
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of the D-acofriose-containing trisaccharide repeating unit of the O-antigen from Azospirillum brasilense JM6B2†
Sanajit
Maiti
 and
Balaram
Mukhopadhyay
*
and
Balaram
Mukhopadhyay
*
Sweet Lab, Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, Nadia 741246, India. E-mail: mbalaram@iiserkol.ac.in
First published on 22nd May 2025
Abstract
The unique D-acofriose (6-deoxy-3-O-methyl-D-mannose) unit present in the target oligosaccharide was synthesized from commercially available D-mannose in six steps with ∼50% overall yield. The synthesis was found to be equally successful at the multigram scale (100 mmol). 2-Bromoethyl α-L-fucopyranoside, required at the reducing end, was prepared exclusively via H2SO4–silica promoted Fischer glycosylation. The influence of the protecting group at the 3-position of the fucosyl moiety on glycosylation at the 4-position was studied and ether protection was found to be essential over ester protection. Further global deprotection gave the target conjugation-ready trisaccharide in 34% overall yield.
Introduction
Selective enrichment of the rhizosphere with adapted microorganisms results in increased plant growth activity and productivity.1,2 Such plant–microbe associations have been extensively studied with Azospirillum bacteria.3 The surface-located glycopolymers produced by the Azospirillum species are key to their rhizosphere adaptation, with lipopolysaccharides (LPSs) playing an important role. The success of the initial stages of bacteria–root association4,5 is governed by bacterial surface polysaccharides that are involved in the adhesion and adsorption of microorganisms to the roots of plants. Furthermore, the O-specific polysaccharide (OPS) portion of the LPS interacts with the environment and possesses antigenic determinants as part of its structure. Owing to the significant influence of Azospirillum species on plant growth activities and productivity, their OPS structures have been the subject of detailed studies. In this context, Fedonenko et al.6 reported the OPS structure of Azospirillum brasilense (A. brasilense) Jm6B2, which has a unique 3-O-methyl-D-rhamnose (D-acofriose) that is not found in other Azospirillum species. In continuation of our effort towards the chemical synthesis of biologically relevant bacterial oligosaccharides,7–9 we herein report the total synthesis of the trisaccharide repeating unit of the OPS from A. brasilense Jm6B2 in the form of its 2-aminoethyl glycoside (Fig. 1). Literature studies on D-acofriose reveal that it is also abundant in some Pseudomonas aeruginosa species.10,11 This information prompted us to develop a convenient route for the gram-scale synthesis of D-acofriose from commercially available D-mannose. | ||
| Fig. 1 Structure of the target trisaccharide repeating unit of the OPS from A. brasilense Jm6B2 in the form of its 2-aminoethyl glycoside. | ||
Results and discussion
Retrosynthetic analysis for the synthesis of the target trisaccharide 1 revealed that sequential glycosylation of the suitably functionalized fucosyl acceptor 18 with the fully protected D-xylose donor 15 and D-acofriose donor 7 would successfully afford the protected trisaccharide. Since no other amino functionalities are present in the structure, an azidoethyl glycoside at the reducing end was considered ideal; this can be subsequently converted to the corresponding 2-aminoethyl glycoside to facilitate further conjugation. Literature reports indicate that insertion of a sugar at the 3-O-position of a fucose moiety significantly reduces the nucleophilicity of the 4-O-position, thereby hindering the introduction of another sugar at that position.11 To address this, we planned to protect the 3-O-position of the fucosyl moiety with a temporary protecting group and introduce a protected D-xylose moiety at the 4-O-position. Furthermore, the removal of the 3-O-protection and introduction of the D-acofriose unit would lead to the protected trisaccharide. Finally, the removal of the protecting groups and reduction of the terminal azide to the corresponding amine would furnish the target molecule (Fig. 2). | ||
| Fig. 2 Retrosynthetic analysis for the synthesis of the target trisaccharide 1. | ||
For the synthesis of D-acofriose, commercially available D-mannose was converted to methyl D-mannoside (2) using H2SO4–silica in dry MeOH at 65 °C.12 Our previously developed Fischer glycosylation strategy worked perfectly to give compound 2 in 95% yield. Further reaction with iodine in the presence of PPh3 using dry DMF13 as solvent gave the 6-iodo derivative, which was filtered through a silica gel column to remove triphenylphosphine oxide, and the iodo-derivative was hydrogenolyzed using H2 in the presence of 10% Pd–C14 to give methyl D-rhamnoside (3)15 in 87% overall yield. Methylation at the 3-O-position was successfully achieved via stannylene chemistry. The formation of the tin ketal using Bu2SnO in refluxing toluene followed by reaction with MeI in the presence of Bu4NI16 gave methyl 3-O-methyl D-acofrioside (4) in 73% yield. It is worth noting that the methylation reaction proceeded rather slowly as the low boiling point of MeI limits its availability as an electrophile and excess MeI is required to drive the reaction to completion with 73% yield. Acetolysis of compound 4 afforded the per-O-acetylated acofriose derivative 5 in 86% yield. Finally, Zemplen de-O-acetylation gave D-acofriose (6) in 96% yield (Scheme 1). Following this route, D-acofriose was obtained in 50% overall yield via six steps from commercially available D-mannose. All reactions were performed at the gram scale without any alteration in yields. It is worth noting that the current method for the preparation of D-acofriose is significantly superior to the methods reported by Sauvageau et al.17,18 and Brimacombe et al.19 (for the L-isomer). Moreover, it is observed that the process is equally efficient at the 100 mmol scale and therefore may be used for large-scale preparation of D-acofriose. To fulfil the requirement of our D-acofriose donor for total synthesis, the per-O-acetylated derivative 5 was reacted with p-thiocresol in the presence of BF3·Et2O at −5 °C to give the corresponding thioglycoside donor 7 in 89% yield (Scheme 1).
 | ||
| Scheme 1 Synthesis of D-acofriose (6) and the thioglycoside donor 7 from commercially available D-mannose. | ||
The initial challenge with the synthesis of the fucosyl acceptor was to install the reducing end α-glycoside. Therefore, the known thioglycoside donor 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 was activated by NIS and TMSOTf in the presence of 2-bromoethanol. Different solvents at varying temperatures were tested to achieve α-selectivity, but with limited success. Glycosylations in pure CH2Cl2 showed no selectivity at 0 °C or −20 °C and only marginal selectivity (3:2) at −40 °C (entries 1, 2 and 3, Table 1). Switching to a CH2Cl2–Et2O mixture21 in varying ratios could achieve only up to a ratio of 2:1 of the desired α-glycoside (entries 4–6). Pre-activation of the donor with DMF22 in CH2Cl2 followed by glycosylation with 2-bromoethanol was also tested, but glycoside 9 was obtained only in a 5:1 (α/β) ratio (entry 7, Table 1). In addition, we were unable to separate the mixture and use the desired isomer further.
