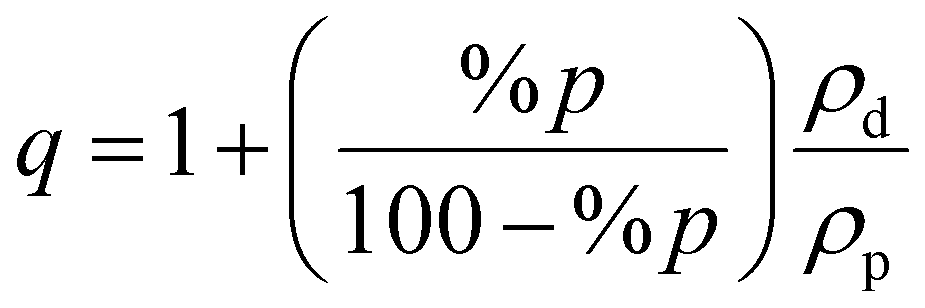DOI:
10.1039/D4PM00301B
(Paper)
RSC Pharm., 2025,
2, 178-185
Efficient development of high drug loaded posaconazole tablets enabled by amorphous solid dispersion
Received
19th October 2024
, Accepted 3rd December 2024
First published on 4th December 2024
Abstract
Determining the upper limits of drug loading in amorphous solid dispersion (ASD) with sufficient physical stability and release performance is critical for developing ASD-enabled tablets for poorly soluble drugs. Recent studies have highlighted the utility of the polymer overlap concentration, c*, in maintaining the physical stability of ASD formulations. The present work demonstrates the feasibility of effectively developing high drug loaded ASD tablets using the c* concept as a guide, with posaconazole as the model drug. By incorporating various material sparing formulation technologies, a record high 50% POS loaded tablet with adequate manufacturability and satisfactory dissolution performance was developed using 1.5 g of POS within 14 days. Physical stabilities of the ASD and tablet were maintained for at least 6 months under ambient conditions and 1 month at 40 °C.
1. Introduction
Drug product development is a complicated process requiring expertise from multiple fields, such as physiology, medicinal chemistry, materials science, process engineering, clinical trial design, project management, intellectual property, regulatory sciences, and manufacturing technology.1,2 To successfully bring any drug product to market, it is essential to address these diverse challenges early in the development process, where efficiency is always emphasized.3 Efficient drug product development helps ensure timely access to cost effective and high quality treatments for patients while maintaining competitive and regulatory standards in the pharmaceutical industry.4–7
The oral route is the most preferred for administering drugs due to the high compliance of patients. Among oral dosage forms, the tablet is especially favored owing to excellent stability, low manufacturing cost, and large production volume.8 However, up to 40% of marketed drugs and about 90% of new chemical entities in the pipeline possess low aqueous solubility, which challenges oral drug delivery by limiting bioavailability.9 Therefore, various enabling formulation strategies, such as amorphous solid dispersion (ASD), have been employed to improve the dissolution and bioavailability of poorly water-soluble drugs.10–12
The solubility advantages of amorphous drugs arise from their high free energies compared to their crystalline counterparts, which lead to reduced physical stability.13 ASDs formulated by admixing drug and a polymer are often used to improve physical stability. Usually, polymers added to ASDs are at a high content, with reduced percent drug loading, ensuring effective inhibition of drug crystallization.14–16 However, this arrangement leads to an increased pill burden. Hence, there is a need to reliably determine the upper limit of drug loading in ASD formulation without sacrificing physical stability.
Recent studies have shown that the overlap concentration of polymer in an ASD, c*, sets the upper limit of drug loading to maintain the physical stability of an ASD.17,18 When the polymer concentration exceeds c* in an ASD, polymer coils overlap and interpenetrate, and the drug–polymer binary system can be regarded as a single homogeneous phase at the molecular level.19,20 It has also been shown that with a polymer concentration exceeding c*, the first nucleation event of amorphous drugs will be delayed, and crystallization will be retarded.21,22 Determination of c* is a first step in quickly identifying the maximum drug loading in an ASD based on the criterion of physical stability.
In addition to physical stability, a quality ASD based immediate release tablet must meet two other performance criteria, namely adequate mechanical strength and rapid dissolution.23,24 Moreover, good flowability of the final blend and low ejection force are required to ensure successful tablet manufacturing. These requirements are typically met by selecting appropriate excipients in optimal amounts within the formulation.25,26 Due to the high cost of investigational drugs in the early stage of drug development, material sparing strategies are sought out to accelerate the process of drug product development while lowering development costs.27
Posaconazole (POS), an antifungal agent, is a fluorine containing triazole molecule with extremely poor aqueous solubility (<1 μg mL−1),28 which makes it a good model drug for examining the drug loading limit in ASD based tablets. Using POS, we aim to demonstrate a strategy for efficiently developing a high drug-loaded ASD-based tablet formulation, guided by the c* concept and enabled by material-sparing formulation techniques.
