
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of drug–drug salt crystals and co-amorphous forms of levofloxacin and 4-aminosalicylic acid for pulmonary applications†
Hiroshi
Ueda
 *a,
Jun Yee
Tse
b,
Tetsuya
Miyano
b,
Yuzuki
Nakayama
c,
Peiwen
Mo
c,
Yuta
Hatanaka
c,
Hiromasa
Uchiyama
c,
Yuichi
Tozuka
c and
Kazunori
Kadota
*cd
*a,
Jun Yee
Tse
b,
Tetsuya
Miyano
b,
Yuzuki
Nakayama
c,
Peiwen
Mo
c,
Yuta
Hatanaka
c,
Hiromasa
Uchiyama
c,
Yuichi
Tozuka
c and
Kazunori
Kadota
*cd
aAnalysis and Evaluation Laboratory, Shionogi & Co., Ltd, Osaka, 561-0825, Japan. E-mail: hiroshi.ueda@shionogi.co.jp
bLaboratory for Medicinal Chemistry Research, Shionogi & Co., Ltd, Osaka, 561-0825, Japan
cDepartment of Formulation Design and Pharmaceutical Technology, Faculty of Pharmacy, Osaka Medical and Pharmaceutical University, Osaka, 569-1094, Japan. E-mail: kazunori.kadota@ompu.ac.jp
dSchool of Pharmaceutical Sciences, Wakayama Medical University, Wakayama, 640-8156, Japan
First published on 3rd February 2025
Abstract
A dry powder inhaler is a viable formulation for pulmonary delivery; however, the co-delivery of multiple drugs requires a specially designed device. This study aimed to design multi-component crystal and amorphous forms for the co-delivery of levofloxacin (LVF) and 4-aminosalicylic acid (ASA). New multi-component crystals of LVF and ASA, crystal-I and crystal-II, were formed by solvent evaporation and slurry conversion. Thermal analysis revealed that crystal-I and crystal-II were the hydrate and anhydrate forms, respectively. Upon heating, each crystal was converted to different crystals. All polymorphs reverted to crystal-I during storage. The co-amorphous (CA) form was obtained by spray drying, which exhibited a relatively high glass transition temperature above 100 °C. Multi-component crystals and CA were estimated as salts by single crystal X-ray diffraction and infrared spectroscopy. An in vitro aerodynamic performance test was performed for LVF, ASA, physical mixture (PM), crystal-I, and CA. The fine particle fraction (FPF, %) of LVF/ASA was 0.9/13.3 for pure drugs and 0.4/14.1 for PM. However, the FPF (%) for crystal-I and CA significantly improved to 25.4/29.9 and 20.0/20.6, respectively, with the co-delivery of LVF and ASA. We conclude that the design of multi-component crystals and co-amorphous forms is an effective strategy for the simultaneous delivery of inhalation drugs.
Introduction
Drug delivery to targeted disease sites is vital for maximizing its pharmacological effect. Peroral administration is convenient and common but it has the disadvantage of poor bioavailability because of the increasing number of poorly water-soluble drugs.1–3 Direct delivery of drugs to the disease site via local administration is a counterpart of systemic drug delivery, which allows a high concentration of the drug at the target site, reducing the therapeutically effective dose of the drug.4–6 Dry powder inhaler (DPI) delivery is one of the most investigated formulations for pulmonary treatment.7,8 Chronic obstructive pulmonary disease (COPD) is a major therapeutic area for DPI application and study.9,10 Self-administration is convenient for patients with COPD requiring chronic drug treatment. Drugs, such as salbutamol, budesonide, and fluticasone, have been developed as DPI formulations. Pulmonary infections, such as those associated with cystic fibrosis, can be caused directly or indirectly by various pathogens. The delivery of antibiotics to the lungs using DPI formulations is an attractive treatment option.11,12 Tuberculosis drugs and conventional antibiotics have often been researched for DPI formulation development. In addition to the efficient use of drugs, DPI formulations are suitable for COPD or tuberculosis drugs that have low solubility via the peroral route, and solubility enhancement investigations have been conducted.13,14 Further efforts such as control of the particle size/morphology and a combination of carrier and storage conditions have been studied to overcome physicochemical issues.7,8 The combination therapy of different drugs for the treatment of COPD and pulmonary infections15–18 is a vital factor in designing an inhalation delivery system. Conventionally designed medical devices have been used for the application of inhaled formulations for human use.19,20 However, the development of a complex medical device for the simultaneous delivery of multiple drugs, each with different physicochemical and particle properties, into the targeted compartments of the lungs is a significant challenge.21,22The modification of the physicochemical and particle properties often focuses on improving the inhalation properties of drugs. Redesigning the chemical structures of compounds can result in different physicochemical properties; however, it may also affect their primary pharmacological, toxicological, and pharmacokinetic aspects.23 Crystal engineering is a representative approach to altering physicochemical properties without modifying the chemical structure. Drug molecules are regularly arranged in a crystal lattice through intermolecular interactions, and different arrangements of these molecules result in polymorphic forms.24 Various polymorphic forms of drug molecules have been discovered where differences in physicochemical properties such as solubility and stability were characterized.25,26 Amorphization is a way to alter the physicochemical properties of a drug molecule through the disruption of a crystal lattice, which often leads to a significant enhancement in solubility.27 Polymorphic forms with different surface morphologies and physicochemical properties exhibit improved inhalation properties.28,29
Further applications of crystal engineering include designing of multi-component crystals composed of different small molecules. Salt is a common crystal form for drug discovery and development. A drug molecule incorporating an ionizable functional group can form ionic interactions with acidic/basic metals or organic molecules through proton transfer.30,31 Salt forms are widely utilized in drug discovery, with approximately 50% of recently approved small molecules being in their salt form.32 However, the formation of salts is limited for acidic/basic drug molecules. Recently, co-crystals with improved physicochemical properties have emerged as a novel pharmaceutical form. Although co-crystals are composed of multi-component molecules without ionic interactions, various intermolecular forces such as hydrogen bonding, π–π interactions, halogen bonding, and van der Waals interactions are observed among different molecules in their crystal form.33 The pKa rule predicts co-crystal formation; molecules with ΔpKa smaller than 2 or 3 constitute an index.34 According to other reports, salt or co-crystal formation can occur when ΔpKa is <4 or ≥1.35,36 Proton transfer can occur in molecules with ΔpKa greater than 3–4, which are then converted to their salt form.
The multi-component amorphous form composed of small molecules is referred to as a co-amorphous (CA) system.37,38 Despite the lower physical stability of the amorphous state than that of the crystalline state, CA is stabilized via intermolecular interactions between the components. While inactive coformers are well combined for the salt/co-crystal/CA form, drug–drug multi-component crystals and amorphous forms have recently been the focus of combination therapy.39 These forms might have the potential for simultaneous delivery of drugs to the target site via the inhalation route without conventionally used complex medical devices and involve the formation of intermolecular interactions between drugs. However, there are only a few studies on the application of drug–drug multi-component crystals and amorphous forms for inhalation delivery. The CA system of ciprofloxacin–quercetin designed by spray drying showed co-deposition of the components deeper into the lungs.40 In our previous study, a levofloxacin (LVF) and theophylline (THE) CA system successfully co-delivered the drugs and showed improvement in inhalation properties.41 In another study, a co-crystal form of favipiravir and THE resulted in improved inhalation properties, but simultaneous delivery was not evaluated.42 Therefore, there is a requirement of the application of drug–drug multi-component crystals and amorphous forms in inhaled formulations for the co-delivery of drugs.
This study aimed to investigate the application of multi-component drug–drug crystal and amorphous forms in an inhaled formulation from the viewpoint of simultaneous delivery. The study focused on tuberculosis drugs as they are commonly used in combination therapy.43 LVF and 4-aminosalicylic acid (ASA) were employed as model tuberculosis drugs; previous studies on the inhalation properties of individual drugs are available.44–49 This study investigated the formation of LVF–ASA salt/co-crystal or co-amorphous forms. The obtained multi-component samples, LVF, and ASA were subjected to physicochemical investigations, such as thermal and spectroscopic analyses, and stability tests. Finally, the inhalation properties of both drugs were evaluated using an in vitro aerodynamic performance test.
