
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst stereoselective approach for structure revision of nagiol and syntheses of 2,3-dihydroxyferruginol and ferruginol†
Sravya
Surendran
,
Arathi
Ramachandran
and
Rajendar
Goreti
 *
*
School of Chemistry, Indian Institute of Science Education and Research, Thiruvananthapuram, Kerala, India-695551. E-mail: rajendar@iisertvm.ac.in
First published on 25th June 2025
Abstract
We report the first enantioselective total synthesis of nagiol, accompanied by a revision of its previously proposed structure. This synthetic strategy has been successfully extended to the total synthesis of (+)-ferruginol as well as to the 2,3-dihydroxyferruginol. Enantiomerically enriched 8,11,13-podocarpatriene-3-ol was utilized as a versatile and efficient precursor to access the abietane diterpenoid framework. Functionalization of the aromatic C-ring on podocarpatriene was achieved through regioselective Friedel–Crafts acylation and further modification. Two distinct strategies were employed to install the 2,3-dihydroxy functionality on the A-ring: (i) selective elimination followed by syn-stereoselective dihydroxylation, and (ii) α-oxygenation of the ketone followed by highly diastereoselective reduction of the diketone intermediates. These methods allowed access to possible C-2/C-3 syn-isomers for comparative spectroscopic analysis. Careful comparison of the NMR data of the synthetic products with the reported values, in conjunction with single-crystal X-ray diffraction studies, led to a revision of the originally proposed structure of nagiol. Furthermore, this study presents the first enantioselective synthesis of 2,3-dihydroxyferruginol, revealing certain structural discrepancies among the isolation reports.
Introduction
Aromatic abietane diterpenoids are widely distributed throughout the plant kingdom and are well recognized for their diverse biological activities.1–7 Remarkably, certain abietane-type diterpenoids exhibit significant stability, having been isolated from geological samples such as fossils, sediments, coals, and petroleum deposits in non-decomposed form.8 These compounds play important roles in everyday life, serving as food additives, perfuming agents, and pharmacological agents. Biogenetically, aromatic abietanes are typically derived from a cationic polycyclization of geranylgeranyl pyrophosphate (GGPP), initially forming miltiradienes that undergo oxidative aromatization to yield C-ring aromatic abietanes.9–11 Subsequent enzymatic oxidations at various carbon atoms within the tricyclic ring system further diversify these structures, resulting in a broad range of derivatives (Fig. 1). Nagiol (2), an abietane mono—norditerpenoid, was isolated as a white amorphous powder from the leaves of Podocarpus nagi by a Chinese research group in 2017.12 | ||
| Fig. 1 Natural aromatic abietane diterpenoids. | ||
Spectroscopic analyses, including NMR, UV, IR, and mass spectrometry, revealed that compound 2 is a tricyclic norditerpenoid bearing a C-15 ketone functionality and three hydroxyl groups, one of which is a phenolic moiety. Structural elucidation further indicated that in compound 2, an acetyl group replaces the isopropyl substituent found in ferruginol (3), accompanied by oxidation at the C-2 and C-3 positions. The relative configuration at C-2 and C-3 in compound 2 was established through analysis of NMR coupling constants, ROESY correlations, and biogenetic considerations.
The parent compound ferruginol (3) is one of the most extensively studied members of the abietane class of diterpenoids. It was first isolated in 1939 from Podocarpus ferrugineus by Brandt and Neubauer.13 Since then, numerous research groups have reported its isolation from a wide range of plant species distributed across the globe.14–18 Notably, ferruginol exhibits a broad spectrum of biological activities, including cardioprotective,14 gastroprotective,19–21 antitumour,22,23 antimalarial,24,25 anti-inflammatory,26 anti-SARS27,28 and several other pharmacological effects.6,14,29 The structural diversification of ferruginol in biological systems through site-selective oxidations has led to the discovery of several analogues with oxygenated functionalities at key positions. For example, the 2,3-dihydroxy-substituted analogue (4) was first identified in 2007 by GC-MS analysis in various members of the Cupressaceae family, including the shoots of Athrotaxis laxifolia and Athrotaxis selaginoides.30 Later, in 2017, Chen and co-workers isolated compound 4 along with nagiol (2) from Podocarpus nagi.12 More recently, in 2022, Liang and co-workers reported the isolation of compound 4 from the aerial parts of Gaultheria leucocarpa var. yunnanensis, alongside several other abietane-type diterpenoids.31
We recently synthesized the possible stereoisomers of isolophanthin E to establish its correct structure, with the support of extensive NMR spectroscopic analysis.32 During this investigation, we undertook a comprehensive analysis of the chemical shift trends in abietane-type diterpenoids featuring a 2,3-dihydroxy substitution pattern on the A-ring. It was observed that the C-2/C-3 anti-isomers typically display relatively upfield chemical shifts (δ < 4.0 ppm for H-2 and δ < 3.0 ppm for H-3), whereas the corresponding syn-isomers exhibit downfield shifts (δ > 4.0 ppm for H-2 and δ > 3.2 ppm for H-3).
