
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModification of fluorene and fluorenone core via C–H functionalization
Shefali
Banga
and
Srinivasarao Arulananda
Babu
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Knowledge City, Sector 81, SAS Nagar, Mohali, Manauli P.O., Punjab 140306, India. E-mail: sababu@iisermohali.ac.in
First published on 30th September 2025
Abstract
We report the modification of fluorene and fluorenone cores, and the assembly of libraries of functionalized fluorenes/fluorenones involving the C(sp2)–H and C(sp3)–H functionalization route. Fluorene/fluorenone motifs are versatile components in materials science, and some are bioactive compounds. In this work, the C–H bonds of fluorene and fluorenone were subjected to C–H arylation, alkylation, benzylation, alkoxylation, and annulation reactions to afford modified fluorenes/fluorenones. Alternatively, various functionalized fluorenes/fluorenones were obtained by subjecting the C–H bonds of aryl or alkyl carboxamides to the C–H arylation process using iodofluorenes or iodofluorenones as the coupling partners. Overall, a comprehensive effort and synthesis of several C(1), C(2), C(3), and C(4) functionalized fluorenes, fluorenones, fluorene–fluorenone couples, fluorene–carbazole couples, π-extended bis fluorenes, and enantioenriched π-extended bis fluorenes are reported. Given the prominence of fluorene/fluorenone motifs, this work contributes to augmenting the library of fluorenes and fluorenones via a C(sp2)–H and C(sp3)–H functionalization strategy.
Introduction
Fluorene and fluorenone are a biaryl class of cyclic molecules that occupy an important place in the family of functional materials and aromatic compounds.1–6 Fluorene and fluorenone motifs have been explored for their physicochemical, optical, and electrochemical properties and in developing fluorophores, organic light-emitting devices (OLEDs),6e liquid crystals, solar cells, oligomers/polymers, etc. (Fig. 1).4,5 | ||
| Fig. 1 Representative fluorene and fluorenone molecules with applications in materials and medicinal chemistry. | ||
While synthetically derived fluorenes and fluorenones are widespread in materials chemistry, several fluorene- and fluorenone-based natural products are also well known (Fig. 1).1 Some fluorene- or fluorenone-based compounds are used as drug molecules (e.g., lumefantrine (1d) and tilorone (1g)). Additionally, fluorene- or fluorenone-based molecules have been reported to exhibit promising biological activities and have been found to be useful in drug discovery/medicinal chemistry research.7,8 (e.g., Hsp90 inhibition,7a tubulin interaction,7b antimyocardial ischemia activities7c,g,h and N-aryl-9-oxo-9H-fluorene-1-carboxamide derivative (1b) was found to induce apoptosis).7d A fluorene motif was shown to reduce the amyloid burden, which initiates neurodegeneration and cognitive deficits in Alzheimer's disease.7e Fluorenone 1f is a probe for studying DNA redox chemistry.7f Notably, 9-fluorenylmethoxycarbonyl (Fmoc)-group-based molecules play a pivotal role in amino acid/peptide chemistry.9
Given their applications and pivotal roles in developing functional materials and medicinally or biologically active agents, various synthetic routes have been designed for synthesizing the core structures of fluorenes and fluorenones.10–19
Functionalized fluorenone- or fluorene-based monomers are generally synthesized via traditional cross-coupling reactions (Scheme 1).19,20 Some reported methods affording fluorenones include: (a) ortho metalation,10 (b) cross-coupling or C–H coupling and Friedel–Crafts reaction,11 (c) radical cyclization,12 (d) CO insertion,13 (e) intra- or intermolecular C–H functionalization and decarboxylative coupling reactions,14 and (f) conversion of fluorene to fluorenones via oxidation.15 Fluorene skeletons have been constructed via the Friedel–Crafts reaction,16 gold-catalyzed C–C bond-forming reactions,17 intramolecular C–H functionalization, etc.18
 | ||
| Scheme 1 Representative commonly employed methods for synthesizing functionalized fluorene and fluorenone molecules. | ||
Some existing methods require the assembly of pre-functionalized organometallic reagents as starting materials for synthesizing fluorene or fluorenone skeletons (Scheme 1). Alternatively, the direct functionalization of C–H bonds of the fluorene or fluorenone skeleton might lead to a step-economical route for building a library of modified fluorene and fluorenone motifs (Scheme 2).
 | ||
| Scheme 2 Modification of fluorene and fluorenone core via C–H functionalization and expansion of their library. | ||
In recent years, the transition-metal-catalyzed site-selective C–H functionalization of small molecules using a directing group has emerged as a practical route for building a library of different classes of functionalized cyclic and aliphatic skeletons.21–24 In particular, transition-metal-catalyzed, directing-group-assisted C–C bond-forming reactions leading to biaryls via direct C–H coupling constitute a powerful alternative to traditional cross-coupling tactics. Taking impetus from the contributions of Pd(II)-catalyzed, directing-group-aided sp2 C–H arylation reactions in expanding the library of privileged skeletons,22–24 we envisaged the direct functionalization of C–H bonds in fluorenes and fluorenones to construct a library of modified fluorene and fluorenone motifs (Scheme 2). This process has enabled the assembly of a variety of fluorene and fluorenone monomeric motifs, which are expected to show scope in the development of materials and medicinal chemistry.
Results and discussion
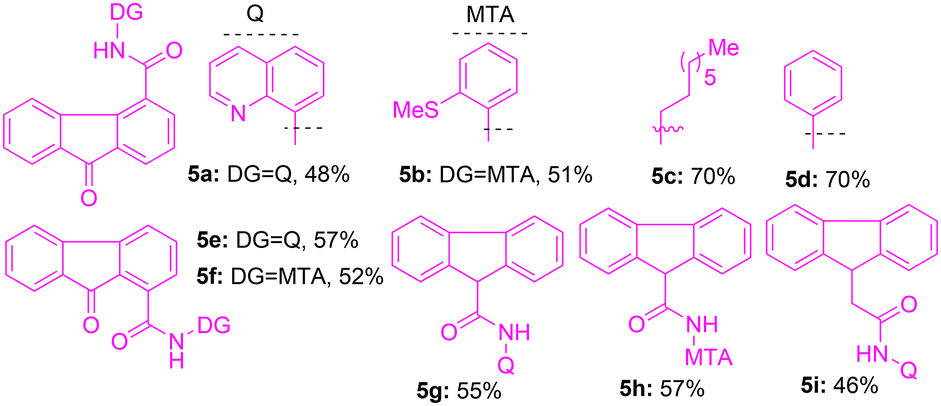
To initiate the modification of the fluorenone and fluorene core via directing-group-aided C–H functionalization, initially, 9-oxo-9H-fluorene-4-carboxylic acid was linked with bidentate directing groups22,23 under standard amide coupling conditions. The preparation of 9-oxo-9H-fluorene-4-carboxamides 5a–d with the corresponding bidentate directing groups (e.g., 8-aminoquinoline (8-AQ), 2-(methylthio)aniline (MTA), and simple amines) was completed (Table 1). Next, we assembled 9-oxo-9H-fluorene-1-carboxamides 5e,f by coupling 9-oxo-9H-fluorene-1-carboxylic acid with 8-AQ and MTA directing groups (Table 1). We also assembled fluorene-based carboxamides 5g–i from their corresponding fluorene carboxylic acid and 8-AQ or MTA using standard procedures (Table 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Additive (0.25 mmol) | Solvent (mL) | T (°C) | 7a: yielda,b (%) |
| a Reaction conditions: 5a (0.1 mmol), 4a (4 equiv.), catalyst, additive, solvent, 100–130 °C, 48 h, sealed tube (filled with N2 atm). b Isolated yield. c In addition to AgOAc, (BnO)2PO2H or PivOH (0.02 mmol) was used, respectively. d ArI (2 equiv.) was used. e Pd(OAc)2 (5 mol%) was added. f Reaction time = 24 h. | |||||
| 1 | Pd(OAc)2 | K2CO3 | o-Xylene (1) | 130 | 48 |
| 2 | Pd(OAc)2 | Ag2CO3 | Neat | 130 | 30 |
| 3 | Pd(OAc)2 | AgOAc | o-Xylene (1) | 130 | 50 |
| 4 | Pd(OAc)2 | AgOAc | Toluene (1) | 110 | 48 |
| 5 | Pd(OAc)2 | AgOAc | HFIP (1) | 100 | 50 |
| 6 | Pd(OAc)2 | AgOAc (BnO)2PO2Hc | Toluene (1) | 110 | 48 |
| 7 | Pd(OAc)2 | AgOAc PivOHc | Toluene (1) | 110 | 45 |
| 8 | Pd(TFA)2 | AgOAc | o-Xylene (1) | 130 | 33 |
| 9 | Pd(MeCN)2Cl2 | Cs2CO3 | o-Xylene (1) | 130 | 20 |
| 10 | Pd(OAc)2 | AgOAc | m-Xylene (1) | 130 | 50 |
| 11 | Pd(OAc)2 | AgOAc | m-Xylene (50 μL) | 130 | 72 |
| 12d | Pd(OAc)2 | AgOAc | m-Xylene (50 μL) | 130 | 32 |
| 13e | Pd(OAc)2 | AgOAc | m-Xylene (50 μL) | 130 | 50 |
| 14f | Pd(OAc)2 | AgOAc | m-Xylene (50 μL) | 130 | 60 |
|
|||||

At the outset, we attempted the π-extension and sp2 C–H arylation of 9-oxo-N-(quinolin-8-yl)-9H-fluorene-4-carboxamide (5a) possessing an 8-aminoquinoline directing group (DG). We aimed at the site-selective β-C(sp2)–H functionalization of the C3 position of 5a under standard conditions.22–24Table 1 reveals optimization studies conducted for the C3 arylation of 5a in the presence of Pd(II) catalysts, silver or alkali metal salt additives, and solvents. Apart from the Pd(II) catalyst, an additive such as a silver salt (AgOAc or Ag2CO3) or an alkali–metal-based salt/base (e.g., Cs2CO3 or K2CO3) is essential for accomplishing the required Pd(II)-catalyzed, 8-aminoquinoline DG-aided C–H arylation reactions.22 The silver or alkali metal salt additive plays the role of a halide ion scavenger, and it helps in the regeneration of the Pd(II) catalyst in the proposed PdII–PdIV catalytic cycle.22,23a
The C(3)–H arylation of fluorene-4-carboxamide 5a was attempted using methyl 4-iodobenzoate (4a, 4 equiv.) in the presence of Pd(OAc)2 (10 mol%) and K2CO3 (2 equiv.) in o-xylene at 130 °C for 48 h (entry 1, Table 1). This reaction afforded C(3)–H arylated fluorenone 7a in 48% yield. Heating a mixture of 5a with 4a in the presence of Pd(OAc)2 and Ag2CO3 (2 equiv.) under neat conditions afforded C(3)–H arylated product 7a in 30% yield (entry 2, Table 1). Performing the same reaction using AgOAc as an additive in o-xylene or toluene or HFIP afforded 7a in 48–50% yields (entries 3–5, Table 1). Next, we heated 5a with 4a in the presence of Pd(OAc)2, AgOAc, and (BnO)2PO2H (20 mol%) or PivOH (20 mol%) as additional additives in toluene at 110 °C, and these reactions gave 7a in 45–48% yields (entries 6 and 7, Table 1).
Next, the same reaction using Pd(TFA)2 or Pd(MeCN)2Cl2 and AgOAc or Cs2CO3 as an additive in o-xylene at 130 °C gave product 7a in a decreased yield (20–33%, entries 8 and 9, Table 1). The reaction using Pd(OAc)2 catalyst and AgOAc in m-xylene solvent afforded product 7a in 50% yield (entry 10, Table 1). Next, the reaction of 5a and 4a in the presence of Pd(OAc)2 and AgOAc in a minimal amount of solvent m-xylene (50 μL), gave 7a in an improved yield (72%, entry 11, Table 1). The arylation of 5a using reduced equiv. of 4a (2 equiv.) in the presence of Pd(OAc)2 and AgOAc in m-xylene (50 μL) gave 7a in low yield (32%, entry 12, Table 1). The arylation of 5a using 4 equiv. of aryl iodide 4a in the presence of 5 mol% of Pd(OAc)2 and AgOAc in m-xylene (50 μL) gave 7a in 50% yield (entry 13, Table 1). The arylation of 5a using 4 equiv. of aryl iodide 4a in the presence of 10 mol% of Pd(OAc)2 and AgOAc in m-xylene (50 μL) for 24 h instead of 48 h gave 7a in 60% yield (entry 14, Table 1). In addition to the 8-AQ DG-assisted C(3)–H arylation of fluorenone 5a, which afforded 7a as the product, there was no indication of C(1)–H arylation directed by the inherent ketone functionality in fluorenone 5a, and the other expected product 7a′ was not obtained (Table 1).
Having examined the arylation of fluorene-4-carboxamide 5a, we intended to explore the competence of different directing groups for accomplishing the C(3)–H arylation of 9-oxo-9H-fluorene-4-carboxamides 5a–d (Scheme 3). Heating a mixture of 5a possessing 8-AQ as the DG with iodofluorene 6a in the presence of Pd(OAc)2 and AgOAc in m-xylene at 130 °C for 48 h afforded fluorenone–fluorene dyad 8a in 72% yield via the C(3)–H arylation of 5a (Scheme 3). The Pd(II)-catalyzed arylation of 5b, possessing MTA as the DG, with 6a, afforded fluorenone–fluorene dyad 8b in 63% yield via the C(3)–H arylation of 5b. The Pd(II)-catalyzed C–H arylation of fluorenone carboxamide 5d or 5c, possessing a simple amide as the DG, with 6a did not afford the corresponding expected fluorenone–fluorene skeleton 8c or 8d (Scheme 3). These attempts suggested that a bidentate directing group is necessary for accomplishing the β-C(sp2)–H arylation of fluorenone carboxamide 5a/5b. This confirmed that the corresponding bidentate directing group 8-AQ in 5a or MTA in 5b provides the required chelation assistance during the β-C(sp2)–H activation in the proposed PdII–PdIV catalytic cycle,22,23a enabling the site-selective C(3)–H arylation of 5a or 5b.
 | ||
Scheme 3 Pd(II)-catalyzed modification of fluorenone/fluorene core via C–H arylation using different directing groups. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Products 8a–d are from the corresponding fluorenone carboxamides 5a–d. bProduct 9a is from 5m using 4a (4 equiv.), Pd(OAc)2 (6 mol%), NMe4Cl (1.3 equiv.), KOAc (1.5 equiv.) and AcOH (1.5 equiv.). cProduct 9b is from 5m using 1-iodo-4-methylbenzene (4b, 4 equiv.), Pd(OAc)2 (2 mol%), Ag2CO3 (1 equiv.), KOAc (1.5 equiv.) and AcOH (3.5 equiv.). dProduct 9c is from 5m using 2-iodo-9,9-dimethyl-9H-fluorene (6a, 4 equiv.), Pd(OAc)2 (6 mol%), NMe4Cl (1.3 equiv.), KOAc (1.5 equiv.) and AcOH (1.5 equiv.). eUsing the conditions of entry 4 of Table 1 and p-anisyl iodide (4c). fUsing 4c, Pd(OAc)2 (10 mol%), Ag2CO3 (1.5 equiv.), KOAc (1.2 equiv.) in HFIP:AcOH (3:7), 110 °C, 24 h, sealed tube (purged with N2 atm). Products 8a–d are from the corresponding fluorenone carboxamides 5a–d. bProduct 9a is from 5m using 4a (4 equiv.), Pd(OAc)2 (6 mol%), NMe4Cl (1.3 equiv.), KOAc (1.5 equiv.) and AcOH (1.5 equiv.). cProduct 9b is from 5m using 1-iodo-4-methylbenzene (4b, 4 equiv.), Pd(OAc)2 (2 mol%), Ag2CO3 (1 equiv.), KOAc (1.5 equiv.) and AcOH (3.5 equiv.). dProduct 9c is from 5m using 2-iodo-9,9-dimethyl-9H-fluorene (6a, 4 equiv.), Pd(OAc)2 (6 mol%), NMe4Cl (1.3 equiv.), KOAc (1.5 equiv.) and AcOH (1.5 equiv.). eUsing the conditions of entry 4 of Table 1 and p-anisyl iodide (4c). fUsing 4c, Pd(OAc)2 (10 mol%), Ag2CO3 (1.5 equiv.), KOAc (1.2 equiv.) in HFIP:AcOH (3:7), 110 °C, 24 h, sealed tube (purged with N2 atm). | ||
Next, we intended to attempt the native carboxylic acid group-mediated C–H arylation of fluorene carboxylic acid 5m or 5n (Scheme 3). We tried the Pd(II)-catalyzed C(3)–H arylation of 5m with aryl iodides 4a or 4b or iodofluorene 6a under different reaction conditions. These reactions did not afford the expected C(3)–H arylated fluorene-4-carboxylic acid 9a–c (Scheme 3). We then attempted the C(1)–H arylation of 9H-fluorene-9-carboxylic acid 5n with p-tolyl iodide in the presence of Pd(OAc)2 and Ag2CO3 in AcOH at 130 °C for 24 h. This reaction did not yield the expected C(1)–H arylated fluorene 10a (Scheme 3). We continued testing the C–H arylation of additional fluorene substrates. The Pd(II)-catalyzed β-C–H arylation of carboxamide 5i, possessing 8-AQ as the DG with p-anisyl iodide, did not yield the expected C(9)–H arylated fluorene 10b (Scheme 3). The γ-C–H arylation of fluorene-9-carboxamide 5g possessing 8-AQ DG with p-anisyl iodide did not give the expected C(1)–H arylated product 10c. Similarly, the γ-C–H arylation of fluorene-9-carboxamide 5h possessing MTA DG did not provide the expected bis C(1)–H arylated product 10d (Scheme 3).