Experimental
Materials
LVF (hemihydrate) and ASA were obtained from Ohara Pharmaceutical Co., Ltd (Shiga, Japan) and Tokyo Chemical Industry Co., Ltd (Tokyo, Japan), respectively. Purified water and acetonitrile (MeCN) were purchased from Otsuka Pharmaceutical Co., Ltd (Tokyo, Japan) and Kanto Chemical Industry Co., Ltd (Tokyo, Japan), respectively. Ethanol (EtOH), isopropyl alcohol (IPA), acetone, tetrahydrofuran (THF), and ethyl acetate (EtOAc) were purchased from Fujifilm Wako Pure Chemical Corporation (Osaka, Japan). Molecular weight and pKa values were calculated using ACD/Percepta software (ver. 14.3.0; Advanced Chemistry Development Inc., Toronto, Canada).Preparation of a physical mixture
A physical mixture (PM) of LVF and ASA in an equivalent molar ratio was prepared by mixing both using a mortar and pestle for 1 min.Jet-milling
To prepare the samples for the in vitro aerodynamic performance test, LVF, ASA, and PM were micronized using A-O jet-milling apparatus (Seishin Enterprise Co., Ltd, Tokyo, Japan). The samples were fed into the milling compartment by injecting and milling at pressures of 0.6 and 0.5 MPa, respectively. The crystal forms of the samples were compared before and after jet-milling. A jet-milled LVF from a previous study41 was used.Spray drying
Particle fabrication via spray drying using a mini spray dryer B-290 (Büchi, Flawil, Switzerland) was conducted to prepare multi-component formulations, according to previously established conditions.41,50,51 In brief, aqueous solutions containing drugs (4 mg mL−1) were introduced into the drying chamber at a rate of 5.5 mL min−1. The inlet temperature was maintained at 130 °C. The atomized nitrogen gas flowed through a 0.7 mm two-fluid spray nozzle, and the drying and airflow rates were set to 473 L h−1 and 35 m3 h−1, respectively. The amorphization of the spray-dried particles (SDPs) was verified using X-ray powder diffraction (XRPD).Screening study of multi-component crystals
The multi-component crystals of LVF–ASA were screened by slurry conversion and solvent evaporation using various organic solvents. Approximately 50 mg of the LVF–ASA mixture was weighed into each 1.6 mL glass vial (No. 01T; Maruemu Corporation, Osaka, Japan), and 1.5 mL each of EtOH, IPA, acetone, MeCN, THF, and EtOAc was added into each vial. Magnetic stirring was performed at 300 rpm and 25 °C for one week. The powder samples were obtained by filtration through an Omnipore® 0.45 μm polytetrafluoroethylene membrane (Merck KGaA, Darmstadt, Germany).The solvent evaporation method was applied as follows: approximately 5 mg of the LVF–ASA mixture was weighed into each 4 mL glass vial (S-1; Nichiden-Rika Glass Co., Ltd, Kobe, Japan), and 3.5 mL each of EtOH, IPA, acetone, MeCN, THF, and EtOAc was added into each vial and magnetically stirred at 600 rpm and 50 °C for 15 min using Cool Stirrer CPS-300 (Scinics Corporation, Tokyo, Japan). The solvent was evaporated at 1700 rpm and 40 °C for 1 h using a Genevac HT-8 Series II evaporation system (Genevac Ltd, Ipswich, UK). The sample prepared using IPA did not evaporate, and a further 1 h of evaporation was performed.
X-ray powder diffraction
The crystal forms of LVF, ASA, and LVF–ASA crystal or amorphous states were determined by XRPD using SmartLab software (Rigaku Corporation, Tokyo, Japan). The samples were placed into a hole (diameter, 2 mm; depth, 0.1 mm) on an aluminum plate. The samples were compressed using a spatula to flatten their surfaces. The instrument was equipped with a 9 kW rotating Cu Kα anode (λ = 0.154186 nm) and a HyPix-3000 detector. The distance between the sample and the detector was 331 mm, and the diffractometer was equipped with a cross-beam optic to provide a parallel beam. A PSC with 2.5° and a slit of 0.05 mm height and 0.5 mm width was used; thus, the beam footprint for all measurement configurations was smaller than that of the sample. No slits were used on the receiver side. The Cu Kα radiation point source was operated at 40 kV and 200 mA. The scan was performed from 3° to 33° (2θ) with β axis rotation (20 rpm) during data collection; the sampling step was 0.02° (2θ), and the count time was 40 s. The data were analyzed using the SmartLab Studio II X64 software (ver. 4.2.111.0; Rigaku Corporation, Tokyo, Japan).Thermogravimetry-differential thermal analysis
The thermal behavior (change in weight against temperature) was evaluated by thermogravimetry-differential thermal analysis (TG-DTA) using STA7200RV (Hitachi High-Tech Science Corporation, Tokyo, Japan). Approximately 1–5 mg of each sample was placed onto an aluminum pan followed by heating under ambient conditions to 350 °C at a rate of 10 °C min−1. The TA7000 standard analysis software (ver. 11.2; Hitachi High-Tech Science Corporation, Tokyo, Japan) was used to analyze the results. The top endothermic peaks were determined as the melting or dehydration point.Differential scanning calorimetry
The melting point (Tm) of the crystalline samples and changes in the heat flow profiles of the PM were measured by differential scanning calorimetry (DSC) using Discovery DSC (TA Instruments Japan, Tokyo, Japan). Nitrogen (50 mL min−1) was used as a purge gas, and the instrument was calibrated using an indium standard. Each sample (approximately 1–7 mg) was weighed onto a Tzero aluminum pan and sealed with a Tzero lid (TA Instruments Japan, Tokyo, Japan). The heat flow profiles were obtained by heating at a rate of 20 °C min−1 from 0 °C to the set point determined from the TG-DTA profile. To determine the glass transition temperature (Tg), modulated DSC (mDSC) was employed for amorphous samples. The sample was prepared according to the method described above and heated at a rate of 2 °C min−1 with a modulation amplitude of 0.2120 °C and a 40 s period. Reversing and non-reversing heat flow signals were obtained. DSC and mDSC data were analyzed using the TRIOS software (ver. 3.3.1; TA Instruments Japan, Tokyo, Japan). The top peaks were considered as the dehydration, melting, and crystallization points because continuous thermal events of LVF made the analysis of onset points difficult. For an amorphous sample, the midpoint Tg was obtained.X-ray powder diffraction under heating
The XRPD measurements under heating were performed using Rigaku RINT 2100 Ultima and XRD-DSC Thermo Plus II (Rigaku Corporation, Tokyo, Japan) instruments simultaneously. Approximately 5 mg of the sample was placed onto an XRD/DSC aluminum pan (7 × 7 × 0.3 mm) (Rigaku Corporation, Tokyo, Japan), and the surface was flattened. The conditions were as follows: heating at a rate of 10 °C min−1 from 30 °C to 250 °C, a voltage of 40 kV, and a current of 40 mA. The X-ray data ranged from 5° to 35° (2-theta) with a scan rate of 60° min−1 and a scan step of 0.02°. The resulting data were analyzed using the XRDDSC (ver. 2. 04; Rigaku Corporation).Single crystal X-ray diffraction
Single crystals of the LVF–ASA multi-component system were prepared by evaporating the solutions. LVF and ASA, each 20 μmol, were added into 3 mL of EtOH or IPA followed by the addition of 300 μL of water. The obtained solutions were placed in a 20 mL glass vial (SV-20; Nichiden Rika Glass Co. Ltd, Kobe, Japan). The crystal structure was analyzed by single crystal X-ray diffraction (SXRD) combined with thin-layer mirror monochromated Cu-Kα radiation (λ = 1.54184 Å). Rigaku XtaLAB P200 system with CrysAlisPro 1.171.39.46e software (Rigaku Oxford Diffraction) was used. The sample was mounted and examined at 230 K under dry nitrogen purge and re-examined after transformation to an anhydrate at 298 K. The direct SHELXT method was used to solve the crystal structure.49 All calculations for observed reflections [I > 2σ(I)] were performed with the CrysAlisPro 1.171.39.46e (Rigaku Oxford Diffraction), except for refinement, which was performed using the SHELXL program.52Fourier-transform infrared spectroscopy
The molecular states of the functional groups in each crystal were determined by Fourier-transform infrared spectroscopy (FT-IR) using VERTEX 70 instrument (Bruker Optics K.K., Tokyo, Japan). The range of wavenumber was 4000–500 cm−1 with 64 scans and 4 cm−1 resolution. Peak positions in the spectra were assigned using an ACD/Spectrus Processor (ver. 2015; Advanced Chemistry Development Inc.).Stability test
Solid-state stability of the LVF–ASA crystal and amorphous forms was studied at 40 °C periodically for one month. The samples were stored in a desiccator with silica-gel or a saturated solution of sodium chloride for maintaining 75% relative humidity (RH).Scanning electron microscopy
Scanning electron microscopy (SEM; Miniscope® TM3030, Hitachi High-Technologies Corporation, Tokyo, Japan) at an acceleration voltage of 15 kV was employed to characterize the surface morphology and particle shapes of the samples. Prior to SEM observation, each sample was sputter-coated with platinum for 2 min using a sputtering equipment set at 40 mA.Particle size distribution
The particle size distributions of the jet-milled samples were analyzed using HEROS & RODOS laser diffraction apparatus (Japan Laser Corporation, Tokyo, Japan) equipped with R3/R1 lens under the following conditions: time base, 100 ms; start, ch.15 ≥ 0.5%; and stop, 2 s ch.15 ≤ 0.5% or 10 s. The distribution methods were cascade (0), distribution, and vacuum pressures of 2 and 0.078 bar.Particle density
The particle densities of the LVF–ASA multi-component crystal and amorphous forms prepared by spray drying were measured using a Quantachrome Ultrapic 1200e (Anton Paar Japan K. K., Tokyo, Japan). The sample (0.15–0.2 g) was weighed into a large cell followed by volume measurement (cm3) under a helium gas flow. The density (g cm−3) was calculated from the values of weight and volume.In vitro aerodynamic performance test
The in vitro inhalation properties of each powder were assessed using a twin impinger (Copley Scientific Ltd, Nottingham, UK) following a previously established method.53,54 Hydroxypropyl methylcellulose capsules (size 3; Qualicaps, Nara, Japan) were used to encapsulate each sample. Inhalation properties were assessed using a Jethaler® device (Tokico System Solutions, Ltd, Kanagawa, Japan),55,56 focusing on the emitted dose (ED) and fine particle fraction (FPF), enabling comparison of the inhalation efficiency and performance among samples. The airflow through the twin impinger was set at 60 L min−1 for 5 s, employing stages 1 and 2 with an appropriate capsule, device, and throat configuration. Briefly, ED and FPF were calculated as previously described.57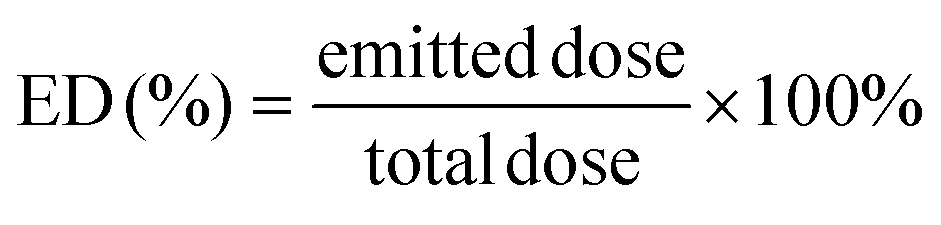 | (1) |
 | (2) |
The samples were evaluated three times, and the mean and standard deviation were calculated. The FPF for the CA system was compared against those of individual LVF and ASA, as well as the PM of ASA/LVF, using one-way analysis of variance followed by Tukey's test (*P < 0.05, **P < 0.01) to determine statistical significance.
