
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tumor immunotherapy by plasmid DNAs encoding adenovirus virus-associated RNA†
Tomoko
Ito
 abc,
Takayuki
Yoshimoto
b,
Izuru
Mizoguchi
b and
Yoshiyuki
Koyama
*abc
abc,
Takayuki
Yoshimoto
b,
Izuru
Mizoguchi
b and
Yoshiyuki
Koyama
*abc
aObara Hospital Research Institute, 3-28-16, Honcho, Nakano-ku, Tokyo 164-0012, Japan
bDepartment of Immunoregulation, Institute of Medical Science, Tokyo Medical University, 6-1-1 Shinjuku, Shinjuku-ku, Tokyo 160-8402, Japan
cGraduate School of Veterinary Science, Osaka Metropolitan University, 1-58 Rinku-oraikita, Izumisano, Osaka 598-8531, Japan
First published on 14th January 2025
Abstract
Immunotherapy has become a most promising weapon for cancer treatment; however, tumor antigens generally exhibit low immunogenicity, limiting its effectiveness. In contrast, viral infections efficiently trigger innate and adaptive immunity. This is attributed to the high immunogenicity of microbial antigens and also to the activation of pattern recognition receptors such as retinoic acid-inducible gene-I (RIG-I). Upon recognizing viral RNA, RIG-I induces secretion of type-I interferons (IFNs). Type I IFNs not only invite antiviral effects but also plays an effective role in cancer immunotherapy. Therefore, activation of RIG-I by the ligands has gained attention as a novel cancer immunotherapy in recent years. Virus-associated RNAs (VA-RNA I and VA-RNA II) are non-coding small RNAs generated from the adenovirus genome. VA-RNA I strongly activates RIG-I, leading to type-I IFN production. In this study, plasmid DNAs encoding both VA-RNA I and II [pDNA(I,II)] or only VA-RNA I [pDNA(I)] were prepared, and their IFN inducing and anti-tumor effects were investigated. In culture cells, introduction of pDNA(I,II) or pDNA(I) effectively induced both IFN-α and IFN-β production. Both plasmids significantly inhibited tumor growth in mice. pDNA(I) exhibited superior IFN-inducing and anti-tumor effects compared to pDNA(I,II). VA-RNA I gene administration holds promise as a novel anti-tumor immunotherapy strategy.
Introduction
In recent years, immunotherapy using immune checkpoint inhibitors has gained attention and emerged as a promising tool for cancer treatment. However, the effectiveness of this therapy is often limited by the low immunogenicity of tumor antigens, posing a challenge to establish effective anti-tumor cellular immunity through immunotherapy.1,2 In contrast, cellular immunity is efficiently induced in response to viral infections, partly attributed to the high immunogenicity of microbial antigens, which allows immune cells to readily identify them as foreign and dangerous. In our previous research, to overcome the difficulty in establishing anti-tumor immunity owing to the low antigenicity of tumor antigens, we introduced plasmid DNAs encoding the strong tuberculosis antigens ESAT-6 or Ag85B into tumors as artificial neoantigens. Those plasmids effectively stimulated the immune system and suppressed tumor growth,3,4 demonstrating significant clinical effects not only in mouse models but also in animal clinical studies involving dogs.In addition to the strong immunogenicity of microorganism antigens, another crucial factor for efficient induction of adaptive immune responses against viral infections is activation of pattern recognition receptors (PRRs), such as retinoic acid-inducible gene-I (RIG-I) and its analogs (RIG-I-like receptors, RLRs), as well as toll-like receptors (TLRs).5 These receptors detect pathogen-associated molecular patterns that trigger innate and adaptive immune responses against microbial infections. RLRs are one of PRRs that detect the pathogenic RNA species generated during infection by RNA viruses, DNA viruses, and certain bacteria. Upon recognizing non-self RNA derived from these microbes, RLRs initiate responses such as secreting type-I interferons (IFNs), activating macrophages (MΦs), dendritic cells (DCs), natural killer cells (NKs).5,6
These RLR-mediated responses not only contribute to the antimicrobial effect but also lead to various effective responses in anti-tumor actions, including tumor cell death and activation of both the innate and adaptive immune systems.7 Based on these responses, novel strategies for anti-tumor immunotherapy involving RLR activation by RIG-I agonists have been proposed in recent years. Jacobson et al. successfully inhibited tumor growth via intratumoral administration of a synthetic RIG-I ligand in a mouse model carrying CT26 colorectal cancer cells.8 Phase I/II trials using a synthetic RNA oligonucleotide, RGT100, which activates RIG-I as RIG-I agonists, are currently underway in patients with advanced or recurrent tumors (NCT03065023).9