2. Materials and methods
2.1 Materials
POS was received from Merck; hydroxypropyl methylcellulose acetate succinate (M grade) (HPMCAS, AquaSolve™; Ashland, Wilmington, DE), microcrystalline cellulose (MCC: Avicel® PH101; International Flavors & Fragrances, Philadelphia, PA), fumed silica (CAB-O-Sil, M5-P, Cabot Corporation, Boston, MA), croscarmellose sodium (Ac-Di-Sol, SD-711, International Flavors & Fragrances, Philadelphia, PA), magnesium stearate (MgSt; Mallinckrodt Inc., St Louis, MO), hydrochloric acid aqueous solution (HCl, 36.5–38%; VWR International LLC., Radnor, PA), sodium phosphate dibasic heptahydrate (Na2HPO4·7H2O, Fisher Scientific, Fair Lawn, NJ), and sodium phosphate monobasic monohydrate (NaH2PO4·H2O, Fisher Scientific, Fair Lawn, NJ), polysorbate 20 (Tween® 20; Amresco LLC., Solon, OH), ethanol (VWR International LLC., Radnor, PA), methanol (MeOH; Sigma-Aldrich, St Louis, MO), dichloromethane (DCM; Sigma-Aldrich, St Louis, MO) were all used as received.
2.2 Methods
2.2.1 Preparation of POS/HPMCAS amorphous solid dispersion.
Amorphous solid dispersions of POS in HPMCAS (M) with 60%, 70%, and 80% drug loadings were prepared by spray drying 2% (w/w) solutions in DCM/MeOH (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, v/v) using a mini spray dryer (Bend Research, Bend, OR). Briefly, a mixture of 300 mg of POS and polymer was dissolved in 18 mL of the cosolvent. The solution was transferred to a 20 mL syringe and injected into the spray dryer. The inlet temperature was 80 °C, and the outlet temperature was approximately 35 °C. Solution flow rate was 0.65 mL min−1, and the nitrogen gas flow rate was 12.8 standard liters per minute (SLPM). Spray dried samples were collected on filter paper, transferred into a scintillation vial, and vacuum dried at ambient temperature overnight to remove residual solvents before further use.
:50, v/v) using a mini spray dryer (Bend Research, Bend, OR). Briefly, a mixture of 300 mg of POS and polymer was dissolved in 18 mL of the cosolvent. The solution was transferred to a 20 mL syringe and injected into the spray dryer. The inlet temperature was 80 °C, and the outlet temperature was approximately 35 °C. Solution flow rate was 0.65 mL min−1, and the nitrogen gas flow rate was 12.8 standard liters per minute (SLPM). Spray dried samples were collected on filter paper, transferred into a scintillation vial, and vacuum dried at ambient temperature overnight to remove residual solvents before further use.
2.2.2 True density measurement.
The true density of the materials was measured using a helium pycnometer (Quantachrome Instruments, Ultrapycnometer 1000e, Byonton Beach, Florida) with 1–2 g of an accurately weighed sample that filled to about 75% of the volume of the sample cell. An analytical balance (Mettler Toledo, Columbus, Ohio, model AG204) was used for weighing. The experiment was stopped when the variation between five consecutive measurements was below 0.005%, and the mean of the last five measurements was taken as the true density.
2.2.3 Viscosity measurement.
Zero shear rate viscosity (η) of pure drug and drug/polymer melts was measured using a rotational rheometer (ARES, TA Instruments, New Castle, DE). A parallel plate geometry with a diameter of 25 mm was employed. Briefly, ∼800 mg of POS/HPMCAS physical mixture powder was placed on the bottom plate after zero torque, normal force, and gap calibrations. The gap between the parallel plates was fixed at approximately 1 mm. Physical mixtures of POS and HPMCAS with various HPMCAS concentrations were first cryomilled (Spex SamplePrep Grinder 6770) in a liquid nitrogen bath. Cryomilling was performed at 10 Hz for five 2-minute milling – 2-minute cooldown cycles. Cryomilled samples were loaded between two plates, heated to 175 °C, and equilibrated for ∼5 min to guarantee complete melting of POS crystals before each measurement. A steady rate sweep test was performed with an initial frequency of 1 Hz and a final frequency of 100 Hz with continuous nitrogen purge at a flow rate of 3 standard cubic feet per minute (SCFM). A viscosity-composition diagram was constructed, from which c* was determined.19
2.2.4 Physical stability by powder X-ray diffractometry.
Spray dried ASD powder consisting of 70% POS in HPMCAS and six formulated tablets containing the same ASD were stored under two conditions: (1) ambient environment and (2) 40 °C oven, both in 20 mL tightly capped glass vials. Samples were examined after 1, 2, and 6 months using a laboratory X-ray diffractometer (PANalytical X'pert pro, Westborough, MA, USA), with Cu Kα radiation (1.54056 Å). Data was collected between 5° and 35° 2θ with step size 0.019699° and dwell time of 0.5 s. Tube voltage and amperage were set at 40 kV and 40 mA, respectively. The tablets were gently ground into powders before PXRD data collection.