High-performance liquid chromatography
The amount of LVF or ASA reaching each twin impinger compartment was determined using an SPD-20 A HPLC detector (Shimadzu Co., Ltd, Kyoto, Japan) equipped with an LC-20AD pump. COSMOSIL 5C18-MS-II was used at 40 °C as an HPLC column (5 μm, 4.6 × 150 mm; Nacalai Tesque Inc., Kyoto, Japan) for drug separation. The mobile phase, comprising methanol and 0.1% trifluoroacetic acid aqueous solution mixed at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75 w/w, was used at a flow rate of 1 mL min−1, and the injection volume was 10 μL. Absorbance was measured at 289 nm using a UV detector. The retention times of LVF and ASA were 7.6 and 3.7 min, respectively.
:75 w/w, was used at a flow rate of 1 mL min−1, and the injection volume was 10 μL. Absorbance was measured at 289 nm using a UV detector. The retention times of LVF and ASA were 7.6 and 3.7 min, respectively.
Results and discussion
Physicochemical properties of LVF and ASA
Fig. 1 presents the chemical structures of LVF and ASA. LVF contains carboxylic acid, carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, halogen connected to an aromatic ring, and methylpiperazine. ASA contains carboxylic acid and amine connected to the aromatic ring. LogP for LVF and ASA was calculated to be 0.17 and 1.46, respectively. LogP of 2–3 is recommended for drug molecules as a higher logP often causes solubility issues.58 Both compounds have preferable logP, and solubility issues have not been reported. The pKa values of LVF were calculated as follows: carboxylic acid (red circle) has a pKa value of 5.3 (acidic), whereas a pKa value of 7.4 (basic) was observed at the nitrogen in methylpiperazine (blue circle) connected to methyl. Both acidic and basic pKa values were also observed for ASA, but the basic pKa corresponding to the amine (blue circle) connected to the aromatic ring was weak at 2.2. The acidic pKa of carboxylic acid was 3.6. The ΔpKa value between the carboxylic acid of ASA and the methylpiperazine of LVF was 3.8. As mentioned in the Introduction, ΔpKa > 3–4 is an index for the formation of salts,34–36 suggesting the possibility of formation of an LVF–ASA salt. However, their multi-component crystal forms have not yet been reported.
O, halogen connected to an aromatic ring, and methylpiperazine. ASA contains carboxylic acid and amine connected to the aromatic ring. LogP for LVF and ASA was calculated to be 0.17 and 1.46, respectively. LogP of 2–3 is recommended for drug molecules as a higher logP often causes solubility issues.58 Both compounds have preferable logP, and solubility issues have not been reported. The pKa values of LVF were calculated as follows: carboxylic acid (red circle) has a pKa value of 5.3 (acidic), whereas a pKa value of 7.4 (basic) was observed at the nitrogen in methylpiperazine (blue circle) connected to methyl. Both acidic and basic pKa values were also observed for ASA, but the basic pKa corresponding to the amine (blue circle) connected to the aromatic ring was weak at 2.2. The acidic pKa of carboxylic acid was 3.6. The ΔpKa value between the carboxylic acid of ASA and the methylpiperazine of LVF was 3.8. As mentioned in the Introduction, ΔpKa > 3–4 is an index for the formation of salts,34–36 suggesting the possibility of formation of an LVF–ASA salt. However, their multi-component crystal forms have not yet been reported.
 | ||
| Fig. 1 Chemical structures of levofloxacin (LVF) and 4-aminosalicylic acid (ASA). The red and blue circles represent the acidic and basic functional groups, respectively. | ||
Fig. 2 shows the XRPD profiles of LVF and ASA. Representative peaks appeared at 6.6°, 9.7°, 13.1°, 15.8°, 19.4°, 20.0°, 26,4°, and 26.7° for LVF, indicating the reported hemihydrate form.59 In the XRPD profile of ASA, the peaks were observed at 7.2°, 12.6°, 13.0°, 14.5°, 16.9°, 25.9°, and 26.7°, which agreed with that of the anhydrate form.60
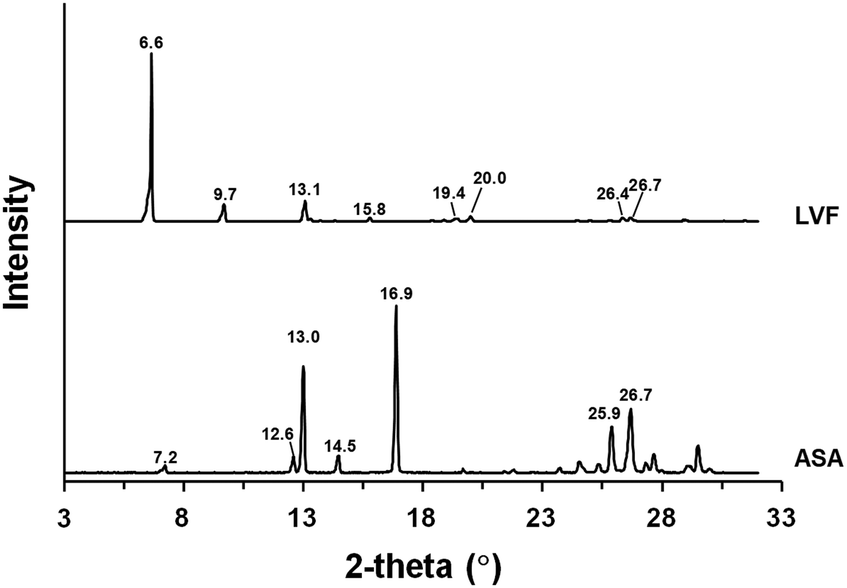 | ||
| Fig. 2 XRPD profiles of (a) levofloxacin (LVF) and 4-aminosalicylic acid (ASA). The values represent the 2-theta (°) of each diffraction peak. | ||
Fig. 3a and b show the TG-DTA profiles of LVF and ASA, respectively. LVF showed two representative endothermic peaks; the first one was at 81.1 °C with weight reduction by 2.5%, corresponding to the loss of half molar ratio of water. The second peak was at 227.1 °C, which reflected the melting event.42 The melting point of ASA was characterized at 143.1 °C.61Fig. 3c shows the DSC profiles of LVF, ASA, and LVF–ASA PM. An extended dehydration event was observed at approximately 50–110 °C for LVF. Complex melting events were observed between 227 °C and 240 °C. These endotherms and exotherms reflected crystal transformation from the anhydrate form following dehydration of the hemihydrate form to metastable and stable forms.62 The melting event of ASA at 145.1 °C was similar to the result of TG-DTA. LVF–ASA PM showed an altered temperature profile compared with that of individual drugs. The dehydration event shifted to a lower temperature. A broad and weak endotherm was observed, with three peaks at 151.7 °C, 164.4 °C, and 172.6 °C. It is well known that changes in temperature profiles, such as reduction/broadening/weakening of melting point and formation of a eutectic mixture by mixing different molecules, are characteristic features of salt/co-crystal formation.63,64
 | ||
| Fig. 3 TG-DTA profiles of (a) levofloxacin (LVF) and (b) 4-aminosalicylic acid (ASA), and (c) DSC profiles of LVF, ASA, and their physical mixture (PM). The values represent the temperature (°C) on endothermic or exothermic peaks. | ||
Screening of LVF–ASA crystals
Solvent-mediated crystal transformation, such as slurry under suspension, is a major method for screening salts or co-crystals.65,66 LVF, ASA, and PM were subjected to slurry conversion in EtOH, IPA, MeCN, acetone, THF, and EtOAc. LVF and ASA showed no change in crystal form after slurry conversion. The XRPD profiles of LVF–ASA after slurry conversion are shown in Fig. 4. The typical XRPD peaks of LVF and ASA (Fig. 2) disappeared for all samples. Novel peaks were observed at 4.3°, 8.6°, 10.1°, 10.7°, 11.8°, 13.2°, 17.3°, 18.2°, and 25.4° in the sample utilizing EtOH. This XRPD pattern was confirmed in samples utilizing MeCN and EtOAc. Another crystal form appeared in samples utilizing IPA, acetone, and THF with new peaks at 6.3°, 7.2/7.3°, 9.0/9.1°, 11.5/11.6°, 14.2°, 14.6°, 15.8/15.9°, 17.8°, 18.2°, 18.5°, 25.4/25.5°, and 26.3°. These XRPD patterns did not agree with the polymorphic forms of each drug,59,60,62 and the results strongly suggest the formation of novel crystal polymorphs of the LVF–ASA multi-component system. The crystal forms obtained from EtOH/MeCN/EtOAc and IPA/acetone/THF were represented as crystal-I and crystal-II, respectively. | ||
| Fig. 4 XRPD profiles of the levofloxacin (LVF)-4-aminosalicylic acid (ASA) mixture after slurry conversion in ethanol (EtOH), isopropyl alcohol (IPA), acetonitrile (MeCN), acetone, tetrahydrofuran (THF), and ethyl acetate (EtOAc). The values represent the 2-theta (°) of each diffraction peak. | ||
Fig. S1† shows the XRPD profiles of LVF–ASA after solvent evaporation, and the screening studies are summarized in Table 1. Solvent evaporation also induced the formation of new LVF–ASA crystals. All samples except IPA resulted in the formation of crystal-I, which is speculated as a relatively stable form. IPA afforded crystal-II from both slurry conversion and solvent evaporation. Solvent-dependent generation of polymorphic forms is well known. In the case of baloxavir marboxil, a metastable form is typically induced by only MeCN aqueous solution after slurry conversion via intermolecular interaction with the solvent; however, stable forms were generated with other solvents.67 LVF–ASA crystal-I and crystal-II should have been induced via different interactions with each organic solvent.