In the originally reported structure of nagiol, the chemical shifts at δ 4.23 ppm for H-2 and δ 3.23 ppm for H-3 were interpreted to support a C-2α/C-3α syn-configuration.12 However, during our studies on the synthesis of isolophanthin E, we observed significant differences in their NMR characteristics; the reported data of nagiol more closely resembled those of the C-2β/C-3β syn-isomer of isolophanthin E synthesized in our laboratory. In particular, the H-3 proton resonance at δ 3.23 ppm is more consistent with an axial orientation. This apparent discrepancy in the assignment of relative configuration prompted us to undertake the total synthesis of nagiol. Moreover, the total synthesis of nagiol has not been reported in the literature. Additionally, the synthetic route developed for nagiol could potentially enable access to structurally related natural products such as 2,3-dihydroxyferruginol (4) and ferruginol (3), further expanding the scope of this abietane-derived chemical space.
Herein, we report an enantioselective synthetic approach to nagiol (2), 2,3-dihydroxyferruginol (4), and ferruginol (3), starting from an enantiomerically pure synthetic 3-hydroxy-podocarpa-8,11,13-triene. To install the 2,3-dihydroxy functionality on the A-ring of nagiol (2), two distinct strategies were developed: (i) elimination of the hydroxyl group followed by syn-selective dihydroxylation, and (ii) α-oxygenation of a ketone followed by stereoselective reduction of the resulting α-diketone. Several synthetic routes to ferruginol (3) have been previously developed, including strategies from relatively abundant diterpenoid precursors, and most of these utilize racemic materials.33 The first total synthesis was reported in 1942 from podocarpic acid.34 Subsequently, the Oishi group utilized dehydroabietic acid,35 while the Marcos36 and Álvarez-Manzaneda37 groups independently developed routes starting from sclareol. González and co-workers employed dehydroabietamine as a starting material.38 In addition, numerous stereoselective approaches have been developed for the synthesis of (+)- and (−)-ferruginol.33 Among the various strategies explored, cationic polyene cyclization has emerged as a powerful tool for constructing the tricyclic core of ferruginol.39,40 Our study presents a new enantioselective route to (+)-ferruginol using synthetic (+)-podocarpatriene-3-ol as the starting material, offering a concise and stereocontrolled access to this important class of abietane diterpenoid.
Results and discussion
The retrosynthetic analysis for our primary target molecule, nagiol (2), is outlined in Scheme 1. We envisioned that the core structure of nagiol (2) could be constructed through a sequence of strategic transformations, starting from a suitably functionalized podocarpatriene-3-ol 8. Key to our strategy was the formation of the advanced intermediate 7, which could be accessed from 8via regioselective Friedel–Crafts acylation, followed by Baeyer–Villiger oxidation and dehydration to introduce the olefinic moiety and install the required oxygen functionality. Intermediate 7 would then serve as a versatile branching point; through asymmetric dihydroxylation and further elaboration, it would furnish compound 2. Similarly, 2,3-dihydroxyferruginol (4) was expected to be obtained from compound 7via selective dihydroxylation followed by functional group modification. Additionally, ferruginol (3) could be accessed from intermediate 7via olefin reduction and isopropyl group installation. | ||
| Scheme 1 Retrosynthetic analysis of nagiol (2), ferruginol (3), and 2,3-dihydroxyferruginol (4). | ||
We initiated our synthesis from the tricyclic compound podocarpatriene-3-ol (8) prepared on a gram scale in our lab.41 Synthesis of 8 is well documented by several groups using cationic polyenecyclization.42–45 We prepared 8 from enantiomerically pure epoxy geranyl acetate46 for this study. To install a C-12 hydroxy group in 8, we employed a regioselective Friedel–Crafts acylation followed by Baeyer–Villiger oxidation and methanolysis reactions (Scheme 2). The Friedel–Crafts reaction of compound 8 resulted in an inseparable mixture of regioisomers, specifically the C-12 and C-13 acylated products (9 and 10), in a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in 90% yield. The formation of the desired isomer in the major amount was further confirmed through NOESY studies. Though the podocarpatriene-3-ol 8 is well known, our approach marks the first report of the regioselective Friedel–Crafts reaction on its aromatic ring. The combined mixture of regioisomers (9 and 10) was then treated with meta-chloroperbenzoic acid to form the corresponding rearranged acetates 11 and 12. The mixture when subjected for Fries rearrangement reactions under different catalysts provided complex mixture of products. Hence, we proceeded with crude mixture for direct methanolysis in the presence of K2CO3 in methanol to form (2S,4aS,10aR)-1,1,4a-trimethyl-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-2,6-diol 13 in 69% and (2S,4aS,10aR)-1,1,4a-trimethyl-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-2,7-diol 14 (isolophanthin C)47 in 17% isolated yield.