20 was activated by NIS and TMSOTf in the presence of 2-bromoethanol. Different solvents at varying temperatures were tested to achieve α-selectivity, but with limited success. Glycosylations in pure CH2Cl2 showed no selectivity at 0 °C or −20 °C and only marginal selectivity (3:2) at −40 °C (entries 1, 2 and 3, Table 1). Switching to a CH2Cl2–Et2O mixture21 in varying ratios could achieve only up to a ratio of 2:1 of the desired α-glycoside (entries 4–6). Pre-activation of the donor with DMF22 in CH2Cl2 followed by glycosylation with 2-bromoethanol was also tested, but glycoside 9 was obtained only in a 5:1 (α/β) ratio (entry 7, Table 1). In addition, we were unable to separate the mixture and use the desired isomer further.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp. | Time |
9α:9βa |
Yield |
| a As obtained from the 1H NMR of the mixture. | |||||
| 1 | Dry CH2Cl2 | 0 °C | 1 h | 1:1 |
81% |
| 2 | −20 °C | 3 h | 1:1 |
72% | |
| 3 | −40 °C | 3 h | 3:2 |
74% | |
| 4 | CH2Cl2–Et2O (3:2) |
−40 °C | 3 h | 3:2 |
76% |
| 5 | CH2Cl2–Et2O (1:1) |
−40 °C | 3 h | 3:2 |
75% |
| 6 | CH2Cl2–Et2O (1:3) |
−40 °C | 3 h | 2:1 |
76% |
| 7 | Dry CH2Cl2 pre-activation with DMF | −20 °C | 3 h | 5:1 |
78% |

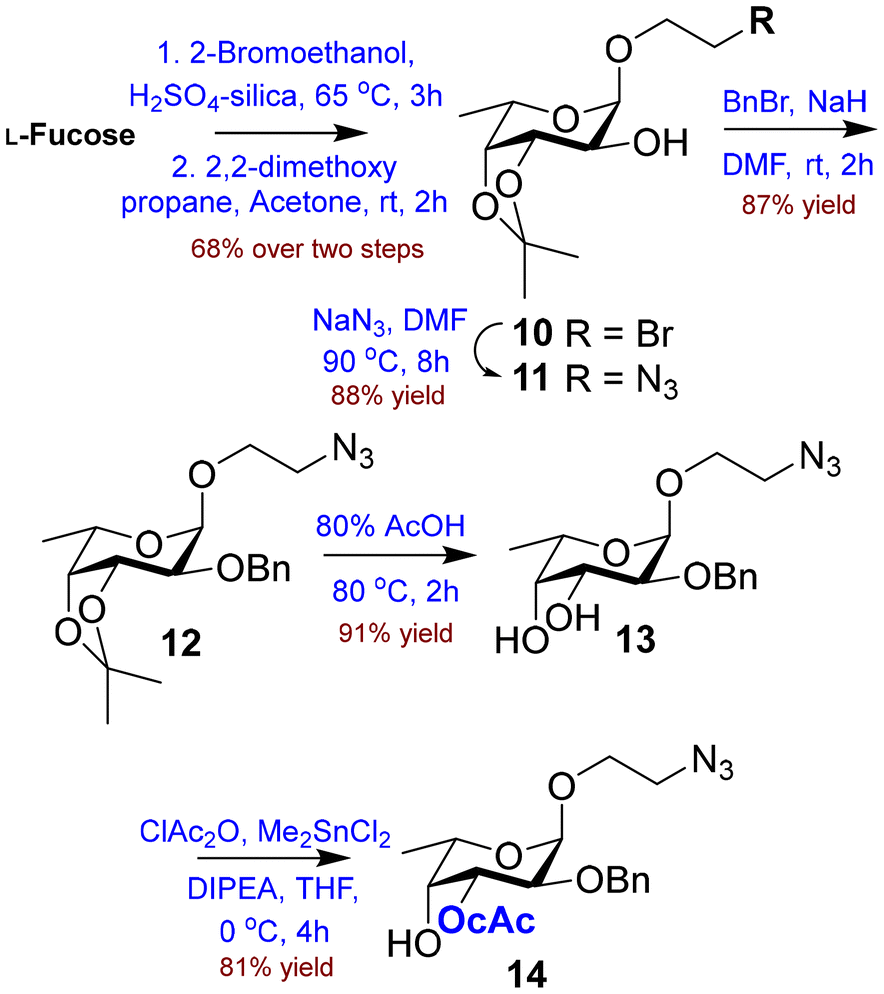
Finally, we resorted to H2SO4–silica promoted Fischer glycosylation12 of free L-fucose with 2-bromoethanol and successfully achieved the desired glycoside. After removal of excess 2-bromoethanol using diethyl ether, the residue was treated with 2,2-dimethoxypropane in acetone to afford the isopropylidene derivative 10 in 68% yield over two steps.
No further acid catalyst was required as H2SO4–silica was present in the mixture and only α-glycoside was isolated in pure form. Furthermore, the terminal bromide was replaced with azide using NaN3 in DMF23 at 90 °C to give the azido derivative 11 in 88% yield. The transformation was confirmed by the IR peak at 2104 cm−1. The free 2-O-position was benzylated using BnBr and NaH24 to afford the fully protected derivative 12 in 87% yield. Next, the hydrolysis of the isopropylidene group using 80% aq. AcOH at 80 °C25 gave diol 13 in 91% yield. Selective chloroacetylation of diol 13 using chloroacetic anhydride in the presence of Me2SnCl226,27 gave the 3-O-chloroacetate derivative 14 in 81% yield (Scheme 2).
 | ||
| Scheme 2 Synthesis of the fucose acceptors 14. | ||
Next, the glycosylation of the fucosyl acceptor 14 with the known xylose donor 1528 was tested through the activation of thioglycoside using NIS in the presence of TfOH.29 However, the desired disaccharide 17 was not formed; instead, the donor was converted to the corresponding hemiacetal. Changing the donor to the known xylose trichloroacetimidate (16)30 led to the same hemiacetal of the donor instead of the desired disaccharide (Scheme 3). It was assumed that the electron-withdrawing chloroacetate group at the neighbouring 3-O-position significantly reduced the nucleophilicity of the 4-O-position of acceptor 14.