| Slurry conversion | Solvent evaporation | |
|---|---|---|
| EtOH | Crystal-I | Crystal-I |
| IPA | Crystal-II | Crystal-II |
| MeCN | Crystal-I | Crystal-I |
| Acetone | Crystal-II | Crystal-I |
| THF | Crystal-II | Crystal-I |
| EtOAc | Crystal-I | Crystal-I |
Thermal properties of LVF–ASA crystals
The physicochemical properties of crystal-I and crystal-II were investigated by thermal analysis. Fig. 5a shows the TG-DTA profile of crystal-I. A small endothermic peak appeared at 56.7 °C for crystal-I, along with weight reduction by 3.4%, suggesting dehydration or desolvation. As shown in Table 1, crystal-I was obtained from various solvents and assumed to be in the hydrate form. A subsequent melting endotherm was observed at 186.0 °C with significant weight reduction corresponding to thermal degradation. In the case of crystal-II, dehydration/desolvation was not suggested, and the Tm was observed at 191.3 °C (Fig. 5b). The difference in the Tm between the crystal forms suggests that crystal-II is more stable than dehydrated crystal-I. | ||
| Fig. 5 TG-DTA profiles of (a) crystal-I and (b) crystal-II, and (c) DSC profiles of crystal-I and crystal-II. The values represent the temperature (°C) on endothermic or exothermic peaks. | ||
Fig. 5c shows the DSC profiles of crystal-I and crystal-II. The temperature profile showed a broad peak from approximately 20 to 100 °C for crystal-I, and the top of peak was at 73.8 °C. The second endotherm, which reflects the melting event, occurred at 185.7/187.8 °C. The Tm of crystal-II was 192.4 °C. The Tm values of crystal-I and crystal-II were similar to the results of the TG-DTA analysis. The exothermic peaks were at 235.4 °C and 234.9 °C for crystal-I and crystal-II, respectively, but thermal degradation was observed at these temperatures in TG-DTA profiles (Fig. 5a and b). The disappearance of the original Tm values of LVF and ASA and the generation of a novel and single Tm for both crystal-I and crystal-II strongly support the possibility of the formation of multi-component crystal forms.
The thermal events observed in the DSC profiles were further investigated using XRPD measurements under heating conditions. Fig. 6a shows the XRPD profiles of crystal-I under heating at approximately 30–240 °C. The profile in the range 30.3–35.8 °C agreed with that of crystal-I shown in Fig. 4, and no change was observed until 77.3–83.2 °C. However, a novel peak at 25.7° was observed at 175.3–180.8 °C. This reflects the transformation of the hydrate to the anhydrate form because dehydration was characterized in TG-DTA and DSC profiles (Fig. 5a and c). This anhydrate form was represented as crystal-III. All the peaks weakened at 186.3–191.5 °C. Different XRPD patterns were observed at 197.4–203.1 °C and further transformed at 219.8–225.6 °C. These patterns were similar to the β and α forms of LVF, respectively.56 In the TG-DTA profile, ASA showed thermal degradation at 143.1 °C after melting, but LVF did not (Fig. 3a and b). It can be considered that crystal-I melted at approximately 180–190 °C according to the DSC profile (Fig. 5c), and ASA rapidly degraded. Subsequently, LVF crystallized to the β form and transformed to a more stable α form. The preparation of crystal-III has also been attempted. Crystal-I was heated at 120 °C for 5 min or 60 °C under reduced pressure under vacuum overnight. Although the TG-DTA profile (Fig. 5a) suggested dehydration of crystal-I over 60 °C, the resulting crystal form showed crystal-I from the XRPD profile. Reversible hydration–rehydration depending on the temperature and/or humidity is well known for crystalline materials.68–70 A crystal lattice of a hydrate is maintained after dehydration by heating and/or drying, and subsequent cooling and/or humidification induces rehydration via intermolecular interaction between water and the host-molecule.69,70
 | ||
| Fig. 6 XRPD profiles of (a) crystal-I and (b) crystal-II under heating conditions. The values represent the 2-theta (°) of each diffraction peak. | ||
The XRPD profile of crystal-II after heating is shown in Fig. 6b. The initial profile measured at 31.4–37.0 °C agreed with that of crystal-II (Fig. 4). However, new peaks at 6.8°, 12.2°, 15.2°, and 19.0° were confirmed at 150.2–155.6 °C. These peaks rapidly grew at 188.6–193.4 °C, suggesting a crystal transformation. All peaks disappeared at 193.4–199.6 °C, which corresponded to the Tm in the DSC profile (Fig. 5c). Further heating induced generation of the β form of LVF at 199.7–205.2 °C and transformation to the α form at 227.5–233.0 °C similar to crystal-I. XRPD measurement under heating could reveal the thermal events observed in the DSC profile, and a new crystal form transformed from crystal-II was discovered, which was represented as crystal-IV.
To obtain crystal-IV, heating of crystal-II at 170 °C for 1 min was attempted. Fig. S2† shows a comparison between the XRPD profiles of crystal-II and crystal-IV. The difference in the peak positions between the polymorphs was characterized in the range of 6–8°. The two peaks appeared at 6.3° and 7.3° for crystal-II, but at 6.8° and 7.2° for crystal-IV. The preparation of crystal-IV was successful because its XRPD pattern agreed with the observed profile of heated crystal-II at 188.6–193.4°.
Preparation of amorphous LVF–ASA
Multi-component amorphous LVF–ASA was prepared by spray drying. The SEM image shows a spherical morphology (Fig. S5†). The particle sizes, D10/D50/D90, were 0.6/2.2/4.8 μm (Table S1†), which were not significantly different from SDPs used for previous inhalation studies.41,53–56 Spray drying successfully led to the amorphization of LVF–ASA, with the disappearance of all XRPD peaks (Fig. 10a and b). The mDSC profiles of SDPs are shown in Fig. 7. In reverse heat flow, a single Tg was observed at 107.8 ± 1.0 °C. It was reported that the Tg of LVF was 83.0 °C.41 Due to the high crystallization tendency, the Tg of ASA could not be determined. Disappearance of native Tg of the components and generation of a single Tg is a characteristic feature of CA formation via intermolecular interactions.37–41 The SDPs of LVF–ASA should form CA. The non-reversing heat flow displayed other thermal events. Exothermic peaks were shown at 112.0 ± 0.1 °C and 126.3 ± 0.0 °C, suggesting crystallization following glass transition. The endothermic and exothermic peaks at 162.6 ± 0.1 °C and 198.3 ± 1.0 °C reflected melting and recrystallization, respectively. | ||
| Fig. 7 mDSC profiles of LVF–ASA spray-dried particles (SDPs). The values in the reversing and non-reversing heat flows represent the glass transition temperature and temperatures on endothermic or exothermic peaks, respectively. | ||
The XRPD profiles of CA under heating conditions are shown in Fig. S3.† The halo pattern of CA was observed at 31.1–36.5 °C; however, generation of small peaks from 10–15° and at 25.7° was characterized at 131.5–137.4 °C. These peaks grew at 154.2–159.6 °C, and the pattern agreed with that of crystal-III generated by the dehydration of crystal-I (Fig. 6a). Further thermal events were similar to those of crystal-I and crystal-II: melting (181.5–187.0 °C), crystallization of the β form of LVF (192.7–198.6 °C), and transformation to the α form (215.2–220.5 °C).