:1 in 90% yield. The formation of the desired isomer in the major amount was further confirmed through NOESY studies. Though the podocarpatriene-3-ol 8 is well known, our approach marks the first report of the regioselective Friedel–Crafts reaction on its aromatic ring. The combined mixture of regioisomers (9 and 10) was then treated with meta-chloroperbenzoic acid to form the corresponding rearranged acetates 11 and 12. The mixture when subjected for Fries rearrangement reactions under different catalysts provided complex mixture of products. Hence, we proceeded with crude mixture for direct methanolysis in the presence of K2CO3 in methanol to form (2S,4aS,10aR)-1,1,4a-trimethyl-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-2,6-diol 13 in 69% and (2S,4aS,10aR)-1,1,4a-trimethyl-1,2,3,4,4a,9,10,10a-octahydrophenanthrene-2,7-diol 14 (isolophanthin C)47 in 17% isolated yield.
 | ||
| Scheme 2 Regioselective aromatic hydroxylation of podocarpatriene-3-ol (8). | ||
Having the diol 13 on a gram scale in hand, we moved ahead, primarily with the selective protection of the phenolic hydroxy group. Methylation using dimethyl sulfate and potassium carbonate produced compound 15 in 97% yield. Further, the secondary alcohol in 15 was subjected to mesylation, followed by elimination in the presence of Li2CO3 and LiBr, heating at 100 °C in DMF to afford the olefin 7. Then, compound 7 underwent citric acid-assisted OsO4-catalyzed syn-dihydroxylation,48 forming syn-diols 16 and 17 in a 1:2.6 ratio in 92% yield.49 It was expected that the exo-addition proceeds through (I) as it prefers the less hindered convex outer face to yield 17 as the major product. Both isomers were separated, and the relative configurations of the two stereogenic centers were confirmed using NOESY data.50 For compound 16, NOE correlations of H-2/H-3 suggested that H-2 and H-3 were co-facial, and NOE correlations of Me-19/H-2, Me-20/H-2 confirmed the β-orientation of H-2 and H-3. In case of compound 17, NOE correlations of H-2/H-3 suggested that both are co-facial, and it shows correlations for Me-18/H-3, H-5/H-3, which confirmed the α-orientation of H-2 and H-3. The required diastereomer, α-diol 16, was obtained in a yield of 26%. Compound 16 was subjected to Friedel–Crafts acylation, and di-O-acetylated and C-acylated product 18 was obtained. After acidic workup, the crude was directly subjected to methanolysis in the presence of K2CO3 to get compound 19 as a single regioisomer in 90% yield over two steps. Finally, O-methyl deprotection was achieved using boron tribromide, resulting in the formation of the reported structure of nagiol 2 in 96% yield.51 Spectral data of synthetic 2 were correlated to the reported data of the natural isomer (Scheme 3).12 Notable differences were found both in 1H and 13C chemical shift values for the C-2, C-3, and C-5 carbon centers.