Having obtained the optimal reaction conditions for the site-selective C–H arylation of fluorenone-4-carboxamide 5a, we then intended to expand the scope of C–H arylation of 5a to prepare a wide range of π-extended fluorenone-4-carboxamides (Scheme 4). The Pd(II)-catalyzed C(3)–H arylation of 5a was attempted using various aryl iodides containing electron-donating or electron-withdrawing substituents at the para or meta position (e.g., OMe, alkyl, COMe, COOR, or NO2) and phenyl iodide in the presence of Pd(OAc)2 and AgOAc in m-xylene (50 μL) at 130 °C for 48 h. These reactions afforded the corresponding C(3)–H arylated, π-extended fluorenones 7a–g and 7k–m in 60–88% yields (Scheme 4). The structure of compound 7c was confirmed by X-ray structure analysis (Fig. 2).
 | ||
| Scheme 4 Expansion of the library of C(3) or C(2) arylated, π-extended fluorenones via Pd(II)-catalyzed C–H arylation of fluorenone-4-carboxamide 5a, 5b and 5e. aThe reactions were carried out using 8-aminoquinoline DG and 2-methylthioaniline DG-linked substrates. bCompounds 7a–y were obtained from 5a. cCompounds 7z and 7za were obtained from 5b. dThe reaction was carried out under neat conditions. eThe reaction was carried out in m-xylene (1 mL). fToluene (1 mL) as solvent at 110 °C. | ||
 | ||
| Fig. 2 X-ray (ORTEP) structures of compounds 7c and 11f. | ||
Next, fluorene-4-carboxamide 5a was subjected to Pd(II)-catalyzed arylation with various disubstituted aryl iodides to afford the corresponding C(3)–H arylated fluorenones 7h–j in 67–71% yields (Scheme 4). The C(3)–H arylation of 5a using heteroaryl iodides, such as iodopyridyls and 5-iodoindole, afforded the corresponding fluorenone–heteroaryl motifs 7n–p in 56–77% yields. Treatment of 5a with azobenzene-based aryl iodide afforded fluorenone–azobenzene motif 7q in 86% yield. Treatment of various biaryl iodides containing electron-donating or electron-withdrawing substituents at the para position (e.g., OMe, alkyl, Br, O-alkyl, OAc) with 5a yielded the corresponding fluorenone–biaryl motifs (π-extended fluorenones) 7r–x in 50–72% yields (Scheme 4). The reaction of 3-iodobiphenyl with 5a afforded the fluorenone–biaryl motif 7y in 60% yield (Scheme 4). Additionally, treatment of fluorene-4-carboxamide 5b possessing MTA as the DG with iodobiaryls yielded the corresponding fluorenone–biaryl motifs 7z and 7za in 49–50% yields (Scheme 4).
Having introduced the aryl group at the C(3)–H of the fluorenone-4-carboxamide motif and obtained the modified fluorenone–aryl systems 7a–z and 7za (Scheme 4), alternatively, we ventured into the synthesis of C(2) arylated π-extended fluorenones 11 and 12via the ortho C–H arylation of aromatic carboxamides (Scheme 5). Accordingly, benzamides 3a–g with 8-AQ DG were subjected to (ortho) C–H arylation with various iodofluorenes/iodofluorenones 6a–d.
 | ||
| Scheme 5 Expansion of the library of C(2) arylated, π-extended fluorenones or fluorenes through the coupling of the C–H bond of benzamides and heteroaryl carboxamides with iodofluorenes or iodofluorenones. | ||
The Pd(II)-catalyzed C–H arylation of benzamides 3a–g with 2-iodo-9,9-dimethyl-9H-fluorene (6a) or 2-bromo-7-iodo-9H-fluoren-9-one (6b) or 2-bromo-7-iodo-9H-fluorene (6d) under the optimized conditions yielded the corresponding fluorene–benzamide or fluorenone–benzamide coupled motifs 11e–m in 62–88% yields (Scheme 5). The structure of compound 11f was confirmed by X-ray structure analysis (Fig. 2). As corresponding meta-substituted benzamides 3e–g contain two ortho β-C–H bonds, arylation occurred selectively at the least hindered ortho β-C–H bond in 3e–g, thereby affording the corresponding mono β-C–H arylation products 11e–g. This observation is in accordance with the earlier reports of directing-group-aided C(sp2)–H arylation of meta-substituted benzamides.22,23h,j,s,t Benzamide substrate 3h possessing ABTD as the DG with 6a under the optimized reaction conditions, gave fluorene–aryl coupled moiety 12 in 81% yield (Scheme 5).
Several functional materials used in organic field effect transistors feature a chemical design based on fluorenone–thiophene coupled motifs. Taking impetus from the applications of fluorenone–thiophene motifs, we attempted the synthesis of fluorenone–thiophene and fluorine–thiophene coupled molecules. Accordingly, Pd(II)-catalyzed reaction of fluorene-1-carboxamide 5e with 2-iodothiophene afforded the fluorenone–thiophene coupled motif 7zb in 65% yield (Scheme 4). Alternatively, thiophene-2-carboxamide 3i, possessing 8-AQ DG, was subjected to Pd(II)-catalyzed C(3)–H arylation with 2-iodofluorene 6a to afford fluorene–thiophene motif 11a in 77% yield (Scheme 5). Thiophene-2-carboxamide 3j, possessing MTA, DG with 6a, also afforded the fluorene–thiophene coupled motif 11b in 51% yield (Scheme 5). Subsequently, we treated furan-2-carboxamide 3k and benzothiophene-2-carboxamide 3l with 8-AQ DG with 6a under standard reaction conditions to afford the corresponding fluorene–furan motif 11c and fluorene–benzothiophene motif 11d in 80–94% yields (Scheme 5).
Next, arylacetamides possessing 8-AQ DG were subjected to the (ortho) γ-C(sp2)–H arylation with iodofluorene 6a. Arylacetamides 3s, 3t, 3v–z and 3aa with substitutions at the ortho- or meta- or para-positions were heated with 6a in the presence of Pd(OAc)2 and AgOAc in toluene. These attempts yielded the corresponding fluorene–arylacetamide motifs (π-extended fluorenes) 13a–g and 13i in 58–85% yields (Scheme 6). Biaryl acetamide 3u with 6a yielded the fluorene-biaryl coupled moiety 13h in 47% yield (Scheme 6). Along these lines, benzylamine substrates possessing 5-methylisoxazole-3-carboxamide DG 3m–o were subjected to Pd(II)-catalyzed (ortho) γ-C(sp2)–H arylation conditions with iodofluorenes 6a, 6b, and 6c, which afforded the corresponding fluorene–benzylamine coupled motifs 13j–l in 69–85% yields (Scheme 6). Further, benzylamines possessing picolinamide DG, 3p and 3q, or naphthylamine 3r with picolinamide DG were treated with 6a in the presence of Pd(OAc)2, CuBr2, and CsOAc in t-amylOH.25a These attempts yielded the corresponding fluorene–benzylamine and fluorene–naphthylamine coupled motifs 13m–o in 70–91% yields (Scheme 6). Additionally, thiophen-2-ylmethanamine substrate 3ab, possessing a picolinamide directing group, was subjected to C(3)–H arylation with 6a, which afforded thiophene–fluorene motif 13p in 82% yield (Scheme 6).
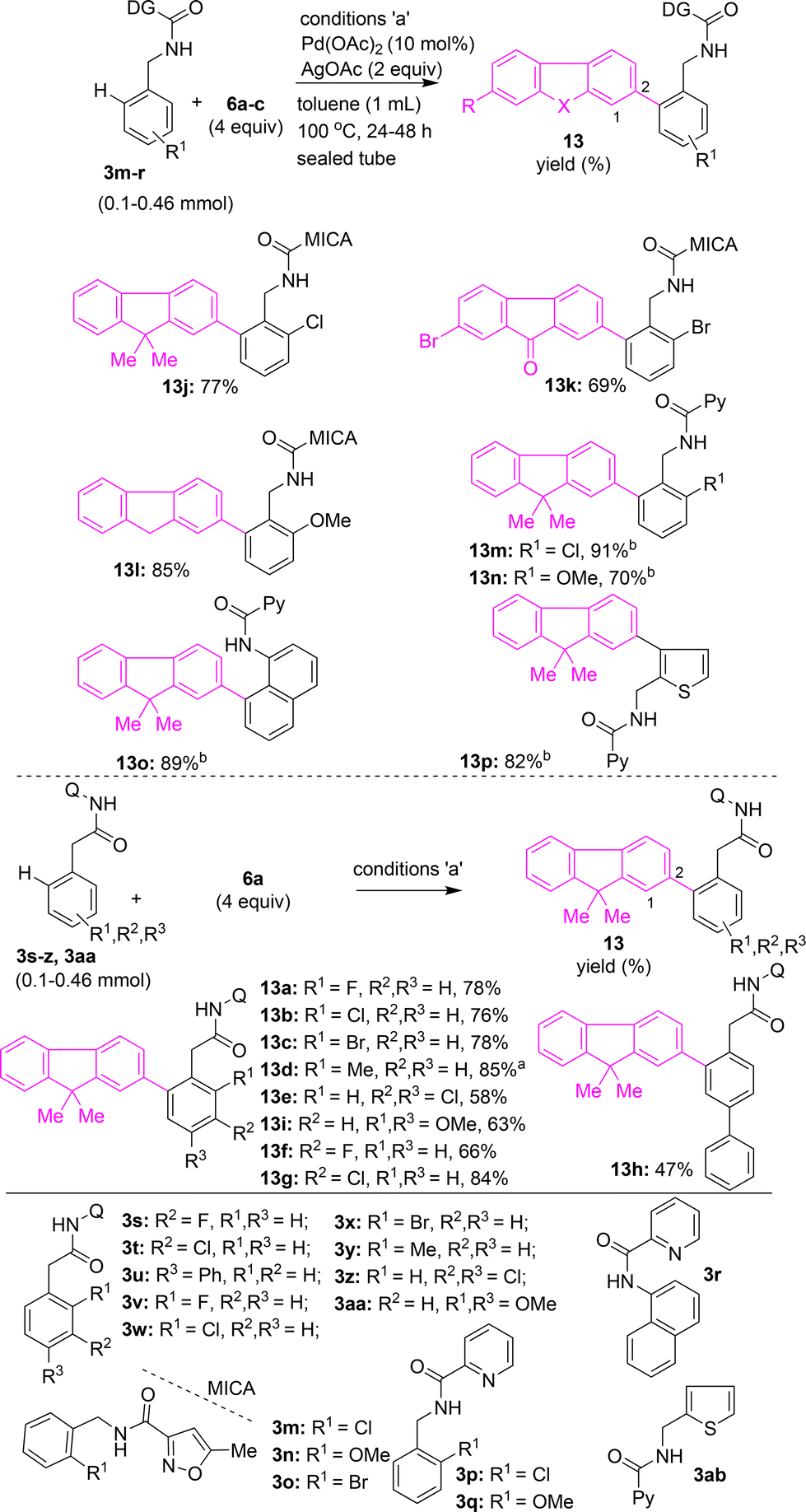 | ||
| Scheme 6 Expansion of the library of C(2) arylated, π-extended fluorenones or fluorenes through the coupling of the ortho γ-C(sp2)–H bond of aryl acetamides and benzylamines with iodofluorenes or iodofluorenones. a13d is from substrate 3y and 6a (4 equiv.), Pd(OAc)2 (10 mol%), K2CO3 (2 equiv.), p-xylene (1 mL), 48 h sealed tube (purged with N2 atm). b13m–p are from their corresponding substrates using conditions 6a (4 equiv.), Pd(OAc)2 (10 mol%), CuBr2 (10 mol%), CsOAc (4 equiv.), t-amylOH (1–3 mL), 130 °C, 36 h sealed tube (purged with N2 atm). | ||
We then intended to expand the scope of this method by synthesizing fluorenones with enhanced conjugation, viz., fluorenone–fluorene motifs (Scheme 7). Towards this, heating fluorene-4-carboxamide 5a with 8-AQ DG with 6a in the presence of Pd(OAc)2 and AgOAc in m-xylene (50 μL) at 130 °C for 48 h afforded fluorenone–fluorene coupled motif 8a in 72% yield via C(3)–H arylation (Scheme 7). Next, the same reaction was performed using fluorene-4-carboxamide 5b with MTA DG, to afford fluorenone–fluorene coupled motif 8b in 63% yield. Further, treatment of 5a with 2-iodofluorene 6c afforded the fluorenone–fluorene coupled motif 8c in 70% yield. Subsequently, the C(2)–H arylation of fluorene-1-carboxamides 5e or 5f with 6a in the presence of Pd(OAc)2 and AgOAc yielded corresponding fluorenone–fluorene coupled motifs 8d and 8e in 76–83% yields (Scheme 7).
 | ||
| Scheme 7 Pd(II)-catalyzed C–H arylation of fluorenone-4-carboxamides 5a, 5b and fluorenone-1-carboxamides 5e and 5f towards the synthesis of bis fluorene (fluorenone–fluorene coupled) motifs. aChlorobenzene (1 mL) was used as a solvent. | ||
Along these lines, we envisioned preparing fluorenone–carbazole and fluorene–carbazole coupled scaffolds. Toward this, we heated fluorene-4-carboxamide 5a with 3-iodocarbazole 6e (4 equiv.) as the coupling partner in the presence of Pd(OAc)2 and AgOAc in m-xylene. This reaction afforded fluorenone–carbazole coupled skeleton 8f in 93% yield (Scheme 8). Alternatively, we treated carbazole-3-carboxamide 5j possessing 8-AQ DG with iodofluorenes 6a, 6c and 6d as the coupling partners in the presence of Pd(OAc)2 and AgOAc in toluene at 110 °C for 48 h. These attempts resulted in the fusion of the C2 position of the fluorene unit with the C2 position of the carbazole unit, affording the corresponding carbazole–fluorene motifs 8g–i in 51–86% yields (Scheme 8).