2.2.5 Dissolution.
Degassed 0.1 M PBS at pH 6.5, containing 0.05% Tween® 20, was used as the dissolution medium. 300 mL of the medium was introduced into a jacketed beaker maintained at 37 °C by a circulating water bath and stirred at 100 rpm with an overhead paddle. The concentration in the medium was monitored in situ by a UV–Vis fiber optic dip probe (Ocean Optics, Dunedin, FL) at λ = 257 nm and converted to a concentration–time profile based on a previously established concentration–absorbance calibration curve. Dissolution profiles of both freshly prepared ASD powders with different drug loadings and formulated ASD tablets (round convex, 6.3 mm diameter), prepared under 3.1 kN compaction force, were evaluated.
2.2.6 Tablet formulation, preparation, and characterization.
A batch of the formulation (Table 1, batch size ∼3 g) was prepared by mixing the ASD (70% POS + 30% HPMCAS) with intra-granular excipients in a plastic bottle at 49 rpm for 5 min, using a Turbula mixer (GlenMills Inc., Clifton, NJ). Approximately 500 mg of the blend was compacted into rectangular (16 mm × 9 mm) flat-faced tablets, i.e., simulated ribbons, using a compaction simulator (STYL'One Evolution; Medelpharm, Beynost, France) under a compaction pressure of 77 MPa. The simulated ribbons were then ground manually using a mortar and pestle and passed through a 500 μm sieve to obtain granules. The granules were mixed with extra-granular excipients (Table 1) for 5 min at 49 rpm to obtain a final blend. Biconvex round tablets of the formulated granules (400 mg) were prepared by the compaction simulator using round concave tooling (6.3 mm in diameter) under different compaction forces.
Table 1 Formulation of 50% drug-loading posaconazole (POS) tablet containing POS/HPMCAS ASD (70% POS loading)
| |
Ingredient |
Function |
wt% |
| Intra-granular |
POS:HPMCAS (7:3, w/w) ASD |
Active |
71.1 |
| MCC PH101 |
Filler |
21.9 |
| Ac-Di-Sol |
Disintegrant |
3 |
| Extra-granular |
Ac-Di-Sol |
Disintegrant |
3 |
| Cab-O-Sil |
Glidant |
0.5 |
| Magnesium stearate |
Lubricant |
0.5 |
| Total |
100 |
2.2.7 Powder flowability and bulk density.
Approximately 3 g of each powder was poured into a graduated 10 mL cylinder, and its volume was recorded. The cylinder was then dropped 50 times onto a padded benchtop from a height of approximately 2 cm before the powder volume was measured. The process was repeated until the difference in tapped volume between two consecutive volume readings was less than 2.0%. Bulk and tapped densities were calculated using the powder weight and the corresponding bulk and tapped volumes. Measurements were in triplicate for each powder. Carr's compressibility index (CI) of the powder was calculated using eqn (1).29| | | CI = 100% × (1 − bulk density/tapped density) | (1) |
2.2.8 Tabletability.
Formulated granules (∼80 mg) were compressed using flat faced round tooling (6 mm) under different pressures (50–450 MPa) using the compaction simulator with a force controlled, symmetrical single compression cycle (2% speed, 2 s compression composed of a 1 s rise and a 1 s fall without holding at the maximum force, followed by 3 s relaxation, and a 2 s ejection step). Tablet diametrical breaking force was determined under ambient conditions using a texture analyzer (TA-XT2i; Texture Technologies Corporation, Scarsdale, NY) with a speed of 0.01 mm s−1, and tablet dimensions were measured using a digital caliper (Mitutoyo Corp, Kanagawa, Japan). Tablet tensile strength (σ, MPa) was calculated according to eqn (2),where F is the breaking force (N), and D and h are the diameter and thickness of the tablet (mm), respectively.30 Tabletability profiles were generated by curve fitting data points to the Vreeman–Sun equation using OriginLab software (version 10.0, Northampton, MA).31
2.2.9 Friability.
The tablet friability profile, i.e., tablet weight loss as a function of compaction force used to prepare tablets, of the final formulation was determined using an expedited method.32 Tablets were produced under different compaction forces in the range of 1.7–10.6 kN in the compaction simulator under a force controlled mode using a symmetrical single compression cycle (see above). Tablets were individually coded and placed into a friabilator (F-2; Pharma Alliance, London, UK), which was run at 25 rpm for 4 min. Before and after the test, tablets were cleaned to remove loose particles on the surface and then weighed to calculate the percent weight loss for individual tablets.