 | ||
| Scheme 3 Preparation of the reported structure of nagiol (2). | ||
Given the structural similarity of compounds 16 and 17 to the target molecule nagiol (2), a detailed NMR analysis was undertaken to determine which compound aligned more closely. The proton and carbon chemical shifts at the C-2 and C-3 positions of compound 16 showed a strong correlation with synthetic 2, indicating that they likely possess the same stereochemistry. In contrast, the NMR shifts for C-2 and C-3 positions of compound 17 were in excellent agreement with the reported values of the natural product.12 This observation strongly suggests that compound 17 likely shares the same C-2/C-3 stereochemistry as natural nagiol. Accordingly, compound 17 was subjected to a sequence of Friedel–Crafts acylation, methanolysis, and demethylation, which led to the formation of compound 22 (Scheme 4). The NMR data (1H and 13C) of compound 22 showed excellent agreement with the reported values for natural nagiol. Specifically, the proton chemical shifts for H-2 and H-3 were observed at δ 4.25 ppm and δ 3.27 ppm, respectively, while the corresponding carbon signals appeared at δ 71.3 ppm (C-2) and δ 78.1 ppm (C-3), all in close agreement with the spectral data of the natural product (Table 1).12
 | ||
| Scheme 4 Conversion of isomer 17 to nagiol 22. | ||
| Carbon number | Natural 2 (reported)12 | Synthetic 2 | Synthetic 22 | |||
|---|---|---|---|---|---|---|
| 1H NMRa | 13C NMRb | 1H NMRa | 13C NMRb | 1H NMRa | 13C NMRb | |
| a Recorded at 500 MHz. b Recorded at 126 MHz. | ||||||
| 1a | 2.70, m | 42.3 | 2.23, m | 42.3 | 2.64, d (1.4) | 42.4 |
| 1b | 1.72, m | 1.83, m | 1.72, (14.1) | |||
| 2a | 4.24, d (3.5) | 71.2 | 4.18, m | 66.9 | 4.25, br s | 71.3 |
| 2b | ||||||
| 3a | 3.27, d (3.5) | 77.9 | 3.53, s | 78.6 | 3.27, br s | 78.1 |
| 3b | ||||||
| 4 | 38.5 | 38.5 | 38.6 | |||
| 5a | 1.35, dd (12.4, 2.4) | 49.1 | 1.83, m | 39.9 | 1.36, d (12.0) | 49.3 |
| 6a | 1.90, m | 18.6 | 2.05, m | 21.8 | 1.92, d (7.6) | 18.7 |
| 6b | 1.85, m | 2.02, m | 1.85, m | |||
| 7a | 2.95, m | 29.6 | 2.94, m | 28.4 | 2.95, m | 29.7 |
| 7b | 2.80, m | 2.83, m | 2.81, d (8.6) | |||
| 8 | 125.4 | 125.8 | 125.5 | |||
| 9 | 159.6 | 158.8 | 159.7 | |||
| 10 | 37.8 | 39.7 | 38.0 | |||
| 11 | 6.86, s | 113.8 | 6.87, s | 113.5 | 6.87, s | 114.0 |
| 12 | 160.1 | 160.4 | 160.3 | |||
| 13 | 117.9 | 118.1 | 118.0 | |||
| 14 | 7.40, s | 131.1 | 7.40, s | 131.1 | 7.41, s | 131.2 |
| 15 | 203.9 | 204.0 | 204.0 | |||
| 16 | 2.57, s | 26.3 | 2.58 s | 25.1 | 2.58, s | 26.4 |
| 17 | ||||||
| 18 | 1.08, s | 29.8 | 0.98, s | 29.2 | 1.09, s | 29.9 |
| 19 | 1.11, s | 17.1 | 1.10, s | 18.5 | 1.11, s | 17.2 |
| 20 | 1.43, s | 26.5 | 1.22, s | 26.6 | 1.43, s | 26.6 |
| OH-12 | 11.90, s | 11.91, s | 11.91, s | |||
| Rotation | [α]25D +6 (c 0.007, MeOH) | [α]25D = +35 (c 0.2, MeOH) | [α]25D = +60 (c 0.3, MeOH) | |||
Specific rotation measurements further differentiated the synthetic isomers: synthetic compound 2 showed a specific rotation of [α]25D +35 (c 0.2, MeOH), while compound 22 exhibited [α]25D +60 (c 0.3, MeOH), both significantly deviating from the reported value of [α]25D +6 (c 0.007, MeOH). Despite this discrepancy in specific rotation value, the complete spectral match, especially in NMR data, confirmed that compound 22 is the natural isomer. Thus, its structure was unambiguously established as 1-((4bS,6S,7R,8aR)-3,6,7-trihydroxy-4b,8,8-trimethyl-4b,5,6,7,8,8a,9,10-octahydrophenanthren-2-yl)ethan-1-one, and it was supported by X-ray crystallographic data.