 | ||
| Scheme 8 Synthesis of fluorenone–carbazole and fluorene–carbazole coupled motifs via the Pd(II)-catalyzed C–H arylation of fluorenone-4-carboxamide 5a and carbazole-3-carboxamide 5j. | ||
We then wished to adopt the transient-directing-group-assisted (TDG) β-C–H functionalization strategy26a to functionalize fluorene moiety 5k and obtain C–H arylated fluorene motif 14a or 14a′. Table 2 describes the screening of reaction conditions for the Pd(II)-catalyzed, transient-directing-group-aided direct β-C(sp2)–H arylation of 2-acetylfluorene (5k) with p-anisyl iodide. We commenced the optimization studies by evaluating the significance of glycine as a TDG to functionalize the C(1)–H or C(3)–H bond of 2-acetylfluorene (5k). We attempted the β-C(sp2)–H arylation of 5k with 4c (3 equiv.) in the presence of glycine (50 mol%), Pd(PPh3)2Cl2, AgOAc or Ag2CO3 as an additive and H2O (4 equiv.) in HFIP:AcOH (9:1). This reaction afforded C(3)–H arylated fluorene 14a in 30–53% yields (entries 1 and 2, Table 2). The same reaction in TFA solvent did not yield 14a (entry 3, Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Additive (x equiv.) | Solvent (mL) | TDG (50 mol%) | 14a: yielda (%) |
| a Isolated yield. b Reaction temperature = 100 °C and H2O (1 mL). c Using 1.5 equiv. of ArI. | |||||
| 1 | Pd(PPh3)2Cl2 | AgOAc (2) | HFIP:AcOH (9:1, 1 mL) |
Glycine | 53 |
| 2 | Pd(PPh3)2Cl2 | Ag2CO3 (2) | HFIP:AcOH (9:1, 1 mL) |
Glycine | 30 |
| 3b | Pd(PPh3)2Cl2 | AgTFA (1.5) | TFA (1 mL) | Glycine | 0 |
| 4 | Pd(OAc)2 | AgOAc (2) | HFIP:AcOH (9:1, 1 mL) |
Glycine | 40 |
| 5 | PdCl2 | AgOAc (2) | HFIP:AcOH (9:1, 1 mL) |
Glycine | 30 |
| 6 | Pd(OAc)2 | AgTFA (2.5) | HFIP:AcOH (9:1, 1 mL) |
Glycine | 55 |
| 7b | Pd(OAc)2 | AgTFA (1.5) | AcOH:H2O (9:1, 1 mL) |
Glycine | 44 |
| 8 | Pd(TFA)2 | AgTFA (1.5) | HFIP:AcOH (9:1, 1 mL) |
Glycine | 50 |
| 9 | Pd(PPh3)2Cl2 | AgOAc (2) | HFIP:AcOH (9:1, 1 mL) |
β-Alanine | 25 |
| 10 | Pd(PPh3)2Cl2 | AgOAc (2) | HFIP:AcOH (9:1, 1 mL) |
Phenylglycine | 10 |
| 11 | Pd(PPh3)2Cl2 | AgOAc (2) | HFIP:AcOH (9:1, 1 mL) |
DL-Alanine | 10 |
| 12 | Pd(OAc)2 | AgTFA (1.5) | HFIP:AcOH (9:1, 1 mL) |
2-Aminoisobutyric acid | 0 |
| 13c | Pd(OAc)2 | AgOAc (2) | HFIP:AcOH (5:1, 1 mL) |
Glycine | 71 |

The C–H arylation of 5k with 4c using Pd(OAc)2 or PdCl2 as the catalyst instead of Pd(PPh3)2Cl2 yielded 14a in 30–40% yields (entries 4 and 5, Table 2). C–H arylation of 5k with 4c in the presence of glycine, Pd(OAc)2, AgTFA as an additive, and H2O in HFIP:AcOH yielded 14a in 55% yield (entry 6, Table 2). The same reaction in AcOH:H2O yielded 14a in 44% yield (entry 7, Table 2). Then, the C–H arylation of 5k with 4c in the presence of glycine, Pd(TFA)2, AgTFA and H2O in HFIP:AcOH yielded 14a in 50% yield (entry 8, Table 2). We then attempted the C–H arylation of 5k with 4c by using other amino acids as the transient directing group (TDG). We attempted the Pd(II)-catalyzed arylation of 5k with 4c in the presence of β-alanine or phenylglycine or DL-alanine or 2-aminoisobutyric acid as the TDG. These reactions were not fruitful, and product 14a was obtained in low yields (entries 9–12, Table 2). This trend suggested that glycine is a suitable TDG to perform the β-C(sp2)–H arylation of 5k with 4c. Accordingly, another reaction condition26b involving the β-C(sp2)–H arylation of 5k with 4c (1.5 equiv.) in the presence of glycine, Pd(OAc)2, AgOAc and H2O (4 equiv.) in HFIP:AcOH afforded 14a with maximum 71% yield (entry 13, Table 2). While the expected product 14a was obtained, we did not obtain 14a′ in characterizable amounts. The C(1)–H bond in 5k is relatively hindered, and presumably C(1)–H arylation is a sluggish process compared to arylation at the C(3)–H position of 5k. This observation is in accordance with earlier work dealing with C–H arylation of a similar type of substrate (e.g., 2-acetylnapthalene), which predominantly afforded the corresponding least hindered ortho C–H functionalized product.26d
With the optimized reaction conditions in hand, we then performed the β-C(3)–H arylation of 2-acetylfluorene 5k with different aryl iodides containing electron-donating or electron-withdrawing substituents (e.g., OMe, COOH, COOMe, COOEt) in the presence of glycine, Pd(OAc)2, AgOAc, H2O (4 equiv.), or HFIP:AcOH. These attempts successfully afforded the corresponding fluorene–aryl coupled skeletons 14a–d in 44–81% yields (Scheme 9). Along these lines, we attempted the transient-directing-group-assisted arylation of methyl C(sp3)–H bond of o-tolualdehyde 5l with iodofluorenes 6a or 6c or iodofluorenone 6b using the reported conditions,26c involving Pd(OAc), glycine, Pd(OAc)2, AgTFA, and AcOH:H2O (9:1). These attempts were successful in affording the fluorene- and fluorenone-based diarylmethanes 14e–g in 80–90% yield using TDG methodology (Scheme 9).
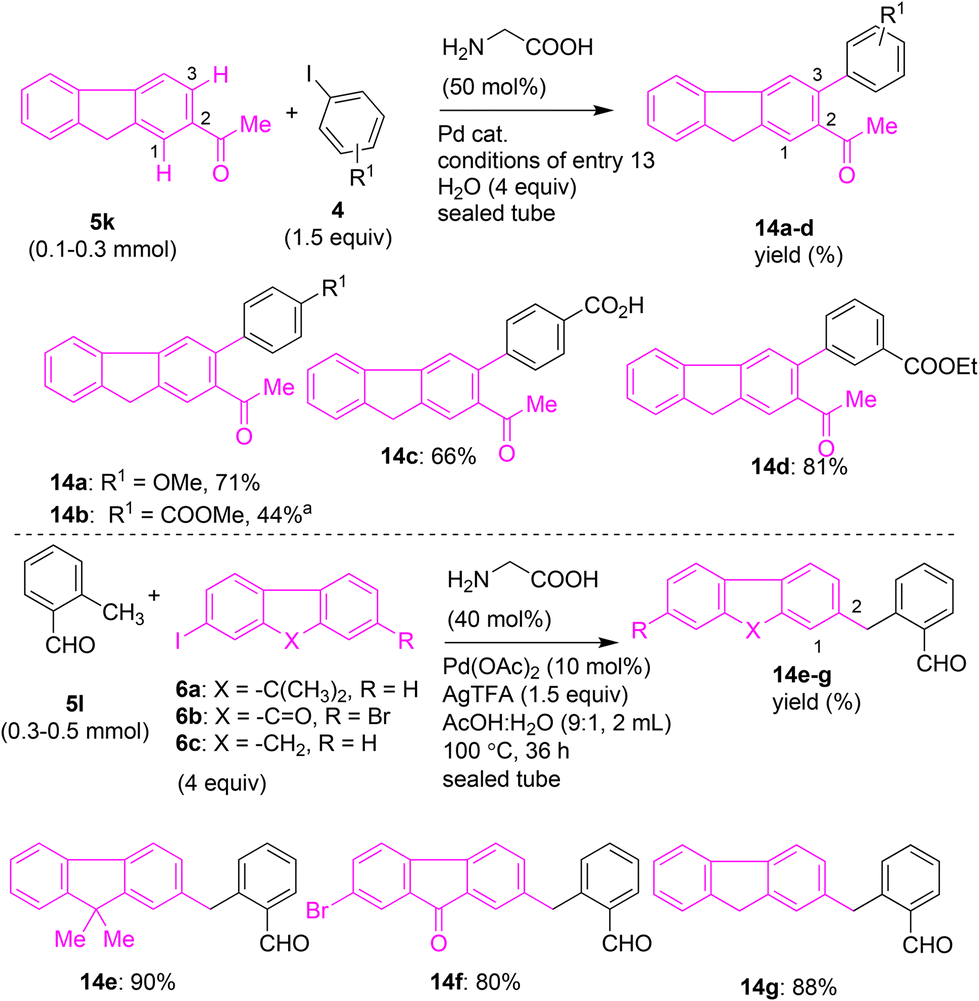 | ||
| Scheme 9 Pd(II)-catalyzed transient directing group (TDG)-promoted C–H arylation of 2-acetylfluorene (5k) and o-tolualdehyde (5l). aCompound 14b was obtained from substrate 5k using reaction conditions including 4a (1.5 equiv.), Pd(PPh3)2Cl2 (10 mol%), AgOAc (2 equiv.), HFIP:HOAc (9:1), 130 °C, 36 h, sealed tube (purged with N2 atm). | ||
We then explored the synthesis of alkylated fluorenones and fluorenes via β-C(sp2)–H alkylation and benzylation of the fluorene core. In this direction, we initially performed the 8-AQ DG-assisted β-C(sp2)–H alkylation of fluorenone-4-carboxamide 5a. Substrate 5a was treated with MeI (4 equiv.) in the presence of Pd(OAc)2, K2CO3, and PivOH in t-amylOH at 130 °C for 48 h. This reaction successfully afforded β-C(3)–H methylated fluorenone scaffold 15a in 67% yield (Scheme 10). Successively, 5a was treated with various primary alkyl iodides (4 equiv.) in the presence of Pd(OAc)2, K2CO3, and PivOH in t-amylOH (2 mL) at 130 °C for 48 h to afford the corresponding β-C(3)–H alkylated fluorenone scaffolds 15b and 15d–g in 43–79% yields (Scheme 10) Compound 15c was obtained from 5a and n-BuI using conditions which included Pd(OAc)2, CuBr2, K2CO3, H2O (1.5 mL), 110 °C, and 48 h. The expected β-C(3)–H alkylated fluorenone scaffolds 15h and 15i were not obtained when alkylation of 5a was attempted using the corresponding secondary alkyl iodides. An inseparable mixture of starting material and unidentified products was obtained in these cases.
 | ||
| Scheme 10 Synthesis of alkylated fluorenones/fluorenes. aCompounds 15a, 15b and 15d–g are from substrate 5a using corresponding R-I (4 equiv.) and conditions Pd(OAc)2 (5 mol%), K2CO3 (2.5 equiv.), PivOH (20 mol%), t-amylOH (2 mL), 130 °C, 48 h, sealed tube (purged with N2 atm). bCompound 15c is from substrate 5a using conditions n-BuI (4 equiv.), Pd(OAc)2 (10 mol%), K2CO3 (2 equiv.), CuBr2 (20 mol%), H2O (1.5 mL), 110 °C, 48 h, sealed tube (filled with ambient air). cCompounds 15h and 15i were detected as an inseparable mixture. dCompounds 16a–d and 16e are from substrates 5a and 5e respectively, using conditions BnBr (3 equiv.), Pd(OAc)2 (5 mol%), KOAc (2 equiv.), 1,4-dioxane (2 mL), 100 °C, 36 h, sealed tube (purged with N2 atm). Product 20 was obtained from the respective carboxamide 19. | ||
Having introduced the alkyl group at the C(3)–H of the fluorenone-4-carboxamide motif and obtained the modified fluorene–alkyl motifs 15a–g (Scheme 10), alternatively, we ventured into the synthesis of fluorene–alkyl motifs 20via arylation of the β-C(sp3)–H of aliphatic/alicyclic carboxamides. Accordingly, aliphatic carboxamides possessing 8-AQ DG 19a–e were subjected to β-C(sp3)–H arylation with various iodofluorenes 6a–d in the presence of Pd(OAc)2 and AgOAc in toluene. These attempts afforded the corresponding fluorene–alkyl motifs 20a–j in 75–93% yields (Scheme 10). Then, alicyclic carboxamides possessing 8-AQ DG, 17 and 18, were subjected to Pd(II)-catalyzed β-C(sp3)–H arylation with iodofluorenes 6a–c. These attempts afforded the corresponding fluorene-appended alicyclic motifs 20m–o in 71–78% yields (Scheme 10). In agreement with reports in the literature,24n,o the corresponding products 20m–o are believed to have cis stereochemistry via arylation of the diastereotopic β-C(sp3)–H bond of the corresponding alicyclic carboxamides (Scheme 10).
Next, fluorene-4-carboxamide 5a was treated with benzyl bromide (3 equiv.) in the presence of Pd(OAc)2 and KOAc in 1,4-dioxane at 100 °C to afford the corresponding β-C(3)–H benzylated fluorenone (fluorenone-based diarylmethane) scaffold 16a in 67% yield (Scheme 10). Along these lines, using the corresponding benzyl bromides, fluorenone-based diarylmethane scaffolds 16b–d were obtained in 68–87% yields from C(3)–H benzylation of 5a. Additionally, the Pd(II)-catalyzed C–H benzylation of fluorene-1-carboxamide 5e with 1-bromo-4-(tert-butyl)benzene afforded the corresponding β-C(2)–H benzylated fluorenone (fluorenone-based diarylmethane) scaffold 16e in 70% yield (Scheme 10). Successively, arylation of the methyl γ-C(sp3)–H bond of thiophene-2-carboxamide or furan-2-carboxamide, 19h and 19i, with iodofluorene 6a in the presence of Pd(OAc)2 and AgOAc in toluene afforded the corresponding fluorenone-based diarylmethane scaffolds 20k and 20l in 69–70% yields (Scheme 10).