In the present study, we focused on “virus-associated RNA-I” (VA-RNA I) as a factor that activates RIG-I. VA-RNAs were first detected in cells infected with adeno-virus (Ad) in 1966;10 they are non-coding small RNAs of approximately 160 nucleotides generated by RNA polymerase III (POL III) from the Ad genome. Most human Ads, including Ad2 and Ad5, encode two distinct VA-RNA transcripts, VA-RNA I and II, which exhibit specific biological activities.11 For example, VA-RNA I binds to the double-stranded RNA-activated protein kinase (PKR) and inhibits its function.12 PKR is a serine/tyrosine kinase that plays an important role in response to viral infection.13 VA-RNA II binds to and inhibits activity of RNA helicase A.14
Besides these characteristic functions, VA-RNA I is known to strongly activate RIG-I, leading to type-I IFN production.15 As mentioned above, the gene of VA-RNA I can be transcribed by POL III without requiring a promoter. DNA coding VA-RNA I is, thus, expected as a novel RIG-I-stimulating cancer immunotherapy agent. In this study, plasmids encoding both VA-RNA I and II [pDNA(I,II)] or only VA-RNA I [pDNA(I)] were constructed, and type-I IFN induction function of the plasmids were investigated in cultured cells. Tumor growth-inhibitory effects of the plasmid DNAs in mouse models was also examined using our DNA/polyethylenimine (PEI)/polyanion “ternary complex transfection system”.3,16 Chondroitin sulfate sodium salt (CS) was used as polyanions in the in vivo experiments. The addition of chondroitin sulfate changed the positive charge on the surface of the DNA/PEI complex into negative, significantly reducing cytotoxicity.3 DNA/PEI/CS ternary complex was very stably dispersed, and they could be freeze-dried and re-hydrated without apparent aggregation or loss of transfection ability. These properties enabled the preparation of a concentrated suspension ([DNA] = 400 μg mL−1) of very small pDNA complex particles (∼200 nm), by preparing the complexes at highly diluted conditions, followed by condensation via lyophilization-and-rehydration procedure. The re-hydrated very small complexes obtained by this method achieved a fairly high level of gene expression within tumors.3 The high tumor-specific gene expression and relatively low toxicity of such DNA ternary complexes make them promising candidates for clinical applications in human cancer therapy. Here we evaluated the potential of the ternary complex comprising plasmid DNA encoding VA-RNA I, PEI, and CS as an anti-tumor immunotherapeutic agent by intratumorally administering it to syngeneic tumor-bearing mice.
Materials and methods
Mice
In this preclinical experimental study, C57BL/6 and BALB/c mice were purchased from Sankyo Labo Service, and all the mice were maintained under pathogen free conditions. The animal experiments were approved by the President and the Institutional Animal Care and Use Committee of Tokyo Medical University and were performed in accordance with institutional, scientific community (no. R5-102 and R6-014), and national guidelines for animal experimentation and the animal research: reporting of in vivo experiments guidelines.Plasmid DNAs
Plasmid DNA, pAdVAntage, encoding both VA-RNA I and II (pDNA(I,II); 4392 bp), which containing base pairs 9831–11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 555 of the adenovirus type 2 genome, was purchased from Promega (Madison, WI, USA). A plasmid DNA encoding only VA-RNA I (pDNA(I); 4229 bp) was constructed by deleting the sequences encoding VA-RNA II in the region from 1036 to 1198 by outsourcing to VectorBuilder Japan Co., Ltd (Kanagawa, Japan). The plasmid DNA coding green fluorescent protein (GFP) (pDNA(GFP)) or mouse interferon-β (pDNA(IFN-β)) was obtained from CLONTECH Laboratories, Inc. (Mountain View, CA) or VectorBuilder Japan Co., Ltd (Kanagawa, Japan), respectively. Plasmid DNA containing firefly luciferase gene (pDNA(Luc)) under the control of a cytomegalovirus promoter was a kind gift from Prof. Kawakami, S. (Nagasaki University, Nagasaki, Japan).
555 of the adenovirus type 2 genome, was purchased from Promega (Madison, WI, USA). A plasmid DNA encoding only VA-RNA I (pDNA(I); 4229 bp) was constructed by deleting the sequences encoding VA-RNA II in the region from 1036 to 1198 by outsourcing to VectorBuilder Japan Co., Ltd (Kanagawa, Japan). The plasmid DNA coding green fluorescent protein (GFP) (pDNA(GFP)) or mouse interferon-β (pDNA(IFN-β)) was obtained from CLONTECH Laboratories, Inc. (Mountain View, CA) or VectorBuilder Japan Co., Ltd (Kanagawa, Japan), respectively. Plasmid DNA containing firefly luciferase gene (pDNA(Luc)) under the control of a cytomegalovirus promoter was a kind gift from Prof. Kawakami, S. (Nagasaki University, Nagasaki, Japan).
Reagents
The Lipofectamine 3000 Transfection Reagent kit and Linear Polyethylenimine (PEI) (PEI “Max” MW 40000 in a hydrochloride salt form, comparable to MW 25000 in a free base form) were purchased from Thermo Fisher Scientific (Waltham, MA, USA) and Polyscience, Inc. (Warrington, PA, USA), respectively. Hyaluronic acid sodium salt (HA) from microorganisms was obtained from Nacalai Tesque, Inc. (Kyoto, Japan). Chondroitin sulfate sodium salt (CS) from shark cartilage (MW 10000) was supplied by Seikagaku Co. (Tokyo, Japan).