Having established the structure of nagiol as the C-2β/C-3β syn-isomer, we subsequently devised an improved synthetic route starting from the key intermediate 15 (Scheme 5). This revised strategy aimed to achieve enhanced diastereoselectivity and improved yields for the desired syn-dihydroxy isomer 17, thereby streamlining access to the correct stereoisomer of nagiol. We subjected compound 15 to oxidation using Dess–Martin periodinane, which afforded the corresponding keto compound 23 in 98% yield.
 | ||
| Scheme 5 Alternate route to the nagiol through α-oxygenation and diastereoselective reduction. | ||
Subsequent oxidation of compound 23 using molecular oxygen in the presence of potassium tert-butoxide in tert-butanol furnished the enol–ketone intermediate 24.52,53 This diketone was then directly subjected to stereoselective reduction using NaBH4, delivering the syn-diol 17 as the sole product in 87% overall yield over two steps.52,53 The spectral data and specific rotation are consistent with those of the previously synthesized compound 17. This highly selective and efficient route enabled a streamlined synthesis of compound 17, which was subsequently transformed into nagiol (22) by following our previously established sequence involving Friedel–Crafts acylation, methanolysis, and demethylation.
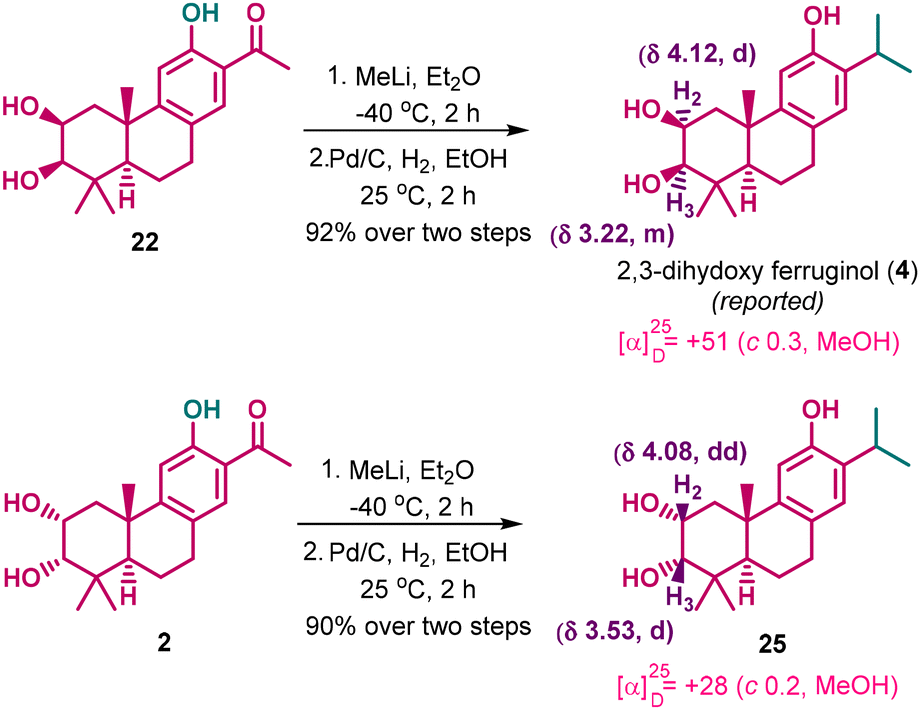
Expanding the scope and application of the strategy, 2,3-dihydroxyferruginol (4) was readily synthesized from nagiol (22). Compound 22 was first subjected to methyl lithium to obtain the corresponding tert-alcohol, which was then subjected to hydrogenolysis using Pd/C under a hydrogen atmosphere to afford the reported structure of 2,3-dihydroxyferruginol (4), in 92% over two steps (Scheme 6). Both the Simoneit30 and Liang31 groups reported the structure of 4 in 2006 and 2022, but neither report provided any spectral data to support the structure. However, when the spectral data of our synthetic compound 4 was compared with the data reported by Chen and co-workers, certain discrepancies were found.12 To further investigate, we also synthesized the C-2/C-3 syn-isomer 25 from synthetic compound 2 using an analogous procedure. Interestingly, the spectral data of compound 25 also deviated from the spectral data reported by Chen and co-workers. Although a racemic synthesis of 4 and 25 was previously reported by Huo et al.,48 our analysis shows that the spectral data they provided do not match well with the natural product 4 reported by Chen and co-workers.12 In contrast, the NMR data of our synthetic compounds 4 and 25 are in good agreement with those reported by Huo et al.48 As we do not have any clarity on the spectral data of the first isolation of 4 by Simoneit and co-workers,30 we conclude that we have synthesized the 2,3-dihydroxy ferruginol. Our study further suggests that the structure of the natural isolate by Chen and co-workers may differ from the originally proposed structure by Simoneit and co-workers. Based on these observations, we believe that the compound referred to as 2,3-dihydroxyferruginol isolated by Chen and co-workers may have a different structure, and we conclude that our synthesis corresponds to the 2,3-dihydroxyferruginol originally proposed by Simoneit and co-workers.