Given the importance of π-extended fluorene motifs in materials science, we envisioned assembling a bis fluorene motif with an aryl ring spacer unit and enantioenriched π-extended fluorene motif (Schemes 11 and 12). Towards this end, we planned to perform the double ortho β-C(sp2)–H arylation of benzamides with iodofluorenes and iodofluorenones to obtain the targeted bis fluorene motifs with an aryl ring spacer unit. At first, benzamide 24a possessing 8-AQ DG was reacted with iodofluorene 6a in the presence of Pd(OAc)2 and AgOAc in toluene. This reaction successfully afforded the expected bis fluorene motif 21a with an aryl ring spacer unit via the double C–H arylation of 24a (see Scheme S2 in SI for the structures of starting materials 24a–w relevant to Scheme 11). Benzamides 24c and 24d were then reacted with 6a to afford bis fluorenes 21b and 21c in 66–71% yields (Scheme 11). Benzamide 24e linked with the ABTD directing group reacted with 6a to afford bis fluorene 21d in 89% yield (Scheme 11). Treatment of azobenzene-based benzamide 24f with 6a yielded the azobenzene-based bis fluorene motif 21e in 75% yield (Scheme 11). Treatment of benzophenone-based carboxamide 24b with 6b yielded the benzophenone-based bis fluorenone motif 21f in 60% yield. Then, arylacetamides 24h–k were subjected to Pd(II)-catalyzed double ortho C(sp2)–H arylation with iodofluorene 6a to afford target bis fluorene motifs 21g–j with an aryl ring spacer unit in 58–78% yields (Scheme 11).
 | ||
| Scheme 11 Construction of π-extended bis fluorenes with an aryl spacer via the ortho C(sp2)–H arylation of benzamide, arylacetamide, benzylamine, amino alcohol and amino acid carboxamides using iodofluorenes and iodofluorenones. See Scheme S2 in SI for the structures of starting materials 24a–w. aCompound 22a was obtained from substrate 24s using 6a (4 equiv.), Pd(OAc)2 (10 mol%), CuBr2 (10 mol%), CsOAc (4 equiv.), t-amylOH (2 mL), 130 °C, 36 h, sealed tube (purged with N2 atm). | ||
 | ||
| Scheme 12 Construction of enantioenriched π-extended bis fluorenes with an aryl spacer via the ortho C(sp2)–H arylation of benzamide, arylacetamide, benzylamine, amino alcohol, and amino acid carboxamides using iodofluorenes and iodofluorenones. See Scheme S2 in SI for the structures of starting materials used in this scheme. | ||
Benzylamine or α-methylbenzylamine substrates 24s, 24u-(RS), 24r-(RS), 24t-(RS) and 24q with picolinamide or 5-methylisoxazole-3-carboxamide directing groups were subjected to Pd(II)-catalyzed double ortho C(sp2)–H arylation with iodofluorenes 6a, 6d or iodofluorenone 6b. These attempts yielded the corresponding targeted benzylamine-based bis fluorene motifs 22a, 22b-(RS), 22e-(RS), 22f-(RS) and 22h in 70–99% yields (Scheme 11). Similarly, the Pd(II)-catalyzed picolinamide DG-assisted double ortho C(sp2)–H arylation of substrates 24v-(RS), 24n-(RS), 24o-(RS) and 24p-(RS) with iodofluorene 6a or 6c was performed. The corresponding phenyl glycinol-based bis fluorene motif 22c-(RS), phenyl alaninol-based bis fluorene motif 23c-(RS) and tyrosinol-based bis fluorene motifs 23a-(RS) and 23d-(RS) were obtained. Furthermore, we subjected aromatic amino acid derivatives, such as phenylglycine, phenylalanine, and tyrosine substrates, to Pd(II)-catalyzed picolinamide DG-assisted double ortho C(sp2)–H arylation with iodofluorenes 6a, 6b and 6d. Accordingly, phenylglycine-based bis fluorene motifs 22g-(DL) and 22i-(DL), phenylalanine-based bis fluorene motif 23b-(DL), β-phenylalanine-based bis fluorene motif 22d-(RS), and tyrosine-based bis fluorene motif 23e-(DL) were obtained from their corresponding substrates 24g-(DL), 24l-(DL), 24w-(RS) and 24m-(DL) (Scheme 11).
Subsequently, we wished to prepare an enantioenriched bis fluorene motif with an aryl ring spacer unit (Scheme 12). Accordingly, enantioenriched α-methylbenzylamine substrates 24u-(R), 24u-(S), 24r-(R), 24r-(S), 24t-(R), and 24t-(S) possessing picolinamide or MICA DG were subjected to Pd(II)-catalyzed double ortho C(sp2)–H arylation with iodofluorenes 6a or iodofluorenone 6b. These attempts yielded the corresponding enantioenriched α-methylbenzylamine-based bis fluorene motifs 22b-(R), 22b-(S), 22e-(R), 22e-(S), 22f-(R) and 22f-(S) in good yields with good enantiopurity (see Scheme S2 in SI for the structures of starting materials relevant to Scheme 12).
We conducted Pd(II)-catalyzed picolinamide DG-assisted double ortho C(sp2)–H arylation of enantioenriched phenyl alaninol substrates 24n-(R) and 24n-(S), tyrosinol substrates 24o-(R) and 24o-(S) with iodofluorene 6a or 6c. These attempts yielded the corresponding enantioenriched phenylalaninol-based bis fluorene motifs 23c-(R) and 23c-(S) and tyrosinol-based bis fluorene motifs 23a-(R) and 23a-(S) in good yields and enantiopurity (Scheme 12). Furthermore, we conducted the Pd(II)-catalyzed picolinamide DG-assisted double ortho C(sp2)–H arylation of enantioenriched aromatic amino acid substrates, such as phenylglycine 24g-(D) and 24g-(L), tyrosine 24m-(D) and 24m-(L) and phenylalanine 24l-(D) and 24l-(L) with iodofluorene 6d or 6b or 6a. These attempts yielded the corresponding enantioenriched phenylglycine-based bis fluorene motifs 22i-(D) and 22i-(L), tyrosine-based bis fluorene motifs 23e-(D) and 23e-(L) and phenylalanine-based bis fluorene motifs 23b-(D) and 23b-(L) in good yields and enantiopurity (Scheme 12).
Having obtained arylated and alkylated fluorene/fluorenone scaffolds, we shifted our attention toward synthesizing modified fluorenones via ring annulation on fluorenones. Taking inspiration from reported cobalt-catalyzed oxidative cyclization, we treated maleimides 6aa–ac with fluorenone-4-carboxamide 5a using the reported conditions (Scheme 13),27a involving Co(OAc)2·H2O, PivOH and Ag2CO3 in 1,2-DCE. These attempts afforded the corresponding fluorenone-based spiro succinimides 25a–c in 61–80% yields (see Scheme S1 in SI for the proposed mechanism of formation of 25a–c). Additionally, we heated acetylenes (symmetrical or unsymmetrical) with fluorene-4-carboxamide 5a under the reported conditions27b involving Co(OAc)2·H2O and PivOH in chlorobenzene. These attempts gave the corresponding fluorenone-based isoquinolone derivatives 27a–d in 51–77% yields (Scheme 13).
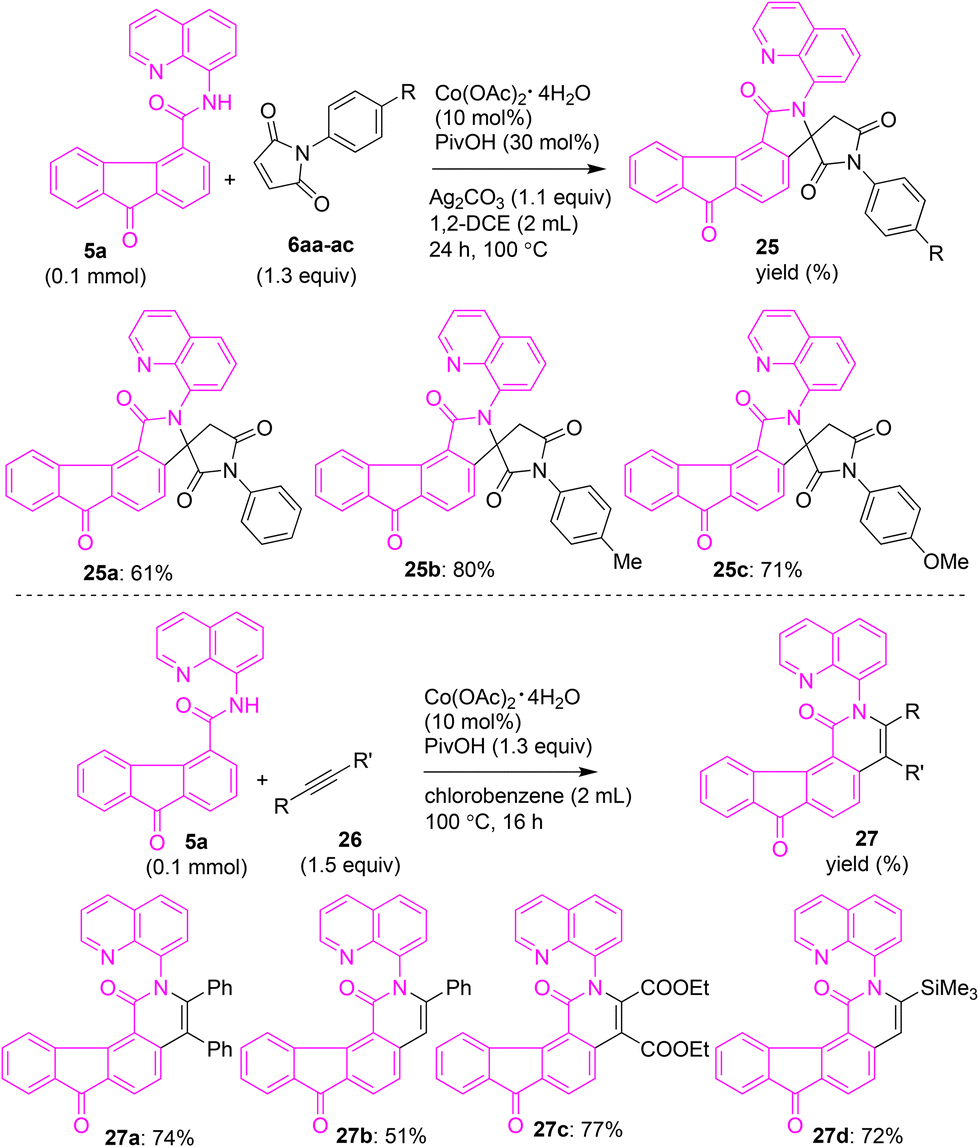 | ||
| Scheme 13 Cobalt-catalyzed 8-aminoquinoline-directed oxidative cyclization of fluorenone carboxamide 5a with maleimides and alkynes. | ||
Next, we intended to show the utility and removal of the bidentate directing group28b after performing Pd(II)-catalyzed C–H arylation reactions affording functionalized fluorenes/fluorenones (Scheme 14). We subjected the C(3)-arylated fluorenone carboxamide 7o or fluorenone-arylacetamide 13f to triflic-acid-mediated amide hydrolysis to afford C(3)-arylated fluorenone carboxylic acid 29a or fluorenone-arylacetic acid 29b (Scheme 14). In another attempt, we succeeded in removing the 8-aminoquinoline DG via the BF3·OEt2-mediated direct amide to ester conversion method. Accordingly, fluorene-arylacetamide 13g, fluorene–benzamide 11n, and aliphatic carboxamide 20f containing 8-AQ DG were subjected to the BF3·OEt2-mediated amide to ester conversion. These reactions afforded the corresponding fluorene motifs 29c, 29d, and 29e (Scheme 14). We also succeeded in removing the picolinoyl group from benzylamine-based bis fluorene motif 22a by using the reported conditions, involving Zn dust and 12 N HCl in a mixture of THF/H2O at rt for 12 h.28a This process gave the benzylamine-based bis fluorene motif 29f (Scheme 14).
 | ||
| Scheme 14 Synthetic transformations. Removal of the directing groups. aConditions (i): Zn dust (15 equiv.), HCl (0.4 mL), THF:H2O (1:1), 12 h, open flask. | ||
Furthermore, the palladium-catalyzed or cobalt-catalyzed C–H alkoxylation25b of the β-C(3)–H bond of fluorene-4-carboxamide 5a using primary alcohols afforded the corresponding β-C(3)–H alkoxylated fluorenone scaffolds 28a–d (Scheme 15). We then attempted a gram-scale synthesis of a modified fluorene scaffold. Accordingly, benzamide 24a was subjected to ortho β-C–H arylation with 6d in the presence of Pd(OAc)2 and AgOAc. This reaction afforded fluorene–aryl motif 11n (20%) and bis fluorene motif 11o (55%). Given that this reaction was performed on a gram scale, it may be sluggish. Hence, we observed fluorene–aryl motif 11n in considerable yield via mono C–H arylation. Notably, on a small scale, C–H arylation of 24a with 6a predominantly afforded bis fluorene motif 21a (Scheme 11). As fluorene and fluorenone compounds are vital components in material chemistry, we wished to further extend the synthetic utility of this work by attempting the synthesis of a few analogues of fluorene and fluorenone, which hold significance in material chemistry. Towards this end, we intended to perform the cross-coupling reaction between the bromo functionality in fluorenone–aryl motifs 7o and 29d with boronic acids 30a and 30b under Suzuki coupling reaction conditions. These attempts enabled the synthesis of π-extended fluorene–aryl motifs 31a and 31b (Scheme 15).
 | ||
| Scheme 15 Synthetic transformations, gram-scale C–H arylation of carboxamide and synthesis of C–H alkoxylated fluorenones and π-extended fluorene motifs via a cross-coupling reaction. aConditions (i): Pd(PPh3)4 (5 mol%), K3CO3 (3 equiv.), toluene:EtOH:H2O (2:1:1), 110 °C, 24 h sealed tube (purged with N2 atm). bCompound 28a is obtained from substrate 5a using Pd(OAc)2 (10 mol%), PhI(OAc)2 (1.5 equiv.), toluene:MeOH (1:1, 2 mL), rt, 48 h, sealed tube (purged with ambient air). cCompounds 28b–d are from substrate 5a using Co(OAc)2·4H2O (20 mol%), NaOAc (2 equiv.), Ag2O (1 equiv.), ROH (2 mL), 80 °C, 24 h, sealed tube (purged with ambient air). | ||
We recorded the UV-vis absorption spectra (λmax (absorption)) of the compounds (see Charts A–V in SI for the plots of UV-vis absorption spectra), and we also obtained the preliminary fluorescence emission spectra of representative modified fluorene scaffolds prepared in this work (see Chart X in SI for the plots of fluorescence emission spectra). Emission spectra of 7q, 8a, 8c, 8f, 8g, 27a, 29f and 31b were recorded in MeCN (concentration of solutions = 27.6 μM) at an excitation wavelength of 260 nm. Two compounds, 29f and 31b, were found to show fluorescence (λmax (emission) (nm) 29f: 342, 31b: 438) (see Chart X in SI). Emission spectra of 31b in different concentrations in MeCN (8.2, 16.5, 22.1, 27.6, and 55.2 μM) were recorded at an excitation wavelength of 260 nm (see Chart W in SI). Preliminary examination of the emission spectra of 31b in different concentrations indicated that emission is constantly increasing with an increase in concentration, and no quenching of fluorescence due to intermolecular interaction was observed (see Chart W in SI). Additional work has yet to be carried out to explore the fluorescence properties and application of other compounds synthesized in this work.
The NMR spectra and HRMS data established the structures of all the modified fluorenes and fluorenones obtained via the C–H functionalization process. In addition to this, the structures of representative C(3)-arylated fluorenone-4-carboxamide motif 7c and fluorene–benzamide motif 11f were unambiguously ascertained from the single-crystal X-ray structure analysis (Fig. 2). The X-ray structures divulged that Pd(II)-catalyzed bidentate-directing-group-aided, chelation-assisted C–H functionalization has occurred at the ortho β-C(sp2)–H bond in the corresponding carboxamides 5a and 3fvia a well-known Pd(II)/Pd(IV) redox cycle.22,23a,26a Based on the X-ray structure of these representative compounds 7c/11f, NMR and HRMS analysis data and in agreement with the reports in the literature22–24 the structures of all other compounds were ascertained.