Cell culture
Colon 26 mouse colon carcinoma cells were kindly provided from Dr Koyanagi, Y. (Tokyo Medical University, Tokyo, Japan). B16 mouse melanoma cells were obtained from RIKEN BioResource Center (Ibaraki, Japan). The cells were seeded onto 24-well plates at 1.0 × 105 cells per well, and cultured for a day in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin G sodium (100 unit per mL), and streptomycin sulfate (0.1 mg mL−1). The primary growth medium was then replaced with 300 μL of fresh DMEM with 10% of FBS and the antibiotics.Gene transfection efficiency in cultured cells
Complexes of pDNAs with a cationic lipid, Lipofectamine 3000 Reagent (LF3000), were prepared as follows: typically, pDNA(GFP) (1.5 μg) in 75 μL of DMEM was added to 3 μL of P3000 Reagent (P3000; a manufacturer's recommended additive), or 7.5 μL of HA solution ([COOH] = 14.5 mM). LF3000 (2.25 μL) was diluted with 75 μL of DMEM, and mixed with the DNA solution prepared above. After standing at room temperature for 10 min, the pDNA complexes were added to the Colon-26 or B16 cells (1.5 μg pDNA per well), and the cells were incubated at 37 °C in a 5% CO2 incubator. The medium was replaced with 0.5 mL fresh one with FBS and the antibiotics 6, 24, 48 and 72 h after the transfection. The fluorescence intensity of the wells was monitored (excitation wave-length: 485 nm, emission wavelength: 535 nm) using a multimode plate reader (Infi-nite200PRO F Plex, TECAN, Switzerland) for 3 days to evaluate gene expression efficiency.Measurement of type I IFN secretion
DNA/LF3000/HA complexes were prepared using pDNA(I,II), pDNA(I), pDNA(IFN-β), or pDNA(Luc) as described above. They were added to the Colon 26 or B16 cells (1.5 μg pDNA per well), and the cells were incubated with the complexes at 37 °C in a 5% CO2 incubator for 6 hours. After removing the transfection medium, the cells were gently washed twice with 0.5 mL of DMEM and 0.3 mL of fresh medium containing FBS and antibiotics was added. After 24 hours, the culture medium was replaced with fresh medium containing FBS and antibiotics. Following an additional 24 hours of incubation, the supernatants were collected, and centrifuged at 3000g for 15 min to remove the cells and debris. The concentrations of IFN-α and IFN-β were measured using corresponding enzyme linked immunosorbent assay (ELISA) kits (PBL Assay Science, NJ, USA).Reverse transcription-PCR (RT-PCR)
B16 cells were transfected with plasmids encoding VA-RNA I, either pDNA(I,II) or pDNA(I). Total RNA was isolated using Sepasol-RNA I Super G (Nacalai Tesque, Inc., Kyoto, Japan), and the amount of VA-RNA I was analyzed by quantitative RT-PCR using the Thermal Cycler Dice Real Time System (Takara Bio Inc., Shiga, Japan) according to the manufacturer's instructions and following the method described in our previous report.17 Hypoxanthine phosphoribosyltransferase (HPRT) mRNA was used as a housekeeping gene for normalization. The ΔCt values were calculated by subtracting the Ct value of HPRT from the Ct value of VA-RNA I for each plasmid. The primers used in PCR for VA-RNA-I were designed based on those reported in Minamitani et al., 2011,18 and their sequences were as follows: 5′-GGGCACTCTTCCGTGGTCTG-3′ and 5′-AGGAGCGCTCCCCCGTTGTC-3′.Evaluation of anti-tumor efficacy in syngeneic mouse models
For in vivo gene delivery, we used a pDNA/PEI/CS ternary complex gene delivery system with high tumor-specific gene expression.16 pDNA/PEI/CS ternary complexes (1:12:8 in charge) containing 50 μg of pDNA were prepared similarly to our previous studies16 as follows: phosphate buffer (PB) (pH 7.2; 7.0 mM, 2370 μL), aqueous solutions of CS (297 μg in 60 μL) and PEI “Max” (147 μg in 30 μL) were sequentially added to an aqueous solution of plasmid DNA (50 μg in 40 μL). After a 20 minute incubation period, 10% aqueous dextran solution (25 μL) was added. The resulting mixture was frozen at −60 °C and freeze-dried at room temperature to obtain a spongy complex, which was rehydrated with 125 μL of water just before use.
Colon 26 or B16 cells were subcutaneously inoculated into female BALB/c or C57BL/6 mice (7 weeks), respectively (1 × 106 cells per mouse). After 4 days, the pDNA/PEI/CS ternary complex containing 50 μg of the pDNA was intratumorally injected twice at 3 day intervals, and the tumor size was monitored.
Results and discussion
Transfection efficiency of pDNA/LF3000/hyaluronic acid (HA) ternary complex
We initially examined whether plasmid DNAs, pDNA(I,II), or pDNA(I) could induce IFN secretion in cells. Before these experiments, we explored efficient methods for introducing plasmid DNA into cultured cells and expressing these genes. Currently, the Lipofectamine 3000 Transfection Kit is widely used as an efficient reagent for gene transfection in cultured cells. It comprises a cationic lipid, LF3000, and a co-reagent, P3000 Reagent (P3000). P3000 is recommended to be added to pDNA to enhance gene expression. However, the composition and structure of P3000 is undisclosed. Unknown content is undesirable for experiments in which the culture supernatant is used later, such as in ELISA. Therefore, we sought an alternative method for efficient gene transfection without using P3000. In previous studies on gene transfection using the cationic polymer PEI instead of the cationic lipids, we found that mixing HA with DNA before mixing with PEI could significantly increase and prolong the expression of introduced genes.19,20 Thus, we investigated whether a similar effect could be achieved by addition of HA to DNA in transfection with LF3000, using a plasmid encoding GFP, pDNA(GFP).First, we prepared the pDNA(GFP)/LF3000/HA complexes by varying the DNA:HA ratio, and examined the expression levels. The amount of LF3000 was fixed at 2.25 μL per 1.5 μg of pDNA, according to the manufacturer's protocol. Cultured Colon 26 and B16 cells were transfected by the DNA/LF3000/HA complexes with P:COOH ratios of 1:12, 1:24, or 1:36, where P represents the phosphorus atoms in DNA and COOH represents the carboxyl groups of HA. As a result, we observed that, on average, the highest expression was achieved with a complex at P:COOH = 1:24 (Fig. S1†). In subsequent experiments, we used DNA/LF3000/HA complexes at P:COOH = 1:24.