 | ||
| Scheme 6 Synthesis of 2,3-dihydroxyferruginol isomers from nagiol isomers. | ||
We further wanted to expand this strategy to the synthesis of ferruginol 3 by the diversification of the A- and C-rings of compound 8. We began modifying intermediate 15, which was first subjected to a Friedel–Crafts acylation reaction, followed by base-mediated methanolysis, to produce the acyl compound 27 in 90% yield over two steps. Subsequently, the secondary alcohol in 27 was mesylated and subjected to elimination using LiBr and Li2CO3. Under reflux conditions in DMF, both elimination and demethylation occurred simultaneously, resulting in the formation of compound 28 with a yield of 70%. Next, the ketone group was treated with methyl lithium to form the corresponding benzylic tertiary alcohol. The crude product obtained was further subjected to H2 in the presence of Pd/C for benzylic deoxygenation and C–C double bond reduction to provide (+)-ferruginol 3 in 92% over two steps (Scheme 7). The spectral data of synthetic 3 was identical to the reported data, and the specific rotation [α]25D +41 (c 0.3, EtOH) is nicely aligned with the reported value {[α]16D +40.6 (c 0.14, EtOH)}.13
 | ||
| Scheme 7 Synthesis of (+)-ferruginol (3). | ||
Conclusion
In conclusion, we have accomplished the first enantioselective total synthesis of nagiol starting from 8,11,13-podocarpatriene-3-ol, along with a revision of its originally proposed structure. This synthetic strategy was further extended to the total synthesis of ferruginol and 2,3-dihydroxyferruginol. The enantiomerically enriched starting podocarpatriene-3-ol, consisting of the desired tricyclic framework, was efficiently synthesized and diversified to different diterpenes. The 2,3-dihydroxy functionality on the A-ring was introduced using two alternative methods: (i) an OsO4-catalyzed stereoselective dihydroxylation of olefin, and (ii) α-oxygenation of a ketone using molecular oxygen and a base, followed by stereocontrolled reduction. Based on a detailed comparison of NMR data and supported X-ray analysis, the C-2β/C-3β syn-dihydroxy isomer was confirmed to be natural nagiol. The synthetic 2,3-dihydroxyferruginol was found to show a discrepancy in the spectral data with the reported data (by Chen and co-workers). However, we declare that we synthesized (+)-2,3-dihydroxyferruginol as per the Simoneit and Liang groups. Overall, the strategy features regioselective Friedel–Crafts acylations, a Baeyer–Villiger rearrangement, and regioselective elimination, stereoselective dihydroxylation, α-oxygenation of ketone, diastereoselective reduction of diketone, one-pot elimination and demethylation and hydrogenolysis reactions as key steps. In total, nagiol was synthesized in a short route in nine steps with seven step purifications in an overall yield of 35.5%.Author contributions
GR and SS devised the idea, conducted experiments with help of AR. GR and SS prepared the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Data availability
Experimental procedures and spectral data of all compounds are available as electronic ESI.†X-Ray crystallographic data of compound 22 CCDC 2457373 is available in the ESI file.†
Acknowledgements
Rajendar acknowledges ANRF India for the SERB-CRG research funding through CRG/2020/003737, and SS is thankful to DST India for INSPIRE fellowship.References
- O. Batista, M. F. Simões, A. Duarte, M. L. Valdeira, M. C. De La Torre and B. Rodríguez, Phytochemistry, 1995, 38, 167–169 CrossRef CAS PubMed.
- H. Achenbach, R. Waibel, H. H. Nkunya and H. Ween, Phytochemistry, 1992, 31, 3781–3784 CrossRef CAS.
- Y. Xiong, K. Wang, Y. Pan, H. Sun and J. Tu, Bioorg. Med. Chem. Lett., 2006, 16, 786–789 CrossRef CAS PubMed.
- J. Gao and G. Han, Phytochemistry, 1997, 44, 759–761 CrossRef CAS.
- P. L. Majumder, D. C. Maiti, W. Kraus and M. Bokel, Phytochemistry, 1987, 26, 3021–3023 CrossRef CAS.
- M. A. González, Nat. Prod. Rep., 2015, 32, 684–704 RSC.
- M. A. González, Eur. J. Med. Chem., 2014, 87, 834–842 CrossRef PubMed.
- A. Otto, J. D. White and B. R. T. Simoneit, Science, 2002, 297, 1543–1545 CrossRef CAS PubMed.
- J. Zi and R. J. Peters, Org. Biomol. Chem., 2013, 11, 7650 RSC.