 | ||
| Scheme 3 Failed synthesis of disaccharide 17 with fucosyl acceptor 14. | ||
Therefore, the diol 13 was converted to the 3-O-allyl derivative 18 in 71% yield via stannylene chemistry, using Bu2SnO in refluxing toluene followed by reaction with AllBr in the presence of Bu4NI.31 Finally, glycosylation of the fucosyl acceptor 18 and the xylosyl thioglycoside 15 using NIS in the presence of TfOH28 at 0 °C gave disaccharide 19 in 81% yield with complete β-selectivity, as confirmed by the peaks at δ 4.74 (d, 1H, J1,2 2.0 Hz, H-1) and 4.68 (d, 1H, J1′,2′ 6.8 Hz, H-1′) in the 1H NMR spectra and at δ 98.2 (C-1) and 100.5 (C-1′) in the 13C NMR spectra. The HRMS peak at 644.2433 [M + Na]+ further confirmed the successful formation of disaccharide 19. In contrast to the chloroacetyl group at the 3-O-position, the allyl ether did not hamper the nucleophilicity of the 4-O-position. Furthermore, the allyl protection was removed using PdCl2 in MeOH32 to afford the disaccharide acceptor 20 in 77% yield. Final glycosylation of the disaccharide acceptor 20 with the acofriose donor 7 using NIS in the presence of TfOH at 0 °C was uneventful and furnished the protected trisaccharide 21 in 83% yield. The successful formation of the trisaccharide was evident from the peaks at δ 97.5 (C-1), 101.4 (C-1′) and 98.9 (C-1′′) in the 13C NMR spectra. The HRMS peak at 848.3061 [M + Na]+ further affirmed the formation of trisaccharide 21. Furthermore, Zemplen de-O-acetylation33 using NaOMe in MeOH gave the de-O-acetylated trisaccharide 22 in 92% yield. Finally, catalytic hydrogenation using 10% Pd–C34 in the presence of H2 gave the target trisaccharide 1 in 71% yield (Scheme 4).
 | ||
| Scheme 4 Synthesis of the target trisaccharide 1. | ||
Conclusions
In conclusion, we have successfully accomplished the synthesis of the target trisaccharide repeating unit of the O-antigen from A. brasilense JM6B2 in the form of its 2-aminoethyl glycoside. In the process, we have developed a concise route for the preparation of D-acofriose from commercially available D-mannose via six steps with ∼50% yield. The route has been tested and found to be effective up to the 100 mmol scale and therefore has the potential to be used for large scale preparation. The 1,2-cis 2-bromoethyl fucoside was obtained exclusively using H2SO4–silica promoted Fischer glycosylation. The target trisaccharide retains the 2-aminoethyl group, allowing for future glycoconjugate formation without affecting the anomeric stereochemistry at the reducing end. The availability of this synthetic variant in its purest possible form will facilitate detailed investigations into the biological roles of this oligosaccharide repeat.Experimental section
General methods
All solvents and reagents were dried before use according to standard methods.35 The commercially purchased reagents were used without any further purification unless mentioned otherwise. Dichloromethane was dried and distilled over P2O5 to make it anhydrous and moisture-free. All reactions were monitored by thin layer chromatography (TLC) on silica-gel 60-F254 with detection by fluorescence, followed by charring after immersion in a 10% ethanolic solution of H2SO4. Flash chromatography was performed with silica gel (100–200 mesh). Optical rotations were measured on the sodium D-line at ambient temperature. IR data were collected on a Bruker Alpha FTIR spectrometer. 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz or 500 MHz and 125 MHz, respectively, on a JEOL JNM ECZL 400S or Bruker Avance Neo 400 or Bruker Avance Neo 500 spectrometer. The 1H and 13C peaks were assigned based on 1H–1H COSY and 1H–13C HSQC spectra. In the target trisaccharide 1, proton signals were denoted as H for the reducing-end L-fucose unit, H′ for D-xylose, and H′′ for D-acofriose. Similarly, the carbon signals are denoted as C for the reducing-end L-fucose unit, C′ for the D-xylose, and C′′ for the D-acofriose unit. HRMS analysis was performed using a XeVO G2-XS Q-TOF (Waters Corporation) instrument in the +ve electrospray ionization mode.:1) showed complete conversion of the starting material to a faster-moving spot, the solvent was evaporated under reduced pressure. The crude product thus obtained was charged on a column of silica gel and eluted with CH2Cl2–MeOH (10:1) to afford the pure compound 4 (1.9 g, 73%) as a yellowish syrup.
[α]25D: + 118 (c 0.8, CHCl3).
IR (cm−1, CHCl3) ν: 3362, 2847, 1248, 1086, 1038, 791.
1H NMR (400 MHz, CDCl3) δ: 4.60 (d, 1H, J1,2 1.6 Hz, H-1), 3.95 (dd, 1H, J1,2 1.6 Hz, J2,3 3.2 Hz, H-2), 3.55–3.51 (m, 1H, H-5), 3.41 (t, 1H, J3,4, J4,5 9.4 Hz, H-4), 3.37 (s, 3H, OCH3), 3.27 (dd, 1H, J2,3 3.2 Hz, J3,4 9.4 Hz, H-3), 3.26 (s, 3H, OCH3), 1.22 (d, 3H, J5,6 6.0 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 100.6 (C-1), 81.3 (C-3), 71.2 (C-4), 67.8 (C-5), 66.8 (C-2), 56.9 (OCH3), 54.7 (OCH3), 17.5 (C-CH3).
HRMS calcd for C8H16O5Na [M + Na]+: 215.0895; found: 215.0891.
:2) showed complete conversion of the starting material to a faster-moving spot. The solution was poured carefully into a solution of Na2CO3 (2.5 g) in 30 mL ice-cold water and stirred well with a glass rod. CH2Cl2 (20 mL) was added and the mixture was extracted twice. The organic layer was further washed with NaHCO3 (30 mL) and brine solution (30 mL), separated, dried over anhydrous Na2SO4, filtered and the solvent was evaporated in vacuo. The crude residue thus obtained was purified by column chromatography using n-hexane–EtOAc (3:1) to give an anomeric mixture (α/β; 5:2) of compound 5 (2.6 g, 86%) as a colourless gel.
α-Product
[α]25D: +29 (c 0.8, CHCl3).IR (cm−1, CHCl3) ν: 2853, 1745, 1252, 1083, 1041, 786.
1H NMR (400 MHz, CDCl3) δ: 6.02 (d, 1H, J1,2 2.0 Hz, H-1), 5.31 (dd, 1H, J1,2 2.0 Hz, J2,3 2.8 Hz, H-2), 5.01 (t, 1H, J3,4, J4,5 10.0 Hz, H-4), 3.90–3.80 (m, 1H, H-5), 3.60 (dd, 1H, J2,3 2.8 Hz, J3,4 10 Hz, H-3), 3.35 (s, 3H, OCH3), 2.14, 2.13, 2.09 (3s, 3 × 3H, 3 × COCH3), 1.20 (d, 3H, J5,6 6.4 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 170.1 (COCH3), 169.9 (COCH3), 168.4 (COCH3), 91.0 (C-1), 76.8 (C-3), 71.9 (C-4), 68.8 (C-5), 66.9 (C-2), 57.8 (OCH3), 20.9 (2) (COCH3), 20.8 (COCH3), 17.5 (C-CH3).
HRMS calcd for C13H20O8Na [M + Na]+: 327.1056; found: 327.1052.