Single crystal X-ray diffraction of LVF–ASA crystals
SXRD was performed to determine the crystal form of LVF–ASA. Crystal-I microcrystals were successfully obtained, but crystal-II microcrystals could not be obtained due to poor crystal growth. Crystal-III was obtained only upon heating of crystal-I, and crystal-IV could only be prepared from crystal-II as described above. The lattice parameters of crystal-I are listed in Table 2. In the asymmetric unit, eight LVF, eight ASA, and twelve water molecules existed, with only the disorder of small parts of LVF. Fig. 8 shows representative intermolecular interactions and bond distances. The carboxylic acid (OH) of ASA interacted with the methylpiperazine (N) of LVF with an OH–N bond distance of 1.72 Å. The bond distances of CO (1.25 Å) and C–O (1.27 Å) in the carboxylic acid of ASA were similar. On the other hand, the bond distances of CO (1.22 Å) and C–O (1.32 Å) in the carboxylic acid of LVF were not equal. In the co-crystal, the ratio of bond distances of CO to C–O in the carboxylic acid was not equal owing to the non-deprotonation of OH.71 SXRD analysis revealed a hydrated salt form with deprotonation of the carboxylic acid of ASA in crystal-I.
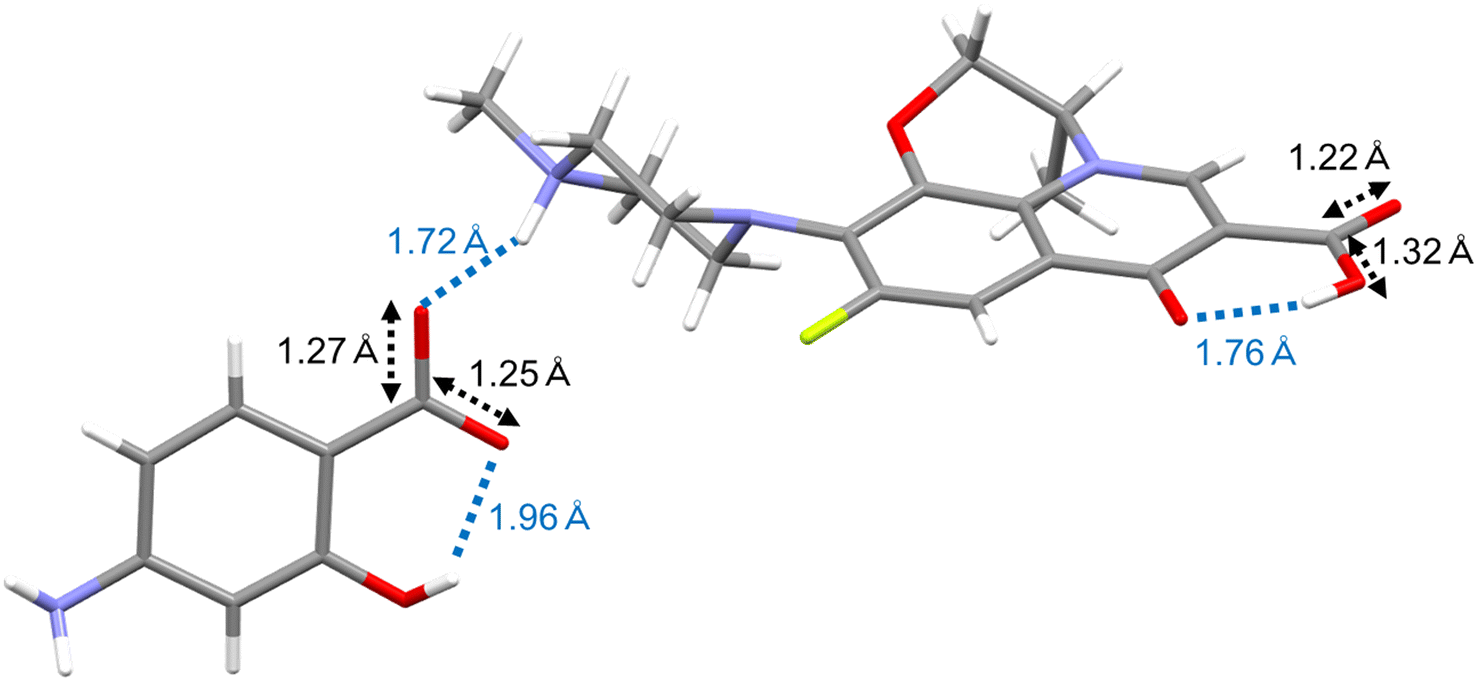 | ||
| Fig. 8 Crystal structure of crystal-I analyzed by single crystal X-ray diffraction. | ||
| Crystal-I | |
|---|---|
| Formula | C25H25.5FN4O8.5 |
| Fw | 531.90 |
| Temperature (K) | 230 |
| Crystal system | Monoclinic |
| Space group | P21 |
| a (Å) | 9.3759(2) |
| b (Å) | 40.6565(10) |
| c (Å) | 26.9361(8) |
| α (°) | 90 |
| β (°) | 91.262(2) |
| γ (°) | 90 |
| V (Å3) | 10265.3(5) |
| Z | 16 |
| ρ calc g cm−3 | 1.39 |
| μ/mm−1 | 0.941 |
| F(000) | 4488 |
| Radiation | CuKα (λ = 1.54184) |
| Reflections collected | 113151 |
| Independent reflections | 38806 |
| Data/restraints/parameters | 38806/1/2717 |
| Goodness-of-fit on F2 | 1.004 |
| Final R indexes [I ≥ 2σ (I)] | R 1 = 0.0699, wR2 = 0.1901 |
| Final R indexes [all data] | R 1 = 0.1156, wR2 = 0.2286 |
| Flack parameter | −0.05(7) |
| CCDC number | 2410276† |
Infrared spectra of the LVF–ASA crystal and amorphous forms
The molecular states of LVF and ASA in each form were also investigated by FT-IR analysis. Fig. 9 shows the FT-IR spectra of LVF, ASA, and the LVF–ASA crystal and CA forms. The FT-IR peaks of LVF were assigned to the functional groups according to a previous report.72 The O–H stretching appeared at 3393 cm−1 and 3246 cm−1. The peaks observed in the range of 2937–2802 cm−1 indicated symmetric or C–H stretching or asymmetric C–H stretching of the methylpiperazine ring. The peak at 1720 cm−1 was characteristic of the CO stretching of the carboxylic acid. The peak at 1618 cm−1 represented CC stretching and C–C stretching of the aromatic ring. The aromatic ring C–C stretching and CH2 scissoring of the methylpiperazine were characterized by peaks at 1539 cm−1/1518 cm−1 and 1468 cm−1/1439 cm−1, respectively.
 | ||
| Fig. 9 FT-IR spectra of levofloxacin (LVF), 4-aminosalicylic acid (ASA), crystal-I, crystal-II, crystal-IV, and co-amorphous (CA) forms. | ||
The FT-IR peaks of ASA were assigned according to previous reports.73,74 The peaks at 3493 cm−1 and 3385 cm−1 were representative of O–H/N–H stretching. Broadening of the spectrum, including the peaks at 2978 cm−1 and 2835 cm−1, was observed in the range of 3200–2700 cm−1, which corresponded to O–H stretching generated from an intramolecular interaction between the hydroxyl and carboxylic acid. The CO stretching of the carboxylic acid appeared as a broadened pattern at approximately 1680–1620 cm−1. The peaks at 1609 cm−1, 1520 cm−1, and 1441 cm−1 corresponded to C–C stretching of the aromatic ring.
The FT-IR spectral patterns of LVF–ASA crystals were different from those of individual LVF and ASA, suggesting a change in the molecular state. The peaks at 3493 cm−1 and 3385 cm−1 of ASA shifted to lower wavenumbers for crystal-I (3447 cm−1 and 3367 cm−1), crystal-II (3460 cm−1 and 3354 cm−1), and crystal-IV (3470 cm−1 and 3354 cm−1). In this range, the peak at 3393 cm−1 of LVF merged with the peaks of ASA, and the peak at 3426 cm−1 showed a smaller shift in crystal-I (3223 cm−1), crystal-II (3225 cm−1), and crystal-IV (3240 cm−1). The peaks in the range of 3200–2700 cm−1 for both LVF and ASA significantly weakened and induced a greater shift in crystal-I, crystal-II, and crystal-IV. These spectral changes suggest changes in the molecular states of the carboxylic acid and methylpiperazine of LVF and the aromatic ring of ASA.
The CO stretching of LVF at 1720 cm−1 showed a smaller shift in crystal-I (1703 cm−1), crystal-II (1713 cm−1), and crystal-IV (1713 cm−1). However, C–C stretching of the aromatic ring at 1618 cm−1 appeared in similar positions at 1620 cm−1 for crystal-I, crystal-II, and crystal-IV. The aromatic ring C–C stretching and CH2 scissoring of the methylpiperazine in the range of 1540–1430 cm−1 significantly weakened or merged with the peaks of ASA in the multi-component crystal forms. The CO stretching of ASA at approximately 1680–1620 cm−1 shortened the broadened range. The peak at 1609 cm−1 of ASA disappeared, and a new peak was observed at 1580 cm−1 (crystal-I), 1582 cm−1 (crystal-II), and 1585 cm−1 (crystal-IV). The other peaks of the aromatic ring (1520 cm−1 and 1441 cm−1) shifted and merged with the peaks of LVF. LVF–ASA crystals showed peaks in the following range: crystal-I (1529/1477/1446 cm−1), crystal-II (1526/1446 cm−1), and crystal-IV (1522/1445 cm−1). These spectral changes suggest changes in the molecular states of the carboxylic acid and aromatic ring of LVF and the aromatic ring of ASA.