In agreement with reports in the literature,22–24 the mechanism of the Pd(II)-catalyzed, bidentate-directing-group-8-AQ-assisted C–H arylation of the ortho C(sp2)–H bond of fluorenone-4-carboxamide 5a is proposed (Scheme 16). The coordination of the 8-AQ moiety in fluorenone-4-carboxamide 5a to the Pd(II) metal center is followed by concerted metalation deprotonation (CMD), generating five-membered Pd(II) species 33via32. Oxidative addition of Pd(II) species 33 with an aryl iodide yields Pd(IV) species 34, which then embarks on reductive elimination to generate a new C–C bond in compound 35. Next, halide removal occurs with the help of a halide ion scavenger, followed by protonolysis, affording C(3)-arylated fluorenone-4-carboxamide motif 7 and the active Pd(II) species is regenerated in the catalytic cycle (Scheme 16). Similarly, based on the reports in the literature,26 a plausible mechanism for the transient directing group (TDG)-assisted arylation of the β-C(sp2)–H bond of 2-acetylfluorene 5k is illustrated. The in situ installation of the glycine directing group with the aromatic ketone generates intermediate 36. The coordination of the TDG in 36 to the Pd(II) metal center is followed by CMD, which affords 38via37. Oxidative addition of 38 with an aryl iodide generates Pd(IV) species 39. Then, intermediate 39 undergoes reductive elimination (generating a new C–C bond in 40) and subsequent halide removal by a halide ion scavenger, elimination of the TDG affords β-C(sp2)–H arylated product 14 and the active Pd(II) species is regenerated.
 | ||
| Scheme 16 The proposed mechanism of Pd(II)-catalyzed, bidentate-directing-group-8-AQ-assisted or transient-directing-group-assisted C–H arylation in agreement with the reports in the literature.22,23a,26a | ||
Conclusions
In summary, we reported the modification of fluorene and fluorenone core through the C–H functionalization route and assembling a library of modified fluorenes and fluorenones. Fluorene and fluorenone motifs are versatile components in functional materials and have found various applications (e.g., in oligomers, polymers, and as fluorophores, components for OLEDs, liquid crystals, bioactive and drug molecules, etc.). Thus, synthesizing modified fluorenes and fluorenones (monomers) is considered a worthwhile effort. In this work, the C–H bonds of the fluorene or fluorenone core were subjected to directing-group-aided arylation, alkylation, benzylation, alkoxylation, and annulation to afford several new examples of modified fluorenes and fluorenones. We also performed cobalt-catalyzed oxidative cyclization of fluorenone substrates, affording the corresponding fluorenone-moiety appended spiro compounds and quinolones. The synthesis of C(1), C(2), C(3), and C(4) functionalized fluorenes or fluorenones was accomplished depending on the starting substrates using bidentate-directing-group- or transient-directing-group-assisted C(sp2)–H and C(sp3)–H functionalization strategies. Alternatively, the functionalized fluorenes or fluorenones were also obtained by reacting the C–H bonds of aryl or alkyl carboxamide substrates with iodofluorenes or iodofluorenones. Overall, we have shown the synthesis of a wide range of fluorene–fluorenone, fluorene–carbazole, π-extended bis fluorenes with an aryl spacer, enantioenriched π-extended bis fluorenes, and arylated or alkylated fluorenes and fluorenones. We have also shown the removal of the directing group after performing C–H functionalization in the fluorenone or fluorene core. We have obtained the UV-vis absorption spectra of the synthesized compounds. Preliminary fluorescence emission spectra of representative modified fluorene scaffolds prepared in this work were ascertained. Given the importance of fluorene- and fluorenone-based motifs in materials and medicinal chemistry, this work contributes to augmenting the library of modified fluorene and fluorenone scaffolds via the C–H activation/functionalization method.Experimental
General
The reagents used are commercially available and used without purification. The TLC analyses were performed on silica gel 60 F254 pre-coated plates or prepared alumina TLC plates and visualized by observation under irradiation with a UV lamp or iodine vapour. Column chromatography separation of crude reaction mixtures/samples was carried out on silica gel (100–200 mesh). 1H NMR and 13C{1H} NMR spectra were recorded on 400 and ∼101 MHz spectrometers, respectively (with TMS as an internal standard). The HRMS analysis data were obtained from a QTOF mass analyzer using the electrospray ionization (ESI) method. The IR spectra were recorded as neat samples/thin films, using KBr to prepare pellets for solid samples, or in a solvent. The required anhydrous solvents were prepared under standard solvent drying procedures, and reactions were carried out under a nitrogen atmosphere or in ambient air in RB flasks or in sealed tubes, as mentioned in the respective schemes/tables. Organic layers obtained from the work-up procedure were dried using anhydrous Na2SO4. Isolated yields of products are reported, and the yields have not been optimized. The UV-vis absorption spectra of the compounds were recorded at a concentration (c) = 0.02 g per 100 mL in CH3CN.We performed HPLC analysis of the starting material (substrates) used and fluorene- and fluorenone-based motifs synthesized in this work via C–H functionalization (see the SI). Initially, the HPLC analysis patterns of the racemic substrates 24g-(DL), 24l-(DL), 24m-(DL), 24n-(RS), 24o-(RS), 24r-(RS), 24t-(RS) and 24u-(RS) were determined. Subsequently, the enantiopurity of substrates 24t-(R) (er 99:2), 24t-(S) (er 98:2), 24u-(R) (er 99:4), 24u-(S) (er 99:4), 24r-(R) (er 99:1), 24r-(S) (er 99:4), 24g-(D) (er 95:5), 24g-(L) (er 92:8), 24l-(D) (er 98:2), 24l-(L) (er 99:1), 24m-(D) (er 98:2), 24m-(L) (er 99:1), 24n-(R) (er 99:1), 24n-(S) (er 99:1), 24o-(R) (er 98:2), and 24o-(S) (er 98:2) were ascertained from HPLC analysis. Next, the HPLC analysis patterns of the synthesized racemic fluorene- and fluorenone-based motifs 22b-(RS), 22e-(RS), 22f-(RS), 22i-(DL), 23a-(RS), 23c-(RS), 23b-(DL) and 23e-(DL) were ascertained. Then, the HPLC analysis patterns of the enantioenriched fluorene- and fluorenone-based motifs 22b-(R), 22b-(S), 22e-(R), 22e-(S), 22f-(R), 22f-(S), 22i-(D), 22i-(L), 23a-(R), 23a-(S), 23c-(R), 23c-(S), 23b-(D), 23b-(L), 23e-(D) and 23e-(L) were ascertained.
General procedure for synthesizing fluorenone carboxamides (5a–f) and fluorene carboxamides (5g–i)
To an RB flask, containing 9-oxo-9H-fluorene-4-carboxylic acid (1–5 mmol, 1 equiv.) or 9-oxo-9H-fluorene-1-carboxylic acid (1 mmol, 1 equiv.) or 9H-fluorene-9-carboxylic acid (1–2 mmol, 1 equiv.) or 2-(9H-fluoren-9-yl)acetic acid (2 mmol, 1 equiv.) in anhydrous DCM (2–10 mL), were added DMF (1 to 2 drops) and (COCl)2 (1.2 equiv.) under a flow of nitrogen atm and the reaction was prolonged for 5 h at rt. After completion of reaction, the solvent was evaporated to afford the corresponding acid chloride of fluorenone or fluorene derivative, and vacuum dried. Next, to an RB flask containing the appropriate amine (8-aminoquinoline or 2-(methylthio)aniline or aniline or 1-octyl amine) in anhydrous DCM (2–10 mL) was added Et3N (1.2 equiv.). Then, to this flask, the requisite acid chloride of fluorenone or fluorene derivative (dissolved in anhydrous DCM) was added dropwise under a nitrogen atm. The resulting reaction mixture was allowed to stir at rt for 36 h. Thereafter, the reaction mixture was diluted with DCM (10–15 mL) and washed with a saturated solution of NaHCO3 (15 mL, three times). The collected organic layers were dried over anhydrous Na2SO4 and concentrated under vacuum to afford the corresponding crude reaction mixture, which was then purified using silica gel column chromatography (EtOAc:hexane) to give the corresponding fluorenone and fluorene carboxamide.
General procedure for the Pd(II)-catalyzed C–H arylation of fluorenone-4-carboxamide 5a,b and fluorenone-1-carboxamide 5e
A mixture of an appropriate fluorenone-4-carboxamide 5a or 5b or fluorenone-1-carboxamide 5e containing a directing group (1 equiv.), an appropriate aryl iodide (4 equiv.), Pd(OAc)2 (10 mol%) and AgOAc (2.5 equiv.) in m-xylene (50 μL) was added to a sealed tube (purged with N2 atm) and the reaction mixture was heated at 130 °C for 48 h. At the end of the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H arylated fluorenone carboxamide (see the respective table/scheme for specific entries).
General procedure for the Pd(II)-catalyzed C–H arylation of carboxamides 3a–o, 3s–x, 3z, 3aa, 5j, 19a–i, 17, 18, 24a–w
A mixture of an appropriate carboxamide containing a directing group (1 equiv.), an appropriate aryl iodide (4 equiv.), Pd(OAc)2 (10 mol%) and AgOAc (2 equiv.) in toluene (1–4 mL) was added to a sealed tube (purged with N2 atm), and the reaction mixture was heated at 130 °C for 48 h. After the completion of the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H arylated fluorenone or fluorene carboxamide (see the respective table/scheme for specific entries).
General procedure for the Pd(II)-catalyzed C–H arylation of carboxamides 3p–r/3ab in the presence of CuBr2
A mixture of an appropriate carboxamide containing a directing group (1 equiv.), an appropriate aryl iodide (4 equiv.), Pd(OAc)2 (10 mol%), CuBr2 (10 mol%), and CsOAc (4 equiv.), in t-amylOH (1–3 mL) was added to a sealed tube (purged with N2 atm) and the reaction mixture was heated at 130 °C for 36 h. At the end of the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H arylated fluorenone or fluorene carboxamide (see the respective table/scheme for specific entries).
General procedure for the Pd(II)-catalyzed C–H arylation of 2-acetylfluorene 5k
To a mixture of substrate 2-acetyl fluorene 5k (1 equiv.) and appropriate aryl iodide (1.5 equiv.) were added Pd(OAc)2 (10 mol%), AgOAc (2 equiv.), glycine (50 mol%), and H2O (4 equiv.) in HFIP:AcOH [5:1] and the reaction was heated at 130 °C for 24 h in a sealed tube (purged with N2 atm). At the end of the reaction, the crude reaction mixture was cooled to rt and the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H arylated fluorene derivative (see the respective table/scheme for specific entries).
General procedure for the Pd(II)-catalyzed C–H arylation of o-tolualdehyde 5l
To a mixture of substrate o-tolualdehyde 5l (1 equiv.) and appropriate aryl iodide (4 equiv.) were added Pd(OAc)2 (10 mol%), AgTFA (1.5 equiv.), glycine (40 mol%), and AcOH:H2O [9:1] and the reaction was heated at 100 °C for 36 h in a sealed tube (purged with N2 atm). At the end of the reaction, the crude reaction mixture was cooled to rt and the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H arylated fluorene derivative (see the respective table/scheme for specific entries).
General procedure for the Pd(II)-catalyzed C–H alkylation of fluorenone-4-carboxamide 5a
To a mixture of an appropriate fluorenone-4-carboxamide 5a containing 8-aminoquinoline as the directing group (1 equiv.), a required alkyl iodide (4 equiv.), Pd(OAc)2 (5 mol%), K2CO3 (2.5 equiv.) and PivOH (20 mol%) in t-amylOH (1–2 mL) were added to a sealed tube (purged with N2 atm) and the reaction mixture was heated at 130 °C for 48 h. At the end of the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H alkylated fluorenone carboxamide (see the respective table/scheme for specific entries).
Typical procedure for the Pd(II)-catalyzed C–H alkylation of fluorenone-4-carboxamide 5a with n-BuI
To a mixture of an appropriate fluorenone-4-carboxamide 5a containing 8-aminoquinoline as the directing group (1 equiv.) and n-BuI (4 equiv.) were added Pd(OAc)2 (10 mol%), K2CO3 (2 equiv.), CuBr2 (20 mol%), and H2O (1.5 mL) at 110 °C for 48 h in a sealed tube (filled with ambient air). At the end of the reaction, the crude reaction mixture was cooled to rt and extracted with EtOAc (5–7 mL). The organic layer was washed with brine, dried over anhydrous Na2SO4, concentrated, and purified by column chromatography on silica gel (EtOAc/hexane as eluent) to afford the corresponding C–H butylated fluorenone carboxamide 15c (see the respective table/scheme for specific entries).General procedure for the Pd(II)-catalyzed C–H benzylation of fluorenone-4-carboxamide 5a or fluorenone-1-carboxamide 5e
To a mixture of an appropriate fluorenone-4-carboxamide 5a or fluorenone-1-carboxamide 5e containing 8-aminoquinoline as the directing group (1 equiv.), a required benzyl bromide (3 equiv.), Pd(OAc)2 (5 mol%) and KOAc (2 equiv.) in 1,4-dioxane (1–2 mL) was added to a sealed tube (purged with N2 atm) and the reaction mixture was heated at 100 °C for 36 h. At the end of the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H benzylated fluorenone carboxamide (see the respective table/scheme for specific entries).
General procedure for the Pd(II)-catalyzed C–H halogenation of fluorenone-4-carboxamide 5a
A mixture of an appropriate fluorenone-4-carboxamide 5a containing 8-aminoquinoline as the directing group (1 equiv.), a required halogen source NIS or NCS (4 equiv.), and Pd(OAc)2 (10 mol%) in toluene or p-xylene was heated at 110 or 130 °C for 48 h in a sealed tube (purged with N2 atm). After the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H halogenated fluorenone carboxamide (see the respective scheme for specific entries).
Typical procedure for the Pd(II)-catalyzed C–H methoxylation of fluorenone-4-carboxamide 5a
A mixture of an appropriate fluorenone-4-carboxamide 5a containing 8-aminoquinoline as the directing group (1 equiv.), Pd(OAc)2 (10 mol%), PhI(OAc)2 (1.5 equiv.), and toluene:MeOH (1:1, 2 mL) was reacted at rt in a sealed tube (purged with ambient air) for 48 h. At the end of the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H methoxylated fluorenone carboxamide 28a (see the respective table/scheme for the specific entry).
General procedure for the Co(II)-catalyzed C–H alkoxylation of fluorenone-4-carboxamides 5a
A mixture of an appropriate fluorenone-4-carboxamide 5a containing 8-aminoquinoline as the directing group (1 equiv.), a required aliphatic alcohol (2 mL), Co(OAc)2·4H2O (20 mol%), Ag2O (1 equiv.) and NaOAc (2 equiv.) was added to a sealed tube (purged with ambient air) and the reaction mixture was heated at 80 °C for 24 h. At the end of the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding C–H alkoxylated fluorenone carboxamide (see the respective table/scheme for specific entries).
General procedure for the Co(II)-catalyzed 8-aminoquinoline-directed oxidative cyclization of fluorenone carboxamide 5a with maleimides 6aa–ac
A mixture of an appropriate fluorenone-4-carboxamide 5a containing 8-aminoquinoline as the directing group (0.1 mmol, 1 equiv.), a required maleimide (1.3 equiv.), Co(OAc)2·4H2O (10 mol%), Ag2CO3 (1.1 equiv.), PivOH (30 mol%), and 1,2-DCE (2 mL) was heated in a sealed tube (filled with ambient air). The reaction mixture was heated at 100 °C for 24 h. At the end of the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding isoindolone-spirosuccinimides unit-linked fluorenone carboxamide (see the respective table/scheme for specific entries).