:1). Solvents were evaporated and the residue was triturated with EtOAc on an ice-bath. It was filtered through a layered pad of Celite® and silica gel (100–200 mesh). The filtrate was evaporated and the residue was dissolved in MeOH (75 mL). 10% Pd–C (5 g) was added followed by DIPEA (20 mL, 0.11 mol) and the mixture was shaken in a Paar hydrogenation assembly at 3 atm H2 for 12 hours. The mixture was filtered through a pad of Celite® and the filtrate was evaporated in vacuo. The residue was suspended in dry toluene (100 mL), Bu2SnO (25 g, 0.1 mmol) was added and the mixture was stirred under reflux for 5 hours. The solution was cooled to room temperature and TBAI (37 g, 0.1 mmol) was added followed by MeI (37 mL, 0.6 mol). The mixture was stirred at 40 °C for 24 hours. Another portion of MeI (18.5 mL, 0.3 mol) was added and the stirring continued for 24 hours. The reaction progress was monitored by TLC (CH2Cl2–MeOH; 7:1). The solvents were evaporated and the residue was dissolved in CH2Cl2 (80 mL), Ac2O (36 mL) was added followed by AcOH (13 mL) and conc. H2SO4 (1.2 mL) and the solution was stirred at room temperature for 3 hours. Then the solution was carefully poured into a solution of Na2CO3 (25 g) in 250 mL ice water and stirred well with a glass rod. CH2Cl2 (70 mL) was added and the organic layer was extracted. The process was repeated one more time with CH2Cl2 (30 mL). The combined organic layer was further washed with aq. NaHCO3 (100 mL) and brine solution (100 mL), separated, dried over anhydrous Na2SO4 (25 g), and filtered and the solvent was evaporated in vacuo. The syrupy residue was purified by column chromatography to remove the tin salts. The light-yellow syrup obtained was redissolved in MeOH (100 mL). Freshly prepared NaOMe in MeOH (0.5 M, 10 mL) was added and the solution was stirred at room temperature for 3 hours. Excess NaOMe was neutralized with DOWEX® 50 W H+ and filtered and the filtrate was evaporated in vacuo to give D-acofriose (8.4 g, 47%) as an off-white sticky mass. It was dissolved in H2O (40 mL) and lyophilized to obtain a white amorphous powder.
:1) indicated complete conversion of the starting material to a faster-moving spot. The solution was then diluted with CH2Cl2 (10 mL) and washed successively with water (30 mL), saturated NaHCO3 solution (2 × 30 mL) and brine (30 mL). The organic layer was separated, dried over anhydrous Na2SO4 and filtered. The solvent was evaporated in vacuo and the crude product thus obtained was purified by column chromatography using n-hexane–EtOAc (3:1) to afford the pure compound 7 (2.8 g, 89%) as a white amorphous powder.
[α]25D: +62 (c 0.9, CHCl3).
IR (cm−1, CHCl3) ν: 2973, 2860, 1740, 1374, 1211, 1040, 804.
1H NMR (400 MHz, CDCl3) δ: 7.34 (d, 2H, ArH), 7.11 (d, 2H, ArH), 5.56 (dd, 1H, J1,2 1.6 Hz, J2,3 3.2 Hz, H-2), 5.35 (d, 1H, J1,2 1.6 Hz, H-1), 5.03 (t, 1H, J3,4, J4,5 9.6 Hz, H-4), 4.33–4.23 (m, 1H, H-5), 3.57 (dd, 1H J2,3 3.2 Hz, J3,4 9.6 Hz, H-3), 3.36 (s, 3H, OCH3), 2.32 (s, 3H, SC6H4CH3), 2.12, 2.11 (2s, 2 × 3H, 2 × COCH3), 1.21 (d, 3H, J5,6 6.4 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 170.3 (COCH3), 170.1 (COCH3), 138.1, 132.3, 130.0, 129.8, 86.4 (C-1), 77.5 (C-3), 72.6 (C-4), 69.6 (C-2), 67.7 (C-5), 57.7 (OCH3), 21.1 (COCH3), 21.0 (COCH3), 20.9 (SC6H4CH3), 17.3 (C-CH3).
HRMS calcd for C18H24O6SNa [M + Na]+: 391.1191; found: 391.1185.
:1) showed complete conversion to a faster running spot. The reaction mixture was quenched with Et3N (1.5 mL), the solvent was evaporated and the residue was purified by column chromatography using n-hexane–EtOAc (3:1) to afford the pure compound 10 (3.9 g, 68%) as a colourless syrup.
[α]25D: −52 (c 0.8, CHCl3).
1H NMR (400 MHz, CDCl3) δ: 4.78 (d, 1H, J1,2 4.0 Hz, H-1), 4.17–4.12 (m, 2H, H-5, H-3), 3.99 (dd, 1H, J3,4 6.0 Hz, J4,5 2.4 Hz, H-4), 3.97–3.92 (m, 1H, OCH2a), 3.81–3.75 (m, 1H, OCH2b), 3.74–3.70 (m, 1H, H-2), 3.49–3.43 (m, 2H, CH2Br), 2.76 (d, 1H, J 6.4 Hz, OH), 1.43 (s, 3H, isopropylidene-CH3), 1.27 (s, 3H, isopropylidene-CH3), 1.23 (d, 3H, J5,6 6.8 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 109.1 (isopropylidene-C), 97.8 (C-1), 75.9 (C-3), 75.5 (C-4), 69.2 (C-2), 67.9 (OCH2), 64.2 (C-5), 30.6 (CH2Br), 27.8 (isopropylidene-CH3), 25.9 (isopropylidene-CH3), 16.2 (C-CH3).
HRMS calcd for C11H19BrO5Na [M + Na]+: 333.0314; found: 333.0317.
:2) showed complete conversion of the starting material to a slower running spot. The solvent was evaporated and the residue was diluted with EtOAc (20 mL) and washed with brine solution (3 × 30 mL). The organic layer was separated, dried over anhydrous Na2SO4, and filtered; the solvent was evaporated under reduced pressure and the crude product thus obtained was purified by column chromatography using n-hexane–EtOAc (2:1) to afford the pure compound 11 (3.0 g, 88%) as a colourless syrup.
[α]25D: −39 (c 0.9, CHCl3).
IR (cm−1, CHCl3) ν: 2991, 2933, 2104, 1381, 1211, 1059, 861.
1H NMR (400 MHz, CDCl3) δ: 4.86 (d, 1H, J1,2 4.0 Hz, H-1), 4.23 (t, 1H, J2,3, J3,4 6.4 Hz, H-3), 4.21–4.16 (m, 1H, H-5), 4.08 (dd, 1H, J3,4 6.4 Hz, J4,5 2.0 Hz, H-4), 4.00–3.95 (m, 1H, OCH2a), 3.82 (dd, 1H, J1,2 4.0 Hz, J2,3 6.4 Hz, H-2), 3.72–3.67 (m, 1H, OCH2b), 3.52–3.46 (m, 1H, CH2aN3), 3.43–3.37 (m, 1H, CH2bN3), 2.43 (bs, 1H, OH), 1.51 (s, 3H, isopropylidene-CH3), 1.36 (s, 3H, isopropylidene-CH3), 1.33 (d, 3H, J5,6 6.8 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 109.2 (isopropylidene-C), 97.8 (C-1), 75.9 (C-3), 75.5 (C-4), 69.2 (C-2), 67.1 (OCH2), 64.3 (C-5), 50.7 (CH2N3), 27.7 (isopropylidene-CH3), 25.9 (isopropylidene-CH3), 16.3 (C-CH3).