3. Results
3.1 Rheological analysis
Fig. 1 displays the effect of polymer concentration on the viscosity of POS/HMPCAS melts. The linear portion of the curve corresponds to the dilute polymer regime, while the nonlinear portion corresponds to the semi-dilute and concentrated regime.33 The overlap concentration, c*, of HPMCAS in molten POS, determined from the boundary between the linear (dilute) and nonlinear (semi-dilute) regimes, is approximately 3%.34 Thus, physically stable POS/HPMCAS ASD can be expected in the vicinity of 97% POS loading.19
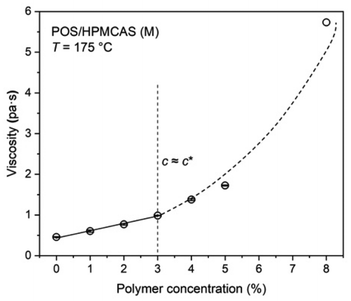 |
| | Fig. 1 Viscosity-composition diagram of POS/HPMCAS at 175 °C. | |
3.2 Impact of ASD drug loading on powder dissolution
Dissolution performance, which is relevant to bioavailability, is crucial for developing high performance oral dosage forms. Therefore, ASD batches of various POS loadings below 97% (1 − c*), i.e., 60%, 70%, and 80% were prepared and powder dissolution studies were performed at a 40 mg dose under non-sink conditions. Only the 60% and 70% ASDs showed appreciable dissolution (Fig. 2), where the concentration of POS in the medium nearly reached the amorphous solubility of POS after ∼20 min.35 The negligible dissolution of the 80% drug loaded ASD is likely due to rapid crystallization of amorphous POS upon contact with the dissolution medium. Therefore, a bulk powder of the 70% POS loaded ASD (∼3 g) was prepared and used for tablet development.
 |
| | Fig. 2 Powder dissolution profiles of POS-HPMCAS ASDs with 60%, 70%, and 80% POS loadings in 0.1 M PBS at pH 6.5 with 0.05% v/v Tween 20 at 37 °C. | |
3.3 Formulation optimization and tablet performance
Effective process optimization and successful selection of suitable excipients to ensure formulation performance depend on a thorough understanding of the structure–property relationships of the materials involved.8 Owing to the low bulk density (0.17 ± 0.02 g cm−3, n = 3) and poor flowability (CI = 40.1 ± 3.1%, n = 3) of the 70% POS loaded ASD, consistent with its fine particle size, granulation was deemed necessary for developing a commercial tablet.36 In this context, dry granulation was employed to prevent potential adverse effects of water on the physical stability and in vivo performance of the POS ASD.37
Due to the inadequate tabletability of pure 70% POS ASD (Fig. 3), the ability of the ASD to form quality ribbons during roller compaction was expected to be poor. Hence, MCC (Avicel PH101), a binder with excellent tabletability, was chosen as an intragranular binder to facilitate ribbon formation. To maintain a 50% drug loading in the tablet to lessen pill burden, 71.1% pure ASD is required. This leaves only 28.9% formulation space for all excipients other than the ASD. To permit incorporation of other functional excipients, 21.9% MCC was used. For the remaining 7% formulation space, 3% of Ac-Di-Sol was used as an intra-granular disintegrant. The pre-granulation blend exhibited substantially improved tabletability over the pure ASD (Fig. 3), and was deemed adequate for ribbon formation.
 |
| | Fig. 3 Tabletability of powders in different stages of tablet formulation design, 70% POS-HPMCAS ASD (red), pregranulation blend (blue), and final blend (black). Lines were obtained by fitting the data to the Vreeman–Sun tabletability equation. | |
To determine a compaction pressure yielding ribbons with a target porosity of 15% to ensure sufficient tabletability, an abbreviated compressibility profile of the pre-granulation blend was obtained using three tablets to save material (Fig. 4). From the fitting curve, based on the Kuentz and Leuenberger (KL) equation,38 a pressure of 77 MPa was predicted to yield ribbons with the target porosity of 15%. The granules obtained from milling ribbons exhibited significantly improved bulk density (0.54 ± 0.008 g cm−3) and flowability (CI = 21.4 ± 1.3%) (Fig. 5), which was expected due to both densification and size enlargement.
 |
| | Fig. 4 Compressibility profile of the pre-granulation blend. The line was obtained by curve fitting with the Kuentz and Leuenberger (KL) equation. | |
 |
| | Fig. 5 Flowability and bulk density of various powders generated in this work. | |
The final tablet formulation was attained by adding the following extra-granular excipients to ensure good manufacturability and performance of the final tablet, (1) 3% Ac-Di-Sol to facilitate the disintegration of tablets during dissolution; (2) 0.5% Cab-O-Sil to further improve flowability; and (3) 0.5% magnesium stearate to reduce ejection force during tablet manufacturing. The amount of each excipient used was based on typical usage in tablet formulation. Formulation optimization was not performed due to the limited amount of ASD available. The final blend exhibited flowability (CI = 26.7 ± 1.6%) comparable to that of MCC PH102 (CI = 25.2 ± 1.7%) (Fig. 5), meeting the criterion for high speed tableting.39
The tablet friability profile of the final blend was determined, using a material sparing and expedited approach, to guide the choice of an appropriate compaction force for tablet manufacturing.32 From the friability profile (Fig. 6), tablets with <0.8% friability could be attained at a compaction force ≥3.0 kN. Using this approach, only 0.7 g of the final blend was used. Importantly, excessive forces that could cause problems, such as slow disintegration, slow dissolution, and lamination of tablets, were avoided.32,40,41
 |
| | Fig. 6 Tablet friability profile of the final blend. | |
3.4 Physical stability
After 6 months of storage at 25 °C under ambient conditions and 1 month of additional storage at 40 °C, no sharp diffraction peaks were detected in the PXRD pattern of the 70% POS ASD (Fig. 7). This suggests adequate physical stability of the ASD powder. Similarly, the absence of diffraction peaks in the PXRD pattern of the stored final tablet also confirms physical stability for at least 6 months in a closed container under ambient conditions.