- J. Guo, X. Ma, Y. Cai, Y. Ma, Z. Zhan, Y. J. Zhou, W. Liu, M. Guan, J. Yang, G. Cui, L. Kang, L. Yang, Y. Shen, J. Tang, H. Lin, X. Ma, B. Jin, Z. Liu, R. J. Peters, Z. K. Zhao and L. Huang, New Phytol., 2016, 210, 525–534 CrossRef CAS PubMed.
- Y. Ma, G. Cui, T. Chen, X. Ma, R. Wang, B. Jin, J. Yang, L. Kang, J. Tang, C. Lai, Y. Wang, Y. Zhao, Y. Shen, W. Zeng, R. J. Peters, X. Qi, J. Guo and L. Huang, Nat. Commun., 2021, 12, 685 CrossRef CAS PubMed.
- H. Zhao, H. Li, G. Huang and Y. Chen, Nat. Prod. Res., 2017, 31, 844–848 CrossRef CAS PubMed.
- C. W. Brandt and L. G. Neubauer, J. Am. Chem. Soc., 1939, 1031 RSC.
- A. Ulubelen, H. Birman, S. Öksüz, G. Topçu, U. Kolak, A. Barla and W. Voelter, Planta Med., 2002, 68, 818–821 CrossRef CAS PubMed.
- A. Koch, J. Orjala, P. C. Mutiso and D. D. Soejarto, Biochem. Syst. Ecol., 2006, 34, 270–272 CrossRef CAS.
- W.-H. Li, S.-T. Chang, S.-C. Chang and H.-T. Chang, Nat. Prod. Res., 2008, 22, 1085–1093 CrossRef CAS PubMed.
- S.-T. Chang, S.-Y. Wang, C.-L. Wu, Y.-C. Su and Y.-H. Kuo, Holzforschung, 1999, 53, 487–490 CAS.
- K. Kuroda, T. Fujiwara, K. Hashida, T. Imai, M. Kushi, K. Saito and K. Fukushima, Ann. Bot., 2014, 113, 1029–1036 CrossRef CAS PubMed.
- M. Espinoza, L. Santos, C. Theoduloz, G. Schmeda-Hirschmann and J. Rodríguez, Planta Med., 2008, 74, 802–808 CrossRef CAS PubMed.
- J. A. Rodríguez, C. Theoduloz, T. Yáñez, J. Becerra and G. Schmeda-Hirschmann, Life Sci., 2006, 78, 2503–2509 CrossRef PubMed.
- C. Areche, C. Theoduloz, T. Yáñez, A. R. M. Souza-Brito, V. Barbastefano, D. De Paula, A. L. Ferreira, G. Schmeda-Hirschmann and J. A. Rodríguez, J. Pharm. Pharmacol., 2008, 60, 245–251 CrossRef CAS PubMed.
- M. Bispo De Jesus, W. F. Zambuzzi, R. R. Ruela De Sousa, C. Areche, A. C. Santos De Souza, H. Aoyama, G. Schmeda-Hirschmann, J. A. Rodríguez, A. R. Monteiro De Souza Brito, M. P. Peppelenbosch, J. Den Hertog, E. De Paula and C. V. Ferreira, Biochimie, 2008, 90, 843–854 CrossRef CAS PubMed.
- K.-H. Son, H.-M. Oh, S.-K. Choi, D. C. Han and B.-M. Kwon, Bioorg. Med. Chem. Lett., 2005, 15, 2019–2021 CrossRef CAS PubMed.
- C. Clarkson, W. E. Campbell and P. Smith, Planta Med., 2003, 69, 720–724 CrossRef CAS PubMed.
- V. Samoylenko, D. C. Dunbar, Md. A. Gafur, S. I. Khan, S. A. Ross, J. S. Mossa, F. S. El-Feraly, B. L. Tekwani, J. Bosselaers and I. Muhammad, Phytother. Res., 2008, 22, 1570–1576 CrossRef CAS PubMed.
- H.-L. Chen, K.-W. Lin, K.-H. Gan, J.-P. Wang, S.-J. Won and C.-N. Lin, Fitoterapia, 2011, 82, 219–224 CrossRef CAS PubMed.
- C.-C. Wen, Y.-H. Kuo, J.-T. Jan, P.-H. Liang, S.-Y. Wang, H.-G. Liu, C.-K. Lee, S.-T. Chang, C.-J. Kuo, S.-S. Lee, C.-C. Hou, P.-W. Hsiao, S.-C. Chien, L.-F. Shyur and N.-S. Yang, J. Med. Chem., 2007, 50, 4087–4095 CrossRef CAS PubMed.