HRMS calcd for C11H19N3O5Na [M + Na]+: 296.1222; found: 296.1225.
:1) showed complete conversion of the starting material to a faster-moving spot. Excess NaH was quenched with MeOH (5 mL) and the solvent was evaporated under reduced pressure. The residue was diluted with EtOAc and washed with brine solution (3 × 30 mL). The organic layer was collected, dried over anhydrous Na2SO4, and filtered and the solvent was evaporated in vacuo. The crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (4:1) to furnish the pure compound 12 (3.5 g, 87%) as a colourless syrup.
[α]25D: −57 (c 0.7, CHCl3).
IR (cm−1, CHCl3) ν: 3068, 2927, 2108, 1513, 1151, 1073, 742.
1H NMR (400 MHz, CDCl3) δ: 7.40–7.27 (m, 5H, ArH), 4.84 (d, 1H, J 12.4 Hz, CH2Ph), 4.77 (d, 1H, J1,2 3.2 Hz, H-1), 4.73 (d, 1H, J 12.4 Hz, CH2Ph), 4.37 (dd, 1H, J2,3 7.6 Hz, J3,4 5.6 Hz, H-3), 4.23–4.17 (m, 1H, H-5), 4.09 (dd, 1H, J3,4 5.6 Hz, J4,5 2.8 Hz, H-4), 3.89–3.83 (m, 1H, OCH2a), 3.61–3.54 (m, 3H, OCH2b, CH2aN3, H-2), 3.37–3.31 (m, 1H, CH2bN3), 1.44 (s, 3H, isopropylidene-CH3), 1.37 (s, 3H, isopropylidene-CH3), 1.35 (d, 3H, J5,6 6.8 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 138.4, 128.4, 127.9, 127.7, 108.8 (isopropylidene-C), 97.5 (C-1), 76.2 (C-2), 76.1 (C-4), 75.7 (C-3), 72.5 (CH2Ph), 67.0 (OCH2), 63.6 (C-5), 50.6 (CH2N3), 28.2 (isopropylidene-CH3), 26.3 (isopropylidene-CH3), 16.3 (C-CH3).
HRMS calcd for C18H25N3O5Na [M + Na]+: 386.1692; found: 386.1688.
:2) showed complete conversion of the starting material to a slower-moving spot. The solvents were evaporated in vacuo and co-evaporated with toluene to ensure complete removal of AcOH. The crude product thus obtained was purified by column chromatography using n-hexane–EtOAc (1:4) to afford the pure compound 13 (2.8 g, 91%) as a colourless syrup.
[α]25D: −18 (c 0.7, CHCl3).
IR (cm−1, CHCl3) ν: 3430, 2918, 2104, 1090, 1040, 742.
1H NMR (400 MHz, CDCl3) δ: 7.39–7.31 (m, 5H, ArH), 4.81 (d, 1H, J1,2 3.6 Hz, H-1), 4.67 (s, 2H, CH2Ph), 4.05–3.98 (m, 2H, H-5, H-3), 3.85 (m, 1H, H-4), 3.83–3.79 (m, 1H, OCH2a), 3.73 (dd, 1H, J1,2 3.6 Hz, J2,3 9.6 Hz, H-2), 3.53–3.47 (m, 2H, OCH2b, CH2aN3), 3.39–3.34 (m, 1H, CH2bN3), 2.55 (d, 1H, J 2.8 Hz, OH), 2.37 (bs, 1H, OH), 1.30 (d, 3H, J5,6 6.8 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 138.2, 128.7, 128.2, 97.1 (C-1), 76.5 (C-2), 72.9 (CH2Ph), 71.6 (C-4), 69.3 (C-5), 66.9 (OCH2), 65.9 (C-3), 50.8 (CH2N3), 16.2 (C-CH3).
HRMS calcd for C15H21N3O5Na [M + Na]+: 346.1379; found: 346.1383.
:1) showed complete conversion of the starting material to a faster running spot. The reaction mixture was then quenched with 3% aqueous HCl (5 mL) and extracted with ethyl acetate (2 × 15 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered, and the solvents were evaporated in vacuo and purified by column chromatography using n-hexane–EtOAc (1:1) to furnish the pure compound 14 (486.0 mg, 81%) as a white amorphous mass.
[α]25D: −72 (c 0.7, CHCl3).
IR (cm−1, CHCl3) ν: 3476, 2918, 2102, 1752, 1158, 1040, 752.
1H NMR (400 MHz, CDCl3) δ: 7.35–7.25 (m, 5H, ArH), 5.25 (dd, 1H, J2,3 10.4 Hz, J3,4 2.8 Hz, H-3), 4.78 (d, 1H, J1,2 3.6 Hz, H-1), 4.65 (d, 1H, J 12.4 Hz, CH2Ph), 4.60 (d, 1H, J 12.4 Hz, CH2Ph), 4.10–4.03 (m, 2H, H-5, CH2aCl), 3.99 (d, 1H, J 14.8 Hz, CH2bCl), 3.94–3.89 (m, 2H, H-4, H-2), 3.81–3.75 (m, 1H, OCH2a), 3.58–3.52 (m, 1H, OCH2b), 3.51–3.44 (m, 1H, CH2aN3), 3.42–3.35 (m, 1H, CH2bN3), 2.81 (bs, 1H, OH), 1.22 (d, 3H, J5,6 6.8 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 166.8 (COCH2Cl), 138.1, 128.5, 128.0, 127.9, 97.5 (C-1), 74.5 (C-3), 73.3 (C-4), 73.1 (CH2Ph), 70.3 (C-2), 66.9 (OCH2), 65.7 (C-5), 50.6 (CH2N3), 40.9 (CH2Cl), 16.0 (C-CH3).
HRMS calcd for C17H22ClN3O6Na [M + Na]+: 422.1095; found: 422.1092.
:1), the reaction mixture was diluted with EtOAc and washed with brine solution (2 × 30 mL). The organic layer was then separated and dried over anhydrous Na2SO4, and the solvent was evaporated in vacuo and the crude product was purified by column chromatography using n-hexane–EtOAc (3:2) to provide the pure compound 18 (387.0 mg, 71%) as a yellowish syrup.
[α]25D: −81 (c 0.7, CHCl3).
IR (cm−1, CHCl3) ν: 3323, 2915, 2101, 1691, 1540, 1270, 1058, 743.