 |
| | Fig. 7 PXRD patterns of the ASD powder and tablet after storage under various conditions. | |
3.5 Tablet dissolution
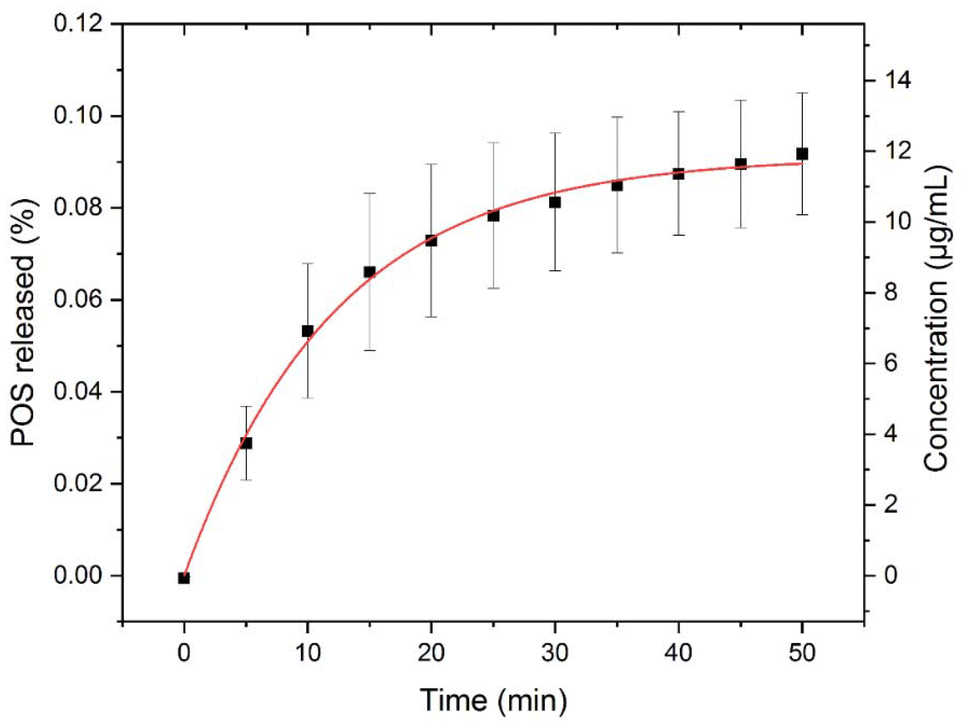
Based on the results obtained so far, we determined that a high drug loaded tablet formulation with 50% POS (Table 1) can be manufactured at 3.1 kN with a 6.3 mm diameter round concave tooling. This POS loading, 70% in ASD and 50% in tablet, is higher than all previously reported POS tablets enabled by ASD (15.9%–40% in tablet).35,42 These 50% POS-loaded tablets also exhibited fast dissolution in 0.1 M PBS at pH 6.5 with 0.05% v/v Tween 20 at 37 °C (Fig. 8). Concentration of POS in the medium reached the amorphous solubility (∼11 μg mL−1) after around 35 min,35 similar to that of 70% POS ASD powder (Fig. 2). The dissolution data is satisfactorily fit by the eqn (3)where C(t) is concentration of released POS in the closed aqueous suspension, Cas = 11.85 ± 0.14 μg ml−1 is the amorphous solubility, and a = 0.0819 ± 0.0032 s−1 is a rate constant determined by the volume of the suspension, the total surface area of the ASD particles in suspension (assumed to be constant at low percent released), and the mass transfer coefficient of the particle/fluid interface. The calculated value of Cas is very close to that reported previously,43 but may be affected by the presence of Tween in the dissolution medium.
 |
| | Fig. 8 Dissolution profile of final tablets in 0.1 M PBS at pH 6.5 with 0.05% v/v Tween 20 at 37 °C (n = 3). The solid curve is the best fit of eqn (3) to the data. | |
4. Discussion
A goal in tablet production for high dose APIs is to minimize the amount of excipients added. One reason is reduced cost, but this is a minor factor since excipients are typically much cheaper than APIs. A more salient reason is that smaller tablets are more convenient for the patient. In our previous investigations, we demonstrated that solid state storage stability of API is greatly enhanced when the ASD is formulated with polymer concentration greater than the overlap concentration, c*, which can be conveniently determined by hot melt rheology. For typically high molecular weight polymers, c* is quite small, which by itself would suggest that minimal amounts of polymer need to be added.
However, physical stability does not necessarily imply good dissolution performance and high bioavailability, likely because the polymer needs to play the dual role of stabilizer and water absorber.19,21,22,42 Indeed, in the present study we showed that POS/HPMCAS ASDs require at least 30% polymer to achieve satisfactory dissolution. How much does this affect tablet size? It is straightforward to show that the volume enhancement, q, due to blending polymer (p) with drug (d) is calculated according to
| |  | (4) |
where %
p is the weight percent of polymer, and the
ρs are the densities of the neat components, assuming negligible volume change upon mixing. From the reported density values,
ρp = 1.26 g cm
−3 and
ρd = 1.27 g cm
−3 (amorphous form) this density ratio is essentially unity.