General procedure for the Co(II)-catalyzed 8-aminoquinoline-directed cyclization of fluorenone carboxamide 5a with substituted alkynes 26
A mixture of an appropriate fluorenone-4-carboxamide 5a containing 8-aminoquinoline as the directing group (0.1 mmol, 1 equiv.), a required mono or disubstituted alkynes (1.3 equiv.), Co(OAc)2·4H2O (10 mol%), PivOH (1.3 equiv.), and chlorobenzene (2 mL) was heated in a sealed tube (filled with ambient air) at 100 °C for 24 h. At the end of the reaction, the crude was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding isoquinolone unit-containing fluorenone carboxamide (see the respective table/scheme for specific entries).
Typical procedure for the triflic-acid-mediated amide hydrolysis of C–H arylated compounds 7o/13f (removal of the 8-aminoquinoline directing group)
To a mixture of a C–H arylated carboxamide 7o or 13f (0.1 mmol, 1 equiv.) was added CF3SO3H (0.2 mL) in toluene:H2O [10:1] and heated at 110 °C for 24 h in a sealed tube (filled with ambient air). At the end of the reaction, the crude reaction mixture was cooled to rt and quenched by slowly adding a saturated solution of Na2CO3 (5 mL) and then washed with ethyl acetate. The aq. layer was acidified with 12 N HCl to pH 2 and extracted with EtOAc (15 mL). The combined organic layers were concentrated and dried over anhydrous Na2SO4 to form the free carboxylic acid containing fluorenone and phenyl acetic acid derivative, which was purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding product (see the respective scheme for the specific entry).
General procedure for removing the 8-aminoquinoline directing group from 13g/11n/20f
A mixture of an appropriate arylated carboxamide (1 equiv.), BF3·Et2O (15 equiv.), and anhydrous MeOH/EtOH (2–4 mL) in a screw-capped sealed tube containing a magnetic bead was stirred and heated at 130 °C for 36 h. Then, the reaction mixture was allowed to attain rt and concentrated under reduced pressure to afford the corresponding crude reaction mixture, which was purified by column chromatography to afford the corresponding ester derivative (see the respective scheme for the specific entry).Typical procedure for the Pd(II)-catalyzed Suzuki–Miyaura cross-coupling reaction on C–H arylated carboxamide 7o/29d
A mixture of a C–H arylated carboxamide or ester derivative 7o or 29d (1 equiv.), a required boronic acid (1.5 equiv.), Pd(PPh3)4 (5 mol%), and K2CO3 (3 equiv.) in toluene:EtOH:H2O [2:1:1] was heated at 110 °C for 24 h in a sealed tube (purged with N2 atm). At the end of the reaction, the crude reaction mixture was concentrated under vacuum and purified by column chromatography on silica gel (EtOAc:hexane) to afford the corresponding product (see the respective scheme for the specific entry).
Characterization (analytical) data of all compounds are provided in the SI. Characterization (analytical) data of representative compounds are given below.
:hexane = 20:80) as a yellow-colored solid (70 mg, 72%, 0.2 mmol scale); Rf (20% EtOAc/hexane) 0.5; mp: 187–189 °C; IR (DCM): 2925, 1672, 750 cm−1; 1H NMR (400 MHz, CDCl3): δH 9.93 (s, 1H), 8.85 (dd, J1 = 7.0, J1 = 1.8 Hz, 1H), 8.59 (dd, J1 = 4.2, J1 = 1.5 Hz, 1H), 8.11 (dd, J1 = 8.3, J1 = 1.5 Hz, 1H), 7.95 (d, J = 8.4 Hz, 2H), 7.83 (d, J = 7.6 Hz, 1H), 7.71–7.65 (m, 4H), 7.59–7.53 (m, 2H), 7.42–7.26 (m, 4H), 3.81 (s, 3H). 13C{1H} NMR (∼101 MHz, CDCl3): δC 192.5, 166.2, 166.0, 148.3, 144.7, 143.5, 142.5, 141.7, 138.2, 136.2, 135.2, 134.4, 134.1, 133.7, 131.2, 130.8, 129.8, 129.7, 129.6, 128.5, 127.8, 127.2, 125.0, 124.3, 123.2, 122.5, 121.7, 116.8, 52.1. HRMS (ESI): m/z [M + H]+ calcd for C31H21N2O4: 485.1501 found, 485.1500.
:hexane = 20:80) as a yellow-colored solid (55 mg, 62%, 0.2 mmol scale); Rf (20% EtOAc/hexane) 0.5; mp: 219–221 °C; IR (DCM): 2921, 1712, 752 cm−1; 1H NMR (400 MHz, CDCl3): δH 9.91 (s, 1H), 8.89 (d, J = 7.3 Hz, 1H), 8.61–8.60 (m, 1H), 8.11 (d, J = 8.2 Hz, 1H), 7.81 (d, J = 7.6 Hz, 1H), 7.67 (dd, J1 = 9.8, J2 = 7.7 Hz, 2H), 7.60–7.55 (m, 2H), 7.47 (d, J = 7.9 Hz, 2H), 7.41–7.24 (m, 4H), 7.07 (d, J = 7.8 Hz, 2H), 2.18 (s, 3H). 13C{1H} NMR (∼101 MHz, CDCl3): δC 192.7, 166.5, 148.2, 146.0, 142.8, 141.6, 138.3, 138.1, 136.1, 136.0, 135.1, 134.6, 134.0, 133.3, 131.3, 130.9, 129.3, 129.1, 128.2, 127.8, 127.2, 124.9, 124.1, 123.1, 122.3, 121.6, 116.7, 21.0. HRMS (ESI): m/z [M + H]+ calcd for C30H21N2O2: 441.1603 found, 441.1616.
:hexane = 20:80) as a yellow-colored solid (78 mg, 72%, 0.2 mmol scale); Rf (20% EtOAc/hexanes) 0.5; mp: 222–224 °C; IR (DCM): 2924, 1670, 750 cm−1; 1H NMR (400 MHz, CDCl3): δH 9.92 (s, 1H), 8.90 (dd, J1 = 7.5, J2 = 1.2 Hz, 1H), 8.55 (dd, J1 = 4.2, J2 = 1.6 Hz, 1H), 8.01 (dd, J1 = 8.3, J2 = 1.6 Hz, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.76–7.63 (m, 4H), 7.59–7.43 (m, 5H), 7.36 (td, J1 = 7.6, J2 = 1.2 Hz, 1H), 7.31–7.22 (m, 5H), 1.22 (s, 6H). 13C{1H} NMR (∼101 MHz, CDCl3): δC 192.8, 166.6, 153.8, 153.7, 148.1, 146.4, 142.8, 141.9, 139.2, 138.4, 138.2, 137.9, 136.1, 135.1, 134.6, 134.0, 133.5, 131.4, 130.9, 129.4, 127.7, 127.4, 127.4, 127.0, 126.8, 124.9, 124.2, 123.2, 122.7, 122.5, 122.3, 121.6, 120.1, 120.0, 116.6, 46.7, 26.8. HRMS (ESI): m/z [M + H]+ calcd for C38H27N2O2: 543.2073 found, 543.2068.
:hexane = 30:70) as a yellow-colored solid (55 mg, 93%, 0.1 mmol scale); Rf (20% EtOAc/hexane) 0.5; mp: 150–152 °C; IR (DCM): 2925, 1671, 747 cm−1; 1H NMR (400 MHz, CDCl3): δH 9.93 (s, 1H), 8.86 (d, J = 7.6 Hz, 1H), 8.38 (d, J = 1.6 Hz, 1H), 8.37 (d, J = 1.4 Hz, 1H), 8.00 (d, J = 7.7 Hz, 1H), 7.93 (d, J = 8.3 Hz, 1H), 7.82 (d, J = 7.7 Hz, 1H), 7.73 (d, J = 7.5 Hz, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.60 (dd, J1 = 8.5, J2 = 1.7 Hz, 1H), 7.52–7.45 (m, 4H), 7.41–7.19 (m, 10H), 7.16–7.13 (m, 1H). 13C{1H} NMR (∼101 MHz, CDCl3): δC 192.8, 166.9, 148.0, 146.7, 142.8, 141.8, 141.0, 140.4, 138.1, 137.1, 135.9, 135.0, 134.6, 134.0, 133.0, 131.5, 131.2, 130.8, 129.7, 129.3, 127.6, 127.4, 127.1, 126.7, 126.4, 126.1, 124.9, 124.1, 123.5, 123.1, 122.1, 121.4, 120.5, 120.2, 120.1, 116.8, 109.7, 109.7. HRMS (ESI): m/z [M + H]+ calcd for C41H26N3O2: 592.2025 found, 592.2026.
:hexane = 10:90) as a colorless solid (120 mg, 72%, 0.35 mmol scale); Rf (10% EtOAc/hexane) 0.5; mp: 201–203 °C; IR (DCM): 2924, 1665, 757 cm−1; 1H NMR (400 MHz, CDCl3): δH 9.73 (s, 1H), 8.79 (d, J = 7.6 Hz, 1H), 8.36 (d, J = 3.7 Hz, 1H), 7.95–7.93 (m, 2H), 7.72 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 7.1 Hz, 1H), 7.57–7.37 (m, 6H), 7.26–7.23 (m, 3H), 7.17 (dd, J1 = 8.2, J2 = 4.2 Hz, 1H), 1.09 (s, 6H). 13C{1H} NMR (∼101 MHz, CDCl3): δC 166.5, 153.9, 153.7, 147.7, 139.1, 138.8, 138.4, 138.2, 137.5, 137.4, 135.9, 134.1, 133.7, 131.9, 130.6, 129.5, 127.9, 127.6, 127.3, 127.1, 126.8, 123.3, 122.4, 121.7, 121.3, 120.1, 119.9, 116.4, 46.6, 26.6. HRMS (ESI): m/z [M + H]+ calcd for C31H24ClN2O: 475.1577 found, 475.1594.
:hexane = 10:90) as a colorless solid (22 mg, 71%, 0.1 mmol scale); Rf (20% EtOAc/hexane) 0.5; mp: 137–139 °C; IR (DCM): 1679, 1609, 731 cm−1; 1H NMR (400 MHz, CDCl3): δH 7.82–7.73 (m, 3H), 7.57 (d, J = 7.1 Hz, 1H), 7.41–7.25 (m, 4H), 6.99 (d, J = 8.5 Hz, 2H), 3.95 (s, 2H), 3.87 (s, 3H), 2.03 (s, 3H); 13C{1H} NMR (∼101 MHz, CDCl3): δC 205.1, 159.5, 144.3, 144.2, 141.7, 140.5, 139.7, 139.3, 133.6, 130.1, 127.7, 127.0, 125.2, 124.6, 121.5, 120.5, 114.1, 55.3, 36.7, 30.5. HRMS (ESI): m/z [M + H]+ calcd for C22H19O2: 315.1385 found, 315.1381.
:hexane = 10:90) as a colorless solid (131 mg, 86%, 0.33 mmol scale); Rf (10% EtOAc/hexane) 0.5; mp: 100–102 °C; IR (DCM): 2924, 1684, 755 cm−1; 1H NMR (400 MHz, CDCl3): δH 9.63 (s, 1H), 8.73 (d, J = 7.5 Hz, 1H), 8.58 (dd, J1 = 4.2, J2 = 1.5 Hz, 1H), 7.98 (d, J = 8.2 Hz, 1H), 7.67–7.64 (m, 2H), 7.46–7.20 (m, 8H), 3.79 (d, J = 21.7 Hz, 1H), 3.72 (d, J = 21.8 Hz, 1H), 3.39–3.32 (m, 1H), 2.91–2.81 (m, 2H), 1.85–1.69 (m, 2H), 1.25–1.18 (m, 10H), 0.82 (t, J = 6.9 Hz, 3H); 13C{1H} NMR (∼101 MHz, CDCl3): δC 170.5, 147.8, 143.6, 143.1, 141.6, 140.0, 138.1, 136.0, 134.3, 127.7, 127.2, 126.5, 126.2, 126.1, 124.8, 124.1, 121.3, 121.2, 119.8, 119.5, 116.3, 46.1, 42.8, 36.7, 36.3, 31.7, 29.5, 29.1, 27.5, 22.5, 14.0. HRMS (ESI): m/z [M + H]+ calcd for C32H35N2O: 463.2749 found, 463.2730.
:hexane = 10:90) as a colorless solid (39 mg, 89%, 0.1 mmol scale); Rf (10% EtOAc/hexane) 0.5; mp: 152–154 °C; IR (DCM): 2924, 1684, 755 cm−1; 1H NMR (400 MHz, CDCl3): δH 9.80 (s, 1H), 8.75 (d, J = 7.3 Hz, 1H), 8.71 (dd, J1 = 4.2, J2 = 1.6 Hz, 1H), 8.10 (dd, J1 = 8.3, J2 = 1.5 Hz, 1H), 7.83–7.20 (m, 15H), 4.90 (t, J = 7.7 Hz, 1H), 3.85 (d, J = 21.9 Hz, 1H), 3.79 (d, J = 21.7 Hz, 1H), 3.40 (d, J = 7.8 Hz, 2H); 13C{1H} NMR (∼101 MHz, CDCl3): δC 169.6, 147.9, 143.9, 143.7, 143.2, 142.4, 141.4, 140.2, 138.1, 136.2, 134.2, 128.6, 127.7, 127.7, 127.3, 126.6, 126.5, 126.4, 124.9, 124.5, 121.4, 121.4, 120.1, 119.9, 119.6, 116.4, 47.3, 44.6, 36.8. HRMS (ESI): m/z [M + H]+ calcd for C31H25N2O: 441.1967 found, 441.1964.
:hexane = 10:90) as a colorless solid (158 mg, 62%, 0.4 mmol scale); Rf (10% EtOAc/hexane) 0.5; mp: 253–257 °C; IR (DCM): 2923, 1672, 764 cm−1; 1H NMR (400 MHz, CDCl3): δH 9.85 (s, 1H), 8.63–8.60 (m, 2H), 7.96 (dd, J1 = 8.3, J2 = 1.5 Hz, 1H), 7.74–7.72 (m, 4H), 7.69–7.63 (m, 7H), 7.38–7.26 (m, 9H), 1.29 (s, 12H); 13C{1H} NMR (∼101 MHz, CDCl3): δC 167.8, 153.7, 153.5, 147.7, 140.8, 139.4, 138.7, 138.3, 138.2, 136.4, 135.9, 134.0, 129.2, 127.6, 127.5, 127.0, 126.9, 126.8, 123.0, 122.4, 121.4, 121.2, 119.9, 119.8, 116.5, 46.6, 26.8. HRMS (ESI): m/z [M + H]+ calcd for C46H37N2O: 633.2906 found, 633.2915.
:hexane = 10:90) as a colorless solid (45 mg, 70%, 0.1 mmol scale); Rf (10% EtOAc/hexane) 0.5; mp: 145–147 °C; IR (DCM): 2923, 1673, 740 cm−1; 1H NMR (400 MHz, CDCl3): δH 8.05 (d, J = 7.8 Hz, 1H), 7.94–7.20 (m, 20H), 5.62–5.55 (m, 1H), 1.60 (s, 12H), 1.47 (d, J = 6.6 Hz, 3H); 13C{1H} NMR (∼101 MHz, CDCl3): δC 162.5, 153.7, 153.5, 149.5, 147.5, 143.9, 140.1, 138.8, 138.4, 137.8, 136.8, 131.2, 130.3, 127.9, 127.3, 127.0, 125.6, 123.5, 122.6, 121.7, 120.0, 119.7, 46.9, 46.3, 27.3, 26.8, 23.6. HRMS (ESI): m/z [M + H]+ calcd for C44H38ClN2O: 645.2673 found, 645.2672. The HPLC of compound 22b-(RS) was determined using a Daicel Chiralpak IB column, hexane/i-PrOH (98:02), flow rate 0.5 mL min−1, UV detection at 254 nm, tR = 15.11 min, tS = 16.54 min.