Then transfection efficiency of DNA/LF3000/HA complex was compared with that of pDNA/LF3000 binary complex or pDNA/LF3000/P3000 system. In Colon 26 cells, the ternary complex transfection system consisting of pDNA(GFP), LF3000, and HA exhibited relatively higher gene expression than the pDNA/LF3000 binary complex or the protocol recommended system using P3000 (Fig. 1(a)). In B16 cells, at 24 hours post-transfection, all three systems showed almost the same level of fluorescence intensities. In the system using pDNA/LF3000/P3000, the fluorescence intensity obviously decreased thereafter. Judging from the fact that a relatively small number of cells were observed under the microscope in that system, it may be due to damage to the cells by the strong transfection reagents. On the other hand, rather higher fluorescence emission was observed at 48–72 hours post-transfection in the other two systems (Fig. 1(b)).
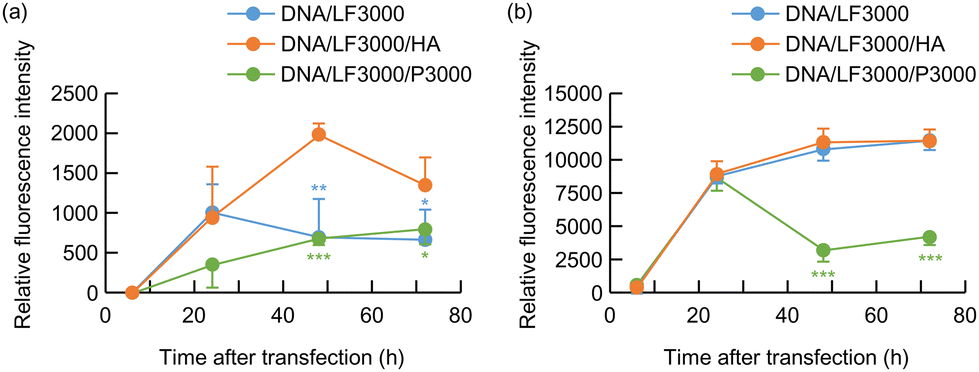 | ||
| Fig. 1 GFP expression by pDNA/LF3000 (blue), pDNA/LF3000/HA (orange), or pDNA/LF3000/P3000 (green) complex in cultured (a) Colon 26 mouse colon carcinoma cells; (b) B16 mouse melanoma cells. (n = 4, mean ± SD, *p < 0.05, **P < 0.01, ***P < 0.001; green asterisk: between pDNA/LF3000/HA and DNA/LF3000/P3000; blue asterisk: between pDNA/LF3000/HA and DNA/LF3000). | ||
We previously reported that HA has multifunctional properties that improve the gene transfection efficiency of the pDNA/PEI complex. HA covers the pDNA complex and recharges its surface potential to reduce the toxicity and undesirable nonspecific interactions.19 Furthermore, the HA coating enhances the transcriptional activity of plasmid/PEI complexes. In transfection mediated by polycations, the low transcription efficiency of DNA/polycation complexes is one of the significant obstacles to achieving high gene expression efficiency. Plasmid DNA forms a very tight complex with polycations such as PEI and polylysine, making it difficult for transcription factors to access them, which results in low transcription efficiency. We demonstrated that HA had the effect of loosening the binding in the DNA/PEI complexes by measuring the recovery of fluorescence intensity of YOYO-1-labeled DNA in complexes with PEI.19 It would facilitate the approach of transcription-factors to the swollen complex, resulting in a enhanced the transcriptional activity of the plasmid/PEI complexes. HA is thought to have a similar effect on gene expression mediated by the cationic lipids.
Type-I IFN production induced by pDNAs encoding VA-RNA I
In this study, we prepared two types of plasmid DNA: one encoding both VA-RNA I and II as pDNA(I,II), and the other containing only the gene for VA-RNA I as pDNA(I), and their ability to induce type-I IFN secretion was investigated using cultured Colon 26 and B16 cells. Soon after adding the DNA complex, non-specific IFN secretion is occasionally observed probably due to the stimulation. Therefore, the medium was replaced with fresh medium 30 hours post-transfection, and after an additional 24 hours of incubation, the supernatant was collected to analyze the concentrations of IFN-α and IFN-β using ELISA. pDNAs encoding IFN-β gene, pDNA(IFN-β), and that with luciferase gene, pDNA(luc), were also similarly evaluated.Both pDNA(I,II) and pDNA(I) effectively induced secretion of significantly higher level of IFN-α and also of IFN-β in Colon 26 and B16 cells compared with the cells treated with PBS or pDNA(luc) (Fig. 2). The IFN-β concentration in the culture medium of the cells transfected by pDNA(IFN-β) was much higher than that of the cells treated with pDNA(I,II) or pDNA(I). This is attributed to the direct induction of IFN production via the IFN-β gene introduced into the cells. In contrast, pDNA(I,II) or pDNA(I) initially produces VA-RNA I, which subsequently activates RIG-I, leading to IFN production through a multistep biological reaction cascade. The need to undergo such complex and numerous reaction steps may contribute to the relatively lower IFN-β production efficiency by pDNA(I,II) and pDNA(I) compared to by pDNA(IFN-β).