1H NMR (400 MHz, CDCl3) δ: 7.39–7.26 (m, 5H, ArH), 6.00–5.90 (m, 1H, AllCH), 5.32 (dd, 1H, Jgem 1.6 Hz, Jtrans 17.2 Hz, AllCH2a), 5.2 (dd, 1H, Jgem 1.6 Hz, Jcis 10.4 Hz, AllCH2b), 4.8 (d, 1H, J 12.0 Hz, CH2Ph), 4.75 (d, 1H, J1,2 2.8 Hz, H-1), 4.64 (d, 1H, J 12.0 Hz, CH2Ph), 4.28–4.18 (m, 2H, OCH2All), 3.98 (q, 1H, J5,6 6.8 Hz, H-5), 3.86 (d, 1H, J3,4 2.4 Hz, H-4), 3.84–3.77 (m, 3H, OCH2a, H-2, H-3), 3.60–3.51 (m, 2H, OCH2b, CH2aN3), 3.37–3.30 (m, 1H, CH2bN3), 2.58 (bs, 1H, OH), 1.29 (d, 3H, J5,6 6.8 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 138.4, 134.6 (AllCH), 128.2, 127.7, 127.5, 117.1 (AllCH2), 97.7 (C-1), 77.1 (C-2), 75.1 (C-3), 73.2 (CH2Ph), 71.3 (OCH2All), 70.0 (C-4), 66.6 (OCH2), 65.5 (C-5), 50.5 (CH2N3), 16.0 (C-CH3).
HRMS calcd for C18H25N3O5Na [M + Na]+: 386.1692; found: 386.1688.
:2) showed complete consumption of the acceptor. The mixture was filtered through Celite® and the filtrate was successively washed with Na2S2O3 solution (2 × 15 mL), saturated NaHCO3 solution (20 mL) and brine solution (20 mL). The organic layer was separated, dried over anhydrous Na2SO4, filtered and evaporated in vacuo. The yellowish syrup thus obtained was purified by column chromatography using n-hexane–EtOAc (2:1) to furnish the pure disaccharide 19 (485.0 mg, 81%) as a colourless syrup.
[α]25D: +121 (c 0.8, CHCl3).
IR (cm−1, CHCl3) ν: 2936, 2103, 1740, 1638, 1369, 1218, 1086, 1037, 733.
1H NMR (400 MHz, CDCl3) δ: 7.39–7.25 (m, 5H, ArH), 6.0–5.90 (m, 1H, AllCH), 5.32 (dd, 1H, Jgem 1.2 Hz, Jtrans 17.2 Hz, AllCH2a), 5.17 (dd, 1H, Jgem 1.2 Hz, Jcis 10.4 Hz, AllCH2b), 5.09 (t, 1H, J2′,3′, J3′,4′ 6.4 Hz, H-3′), 5.01 (dd, 1H, J1′,2′ 6.8 Hz, J2′,3′ 6.4 Hz, H-2′), 4.95–4.90 (m, 1H, H-4′), 4.87 (d, 1H, J 12.8 Hz, CH2Ph), 4.74 (d, 1H, J1,2 2.0 Hz, H-1), 4.68 (d, 1H, J1′,2′ 6.8 Hz, H-1′), 4.66 (d, 1H, J 12.8 Hz, CH2Ph), 4.40 (dd, 1H, Jgem 12.4 Hz, J4′,5′ 4.0 Hz, H-5′a), 4.23 (m, 1H, OCH2aAll), 4.12 (m, 1H, OCH2bAll), 3.96 (q, 1H, J5,6 6.8 Hz, H-5), 3.87 (s, 1H, H-4), 3.83–3.77 (m, 3H, H-3, H-2, OCH2a), 3.64–3.53 (m, 2H, OCH2b, CH2aN3), 3.43 (dd, 1H, Jgem 12.4 Hz, J4′,5′ 6.4 Hz, H-5′b), 3.35–3.30 (m, 1H, CH2bN3), 2.09 (s, 3H, COCH3), 2.08 (s, 3H, COCH3), 1.95 (s, 3H, COCH3), 1.23 (d, 3H, J5,6 6.8 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 169.9 (COCH3), 169.8 (COCH3), 169.3 (COCH3), 138.8, 134.9 (AllCH), 128.2, 127.6, 127.5, 116.7 (AllCH2), 100.5 (C-1′), 98.2 (C-1), 78.2 (C-4), 76.9 (C-3), 75.0 (C-2), 73.5 (CH2Ph), 71.1 (OCH2All), 69.7 (C-3′), 69.6 (C-2′), 68.4 (C-4′), 66.8 (OCH2), 66.2 (C-5), 61.0 (C-5′), 50.6 (CH2N3), 20.8 (COCH3), 20.7 (COCH3), 20.6 (COCH3), 16.3 (C-CH3).
HRMS calcd for C29H39N3O12Na [M + Na]+: 644.2431; found: 644.2433.
:2) showed complete conversion of the starting material to a slower running spot. The solvent was removed under reduced pressure and the crude product was immediately charged into a column of silica gel and eluted with n-hexane–EtOAc (2:3) to afford the pure compound 20 (325.0 mg, 77%) as a colourless syrup.
[α]25D: +73 (c 0.8, CHCl3).
IR (cm−1, CHCl3) ν: 3342, 2105, 1742, 1373, 1216, 1082, 1040, 753.
1H NMR (400 MHz, CDCl3) δ: 7.38–7.23 (m, 5H, ArH), 5.18 (t, 1H, J2′,3′, J3′,4′ 9.2 Hz, H-3′), 5.04–4.96 (m, 2H, H-2′, H-4′), 4.83 (d, 1H, J 12.0 Hz, CH2Ph), 4.68 (d, 1H, J1,2 3.6 Hz, H-1), 4.60 (d, 1H, J 12.0 Hz, CH2Ph), 4.49 (d, 1H, J1′,2′ 7.6 Hz, H-1′), 4.15 (dd, 1H, Jgem 11.6 Hz, J4′,5′ 5.2 Hz, H-5′a), 4.03–3.96 (m, 2H, H-5, H-3), 3.76 (d,1H, J4,5 2.8 Hz, H-4), 3.73–3.68 (m, 1H, OCH2a), 3.57 (dd, 1H, J1,2 3.6 Hz, J2,3 10.0 Hz, H-2), 3.51–3.44 (m, 2H, OCH2b, CH2aN3), 3.36–3.25 (m, 2H, CH2bN3, H-5′b), 3.21 (d, J 8.8 Hz, OH), 2.02 (s, 6H, 2 × COCH3), 2.01 (s, 3H, COCH3), 1.17 (d, 3H, J5,6 6.8 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 170.1 (COCH3), 169.6 (COCH3), 169.1 (COCH3), 138.6, 128.2, 127.8, 127.6, 102.2 (C-1′), 97.8 (C-1), 83.5 (C-4), 76.9 (C-2), 73.3 (CH2Ph), 71.9 (C-3′), 71.4 (C-2′), 68.5 (C-4′, C-3), 66.8 (OCH2), 65.7 (C-5), 62.5 (C-5′), 50.6 (CH2N3), 20.6 (COCH3), 20.5 (2 × COCH3), 16.0 (C-CH3).