:hexane = 10:90) as a colorless solid (45 mg, 70%, 0.1 mmol scale); Rf (10% EtOAc/hexane) 0.5; mp: 141–143 °C; IR (DCM): 2923, 1680, 740 cm−1; 1H NMR (400 MHz, CDCl3): δH 8.05 (d, J = 7.8 Hz, 1H), 7.93–7.19 (m, 20H), 5.61–5.54 (m, 1H), 1.59 (s, 12H), 1.46 (d, J = 6.4 Hz, 3H); 13C{1H} NMR (∼101 MHz, CDCl3): δC 162.6, 153.7, 153.5, 149.5, 147.5, 143.9, 140.1, 138.8, 138.4, 137.7, 136.8, 131.2, 130.3, 127.9, 127.3, 127.0, 125.6, 123.5, 122.6, 121.7, 120.0, 119.7, 46.9, 46.3, 27.3, 26.7, 23.6. HRMS (ESI): m/z [M + H]+ calcd for C44H38ClN2O: 645.2673 found, 645.2678. [α]25D = −35.00 (c = 0.02 g mL−1, CHCl3). The enantiomeric ratio (er = >95:5) of compound 22b-(R) was determined by HPLC using a Daicel Chiralpak IB column, hexane/i-PrOH (98:02), flow rate 0.5 mL min−1, UV detection at 254 nm, tR = 15.06 min, tS = 16.70 min.
:hexane = 10:90) as a colorless solid (44 mg, 68%, 0.1 mmol scale); Rf (10% EtOAc/hexane) 0.5; mp: 142–144 °C; IR (DCM): 2923, 1679, 740 cm−1; 1H NMR (400 MHz, CDCl3): δH 8.05 (d, J = 7.8 Hz, 1H), 7.93–7.19 (m, 20H), 5.61–5.54 (m, 1H), 1.59 (s, 12H), 1.46 (d, J = 6.5 Hz, 3H); 13C{1H} NMR (∼101 MHz, CDCl3): δC 162.5, 153.7, 153.5, 149.5, 147.5, 143.9, 140.2, 138.8, 138.5, 137.8, 136.8, 131.2, 130.3, 128.0, 127.3, 127.0, 125.6, 123.5, 122.6, 121.7, 120.0, 119.7, 46.9, 46.3, 27.3, 26.7, 23.6. HRMS (ESI): m/z [M + H]+ calcd for C44H38ClN2O: 645.2673 found, 645.2676. [α]25D = +33.00 (c = 0.02 g mL−1, CHCl3). The enantiomeric ratio (er = 98:2) of compound 22b-(S) was determined by HPLC using a Daicel Chiralpak IB column, hexane/i-PrOH (98:02), flow rate 0.5 mL min−1, UV detection at 254 nm, tR = 15.49 min, tS = 16.83 min.
:hexane = 90:10) as a yellow-colored solid (29 mg, 76%, 0.1 mmol scale); Rf (EtOAc) 0.5; mp: 197–199 °C; IR (DCM): 2925, 1715, 771 cm−1; 1H NMR (400 MHz, DMSO-d6): δH 8.79 (d, J = 2.2 Hz, 1H), 8.21 (dd, J1 = 8.4, J2 = 2.3 Hz, 1H), 7.79–7.64 (m, 6H), 7.46 (t, J = 7.4 Hz, 1H), 3.55 (br. s, 1H). (The proton signal corresponding to COOH is broad.) 13C{1H} NMR (∼101 MHz, DMSO-d6): δC 192.1, 169.5, 155.1, 150.2, 142.6, 142.4, 140.4, 136.1, 134.2, 134.2, 130.9, 130.5, 129.8, 125.0, 124.9, 124.7, 123.3, 120.7. HRMS (ESI): m/z [M + H]+ calcd for C19H11BrNO3: 379.9922 found, 379.9912.
:hexane = 90:10) as a colorless solid (15 mg, 43%, 0.1 mmol scale); Rf (EtOAc) 0.5; mp: 86–88 °C; IR (DCM): 2924, 1708, 757 cm−1; 1H NMR (400 MHz, CDCl3): δH 7.74 (d, J = 7.6 Hz, 2H), 7.45–7.43 (m, 1H), 7.37–7.31 (m, 4H), 7.25–7.22 (m, 1H), 7.13–7.04 (m, 2H), 3.64 (s, 2H), 1.47 (s, 6H). (The proton signal corresponding to COOH is not identified.) 13C{1H} NMR (∼101 MHz, CDCl3): δC 177.2, 161.9 (d, JC–F = 247.3), 153.8, 153.7, 139.0 (d, JC–F = 3.1 Hz), 138.8, 138.7, 138.5, 133.1 (d, JC–F = 7.9 Hz), 131.7 (d, JC–F = 8.2 Hz), 128.1, 127.4, 127.0, 123.7, 122.6, 120.1, 119.8, 117.5 (d, JC–F = 21.7 Hz), 114.4 (d, JC–F = 20.8 Hz), 46.9, 38.6, 27.0. 19F{1H} NMR (∼376 MHz, CDCl3): δF = −115.12. HRMS (ESI): m/z [M − H]+ calcd for C23H18FO2: 345.1291 found, 345.1289.
:hexane = 10:90) as a colorless solid (71 mg, 94%, 0.2 mmol scale); Rf (10% EtOAc/hexane) 0.5; mp: 70–72 °C; IR (DCM): 2925, 1736, 767 cm−1; 1H NMR (400 MHz, CDCl3): δH 7.75–7.74 (m, 2H), 7.46–7.44 (m, 1H), 7.38–7.22 (m, 7H), 3.64 (s, 3H), 3.60 (s, 2H), 1.49 (s, 6H); 13C{1H} NMR (∼101 MHz, CDCl3): δC 171.7, 153.7, 153.7, 141.3, 138.8, 138.6, 138.5, 133.5, 133.1, 131.4, 130.4, 127.9, 127.4, 127.3, 127.0, 123.5, 122.6, 120.1, 119.8, 52.0, 46.8, 38.7, 27.0. HRMS (ESI): m/z [M + Na]+ calcd for C24H21ClNaO2: 399.1128 found, 399.1113.
Conflicts of interest
There are no conflicts to declare.Data availability
The data are available within the article or its supplementary information (SI). Brief crystal data of compounds 7c/11f, a proposed mechanism relevant to formation of compound 25 and structures of relevant starting materials, experimental proceduers, characterization data of all compounds, plots of absorption and emission spectra of compounds, copies of 1H and 13C NMRs and HPLC charts are provided in supplementary information. See DOI: https://doi.org/10.1039/d5ob01242b.CCDC 2416200 (7c) and 2416201 (11f) contain the supplementary crystallographic data for this paper.29a,b
Acknowledgements
S. A. B. thanks IISER Mohali for funding this research work. We thank IISER Mohali for its analytical (NMR, HRMS, and X-ray) facilities. S. B. thanks IISER Mohali for the PhD fellowship. The authors also thank the Departmental NMR facility supported by DST-FIST (SR/FST/CS-II/2019/94 (TPN No. 32545)).References
- Fluorene/fluorenone natural products: (a) S. K. Talapatra, S. Bose, A. K. Mallik and B. Talapatra, Tetrahedron, 1985, 41, 2765 CrossRef CAS; (b) Y. Shi and S. Gao, Tetrahedron, 2016, 72, 1717 CrossRef CAS; (c) X. Y. Wu, G. W. Qin, D. J. Fan and R. S. Xu, Phytochemistry, 1994, 36, 477 CrossRef CAS; (d) M. V. Sarget, J. Chem. Soc., Perkin Trans. 1, 1987, 2553 RSC; (e) Q.-F. Hu, B. Zhou, J.-M. Huang, X.-M. Gao, L.-D. Shu, G.-Y. Yang and C.-T. Che, J. Nat. Prod., 2013, 76, 292 CrossRef CAS PubMed.
- Reviews on the synthesis of fluorenes/fluorenones: (a) R. P. Kaiser, I. Caivano and M. Kotora, Tetrahedron, 2019, 75, 2981 CrossRef CAS; (b) B. Large and D. Prim, Eur. J. Org. Chem., 2022, e202101032 CrossRef CAS; (c) S. Patel, B. Rathod, S. Regu, S. Chak and A. Shard, ChemistrySelect, 2020, 5, 10673 CrossRef CAS; (d) A.-H. Zhou, F. Pan, C. Zhu and L.-W. Ye, Chem. – Eur. J., 2015, 21, 10278 CrossRef CAS PubMed.
- Reviews on applications of fluorenes/fluorenones: (a) M.-S. Yuan, X. Du, F. Xu, D.-E. Wang, W.-J. Wang, T.-B. Li, Q. Tu, Y. Zhang, Z. Du and J. Wang, Dyes Pigm., 2015, 123, 355 CrossRef CAS; (b) Y. Wei, Y. Yan, X. Li, L. Xie and W. Huang, Org. Biomol. Chem., 2022, 20, 73 RSC; (c) B. Hu, P. Lu and Y. Wang, New J. Chem., 2013, 37, 1645 RSC; (d) S. Semin, X. Li, Y. Duan and T. Raising, Adv. Opt. Mater., 2021, 9, 2100327 CrossRef CAS; (e) R. Abbel, A. P. H. J. Schenning and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4215 CrossRef CAS; (f) M.-n. Yu, C.-j. Ou, B. Liu, D.-q. Lin, Y.-Y. Liu, W. Xue, Z.-q. Lin, J.-y. Lin, Y. Qian, S.-s. Wang, H.-t. Cao, L.-y. Bian, L.-h. Xie and W. Huang, Chin. J. Polym. Sci., 2017, 35, 155 CrossRef CAS; (g) Y. Han, L. Bai, J. Lin, X. Ding, L. Xie and W. Huang, Adv. Funct. Mater., 2021, 31, 2105092 CrossRef CAS.
- Application of fluorenes/fluorenones: (a) F. Uckert, Y.-H. Tak, K. Müllen and H. Bässler, Adv. Mater., 2000, 12, 905 CrossRef CAS; (b) P. C. Rodrigues, B. D. Fontes, B. B. M. Torres, W. S. Sousa, G. C. Faria, D. T. Balogh, R. M. Faria and L. Akcelrud, J. Appl. Polym. Sci., 2015, 132, 42579 CrossRef; (c) X. Gong, D. Moses, A. J. Heeger and S. Xiao, J. Phys. Chem. B, 2004, 108, 8601 CrossRef CAS; (d) S. Fratiloiu, S. M. Fonseca, F. C. Grozema, H. D. Burrows, M. L. Costa, A. Charas, J. Morgado and L. D. A. Siebbeles, J. Phys. Chem. C, 2007, 111, 5812 CrossRef CAS; (e) K. M. Omer, S.-Y. Ku, K.-T. Wong and A. J. Bard, J. Am. Chem. Soc., 2009, 131, 10733 CrossRef CAS PubMed; (f) S. Hayashi, S. Inagi and T. Fuchigami, Macromolecules, 2009, 42, 3755 CrossRef CAS; (g) O. A. Kucherak, P. Didier, Y. Mély and A. S. Klymchenko, J. Phys. Chem. Lett., 2010, 1, 616 CrossRef CAS; (h) J. A. McCubbin, X. Tong, R. Wang, Y. Zhao, V. Snieckus and R. P. Lemieux, J. Am. Chem. Soc., 2004, 126, 1161 CrossRef CAS PubMed; (i) H. Thakellapalli, S. Li, B. Farajidizaji, N. N. Baughman, N. G. Akhmedov, B. V. Popp and K. K. Wang, Org. Lett., 2017, 19, 2674 CrossRef CAS PubMed.
- (a) A. Goel, S. Chaurasia, M. Dixit, V. Kumar, S. Prakash, B. Jena, J. K. Verma, M. Jain, R. S. Anand and S. S. Manoharan, Org. Lett., 2009, 11, 1289 CrossRef CAS PubMed; (b) X.-H. Zhou, J.-C. Yan and J. Pei, Org. Lett., 2003, 5, 3543 CrossRef CAS PubMed; (c) Z. Ma, Y.-Y. Wang, P. Wang, W. Huang, Y.-B. Li, S.-B. Lei, Y.-L. Yang, X.-L. Fan and C. Wang, ACS Nano, 2007, 1, 160 CrossRef CAS PubMed; (d) K.-T. Wong, Y.-M. Chen, Y.-T. Lin, H.-C. Su and C.-c. Wu, Org. Lett., 2005, 7, 5361 CrossRef CAS PubMed; (e) F. Lincker, B. Heinrich, R. D. Bettignies, P. Rannou, J. Pécaut, B. Grévin, A. Pron, B. Donnio and R. Demadrille, J. Mater. Chem., 2011, 21, 5238 RSC.
- (a) K. D. Belfield, A. R. Morales, B.-S. Kang, J. M. Hales, D. J. Hagan, E. W. V. Stryland, V. M. Chapela and J. Percino, Chem. Mater., 2004, 16, 4634 CrossRef CAS; (b) A. P. Kulkarni, X. Kong and S. A. Jenekhe, J. Phys. Chem. B, 2004, 108, 8689 CrossRef CAS; (c) C. Qin, A. Islam and L. Han, J. Mater. Chem., 2012, 22, 19236 RSC; (d) W. Porzio, S. Destri, M. Pasini, U. Giovanella, M. Ragazzi, G. Scavia, D. Kotowski, G. Zotti and N. Vercelli, New J. Chem., 2010, 34, 1961 RSC; (e) G. Mu, S. Zhuang, C. Ren and P. Xu, Patent application, CN109574917B, 2020.
- Bio-active fluorenes/fluorenones: (a) F. Vallée, C. Carrez, F. Pilorge, A. Dupuy, A. Parent, L. Bertin, F. Thompson, P. Ferrari, F. Fassy, A. Lamberton, A. Thomas, R. Arrebola, S. Guerif, A. Rohaut, V. Certal, J.-M. Ruxer, T. Gouyon, C. Delorme, A. Jouanen, J. Dumas, C. Grépin, C. Combeau, H. Goulaouic, N. Dereu, V. Mikol, P. Mailliet and H. Minoux, J. Med. Chem., 2012, 55, 564 CrossRef; (b) F. Calogero, S. Borrelli, G. Speciale, M. S. Christodoulou, D. Cartelli, D. Ballinari, F. Sola, C. Albanese, A. Ciavolella, D. Passarella, G. Cappelletti, S. Pieraccini and M. Sironi, ChemPlusChem, 2013, 78, 663 CrossRef CAS PubMed; (c) K.-W. Si, J.-T. Liu, L.-C. He, X.-K. Li, W. Gou, C.-h. Liu and X.-Q. Li, Basic Clin. Pharmacol. Toxicol., 2010, 107, 976 CrossRef CAS PubMed; (d) W. Kemnitzer, N. Sirisoma, S. Jiang, S. Kasibhatla, C. Crogan-Grundy, B. Tseng, J. Drewe and S. X. Cai, Bioorg. Med. Chem. Lett., 2010, 20, 1288 CrossRef CAS PubMed; (e) H.-S. Hong, I. Maezawa, M. Budamagunta, S. Rana, A. Shi, R. Vassar, R. Liu, K. S. Lam, R. H. Cheng, D. H. Hua, J. C. Voss and L.-W. Jin, Neurobiol. Aging, 2010, 31, 1690 CrossRef CAS PubMed; (f) M. T. Tierney and M. W. Grinstaff, J. Org. Chem., 2000, 65, 5355 CrossRef CAS PubMed; (g) S. Wang, B. Wen, N. Wang, J. Liu and L. He, Arch. Pharmacal Res., 2009, 32, 521 CrossRef CAS PubMed; (h) K. Si, J. Liu, L. He, X. Li, W. Gou, C. Liu and X. Li, J. Pharmacol. Sci., 2010, 113, 368 CrossRef CAS PubMed.