44 For 3% HPMCAS, we estimate
q ≈ 1.03 while for 30% HPMCAS
q ≈ 1.43, representing an approximately 40% increase in volume. Notice that the above equation predicts a strong increase in volume when the polymer component exceeds the drug component, as occurs in many ASDs. We conclude by noting that the amounts of the other excipients to be added are proportional to the mass of the ASD; therefore, reducing the polymer load also reduces the amounts of the other excipients.
A recent study published by Mudie et al. reported a physically stable ASD POS/Eudragit L100 containing 75 wt% drug, which is slightly superior to our 70% wt% drug ASD.35 Aside from the difference in polymer used, we probably could increase the drug loading in our ASD above 70% but not as far as 80%. Mudie et al. also incorporated extra polymer (HPMCAS-H) in their tablet formulation in order to prevent precipitation of released drug. As a result, their final drug loading was 40%, less than our 50%. In the present study we did not carry out dissolution to the point where precipitation could occur, nor did we add extra polymer to prevent it. Due to differences in the dissolution conditions used in the two studies, one cannot draw a definitive conclusion on the dissolution performance of the two formulations. As noted by Mudie et al., amorphous solubility of POS increases greatly upon addition of FaSSIF. Moreover, in the in vivo case, rapid absorption of POS should reduce the driving force for precipitation. Recently, in vitro techniques have been developed to simulate dissolution in the presence of absorption, and these techniques could be used to determine the minimal amount, if any, of polymer that should be added to prevent precipitation.43,45
5. Conclusions
The identification of an upper limit of drug loading in ASD based on the c* concept was instrumental in identifying the upper limit of POS loading with adequate physical stability. By using material-sparing techniques, we subsequently developed a record high (50%) POS-loaded tablet, exhibiting adequate manufacturability and good dissolution performance. The entire development process utilized approximately 1.5 grams of POS and spanned 14 days. This material-sparing and fast tablet development strategy, demonstrated with POS in this work, has the potential to accelerate tablet development during the preclinical phase when only minimal amounts of active pharmaceutical ingredient are available.
Data availability
All data supporting the conclusion of this article have been included in the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the National Science Foundation for support through the Industry University Collaborative Research Center (IUCRC) grant IIP-2137264, Center for Integrated Materials Science and Engineering for Pharmaceutical Products (CIMSEPP).
References
- A. S. Rathore and H. Winkle, Quality by Design for Biopharmaceuticals, Nat. Biotechnol., 2009, 27(1), 26–34, DOI:10.1038/nbt0109-26.
- J. M. Twombly, J. Fälting, M. Giorgetti, A. C. Maroney and G. Osswald, How Partnership Should Work to Bring Innovative Medicines to Patients, Drug Discovery Today, 2020, 25(6), 965–968 Search PubMed.
- D. R. Kalaria, K. Parker, G. K. Reynolds and J. Laru, An Industrial Approach towards Solid Dosage Development for First-in-Human Studies: Application of Predictive Science and Lean Principles, Drug Discovery Today, 2020, 25(3), 505–518 CrossRef CAS PubMed.
- A. Schuhmacher, O. Gassmann and M. Hinder, Changing R&D Models in Research-Based Pharmaceutical Companies, J. Transl. Med., 2016, 14(1), 105, DOI:10.1186/s12967-016-0838-4.
- J. A. DiMasi, R. W. Hansen and H. G. Grabowski, The Price of Innovation: New Estimates of Drug Development Costs, J. Health Econ., 2003, 22(2), 151–185, DOI:10.1016/S0167-6296(02)00126-1.
- S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, How to Improve R&D Productivity: The Pharmaceutical Industry's Grand Challenge, Nat. Rev. Drug Discovery, 2010, 9(3), 203–214, DOI:10.1038/nrd3078.
- K. I. Kaitin, Deconstructing the Drug Development Process: The New Face of Innovation, Clin. Pharmacol. Ther., 2010, 87(3), 356–361 CrossRef CAS PubMed.
- C. C. Sun, Materials Science Tetrahedron—A Useful Tool for Pharmaceutical Research and Development, J. Pharm. Sci., 2009, 98(5), 1671–1687, DOI:10.1002/jps.21552.
- S. V. Bhujbal, B. Mitra, U. Jain, Y. Gong, A. Agrawal, S. Karki, L. S. Taylor, S. Kumar and Q. (Tony) Zhou, Pharmaceutical Amorphous Solid Dispersion: A Review of Manufacturing Strategies, Hot Top. Rev. Drug Delivery, 2021, 11(8), 2505–2536, DOI:10.1016/j.apsb.2021.05.014.
- F. Qian, J. Huang and M. A. Hussain, Drug–Polymer Solubility and Miscibility: Stability Consideration and Practical Challenges in Amorphous Solid Dispersion Development, J. Pharm. Sci., 2010, 99 (7), 2941–2947, DOI:10.1002/jps.22074.