HRMS calcd for C26H35N3O12Na [M + Na]+: 604.2118; found: 604.2114.
:3) confirmed complete consumption of the acceptor. The mixture was filtered through Celite® and the filtrate was successively washed with Na2S2O3 solution (15 mL × 2), saturated NaHCO3 solution (30 mL) and brine solution (30 mL). The organic layer was separated, dried over anhydrous Na2SO4, filtered and evaporated in vacuo. The crude product thus obtained was purified by column chromatography using n-hexane–EtOAc (2:3) to afford the pure trisaccharide 21 (355.0 mg, 83%) as a yellowish syrup.
[α]25D: +135 (c 0.9, CHCl3).
IR (cm−1, CHCl3) ν: 2872, 2101, 1744, 1371, 1216, 1082, 1041, 746.
1H NMR (400 MHz, CDCl3) δ: 7.36–7.28 (m, 5H, ArH), 5.48 (s, 1H, H-2′′), 5.24–5.17 (m, 2H, H-3′, H-1′′), 5.11 (t, 1H, J1′,2′, J2′,3′ 8.0 Hz, H-2′), 5.0–4.9 (m, 2H, H-4′, H-4′′), 4.78 (d, 1H, J 12.0 Hz, CH2Ph), 4.66 (d, 1H, J1,2 2.8 Hz, H-1), 4.62–4.53 (m, 2H, CH2Ph, H-1′), 4.34 (dd, 1H, Jgem 11.2 Hz, J4′,5′ 4.4 Hz, H-5′a), 4.08 (bd, 1H, J2,3 10.0 Hz, H-3), 4.04–3.92 (m, 2H, H-5, H-5′′), 3.82 (d, 1H, J1,2 2.8 Hz, J2,3 10.0 Hz, H-2), 3.8–3.7 (m, 3H, H-4, OCH2a, H-3′′), 3.64–3.48 (m, 2H, OCH2b, CH2aN3), 3.43 (s, 3H, OCH3), 3.39–3.29 (m, 2H, CH2bN3, H-5′b), 2.11 (s, 3H, COCH3), 2.09 (s, 3H, COCH3), 2.07 (s, 6H, 2 × COCH3), 2.05 (s, 3H, COCH3), 1.24 (d, 3H, J5,6 6.4 Hz, C-CH3) 1.18 (d, 3H, J5′′,6′′ 6.0 Hz, C-CH3).
13C NMR (100 MHz, CDCl3) δ: 170.1 (COCH3), 170.0 (COCH3), 169.9 (COCH3), 169.8 (COCH3), 169.4 (COCH3), 138.3, 128.4, 128.0, 127.8, 101.4 (C-1′), 98.9 (C-1′′), 97.5 (C-1), 79.8 (C-4), 77.2 (C-3′′), 76.5 (C-2), 73.2 (CH2Ph), 73.0 (C-4′), 72.3 (C-3), 71.2 (C-3′), 71.1 (C-2′), 69.2 (C-4′′), 68.1 (C-2′′), 66.9 (OCH2), 66.6 (C-5), 66.2 (C-5′′), 61.6 (C-5′), 57.9 (OCH3), 50.6 (CH2N3), 21.0 (COCH3), 20.9 (COCH3), 20.8 (COCH3), 20.7 (2 × COCH3), 17.5 (C-CH3), 16.2 (C-CH3).
HRMS calcd for C37H51N3O18Na [M + Na]+: 848.3065; found: 848.3061.
[α]25D: + 98 (c 0.7, CHCl3).
IR (cm−1, CHCl3) ν: 3432, 2921, 2105, 1092, 1037, 743.
1H NMR (500 MHz, CD3OD) δ: 7.42–7.28 (m, 5H, ArH), 5.13 (s, 1H, H-1′′), 4.81 (d, 1H, J1,2 3.5 Hz, H-1), 4.70 (d, 1H, J 12.0 Hz, CH2Ph), 4.64 (d, 1H, J 12.0 Hz, CH2Ph), 4.31 (d, 1H, J1′,2′ 6.5 Hz, H-1′), 4.11–4.06 (m, 4H, H-3, H-2′′, H-5′a, H-5), 3.95–3.88 (m, 3H, H-4′′, H-2, H-5′′), 3.82–3.78 (m, 1H, OCH2a), 3.56–3.45 (m, 8H, H-4′, H-4, H-3′, OCH2b, CH2aN3, OCH3), 3.42–3.37 (m, 1H, CH2bN3), 3.37–3.29 (m, 2H, H-2′, H-3′′), 3.15 (t, 1H, Jgem, J4′,5′ 11.0 Hz, H-5′b), 1.32 (d, 3H, J5,6 6.5 Hz, C-CH3), 1.24 (d, 3H, J5′′,6′′ 6.0 Hz, C-CH3).
13C NMR (125 MHz, CD3OD) δ: 138.5, 128.1, 127.8, 127.5 (ArC), 104.8 (C-1′), 101.5 (C-1′′), 97.4 (C-1), 80.3 (C-4), 79.4 (C-2), 76.5 (C-4′′), 76.4 (C-3′′), 74.1 (C-2′), 72.4 (C-3), 72.3 (CH2Ph), 71.5 (C-4′), 69.9 (C-3′), 68.8 (C-5′′), 67.1 (C-2′′), 66.9 (OCH2), 66.8 (C-5), 65.4 (C-5′), 56.1 (OCH3), 50.4 (CH2N3), 16.6 (C-CH3), 15.2 (C-CH3).
HRMS calcd for C27H41N3O13Na [M + Na]+: 638.2537; found: 638.2532.
[α]25D: +102 (c 0.4, CH3OH).
1H NMR (400 MHz, CD3OD) δ: 5.08 (s, 1H, H-1′′), 4.91 (s, 1H, H-1), 4.32 (d, 1H, J1′,2′ 7.2 Hz, H-1′), 4.20 (s, 1H, H-2′′), 4.11–4.01 (m, 3H, H-3, H-5, H-5′a), 3.99–3.93 (m, 3H, H-5′′, OCH2a, H-4′′), 3.89 (m, 1H, H-2), 3.80–3.74 (m, 1H, OCH2b), 3.50–3.46 (m, 6H, H-4′, H-4, H-3′, OCH3), 3.34–3.28 (m, 4H, H-3′′, CH2N, H-2′), 3.16 (t, 1H, Jgem, J4′,5′ 10.8 Hz, H-5′b), 1.36 (d, 3H, J5,6 6.4 Hz, C-CH3), 1.26 (d, 3H, J5′′,6′′ 6.0 Hz, C-CH3).
13C NMR (100 MHz, CD3OD) δ: 104.3 (C-1′), 101.6 (C-1′′), 98.6 (C-1), 79.9 (C-4), 79.3 (C-2), 75.7 (C-3′′), 73.6 (2C, C-2′, C-4′′), 70.8 (C-4′), 69.4 (C-3′), 69.0 (C-5′′), 68.5 (C-3), 67.5 (C-5), 65.5 (C-2′′), 64.9 (C-5′), 62.5 (OCH2), 56.0 (2C, OCH3, CH2N), 16.6 (C-CH3), 15.0 (C-CH3).