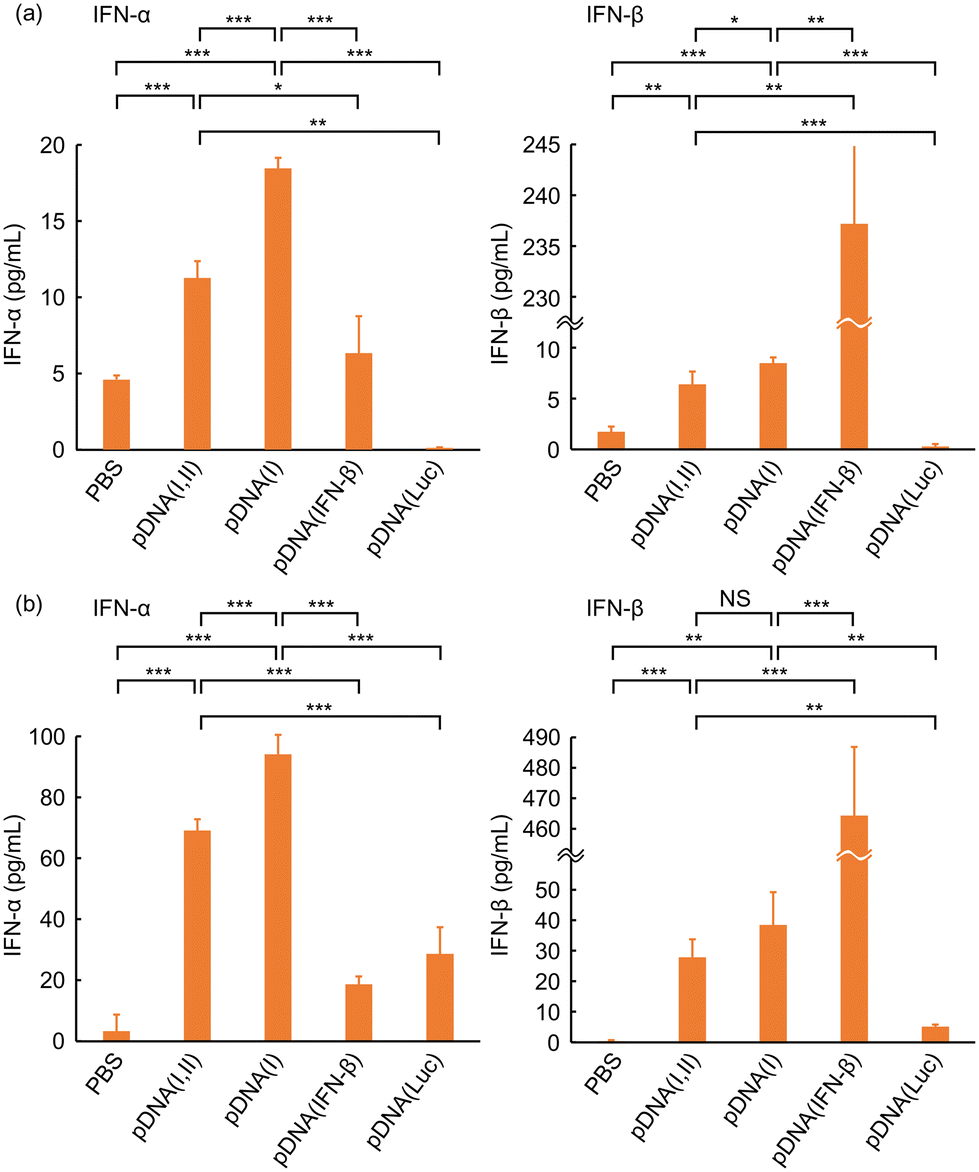 | ||
| Fig. 2 Type-I IFN production by transfection with pDNA encoding VA-RNA I and VA-RNA II (pDNA(I,II)), VA-RNA I (pDNA(I)), IFN-β (pDNA(IFN-β)), or luciferase (pDNA(Luc)) in cultured (a) Colon 26 or (b) B16 cells. (n ≧ 3, mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001). | ||
pDNA(I) always introduced higher level of both IFN-α and IFN-β secretion than pDNA(I,II). The size of pDNA(I) was 163 bp (approximately 4%) smaller than that of pDNA(I,II). Plasmid size affects nuclear uptake efficiency and gene expression.21 The smaller pDNA(I) size may result in higher VA-RNA I expression. Another essential difference between pDNA(I,II) and pDNA(I) lies in that pDNA(I,II) encodes genes for both VA-RNA I and II, whereas pDNA(I) encodes only for VA-RNA I. VA-RNA II produced in the cells transfected by pDNA(I,II) might have interfered with the synthesis of IFN, leading to a reduced secretion level. Biological functions of VA-RNA II are not fully understood. Liao et al. reported that VA-RNA II binds to an RNA helicase A, and inhibits the helicase activity.14 This interaction may potentially suppress the translation of IFN mRNA by sequestering RNA helicase A, which is known to facilitate efficient translation through its involvement in ribosome assembly and mRNA stabilization. RIG-I has a helicase domain and it is essential for recognition of dsRNA and the downstream signals. VA-RNA II may bind to the domain and interfere with its functionality. Wakabayashi et al. reported that mivaRNAII, a microRNA produced through intracellular processing of VA-RNA II, targets and knocks down the cullin 4A (CUL4A) gene.22 CUL4A is a component of the ubiquitin ligase complex, and suppression of CUL4A expression may lead to an increase in Jun-N-terminal kinase signalling, which plays a crucial role in the regulation of cellular processes, including proliferation, and apoptosis.