- N. J. Babu and A. Nangia, Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals, Cryst. Growth Des., 2011, 11(7), 2662 CAS.
- S. Jain, N. Patel and S. Lin, Solubility and Dissolution Enhancement Strategies: Current Understanding and Recent Trends, Drug Dev. Ind. Pharm., 2015, 41 (6), 875–887, DOI:10.3109/03639045.2014.971027.
- P. Gupta, R. Thilagavathi, A. K. Chakraborti and A. K. Bansal, Role of Molecular Interaction in Stability of Celecoxib−PVP Amorphous Systems, Mol. Pharm., 2005, 2(5), 384–391, DOI:10.1021/mp050004g.
- G. V. Mooter, M. den Wuyts, N. Blaton, R. Busson, P. Grobet, P. Augustijns and R. Kinget, Physical Stabilisation of Amorphous Ketoconazole in Solid Dispersions with Polyvinylpyrrolidone K25, Eur. J. Pharm. Sci., 2001, 12(3), 261–269, DOI:10.1016/S0928-0987(00)00173-1.
- T. Miyazaki, S. Yoshioka, Y. Aso and S. Kojima, Ability of Polyvinylpyrrolidone and Polyacrylic Acid to Inhibit the Crystallization of Amorphous Acetaminophen, J. Pharm. Sci., 2004, 93(11), 2710–2717, DOI:10.1002/jps.20182.
- W. Zhang, R. Noland, S. Chin, M. Petkovic, R. Zuniga, B. Santarra, B. Conklin, H. H. Hou, K. Nagapudi, J. A. Gruenhagen, P. Yehl and T. Chen, Impact of Polymer Type, ASD Loading and Polymer-Drug Ratio on ASD Tablet Disintegration and Drug Release, Int. J. Pharm., 2021, 592, 120087, DOI:10.1016/j.ijpharm.2020.120087.
- A. Sahoo, R. Suryanarayanan and R. A. Siegel, Stabilization of Amorphous Drugs by Polymers: The Role of Overlap Concentration (c*), Mol. Pharm., 2020, 17(11), 4401–4406, DOI:10.1021/acs.molpharmaceut.0c00576.
- A. Sahoo and R. A. Siegel, Drug-Polymer Miscibility and the Overlap Concentration (c*) as Measured by Rheology: Variation of Polymer Structure, Pharm. Res., 2023, 40(9), 2229–2237 CrossRef CAS.
- S. Song, C. Wang, B. Zhang, C. C. Sun, T. P. Lodge and R. A. Siegel, A Rheological Approach for Predicting Physical Stability of Amorphous Solid Dispersions, J. Pharm. Sci., 2023, 112(1), 204–212, DOI:10.1016/j.xphs.2022.08.028.
- S. Song, J. Xu, Z. Chen, C. C. Sun, E. J. Munson and R. A. Siegel, Miscibility of Amorphous Solid Dispersions: A Rheological and Solid-State NMR Spectroscopy Study, J. Pharm. Sci., 2024 DOI:10.1016/j.xphs.2024.05.017.
- S. Song, X. Yao, C. Wang, C. C. Sun and R. A. Siegel, Delaying the First Nucleation Event of Amorphous Solid Dispersions above the Polymer Overlap Concentration (c*): PVP and PVPVA in Posaconazole, J. Pharm. Sci., 2024 DOI:10.1016/j.xphs.2024.04.026.
- S. Song, S. Cui, C. C. Sun, T. P. Lodge and R. A. Siegel, Crystallization Inhibition in Molecular Liquids by Polymers above the Overlap Concentration (c*): Delay of the First Nucleation Event, J. Pharm. Sci., 2024 DOI:10.1016/j.xphs.2024.02.011.
- C. C. Sun, H. Hou, P. Gao, C. Ma, C. Medina and F. J. Alvarez, Development of a High Drug Load Tablet Formulation Based on Assessment of Powder Manufacturability: Moving towards Quality by Design, J. Pharm. Sci., 2009, 98(1), 239–247 CAS.
- S. Paul, Y. Guo, C. Wang, J. Dun and C. C. Sun, Enabling, Direct Compression Tablet Formulation of Celecoxib by Simultaneously Eliminating Punch Sticking, Improving Manufacturability, and Enhancing Dissolution through Co-Processing with a Mesoporous Carrier, Int. J. Pharm., 2023, 641, 123041, DOI:10.1016/j.ijpharm.2023.123041.
- C. C. Sun, Dependence of Ejection Force on Tableting Speed—A Compaction Simulation Study, Powder Technol., 2015, 279, 123–126, DOI:10.1016/j.powtec.2015.04.004.
- K. T. Mitrevej and L. Augsburger, Adhesion of Tablets in a Rotary Tablet Press II. Effects of Blending Time, Running Time, and Lubricant Concentration, Drug Dev. Ind. Pharm., 1982, 8(2), 237–282 CrossRef CAS.
- Innovation or Stagnation? Challenge and Opportunity on the Critical Path to New Medical Products, 2004.