It is well known that the FT-IR spectra are different among polymorphs and salt/co-crystals formed via changes in intramolecular/intermolecular interactions.75,76 As described in “Physicochemical properties of LVF and ASA”, ΔpKa between the carboxylic acid of ASA and methylpiperazine of LVF suggests the possibility of salt formation. From this viewpoint, the changes in the FT-IR spectra of LVF/ASA were discussed. Significant spectral changes such as peak shift and shape change of the methylpiperazine in 3400–2700 cm−1 of LVF were caused by the formation of salt with citric acid.75 In addition, ASA alginate showed changes from native ASA: the shape change of the CO of ASA at approximately 1680–1620 cm−1 and a smaller peak shift of the aromatic ring at 1609 cm−1. These changes are common in LVF–ASA crystals prepared in this study. Furthermore, SXRD analysis revealed that crystal-I was in its salt form via ionic interaction between ASA and LVF. Hence, it is likely that the carboxylic acid (OH) of ASA and the methylpiperazine (N) of LVF also formed salt in crystal-II and crystal-IV via ionic interactions. The CA spectrum showed a pattern similar to that of salt crystals, although some peaks changed in shape, suggesting that the CA state was stabilized by salt formation. Crystal-III could not be obtained under ambient conditions; however, it was obtained by heating crystal-I (Fig. 6a). Furthermore, crystal-III was reversibly transformed to crystal-I, suggesting a salt form. Hence, LVF–ASA crystal-I, crystal-II, crystal-III, and crystal-IV were defined as salt crystal (SC)-I (hydrate), SC-II, SC-III, and SC-IV, respectively.
Stability of LVF and ASA salt forms
The isothermal stabilities of LVF–ASA salt crystals and CA were investigated. Fig. 10a shows the XRPD profiles before and after storage at 40 °C with silica gel. LVF–ASA SC-I (hydrate), SC-IV, and CA maintained their forms after storage. However, the peak at 6.3° of SC-II split after storage, suggesting the generation of SC-IV. In contrast to stability under dry conditions, humidity induced transformation of the samples. Fig. 10b shows the XRPD profiles before and after storage at 40 °C/75% RH. SC-I (hydrate) showed higher stability without transformation, but all other forms rapidly converted to SC-I (hydrate) on day 1, suggesting that humidity strongly induced hydration of both salt crystals and CA. SC-I (hydrate) was only stable under both dry and humidified conditions and subjected to further study. | ||
| Fig. 10 XRPD profiles of salt crystal (SC)-I, SC-II, SC-IV, and co-amorphous (CA) forms after storage at (a) 40 °C with silica-gel and (b) 40 °C/75% RH. | ||
The phase diagram of the crystal transformation among LVF–ASA salt crystals and CA is shown in Fig. 11.
 | ||
| Fig. 11 Phase diagram of crystal transformation for salt crystals (SCs) and co-amorphous (CA) forms. | ||
Jet-milling of LVF and ASA
LVF and ASA were micronized by jet-milling prior to the in vitro aerodynamic test. The crystal forms of LVF–ASA did not change after jet-milling (Fig. S4†). LVF–ASA SC-I (hydrate) was obtained from the storage of SDPs at CA at 40 °C/75% RH overnight. Fig. S5† shows SEM images of the micronized LVF, ASA, and SC-I (hydrate). The particle sizes, D10/D50/D90, of LVF, ASA, and SC-I (hydrate) were 0.6/2.5/5.0 μm, 0.7/2.4/4.7 μm, and 0.8/4.3/7.0 μm, respectively. Their particle sizes were comparable to that of CA. The span was calculated in the range of 1.5–1.9, which was similar among the samples, indicating their appropriateness as dry powder-inhaled particles.41,53–56 Jet-milling reduces the geometric particle size of drugs; however, it is generally known that in dry milling methods like jet-milling, particle aggregation tends to occur below 5 μm.77,78 As a result, the current particle size is close to the lower limit achievable by this process for dry powder inhaler formulations.In vitro aerodynamic properties of LVF–ASA crystal and amorphous forms
The inhalation properties of each sample were compared using twin impinger. The aerodynamic diameter (da) was calculated using the following equation:79,80 | (3) |
The deposition rate of each drug in PM varied significantly in CA and SC-I (hydrate) (Fig. 12a, b and Table S2†). The FPF values are shown in Fig. 13a, b and Table S3.† The FPF value of untreated ASA was 13.3 ± 3.4%, whereas that of untreated LVF was 1.1 ± 0.2%. The FPF value of LVF and ASA in PM was 0.8 ± 0.2% and 14.1 ± 0.8%, respectively. In both samples, the FPF value of ASA was higher than that of LVF. Interestingly, LVF and ASA in both LVF–ASA CA and SC-I (hydrate) demonstrated synchronized delivery performance; the FPF values of LVF/ASA in CA and SC-I (hydrate) were 20.0 ± 4.7/20.6 ± 4.5% and 25.4 ± 4.8/29.9 ± 2.9%, respectively. Notably, the FPF value of LVF significantly improved when it was co-formulated with ASA. A comparison of the FPF values of CA and SC-I (hydrate) formulations revealed a notable improvement in the FPF values of the crystallized salt. In a previous study, the inhalation properties of THE-LVF co-amorphous inhalation formulations also achieved synchronized delivery to the lungs.41 LVF, with its relatively hydrophobic properties, showed improvement in inhalation performance. A similar trend was observed for LVF–ASA salt forms. The inhalation properties of SC-I (hydrate) were superior to those of CA. This may be attributed to the fact that the crystalline structure facilitates better dispersibility in the inhalation device and inhibits aggregation between particles compared with amorphous powders.81,82
 | ||
| Fig. 12 In vitro aerodynamic performance of levofloxacin (LVF)/4-aminosalicylic acid (ASA), physical mixture (PM), co-amorphous (CA) forms, and salt crystal (SC)-I (hydrate): (a) LVF and (b) ASA. The error bars represent the standard deviation (n = 3). | ||
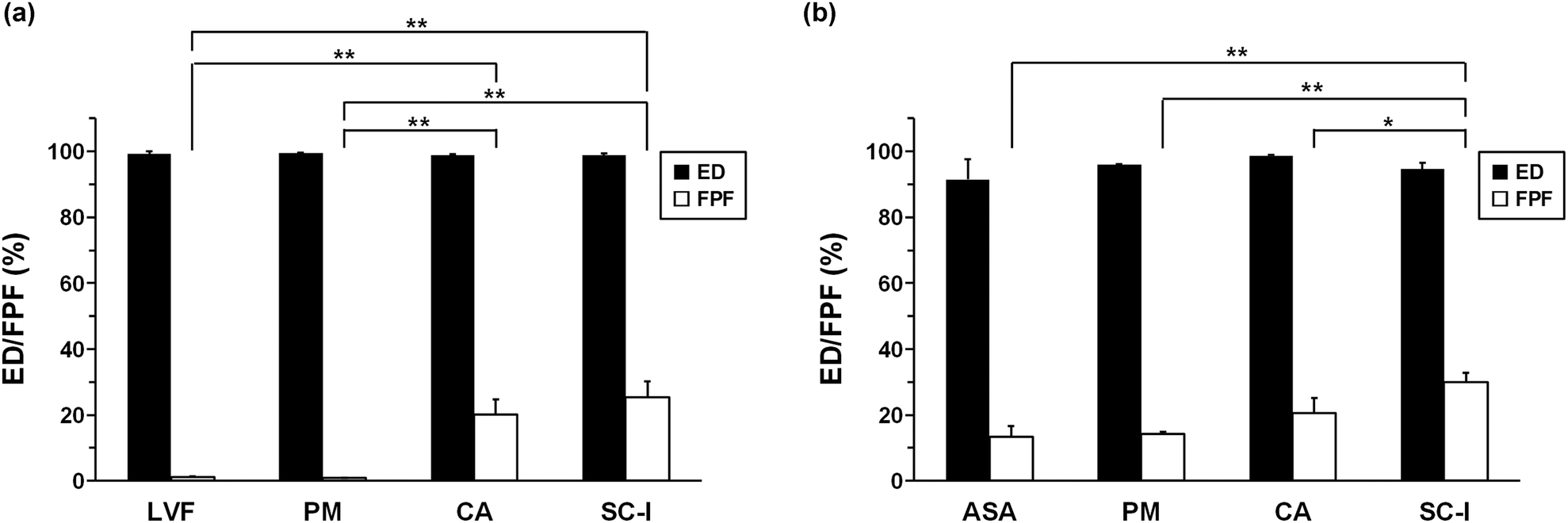 | ||
| Fig. 13 ED (%) and FPF (%) of levofloxacin (LVF)/4-aminosalicylic acid (ASA), physical mixture (PM), co-amorphous (CA) forms, and salt crystal (SC)-I (hydrate): (a) LVF and (b) ASA. The error bars represent standard deviation (n = 3). The FPF data of SDPs were compared with that of PM by Tukey's test (*P < 0.05, **P < 0.01). | ||
Conclusions
Multi-component crystal and amorphous forms were designed by combining LVF and ASA for co-delivery to the lungs. Solvent evaporation and slurry conversion led to the formation of LVF–ASA SC polymorphs. Anhydrate metastable and stable forms were formed, which were converted to the hydrate form after storage. The CA form of LVF–ASA was obtained by spray drying. The SC polymorphs and CA showed similar FT-IR profiles, suggesting that LVF and ASA formed salts in crystal and CA forms. Both SC and CA significantly improved the FPF values compared with individual drugs or PM as determined by in vitro aerodynamic performance. Furthermore, LVF and ASA were successfully co-delivered to the lung compartments. This study demonstrated that multi-component drug–drug crystal and amorphous forms are promising as inhalation systems for simultaneous delivery without utilizing any specialized device. The limitation of using a salt/co-crystal is that the stoichiometry of components is fixed, and the administration of LVF and ASA to the lungs is not approved. Hence, further investigation is required to overcome this issue based on dose adjustment from the viewpoint of pharmacological and toxicological effects, such as non-clinical and clinical trials.Author contributions
H.U.: conceptualization, methodology, data curation, formal analysis, writing – original draft, writing – editing & reviewing, project administration, and supervision; J.T.: methodology, data curation, and formal analysis; T.M.: methodology, data curation, and formal analysis; Y.N.: data curation and investigation; P.M.: data curation and investigation; Y.H.: methodology, data curation, and investigation; H.U.: writing – editing & reviewing; Y.T.: writing – editing & reviewing; K.K.: conceptualization, formal analysis, writing – editing & reviewing, project administration, and supervision.Data availability
The data included in this article will be available on request to the corresponding authors.Conflicts of interest
The authors H.U., J.T., and T.M. are employed by Shionogi & Co., Ltd.Acknowledgements
The authors thank Tokico System Solutions, Ltd for lending the Jethaler® device for evaluating inhalation properties. This study was partially supported by the JSPS KAKENHI grant (no. 23K06245, K. K.).References
- N. Fine-Shamir and A. Dahan, Expert Opin. Drug Delivery, 2024, 21, 13–29, DOI:10.1080/17425247.2023.2298247.