If the lower induction of IFN by pDNA(I,II) compared to pDNA(I) is primarily due to the inhibition of IFN production by VA-RNA II generated from pDNA(I,II), it can be assumed that the amount of VA-RNA I produced intracellularly would be the same between cells transfected with pDNA(I,II) and those transfected with pDNA(I). Then, we measured and compared the levels of VA-RNA I in B16 cells transfected with pDNA(I,II) or pDNA(I) using RT-PCR. The ΔCt values of VA-RNA I generated by transfection with pDNA(I,II) and pDNA(I) were 6.90 ± 0.11 and 7.18 ± 0.27, respectively, with no significant difference between the two plasmids (P = 0.18). These values are within the range of experimental error, indicating that the amount of VA-RNA I produced by the two plasmids is essentially the same. Therefore, the difference in IFN expression efficiency in cells transfected with pDNA(I,II) or pDNA(I) is unlikely to be solely attributable to the amount of VA-RNA I produced. Whether the difference in IFN-inducing activity between pDNA(I,II) and pDNA(I) is linked to the function of VA-RNA II or its degradation product, mivaRNAII, is still unclear and remains to be investigated.
Tumor growth suppression by the VA-RNA I-encoding pDNAs
We then investigated the tumor growth-inhibitory functions of pDNA(I,II) and pDNA(I) using syngeneic mouse models. For in vivo gene delivery, we developed a pDNA/PEI/CS ternary complex gene delivery system, demonstrating high gene delivery and expression efficiency, specifically within tumor tissues over 24–72 h when administered to tumors.16 Colon 26 or B16 cells, in which induction of IFN synthesis by pDNA(I,II) and pDNA(I) was confirmed, were transplanted subcutaneously into BALB/c or C57BL/6 mice, respectively to generate syngeneic tumor models. Four days after inoculation, the ternary complex of pDNA(I,II) or pDNA(I) with PEI and CS was injected twice at 3 day intervals into the tumor, and changes in tumor size were monitored.In both Colon 26- and B16-bearing mice, pDNA(I,II) and pDNA(I) exhibited remarkable inhibition of tumor growth, as shown in Fig. 3. In mice transplanted with Colon 26, these plasmids encoding VA-RNA I suppressed the tumor size to be significantly smaller than that of mice treated with saline throughout the period from day 1 to day 16 after administration (P < 0.01). Similarly, in B16-bearing mice, these plasmids also resulted in significant tumor suppression, observed from day 3 to day 13 (P < 0.05). The complex of pDNA(Luc), which does not encode any cytokines, slightly reduced the tumor growth. The effect of pDNA(Luc) may be attributable to the “danger signal effect” of the immunogenicity of the heterologous protein, luciferase. As mentioned in the introduction, we reported that intratumoral administration of ESAT-6 gene, a pathogenic antigen from Mycobacterium tuberculosis, has been found to demonstrate highly effective tumor growth suppression in syngeneic tumor-bearing mice.3,4 Administration of such heterologous animal antigen proteins into tumors should stimulate the immune system by recognizing them as “foreign danger signals”, leading to the activation of an anti-tumor immune response. Since luciferase protein is also an antigen from a heterologous species for mice, it may have had a similar effect. Alternatively, the anti-tumor effect of these plasmid DNA complexes may be partly due to the toxicity of the DNA complexes themselves. However, the efficacy of pDNA(Luc) was generally lower than that of pDNA(I) or pDNA(I,II). While no significant difference was observed in B16 cells, significant differences were detected on multiple days post-administration in Colon 26 cells, suggesting that the superior anti-tumor effect of these plasmids encoding VA-RAN I was most likely due to the anti-tumor function of the non-coding small RNA, rather than the cytotoxic effects of the pDNA complexes.
 | ||
| Fig. 3 Anti-tumor effect of pDNA encoding VA-RNA I and II (pDNA(I,II); GREEN line), VA-RNA I (pDNA(I); orange line), IFN-β (pDNA(IFN-β); blue line), or luciferase (pDNA(Luc); gray line) in mice bearing (a) Colon 26 colorectal cancer cells or (b) B16 melanoma cells. Colon 26 cells or B16 cells were injected on day −4 into BALB/c mice or C57BL/6 mice, respectively. On days 0 and 4, complexes containing 50 μg of the pDNAs were injected into mice. (n > 3, mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001; black asterisk: between pDNA(I) and saline; blue asterisk: between pDNA(I) and pDNA(IFN-β); gray asterisk: between pDNA(I) and pDNA(Luc); green asterisk: between pDNA(I) and pDNA(I,II)). | ||
pDNA(IFN-β) also demonstrated a slight anti-tumor effects. Although pDNA(IFN-β) induced much higher IFN-β expression in both B16 and Colon 26 cultured cells as shown in Fig. 2, its in vivo anti-tumor efficacy was much lower than that of pDNA(I) or pDNA(I,II). For B16-bearing mice, significant differences were rarely observed. On the other hand, for Colon 26-bearing mice, pDNA(I) or pDNA(I,II) consistently outperformed pDNA(IFN-β) across all days from day 1 to day 16. This can be attributed to the fact that pDNA(IFN-β) produces solely IFN-β, whereas pDNA(I) and pDNA(I,II) induce various other subtypes of type-I IFNs. In mice, 14 subtypes of IFN-α and a single type of IFN-β have been identified. Although the roles of each IFN in the anti-tumor effects are not fully understood, the higher anti-tumor function of pDNA(I) and pDNA(I,II) than of pDNA(IFN-β) may be attributed to the coordinated expression and cooperative action of these IFNs.