- Y. B. Ryu, H. J. Jeong, J. H. Kim, Y. M. Kim, J.-Y. Park, D. Kim, T. T. H. Naguyen, S.-J. Park, J. S. Chang and K. H. Park, Bioorg. Med. Chem., 2010, 18, 7940–7947 CrossRef CAS PubMed.
- Y. Wei, J. He, H. Qin, X. Wu and X. Yao, Biomed. Chromatogr., 2009, 23, 1116–1120 CrossRef CAS PubMed.
- R. E. Cox, S. Yamamoto, A. Otto and B. R. T. Simoneit, Biochem. Syst. Ecol., 2007, 35, 342–362 CrossRef CAS.
- Y.-J. Hu, M.-L. Chen and D. Liang, Fitoterapia, 2022, 162, 105293 CrossRef CAS PubMed.
- S. Surendran, Chandrendu K. C. and G. Rajendar, J. Nat. Prod., 2025, 88, 502–512 CrossRef CAS PubMed.
- M. Li, P. Chen, H. Liu, J. Huang and Y. Chen, Synthesis, 2024, 1355–1368 CAS.
- F. E. King, T. J. King and J. G. Topliss, J. Chem. Soc., 1957, 573–577 RSC.
- M. Tada and K. Ishimaru, Chem. Pharm. Bull., 2006, 54, 1412–1417 CrossRef CAS PubMed.
- I. S. Marcos, A. Beneitez, R. F. Moro, P. Basabe, D. Díez and J. G. Urones, Tetrahedron, 2010, 66, 7773–7780 CrossRef CAS.
- E. Alvarez-Manzaneda, R. Chahboun, E. Cabrera, E. Alvarez, R. Alvarez-Manzaneda, M. Hmamouchi and H. Es-Samti, Tetrahedron Lett., 2007, 48, 8930–8934 CrossRef CAS.
- M. A. González, J. Clark, M. Connelly and F. Rivas, Bioorg. Med. Chem. Lett., 2014, 24, 5234–5237 CrossRef PubMed.
- M. Majhi, S. Kundu, D. Jana, S. Sadhukhan and A. Bisai, J. Org. Chem., 2025, 90, 2077–2092 CrossRef CAS PubMed.
- Z. Tao, K. A. Robb, K. Zhao and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 3569–3573 CrossRef CAS PubMed.
- See ESI† for the preparation of compound 8.
- V. Rosales, J. Zambrano and M. Demuth, Eur. J. Org. Chem., 2004, 2004, 1798–1802 CrossRef.
- J.-F. Zhao, Y.-J. Zhao and T.-P. Loh, Chem. Commun., 2008, 1353 RSC.
- A. Castillo, J. F. Q. Del Moral and A. F. Barrero, Nat. Prod. Commun., 2017, 12, 1934578X1701200503 CrossRef CAS.
- Y. Tian, X. Xu, L. Zhang and J. Qu, Org. Lett., 2016, 18, 268–271 CrossRef CAS PubMed.
- R. R. S. S. Surendran, G. James and G. Rajendar, Eur. J. Org. Chem., 2023, 26, e202300748 CrossRef.
- L. Yang, L. Li, S. Huang, J. Pu, Y. Zhao, Y. Ma, J. Chen, C. Leng, Z. Tao and H. Sun, Chem. Pharm. Bull., 2011, 59, 1102–1105 CrossRef CAS PubMed.
- C.-Y. Huo, T.-L. Zheng, W.-H. Dai, Z.-H. Zhang, J.-D. Wang, D.-Y. Zhu, S.-H. Wang, X.-M. Zhang and X.-T. Xu, Chem. Sci., 2022, 13, 13893–13897 RSC.
- P. Dupau, R. Epple, A. Thomas, V. Fokin and K. Barry Sharpless, Adv. Synth. Catal., 2002, 344, 421–433 CrossRef CAS.
- See ESI† for the NOESY data.
- R. Sunnapu and G. Rajendar, Eur. J. Org. Chem., 2021, 2021, 1637–1642 CrossRef CAS.
- H. Zhang, P. Zhu, J. Liu, X. Yang, S. Xu, H. Yao, J. Jiang, W. Ye, X. Wu and J. Xu, Eur. J. Med. Chem., 2014, 87, 159–167 CrossRef CAS PubMed.
- T. C. Denner, N. V. Heise, S. Hoenke and R. Csuk, Molecules, 2024, 29, 2346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2457373. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ob00946d |
| This journal is © The Royal Society of Chemistry 2025 |