- J. Li, Y. Wang and D. Yu, AAPS PharmSciTech, 2023, 24, 175, DOI:10.1208/s12249-023-02622-8.
- B. Rocha, L. A. de Morais, M. C. Viana and G. Carneiro, Expert Opin. Drug Discovery, 2023, 18, 615–627, DOI:10.1080/17460441.2023.2211801.
- S. S. Gaikwad, A. L. Zanje and J. D. Somwanshi, Int. J. Pharm., 2024, 652, 123856, DOI:10.1016/j.ijpharm.2024.123856.
- J. Li, R. Ge, K. Lin, J. Wang, Y. He, H. Lu and H. Dong, Small, 2024, 20, 2306222, DOI:10.1002/smll.202306222.
- A. Biswas, A. D. Choudhury, A. C. Bisen, S. Agrawal, S. N. Sanap, S. K. Verma, A. Mishra, S. Kumar and R. S. Bhatta, AAPS PharmSciTech, 2023, 24, 217, DOI:10.1208/s12249-023-02673-x.
- S. Magramane, K. Vlahović, P. Gordon, N. Kállai-Szabó, R. Zelkó, I. Antal and D. Farkas, Pharmaceuticals, 2023, 16, 1658, DOI:10.3390/ph16121658.
- S. Stegemann, E. Faulhammer, J. T. Pinto and A. Paudel, Int. J. Pharm., 2022, 614, 121445, DOI:10.1016/j.ijpharm.2021.121445.
- M. T. Leving, S. Bosnic-Anticevich, J. van Cooten, J. C. de Sousa, B. Cvetkovski, R. Dekhuijzen, L. Dijk, M. G. Pardo, A. Gardev, R. Gawlik, I. van der Ham, Y. Janse, F. Lavorini, T. Maricoto, J. Meijer, B. Metz, D. Price, M. Roman-Rodriguez, K. Schuttel, N. Stoker, I. Tsiligianni, O. Usmani, R. Emerson-Stadler and J. W. H. Kocks, NPJ Prim. Care Respir. Med., 2022, 32, 59, DOI:10.1038/s41533-022-00318-3.
- B. Chaurasiya and Y. Y. Zhao, Pharmaceutics, 2020, 13, 31, DOI:10.3390/pharmaceutics13010031.
- D. de la Rosa-Carrillo, G. Suárez-Cuartín, O. Sibila, R. Golpe, R. M. Girón and M. A. Martínez-García, J. Clin. Med., 2023, 12, 3577, DOI:10.3390/jcm12103577.
- N. Nainwal, Y. Sharma and V. Jakhmola, Tuberculosis, 2022, 135, 102228, DOI:10.1016/j.tube.2022.102228.
- A. G. Dos Santos, J. C. Bayiha, G. Dufour, D. Cataldo, B. Evard, L. C. Silva, M. Deleu and M. P. Mingeot-Leclercq, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1930–1940, DOI:10.1016/j.bbamem.2017.06.010.
- N. J. Caggiano, M. S. Armstrong, J. S. Georgiou, A. Rawal, B. K. Wilson, C. E. White, R. D. Priestley and R. K. Prud'homme, Mol. Pharm., 2023, 20, 4546–4558, DOI:10.1021/acs.molpharmaceut.3c00240.
- W. H. van Geffen, D. J. Tan, J. A. Walters and E. H. Walters, Cochrane Database Syst. Rev., 2023, 12, CD011600, DOI:10.1002/14651858.CD011600.pub3.
- C. Zhang, D. D'Angelo, F. Buttini and M. Yang, Adv. Drug Delivery Rev., 2024, 204, 115146, DOI:10.1016/j.addr.2023.115146.
- M. Mahmoud and Y. Tan, Expert Rev. Anti-Infect. Ther., 2023, 21, 863–870, DOI:10.1080/14787210.2023.2240022.
- T. T. Brehm, N. Köhler, S. Schmiedel, E. Terhalle, J. Martensen, B. Kalsdorf, J. Kandulla, J. Heyckendorf, M. Kuhns, I. Friesen and C. Lange, Inn. Med., 2023, 64, 701–707, DOI:10.1007/s00108-023-01523-z.
- D. Brocklebank, F. Ram, J. Wright, P. Barry, C. Cates, L. Davies, G. Douglas, M. Muers, D. Smith and J. White, Health Technol. Assess., 2001, 5, 1–149, DOI:10.3310/hta5260.
- J. Sanchis, C. Corrigan, M. L. Levy, J. L. Viejo and ADMIT Group, Respir. Med., 2013, 107, 495–502, DOI:10.1016/j.rmed.2012.12.007.
- H. Chrystyn, Int. J. Clin. Pract., 2007, 61, 1022–1036, DOI:10.1111/j.1742-1241.2007.01382.x.
- A. C. Grant, R. Walker, M. Hamilton and K. Garrill, J. Aerosol Med. Pulm. Drug Delivery, 2015, 28, 474–485, DOI:10.1089/jamp.2015.1223.
- N. A. Meanwell, Chem. Res. Toxicol., 2011, 24, 1420–1456, DOI:10.1021/tx200211v.
- Q. Shi, H. Chen, Y. Wang, J. Xu, Z. Liu and C. Zhang, Int. J. Pharm., 2022, 611, 121320, DOI:10.1016/j.ijpharm.2021.121320.
- L. Yang, Z. Yan, J. Cheng and C. Yang, J. Mol. Liq., 2019, 295, 11717, DOI:10.1016/j.molliq.2019.111717.
- R. Censi and P. Di Martino, Molecules, 2015, 20, 18759–18776, DOI:10.3390/molecules201018759.
- S. V. Bhujbal, B. Mitra, U. Jain, Y. Gong, A. Agrawal, S. Karki, L. S. Taylor, S. Kumar and Q. T. Zhou, Acta Pharm. Sin. B, 2021, 11, 2505–2536, DOI:10.1016/j.apsb.2021.05.014.
- B. R. Adhikari, K. C. Gordon and S. C. Das, Adv. Drug Delivery Rev., 2022, 189, 114468, DOI:10.1016/j.addr.2022.114468.
- A. Altay Benetti, A. Bianchera, F. Buttini, L. Bertocchi and R. Bettini, Pharmaceutics, 2021, 13, 1113, DOI:10.3390/pharmaceutics13081113.
- D. Gupta, D. Bhatia, V. Dave, V. Sutariya and S. V. Gupta, Molecules, 2018, 23, 1719, DOI:10.3390/molecules23071719.
- M. S. Hossain Mithu, S. Economidou, V. Trivedi, S. Bhatt and D. Douroumis, Cryst. Growth Des., 2021, 21, 1358–1374, DOI:10.1021/acs.cgd.0c01427.
- S. S. Bharate, Drug Discovery Today, 2021, 26, 384–398, DOI:10.1016/j.drudis.2020.11.016.
- J. Xu, Q. Shi, Y. Wang, X. Junbo, C. Jin and L. Fang, Molecules, 2023, 28, 613, DOI:10.3390/molecules28020613.
- M. Singh, H. Barua, V. G. S. S. Jyothi, M. R. Dhondale, A. G. Nambiar, A. K. Agrawal, P. Kumar, N. R. Shastri and D. Kumar, Pharmaceutics, 2023, 15, 1161, DOI:10.3390/pharmaceutics15041161.
- S. Das, S. M. Pratik and A. Datta, J. Phys. Chem. C, 2024, 128(40), 17175–17183, DOI:10.1021/acs.jpcc.4c05301.
- A. J. Cruz-Cabeza, M. Lusi, H. P. Wheatcroft and A. D. Bond, Faraday Discuss., 2022, 235, 446–466, 10.1039/D1FD00081K.
- Q. Shi, Y. Wang, S. M. Moinuddin, X. Feng and F. Ahsan, AAPS PharmSciTech, 2022, 23, 259, DOI:10.1208/s12249-022-02421-7.
- J. Liu, H. Grohganz, K. Löbmann, T. Rades and N. J. Hempel, Pharmaceutics, 2021, 13, 389, DOI:10.3390/pharmaceutics13030389.