The effect of pDNA(I), which does not express VA-RNA II, was slightly greater than that of pDNA(I,II), possibly due to the higher IFN expression efficiency of pDNA(I) compared to pDNA(I,II) (Fig. 2) or the potential unfavorable effects of VA-RNA II. As discussed earlier, VA-RNA II has been reported to bind to RNA helicase A and inhibit its helicase activity, potentially suppressing mRNA translation.14 These and other potential mechanisms previously mentioned may underlie the reduced efficacy of pDNA(I,II). VA-RNA I not only induces secretion of type-I IFNs but also inhibits PKR. PKR is suggested to have both tumor-suppressive and tumor-promoting functions.23 Whether the PKR inhibitory effect of VA-RNA I positively or negatively influenced tumor growth inhibition remained unclear.
Almost all mammalian cells secrete extracellular vesicles (EVs), which act as carriers transporting various substances, including RNA, DNA, lipids, and proteins.24 Groot reported that RNAs were selectively incorporated into EVs through mechanisms involving RNA-binding proteins and membrane proteins.25 Brachtlova et al. found that mivaRNAI, a microRNA generated from VA-RNA I by Dicer, is present in EVs secreted by adenovirus-infected cells.26 Given that mivaRNAI is found in EVs, it is plausible that VA-RNA I itself might also be packaged into EVs, potentially through similar or related mechanisms. Therefore, it is expected that intact VA-RNA I molecule would also be present in EVs secreted by the cells into which its gene has been introduced. In our preliminary experiments, a significant enhancement of type I IFN secretion was observed in the cultured cells treated with EVs secreted from the cells transfected with VA-RNA I gene. These EVs could potentially be taken up by neighboring tumor cells, and the bystander effect is anticipated to lead to an additional increase in tumor suppression. Research on cancer therapy using EVs containing VA-RNA I is currently ongoing, and will be presented in a separate paper in the near future. The mechanism underlying the anti-tumor effects of VA-RNA I has not yet been elucidated. However, it is clear that the VA-RNA-I gene generates a distinct anti-tumor effect; therefore, VA-RNA I gene administration is expected to be a promising new therapeutic approach.
Conclusions
Transfection with pDNAs encoding VA-RNA I effectively induced IFN-α and IFN-β expression in cultured Colon 26 and B16 cells. These pDNAs significantly suppressed tumor growth in syngeneic tumor-bearing mouse models. VA-RNA I gene administration can serve as a novel strategy for anti-tumor immunotherapy.Author contributions
Tomoko Ito: conceptualization, formal analysis, funding acquisition, investigation, methodology, writing – original draft. Takayuki Yoshimoto: methodology, writing – review & editing. Izuru Mizoguchi: methodology, writing – review & editing. Yoshiyuki Koyama: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – original draft, writing – review & editing.Data availability
All data supporting the findings of this study were generated during our experiments and are available in the ESI (Fig. S1†) submitted alongside the manuscript. No external data or repositories were used.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We would like to express our sincere gratitude to Dr Yasuhiro Katahira of the Institute of Medical Science, Tokyo Medical University, for his invaluable assistance with the RT-PCR measurements conducted in this study. This research was supported by JSPS KAKENHI Grant Numbers 19K12806, 21K05279, and 23K11854.References
- F. M. Foss, Immunologic mechanisms of antitumor activity, Semin. Oncol., 2002, 29, 5–11 CrossRef CAS.
- S. Wang, Z. He, X. Wang, H. Li and X.-S. Liu, Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction, eLife, 2019, 8, e49020 CrossRef CAS PubMed.
- Y. Koyama, C. Yoshihara and T. Ito, Novel antitumor strategy utilizing a plasmid expressing a Mycobacterium tuberculosis antigen as a “danger signal” to block immune escape of tumor cells, Pharmaceutics, 2015, 7, 165–174 CrossRef CAS PubMed.
- T. Ushigusa, Y. Koyama, T. Ito, K. Watanabe, J. K. Chambers, A. Hasegawa, K. Uchida, R. Kanegi, S. Hatoya, T. Inaba and K. Sugiura, Innate immunity mediated by dendritic cells/macrophages plays a central role in the early period in tumor treatment using gene of Mycobacterium tuberculosis antigen, J. Vet. Med. Sci., 2018, 80, 190–196 CrossRef CAS.
- R. Medzhitov, Recognition of microorganisms and activation of the immune response, Nature, 2007, 449, 819–826 CrossRef CAS PubMed.
- J. R. Patel and A. García-Sastre, Activation and regulation of pathogen sensor RIG-I, Cytokine Growth Factor Rev., 2014, 25, 513–523 CrossRef CAS PubMed.
- Y. Jiang, H. Zhang, J. Wang, J. Chen, Z. Guo, Y. Liu and H. Hua, Exploiting RIG-I-like receptor pathway for cancer immunotherapy, J. Hematol. Oncol., 2023, 16, 8 CrossRef CAS.