- J. Walravens, J. Brouwers, I. Spriet, J. Tack, P. Annaert and P. Augustijns, Effect of pH and Comedication on Gastrointestinal Absorption of Posaconazole, Clin. Pharmacokinet., 2011, 50(11), 725–734, DOI:10.2165/11592630-000000000-00000.
- W. Tharanon, Y. Guo, J. Peerapattana and C. C. Sun, A Systematic Comparison of Four Pharmacopoeial Methods for Measuring Powder Flowability, Int. J. Pharm., 2024, 661, 124454, DOI:10.1016/j.ijpharm.2024.124454.
- J. Fell and J. Newton, Determination of Tablet Strength by the Diametral-Compression Test, J. Pharm. Sci., 1970, 59(5), 688–691 CrossRef CAS PubMed.
- G. Vreeman and C. C. Sun, A Powder Tabletability Equation, Powder Technol., 2022, 408, 117709, DOI:10.1016/j.powtec.2022.117709.
- F. Osei-Yeboah and C. C. Sun, Validation and Applications of an Expedited Tablet Friability Method, Int. J. Pharm., 2015, 484(1), 146–155, DOI:10.1016/j.ijpharm.2015.02.061.
-
M. Doi, S. F. Edwards and S. F. Edwards, The Theory of Polymer Dynamics, Oxford University Press, 1988, vol. 73 Search PubMed.
-
P.-G. De Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, 1979 Search PubMed.
- D. M. Mudie, A. M. Stewart, N. Biswas, T. J. Brodeur, K. B. Shepard, A. Smith, M. M. Morgen, J. M. Baumann and D. T. Vodak, Novel High-Drug-Loaded Amorphous Dispersion Tablets of Posaconazole; In Vivo and In Vitro Assessment, Mol. Pharm., 2020, 17(12), 4463–4472, DOI:10.1021/acs.molpharmaceut.0c00471.
- C. C. Sun, Setting the Bar for Powder Flow Properties in Successful High Speed Tableting, Powder Technol., 2010, 201(1), 106–108 CAS.
- B. Démuth, Z. K. Nagy, A. Balogh, T. Vigh, G. Marosi, G. Verreck, I. Van Assche and M. Brewster, Downstream Processing of Polymer-Based Amorphous Solid Dispersions to Generate Tablet Formulations, Int. J. Pharm., 2015, 486(1–2), 268–286 Search PubMed.
- M. Kuentz and H. Leuenberger, Pressure Susceptibility of Polymer Tablets as a Critical Property: A Modified Heckel Equation, J. Pharm. Sci., 1999, 88(2), 174–179 CAS.
- S. Chattoraj, L. Shi and C. C. Sun, Profoundly Improving Flow Properties of a Cohesive Cellulose Powder by Surface Coating with Nano–silica through Comilling, J. Pharm. Sci., 2011, 100(11), 4943–4952, DOI:10.1002/jps.22677.
- S. G. Late, Y.-Y. Yu and A. K. Banga, Effects of Disintegration-Promoting Agent, Lubricants and Moisture Treatment on Optimized Fast Disintegrating Tablets, Int. J. Pharm., 2009, 365(1–2), 4–11 CrossRef CAS.
- V. Mazel and P. Tchoreloff, Lamination of Pharmaceutical Tablets: Classification and Influence of Process Parameters, J. Pharm. Sci., 2022, 111(5), 1480–1485, DOI:10.1016/j.xphs.2021.10.025.
- Y. Chen, S. Wang, S. Wang, C. Liu, C. Su, M. Hageman, M. Hussain, R. Haskell, K. Stefanski and F. Qian, Sodium Lauryl Sulfate Competitively Interacts with HPMC-AS and Consequently Reduces Oral Bioavailability of Posaconazole/HPMC-AS Amorphous Solid Dispersion, Mol. Pharm., 2016, 13 (8), 2787–2795, DOI:10.1021/acs.molpharmaceut.6b00391.
- K. M. H. Jain, H. H. Hou and R. A. Siegel, An Artificial Gut/Absorption Simulator: Simultaneous Evaluation of Desupersaturation and Absorption from Ketoconazole Supersaturated Solutions, J. Pharm. Sci., 2023, 112(8), 2212–2222, DOI:10.1016/j.xphs.2022.09.017.
- Y. Li, J. Yu, S. Hu, Z. Chen, M. Sacchetti, C. C. Sun and L. Yu, Polymer Nanocoating of Amorphous Drugs for Improving Stability, Dissolution, Powder Flow, and Tabletability: The Case of Chitosan-Coated Indomethacin, Mol. Pharm., 2019, 16(3), 1305–1311, DOI:10.1021/acs.molpharmaceut.8b01237.
- K. M. H. Jain, H. H. Hou and R. A. Siegel, An Artificial Gut/Absorption Simulator: Understanding the Impact of Absorption on In Vitro Dissolution, Speciation, and Precipitation of Amorphous Solid Dispersions, Mol. Pharm., 2024, 21(4), 1884–1899, DOI:10.1021/acs.molpharmaceut.3c01180.
Footnote |
| † Authors contributed to this work equally. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Sichen
Song†
,
Sichen
Song†