- J. Li, X. Wang, D. Yu, Y. Zhoujin and K. Wang, Int. J. Pharm., 2023, 648, 123555, DOI:10.1016/j.ijpharm.2023.123555.
- N. Alhajj, N. J. O'Reilly and H. Cathcart, Int. J. Pharm., 2022, 618, 121657, DOI:10.1016/j.ijpharm.2022.121657.
- H. Ueda, Y. Hirakawa, T. Miyano, Y. Nakayama, Y. Hatanaka, H. Uchiyama, Y. Tozuka and K. Kadota, Mol. Pharm., 2023, 20, 6368–6379, DOI:10.1021/acs.molpharmaceut.3c00756.
- S. N. Wong, J. Weng, I. Ip, R. Chen, R. Lakerveld, R. Telford, N. Blagden, I. J. Scowen and S. F. Chow, Pharmaceutics, 2022, 14, 300, DOI:10.3390/pharmaceutics14020300.
- A. Alsultan and C. A. Peloquin, Drugs, 2014, 74, 839–854, DOI:10.1007/s40265-014-0222-8.
- T. Al Hagbani, B. Vishwa, A. S. Abu Lila, H. F. Alotaibi, E. S. Khafagy, A. Moin and D. V. Gowda, Pharmaceuticals, 2022, 15, 560, DOI:10.3390/ph15050560.
- N. Tsapis, D. Bennett, K. O'Driscoll, K. Shea, M. M. Lipp, K. Fu, R. W. Clarke, D. Deaver, D. Yamins, J. Wright, C. A. Peloquin, D. A. Weitz and D. A. Edwards, Tuberculosis, 2023, 83, 379–385, DOI:10.1016/j.tube.2003.08.016.
- A. Salem, E. Khanfar, S. Nagy and A. Széchenyi, Int. J. Pharm., 2022, 623, 121924, DOI:10.1016/j.ijpharm.2022.121924.
- A. Pacławski, S. Politis, E. Balafas, E. Mina, P. Papakyriakopoulou, E. Christodoulou, N. Kostomitsopoulos, D. M. Rekkas, G. Valsami and S. Giovagnoli, Pharmaceutics, 2022, 14, 2819, DOI:10.3390/pharmaceutics14122819.
- P. A. Gurbel, K. P. Bliden, R. Chaudhary and U. S. Tantry, Circulation, 2020, 142, 1305–1307, DOI:10.1161/CIRCULATIONAHA.120.047477.
- S. N. Wong, K. H. Low, Y. L. Poon, X. Zhang, H. W. Chan and S. F. Chow, Int. J. Pharm., 2023, 640, 122983, DOI:10.1016/j.ijpharm.2023.122983.
- J. Y. Tse, K. Kadota, T. Nakajima, H. Uchiyama, S. Tanaka and Y. Tozuka, Cryst. Growth Des., 2022, 22, 1143–1154, DOI:10.1021/acs.cgd.1c01091.
- K. Kadota, J. Y. Tse, S. Fujita, N. Suzuki, H. Uchiyama, Y. Tozuka and S. Tanaka, ACS Appl. Bio Mater., 2023, 6, 3451–3462, DOI:10.1021/acsabm.3c00162.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112–122, DOI:10.1107/S0108767307043930.
- Y. Miyazaki, H. Sugihara, A. Nishiura, K. Kadota, Y. Tozuka and H. Takeuchi, Drug Dev. Ind. Pharm., 2017, 43, 30–41, DOI:10.1080/03639045.2016.1201099e.
- K. Kadota, M. Tanaka, H. Nishiyama, J. Y. Tse, H. Uchiyama, Y. Shirakawa and Y. Tozuka, Powder Technol., 2020, 376, 517–526, DOI:10.1016/j.powtec.2020.08.063.
- K. Kadota, A. Senda, T. Ito and Y. Tozuka, Eur. J. Pharm. Sci., 2015, 79, 79–86, DOI:10.1016/j.ejps.2015.09.006.
- Y. Tse, A. Koike, K. Kadota, H. Uchiyama, K. Fujimori and Y. Tozuka, Eur. J. Pharm. Biopharm., 2021, 167, 116–126, DOI:10.1016/j.ejpb.2021.07.017.
- M. D. A. Muhsin, G. George, K. Beagley, V. Ferro, H. Wang and N. Islam, Mol. Pharm., 2016, 13, 1455–1466, DOI:10.1021/acs.molpharmaceut.5b00859.
- C. A. S. Bergström and P. Larsson, Int. J. Pharm., 2018, 540, 185–193, DOI:10.1016/j.ijpharm.2018.01.044.
- N. Wei, L. Jia, Z. Shang, J. Gong, S. Wu, J. Wang and W. Tang, CrystEngComm, 2019, 21, 6196–6207, 10.1039/C9CE00847K.
- V. André, D. Braga, F. Grepioni and M. T. Duarte, Cryst. Growth Des., 2009, 9(12), 5108–5116, DOI:10.1021/cg900495s.
- B. Baul, A. Ledeţi, D. Cîrcioban, A. Ridichie, T. Vlase, G. Vlase, F. Peter and L. Ledeţi, Processes, 2023, 11, 1738, DOI:10.3390/pr11061738.
- H. Kitaoka, C. Wada, R. Moroi and H. Hakusui, Chem. Pharm. Bull., 1995, 43, 649–653, DOI:10.1248/cpb.43.649.
- H. Yamashita, Y. Hirakura, M. Yuda and K. Terada, Pharm. Res., 2014, 31, 1946–1957, DOI:10.1007/s11095-014-1296-4.
- R. D. Patel and M. K. Raval, Results Chem., 2022, 4, 100315, DOI:10.1016/j.rechem.2022.100315.
- M. M. Haskins and M. J. Zaworotko, Cryst. Growth Des., 2021, 21(7), 4141–4150, DOI:10.1021/acs.cgd.1c00418.
- T. S. Wiedmann and A. Naqwi, Asian J. Pharm. Sci., 2016, 11, 722–734, DOI:10.1016/j.ajps.2016.07.002.
- H. Ueda, H. Osaki and T. Miyano, J. Pharm. Sci., 2023, 112, 158–165, DOI:10.1016/j.xphs.2022.07.004.
- A. Saleki-Gerhardt, J. G. Stowell, S. R. Byrn and G. Zografi, J. Pharm. Sci., 1995, 84, 318–323, DOI:10.1002/jps.2600840311.
- T. Miyano, K. Sugita and H. Ueda, Pharmaceutics, 2024, 16, 949, DOI:10.3390/pharmaceutics16070949.
- M. Takahashi and H. Uekusa, J. Pharm. Sci., 2022, 111, 618–627, DOI:10.1016/j.xphs.2021.10.033.
- T. Miyano, S. Ando, D. Nagamatsu, Y. Watanabe, D. Sawada and H. Ueda, Mol. Pharm., 2024, 21, 6473–6483, DOI:10.1021/acs.molpharmaceut.4c01108.
- S. Gunasekaran, K. Rajalakshmi and S. Kumaresan, Spectrochim. Acta, Part A, 2013, 112, 351–363, DOI:10.1016/j.saa.2013.04.074.
- Y. Akkaya and S. Akyuz, Vib. Spectrosc., 2006, 42, 292–301, DOI:10.1016/j.vibspec.2006.05.011.
- R. Anitha, M. Gunasekaran, S. S. Kumar, S. Athimoolam and B. Sridhar, Spectrochim. Acta, Part A, 2015, 150, 488–498, DOI:10.1016/j.saa.2015.05.091.
- I. Nugrahani, A. N. Laksana, H. Uekusa and H. H. Oyama, Molecules, 2022, 27, 2166, DOI:10.3390/molecules27072166.
- D. R. Chejara, M. Mabrouk, P. Kumar, Y. E. Choonara, P. P. D. Kondiah, R. V. Badhe, L. C. du Toit, D. Bijukumar and V. Pillay, Mar. Drugs, 2017, 15, 257, DOI:10.3390/md15080257.
- H. Zhao, Y. Le, H. Liu, T. Hu, Z. Shen, J. Yun and J. F. Chen, Powder Technol., 2009, 194, 81–86, DOI:10.1016/j.powtec.2009.03.031.
- K. Kadota, T. R. Sosnowski, S. Tobita, I. Tachibana, J. Y. Tse, H. Uchiyama and Y. Tozuka, Adv. Powder Technol., 2020, 31, 219–226, DOI:10.1016/j.apt.2019.10.013.
- W.-R. Ke, P. C. L. Kwow, D. Khanal, R. Y. K. Chang and H.-K. Chan, Int. J. Pharm., 2021, 602, 120608, DOI:10.1016/j.ijpharm.2021.120608.
- K. Rode, I. Maji, S. Mahajan and P. K. Singh, Drug Discovery Today, 2024, 29, 104050, DOI:10.1016/j.drudis.2024.104050.
- M. S. Hassan and R. W. M. Lau, AAPS PharmSciTech, 2009, 10, 1252–1262, DOI:10.1208/s12249-009-9313-3.
- K. Kadota, T. Nishimura, Y. Nakatsuka, K. Kubo and Y. Tozuka, J. Pharm. Innov., 2017, 12, 249–259, DOI:10.1007/s12247-017-9285-x.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2410276. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4pm00250d |
| This journal is © The Royal Society of Chemistry 2025 |