- M. E. Jacobson, L. Wang-Bishop, K. W. Becker and J. T. Wilson, Delivery of 5′-triphosphate RNA with endosomolytic nanoparticles potently activates RIG-I to improve cancer immunotherapy, Biomater. Sci., 2019, 7, 547–559 RSC.
- L. Horvath, B. Thienpont, L. Zhao, D. Wolf and A. Pircher, Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC) - novel approaches and future outlook, Mol. Cancer, 2020, 19, 141 CrossRef CAS PubMed.
- P. R. Reich, B. G. Forget, S. M. Weissman and J. A. Rose, RNA of low molecular weight in KB cells infected with adenovirus type 2, J. Mol. Biol., 1966, 17, 428–439 CrossRef CAS PubMed.
- V. K. Vachon and G. L. Conn, Adenovirus VA RNA: an essential pro-viral non-coding RNA, Virus Res., 2016, 212, 39–52 CrossRef CAS PubMed.
- I. V. Hood, J. M. Gordon, C. Bou-Nader, F. E. Henderson, S. Bahmanjah and J. Zhang, Crystal structure of an adenovirus virus-associated RNA, Nat. Commun., 2019, 10, 2871 CrossRef PubMed.
- S. Balachandran, P. C. Roberts, L. E. Brown, H. Truong, A. K. Pattnaik, D. R. Archer and G. N. Barber, Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection, Immunity, 2000, 13, 129–141 CrossRef CAS.
- H. J. Liao, R. Kobayashi and M. B. Mathews, Activities of adenovirus virus-associated RNAs: Purification and characterization of RNA binding proteins, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8514–8519 CrossRef CAS.
- T. Minamitani, D. Iwakiri and K. Takada, Adenovirus Virus-Associated RNAs Induce Type I Interferon Expression through a RIG-I-Mediated Pathway, J. Virol., 2011, 85, 4035–4040 CrossRef CAS PubMed.
- Y. Koyama, K. Sugiura, C. Yoshihara, T. Inaba and T. Ito, Highly effective non-viral antitumor gene therapy system comprised of biocompatible small plasmid complex particles consisting of pDNA, anionic polysaccharide, and fully deprotected linear polyethylenimine, Pharmaceutics, 2015, 7, 152–164 CrossRef CAS PubMed.
- I. Mizoguchi, Y. Katahira, S. Inoue, E. Sakamoto, A. Watanabe, Y. Furusaka, A. Irie, S. Senju, Y. Nishimura, S. Mizukami, K. Hirayama, S. Nakamura, K. Eto, H. Hasegawa and T. Yoshimoto, A novel coculture system for assessing respiratory sensitizing potential by IL-4 in T cells, ALTEX, 2023, 40, 204–216 Search PubMed.
- T. Minamitani, D. Iwakiri and K. Takada, Adenovirus virus-associated RNAs induce type I interferon expression through a RIG-I-mediated pathway, J. Virol., 2011, 85, 4035–4040 CrossRef CAS PubMed.
- T. Ito, N. Iida-Tanaka, T. Niidome, T. Kawano, K. Kubo, K. Yoshikawa, T. Sato, Z. Yang and Y. Koyama, Hyaluronic acid and its derivative as a multi-functional gene expression enhancer: protection from non-specific interactions, adhesion to targeted cells, and transcriptional activation, J. Controlled Release, 2006, 112, 382–388 Search PubMed.
- T. Ito, Y. Koyama and M. Otsuka, Preparation of Calcium Phosphate Nanocapsule Including Deoxyribonucleic Acid-Polyethyleneimine-Hyaluronic Acid Ternary Complex for Durable Gene Delivery, J. Pharm. Sci., 2014, 103, 179–184 Search PubMed.
- S. Ribeiro, J. Mairhofer, C. Madeira, M. M. Diogo, C. Lobato Da Silva, G. Monteiro, R. Grabherr and J. M. Cabral, Plasmid DNA Size Does Affect Nonviral Gene Delivery Efficiency in Stem Cells, Cell. Reprogram., 2012, 14, 130–137 Search PubMed.
- K. Wakabayashi, M. Machitani, M. Tachibana, F. Sakurai and H. Mizuguchi, A MicroRNA Derived from Adenovirus Virus-Associated RNAII Promotes Virus Infection via Posttranscriptional Gene Silencing, J. Virol., 2019, 93, e01265–e01218 Search PubMed.
- S. Gal-Ben-Ari, I. Barrera, M. Ehrlich and K. Rosenblum, PKR: a kinase to remember, Front. Mol. Neurosci., 2019, 11, 480 Search PubMed.
- K. O'Brien, K. Breyne, S. Ughetto, C. L. Louise and O. B. Xandra, RNA delivery by extracellular vesicles in mammalian cells and its applications, Nat. Rev. Mol. Cell Biol., 2020, 21, 585–606 Search PubMed.
- M. Groot and H. Lee, Sorting Mechanisms for MicroRNAs into Extracellular Vesicles and Their Associated Diseases, Cells, 2020, 9, 1044 Search PubMed.
- T. Brachtlova, J. Li, I. H. van der Meulen-Muileman, F. Sluiter, W. von Meijenfeldt, I. Witte, S. Massaar, R. van den Oever, J. de Vrij and V. W. van Beusechem, Quantitative Virus-Associated RNA Detection to Monitor Oncolytic Adenovirus Replication, Int. J. Mol. Sci., 2024, 25, 6551 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00219a |
| This journal is © The Royal Society of Chemistry 2025 |