- Bio-active fluorenes/fluorenones: (a) P. J. Perry, M. A. Read, R. T. Davies, S. M. Gowan, A. P. Reszka, A. A. Wood, L. R. Kelland and S. Neidle, J. Med. Chem., 1999, 42, 2679 CrossRef CAS PubMed; (b) C.-C. Lee, D.-M. Chang, K.-F. Huang, C.-L. Chen, T.-C. Chen, Y. Lo, J.-H. Guh and H.-S. Huang, Bioorg. Med. Chem. Lett., 2013, 21, 7125 CrossRef CAS PubMed; (c) D. Zhou, W. Tuo, H. Hu, J. Xu, H. Chen, Z. Rao, Y. Xiao, X. Hu and P. Liu, Eur. J. Med. Chem., 2013, 64, 432 CrossRef CAS PubMed; (d) T. Akiyama, S. Harada, F. Kojima, Y. Takahashi, C. Imada, Y. Okami, Y. Muraoka, T. Aoyagi and T. Takeuchi, J. Antibiot., 1998, 51, 553 CrossRef CAS PubMed; (e) S.-r. Choi, M. A. Larson, S. H. Hinrichs and P. Narayanasamy, Bioorg. Med. Chem. Lett., 2016, 26, 1997 CrossRef CAS PubMed; (f) S. Alcaro, A. Adriana, R. D. Bella, S. Neri, R. Ottanà, F. Ortuso, B. Pavone and M. G. Vigorita, ARKIVOC, 2004, 2004, 334 Search PubMed.
- (a) W. Li, N. M. O'Brien-Simpson, M. A. Hossain and J. D. Wade, Aust. J. Chem., 2020, 73, 271 CrossRef CAS; (b) F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232 CrossRef CAS PubMed; (c) K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935 RSC; (d) R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4 CrossRef CAS PubMed.
- Fluorenones via metallation: (a) W. Wang and V. Snieckus, J. Org. Chem., 1992, 57, 424 CrossRef CAS; (b) D. Tilly, S. S. Samanta, A. De, A.-S. Catanet and J. Mortier, Org. Lett., 2005, 7, 827 CrossRef CAS PubMed.
- Fluorenones via cross-coupling and/or Friedel–Crafts reaction: (a) T. Alves, A. B. de Oliveira and V. Snieckus, Tetrahedron Lett., 1988, 29, 2135 CrossRef CAS; (b) R. K. Chinnagolla and M. Jeganmohan, Org. Lett., 2012, 14, 5246 CrossRef CAS PubMed; (c) D. Shabashov, J. R. M. Maldonado and O. Daugulis, J. Org. Chem., 2008, 73, 7818 CrossRef CAS PubMed; (d) S. Reim, M. Lau and P. Langer, Tetrahedron Lett., 2006, 47, 6903 CrossRef CAS; (e) M. Alessi, A. L. Larkin, K. A. Ogilvie, L. A. Green, S. Lai, S. Lopez and V. Snieckus, J. Org. Chem., 2007, 72, 1588 CrossRef CAS PubMed; (f) D. R. Kumar, D. R. Kishore and G. Satyanarayana, SynOpen, 2018, 2, 268 CrossRef CAS; (g) D. E. Ames and A. Opalko, Tetrahedron, 1984, 40, 1919 CrossRef CAS; (h) L. G. Wade Jr., K. J. Acker, R. A. Earl and R. A. Osteryoung, J. Org. Chem., 1979, 44, 3724 CrossRef.
- Fluorenones via radical cyclization: (a) Z. Shi and F. Glorius, Chem. Sci., 2013, 4, 829 RSC; (b) S. Wertz, D. Leifert and A. Studer, Org. Lett., 2013, 15, 928 CrossRef CAS PubMed; (c) M. A. Campo and R. C. Larock, J. Org. Chem., 2002, 67, 5616 CrossRef CAS PubMed; (d) I. A. P. Jourjine, L. Zeisel, J. Krauß and F. Bracher, Beilstein J. Org. Chem., 2021, 17, 2668 CrossRef CAS PubMed; (e) J. N. Moorthy and S. Samanta, J. Org. Chem., 2007, 72, 9786 CrossRef CAS PubMed.
- Fluorenones via CO insertion: M. R. Campo and R. C. Larock, Org. Lett., 2000, 2, 3675 CrossRef CAS PubMed.
- Fluorenones via inter- or intramolecular C–H functionalization: (a) C. Wang, I. Piel and F. Glorius, J. Am. Chem. Soc., 2009, 131, 4194 CrossRef CAS PubMed; (b) H. Li, R.-Y. Zhu, W.-J. Shi, K.-H. He and Z.-J. Shi, Org. Lett., 2012, 14, 4850 CrossRef CAS PubMed; (c) J.-C. Wan, J.-M. Huang, Y.-H. Jhan and J.-C. Hsieh, Org. Lett., 2013, 15, 2742 CrossRef CAS PubMed.
- Fluorenones via oxidation of fluorenes: (a) G. Yang, Q. Zhang, H. Miao, X. Tong and J. Xu, Org. Lett., 2005, 7, 263 CrossRef CAS PubMed; (b) X. Zhang, X. Ji, S. Jiang, L. Liu, B. L. Weeks and Z. Shang, Green Chem., 2011, 13, 1891 RSC.
- Synthesis of fluorenes via Friedel–Crafts reaction: S.-G. Wang, L. Han, M. Zeng, F.-L. Sun, W. Zhang and S.-L. You, Org. Biomol. Chem., 2012, 10, 3202 RSC.
- Synthesis of fluorenes using gold-catalysis: J. Bucher, T. Wurm, S. Taschinski, E. Sachs, D. Ascough, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2017, 359, 225 CrossRef CAS.
- Fluorenes via C–H functionalization: S. Pan, Q. Zhu and Y. Zhang, Synlett, 2022, 1826 CAS.
- Modification of fluorene or fluorenone core via cross-coupling: (a) See ref. 4b, 5e and 6a, c, d; (b) L. C. R. M. da Frota, C. Schneider, M. B. De Amorim, A. J. M. Da Silva and V. Snieckus, Synlett, 2017, 28, 2587 CrossRef CAS; (c) F. Jaramillo-Isaza and M. L. Turner, J. Mater. Chem., 2006, 16, 83 RSC; (d) N. Agarwal, P. K. Nayak and N. Periasamy, J. Chem. Sci., 2008, 120, 355 CrossRef CAS; (e) Z. Zhou, Y. Yuan, Y. Xie and M. Li, Catal. Lett., 2018, 148, 2696 CrossRef CAS.
- Selected reviews/articles on biaryls: (a) J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651 CrossRef CAS PubMed; (b) Metal-Catalyzed Cross-Coupling Reactions, ed. F. Dieterich and P. J. Stang, Wiley-VCH, New York, 1998 Search PubMed; (c) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004, vol. 1–2 Search PubMed; (d) P. J. Hajduk, M. Bures, J. Praestgaard and S. W. Fesik, J. Med. Chem., 2000, 43, 3443 CrossRef CAS PubMed; (e) F. Kloss, T. Neuwirth, V. G. Haensch and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 14476 CrossRef CAS PubMed.
- Selected reviews on C–H functionalization: (a) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826 CrossRef CAS PubMed; (b) M. Miura, T. Satoh and K. Hirano, Bull. Chem. Soc. Jpn., 2014, 87, 751 CrossRef CAS; (c) S. Rej, A. Das and N. Chatani, Coord. Chem. Rev., 2021, 431, 213683 CrossRef CAS; (d) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (f) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed; (g) B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2013, 42, 5744 RSC; (h) M.-Z. Lu, J. Goh, M. Maraswami, Z. Jia, J.-S. Tian and T.-P. Loh, Chem. Rev., 2022, 122, 17479 CrossRef CAS PubMed; (i) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (j) R. L. de Carvalho, E. B. T. Diogo, S. L. Homölle, S. Dana, E. N. da Silva Júnior and L. Ackermann, Chem. Soc. Rev., 2023, 52, 6359 RSC; (k) X. Chen, K. M. Engle, D. H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (l) T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830 CrossRef CAS PubMed; (m) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682 CrossRef CAS PubMed; (n) R. Manoharan and M. Jeganmohan, Asian J. Org. Chem., 2019, 8, 1949 CrossRef CAS; (o) L. McMurray, F. O'hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (p) A. Mandal, S. Dana, D. Choudhury and M. Baidya, Chem. – Asian J., 2019, 14, 4074 CrossRef CAS PubMed; (q) S. A. Babu, A. Dalal and S. Bodak, Chemistry, 2023, 5, 2713 CrossRef.
- Selected reviews on bidentate directing group-assisted C–H functionalization: (a) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (b) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed; (c) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (d) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635 CrossRef CAS PubMed; (e) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191 RSC; (f) R. K. Rit, M. R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450 CrossRef CAS; (g) B. Liu, A. M. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957 CrossRef CAS PubMed; (h) S.-F. Ni, G. Huang, Y. Chen, J. S. Wright, M. Li and L. Dang, Coord. Chem. Rev., 2022, 455, 214255 CrossRef CAS.
- Selected reports on bidentate directing group-assisted C–H functionalization: (a) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed; (b) C. Reddy, N. Bisht, R. Parella and S. A. Babu, J. Org. Chem., 2016, 81, 12143 CrossRef CAS PubMed; (c) J. Bolsakova, L. Lukasevics and L. Grigorjeva, J. Org. Chem., 2020, 85, 4482 CrossRef CAS PubMed; (d) S. Martínez-Flores, C. A. Mujica-Martinez and L. A. Polindara-García, Eur. J. Org. Chem., 2022, e202101517 CrossRef; (e) Y. Zhao, G. He, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2948 CrossRef CAS PubMed; (f) C. Kuai, L. Wang, B. Li, Z. Yang and X. Cui, Org. Lett., 2017, 19, 2102 CrossRef CAS PubMed; (g) A. Seki and Y. Takahashi, Tetrahedron Lett., 2021, 74, 153130 CrossRef CAS; (h) J. Xu, L. Qiao, J. Shen, K. Chai, C. Shen and P. Zhang, Org. Lett., 2017, 19, 5661 CrossRef CAS PubMed; (i) K. S. Kanyiva, Y. Kuninobu and M. Kanai, Org. Lett., 2014, 16, 1968 CrossRef CAS PubMed; (j) J. Han, P. Liu, C. Wang, Q. Wang, J. Zhang, Y. Zhao, D. Shi, Z. Huang and Y. Zhao, Org. Lett., 2014, 16, 5682 CrossRef CAS PubMed; (k) R. Parella and S. A. Babu, J. Org. Chem., 2017, 82, 6550 CrossRef CAS PubMed; (l) A. García-Rubia, E. Laga, C. Cativiela, E. P. Urriolabeitia, R. Gomez-Arrayás and J. C. Carretero, J. Org. Chem., 2015, 80, 3321 CrossRef PubMed; (m) P. Singh and S. A. Babu, Synthesis, 2023, 4113 CAS; (n) R. Padmavathi, R. Sankar, B. Gopalakrishnan, R. Parella and S. A. Babu, Eur. J. Org. Chem., 2015, 3727 CrossRef CAS; (o) P. Singh and S. A. Babu, Eur. J. Org. Chem., 2023, e202300440 CrossRef CAS; (p) S. Banga and S. A. Babu, Eur. J. Org. Chem., 2024, e202400272 CrossRef CAS; (q) R. Parella and S. A. Babu, J. Org. Chem., 2015, 80, 12379 CrossRef CAS PubMed; (r) S. Banga, R. Kaur and S. A. Babu, Eur. J. Org. Chem., 2021, 3641 CrossRef CAS; (s) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (t) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689 CrossRef CAS PubMed.
- Selected reports on bidentate directing group-assisted C–H functionalization: (a) R. Odani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2013, 78, 11045 CrossRef CAS PubMed; (b) T. Uemura, M. Yamaguchi and N. Chatani, Angew. Chem., Int. Ed., 2016, 55, 3162 CrossRef CAS PubMed; (c) X. Wang, S. Niu, L. Xu, C. Zhang, L. Meng, X. Zhang and D. Ma, Org. Lett., 2017, 19, 246 CrossRef CAS PubMed; (d) W. Zeng, M. Nukeyeva, Q. Wang and C. Jiang, Org. Biomol. Chem., 2018, 16, 598 RSC; (e) S. Roy, S. Pradhan and T. Punniyamurthy, Chem. Commun., 2018, 54, 3899 RSC; (f) U. Narang, P. Singh and S. A. Babu, Eur. J. Org. Chem., 2023, e202300463 CrossRef CAS; (g) T. Xiang, Y. Liu, Q. Xu, K. He and F. Pan, J. Org. Chem., 2022, 87, 3135 CrossRef CAS PubMed; (h) C. Li, D. Zhang, W. Zhu, P. Wan and H. Liu, Org. Chem. Front., 2016, 3, 1080 RSC; (i) Naveen, V. Rajkumar, S. A. Babu and B. Gopalakrishnan, J. Org. Chem., 2016, 81, 12197 CrossRef CAS PubMed; (j) A. Dalal, S. Bodak and S. A. Babu, Org. Biomol. Chem., 2024, 22, 1279 RSC; (k) R. Kaur, H. Dilip, S. Kirubakaran and S. A. Babu, Org. Biomol. Chem., 2024, 22, 8916 RSC; (l) S. Suwasia, S. Venkataramani and S. A. Babu, Org. Biomol. Chem., 2023, 21, 1793 RSC; (m) R. Padmavathi and S. A. Babu, Org. Biomol. Chem., 2023, 21, 2689 RSC; (n) R. Parella, B. Gopalakrishnan and S. A. Babu, J. Org. Chem., 2013, 78, 11911 CrossRef CAS PubMed; (o) R. Parella and S. A. Babu, J. Org. Chem., 2015, 80, 2339 CrossRef CAS PubMed.
- (a) C. E. Coomber, L. Benhamou, D. K. Bučar, P. D. Smith, M. J. Porter and T. D. Sheppard, J. Org. Chem., 2018, 83, 2495 CrossRef CAS PubMed; (b) L.-B. Zhang, X.-Q. Hao, S.-K. Zhang, Z.-J. Liu, X.-X. Zheng, J.-F. Gong, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2015, 54, 272 CrossRef CAS PubMed.
- (a) J. I. Higham and J. A. Bull, Org. Biomol. Chem., 2020, 18, 7291 RSC; (b) J. Xu, Y. Liu, Y. Wang, Y. Li, X. Xu and Z. Jin, Org. Lett., 2017, 19, 1562 CrossRef CAS PubMed; (c) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed; (d) Y. Cheng, J. Zheng, C. Tian, Y. He, C. Zhang, Q. Tan, G. An and G. Li, Asian J. Org. Chem., 2019, 8, 526 CrossRef CAS.
- (a) R. Manoharan and M. Jeganmohan, Org. Lett., 2017, 19, 5884 CrossRef CAS PubMed; (b) R. Manoharan and M. Jeganmohan, Org. Biomol. Chem., 2018, 16, 8384 RSC.
- (a) D. H. O’ Donovan, C. D. Fusco and D. R. Spring, Tetrahedron Lett., 2016, 57, 2962 CrossRef; (b) L. S. Fitzgerald and M. L. O'Duill, Chem. – Eur. J., 2021, 27, 8411 CrossRef CAS PubMed.
- (a) CCDC 2416200: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2m37yy; (b) CCDC 2416201: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2m37zz.
| This journal is © The Royal Society of Chemistry 2025 |