HRMS calcd for C20H37NO13Na [M + Na]+: 522.2163; found: 522.2157.
Author contributions
SM performed the experiments and collected analytical data. BM and SM contributed equally towards the analysis of the data and preparation of the manuscript and its associated content.Data availability
Necessary data are available in the ESI† associated with the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SM is thankful to DST INSPIRE for a Senior Research fellowship (SRF). The analytical facility of IISER Kolkata is gratefully acknowledged. This work is supported by the research grant CRG/2022/005805 from the Science and Engineering Research Board (SERB), New Delhi to BM.References
- G. Berg, Plant-microbe interactions promoting plant growth and health: perspectives for controlled use of microorganisms in agriculture, Appl. Microbiol. Biotechnol., 2009, 84, 11–18 Search PubMed.
- M. S. Zawoznik, M. Ameneiros, M. P. Benavides, S. Vazquez and M. D. Groppa, Response to saline stress and aquaporin expression in Azospirillum-inoculated barley seedlings, Appl. Microbiol. Biotechnol., 2011, 90, 1389–1397 CrossRef CAS PubMed.
- S. Fibach-Paldi, S. Burdman and Y. Okon, Key physiological properties contributing to rhizosphere adaptation and plant growth promotion abilities of Azospirillum brasilense, FEMS Microbiol. Lett., 2012, 326, 99–108 CrossRef CAS PubMed.
- O. Steenhoudt and J. Vanderleyden, Azospirillum, a free-living nitrogen-fixing bacterium closely associated with grasses: genetic, biochemical and ecological aspects, FEMS Microbiol. Rev., 2000, 24, 487–506 CrossRef CAS PubMed.
- I. V. Egorenkova, S. A. Konnova, V. N. Sachuk and V. V. Ignatov, Azospirillum brasilense colonisation of wheat roots and the role of lectin–carbohydrate interactions in bacterial adsorption and root-hair deformation, Plant Soil, 2001, 231, 275–282 CrossRef.
- A. S. Boyko, A. S. Dmitrenok, Y. P. Fedonenko, E. L. Zdorovenko, S. A. Konnova, Y. A. Knirel and V. V. Ignatov, Structural analysis of the O-polysaccharide of the lipopolysaccharide from Azospirillum brasilense Jm6B2 containing 3-O-methyl-D-rhamnose (D-acofriose), Carbohydr. Res., 2012, 355, 92–95 Search PubMed.
- A. Adak, M. Bera and B. Mukhopadhyay, Synthesis of the Hexasaccharide Related to the Exopolysaccharide from Lactobacillus mucosae VG1 through Regioselective Glycosylation, Org. Lett., 2023, 25, 4711–4714 CrossRef CAS PubMed.
- S. Maiti and B. Mukhopadhyay, Synthesis of the conjugation-ready β-mannosamine-containing O-antigen repeat from Vibrio cholerae, O14, Org. Biomol. Chem., 2025, 23, 1866–1873 RSC.
- S. Das, S. Maiti and B. Mukhopadhyay, Chemical synthesis of 6-deoxy-d-talose containing a tetrasaccharide repeating unit of the O-specific polysaccharide from Enterobacter cloacae, G3422 in the form of its 2-aminoethyl glycoside, Org. Biomol. Chem., 2024, 22, 2414–2422 RSC.
- C. M. Cairns, F. S. Michael, M. Jamshidi, H. van Faassen, Q. Yang, K. A. Henry, G. Hussack, J. Sauvageau, E. V. Vinogradov and A. D. Cox, Structural Characterization and Evaluation of an Epitope at the Tip of the A-Band Rhamnan Polysaccharide of Pseudomonas aeruginosa, ACS Infect. Dis., 2022, 8, 1336–1346 CrossRef CAS PubMed.
- C. Qin, B. Schumann, X. Zou, C. L. Pereira, G. Tian, J. Hu, P. H. Seeberger and J. Yin, Total synthesis of a densely functionalized Plesiomonas shigelloides serotype 51 aminoglycoside trisaccharide antigen, J. Am. Chem. Soc., 2018, 140, 3120–3127 CrossRef CAS PubMed.
- B. Roy and B. Mukhopadhyay, Sulfuric acid immobilized on silica: an excellent catalyst for Fischer type glycosylation, Tetrahedron Lett., 2007, 48, 3783–3787 CrossRef CAS.
- P. Leon-Ruaud and D. Plusquellec, Synthesis of new surfactant glycosides via sugar halides. Methyl α-D-glucoside and methyl α-D-mannoside thioethers and sulfones, Tetrahedron, 1991, 47, 5185–5192 CrossRef CAS.
- C. L. Stevens, R. P. Glinski, K. G. Taylor, P. Blumbergs and F. Sirokman, New rearrangements of hexose 4- and 5-O-sulfonates, J. Am. Chem. Soc., 1966, 88, 2073–2074 CrossRef CAS.
- E. Kaufmann, H. Hattori, H. Miyatake-Ondozabal and K. Gademann, Total Synthesis of the Glycosylated Macrolide Antibiotic Fidaxomicin, Org. Lett., 2015, 17, 3514–3517 CrossRef CAS PubMed.
- L. Xia, R. B. Zheng and T. L. Lowary, Revisiting the Specificity of an α-(1→4)-Mannosyltransferase Involved in Mycobacterial Methylmannose Polysaccharide Biosynthesis, ChemBioChem, 2012, 13, 1139–1151 CrossRef CAS PubMed.
- M. P. Jamshidi, C. Cairns, N. H. Khieu, K. Chan, F. St. Michael, A. Cox and J. Sauvageau, Optimization of the Synthesis and Conjugation of the Methyl Rhamnan Tip of Pseudomonas aeruginosa A–B and Polysaccharide and Immunogenicity Evaluation for the Continued Development of a Potential Glycoconjugate Vaccine, ACS Infect. Dis., 2024, 10, 1361–1369 CrossRef CAS PubMed.
- M. P. Jamshidi, C. Cairns, S. Chong, F. St. Michael, E. V. Vinogradov, A. D. Cox and J. Sauvageau, Synthesis and Immunogenicity of a Methyl Rhamnan Pentasaccharide Conjugate from Pseudomonas aeruginosa A–B and Polysaccharide, ACS Infect. Dis., 2022, 8, 1347–1355 CrossRef CAS PubMed.
- J. S. Brimacombe, N. Robinson and J. M. Webber, Syntheses of 6-Deoxy-3-O-methyl-D-gulose and 6-Deoxy-3-O-methyl-L-mannose (L-Acofriose), J. Chem. Soc. C, 1971, 613–618 RSC.
- D. Budhadev and B. Mukhopadhyay, Chemical synthesis of a tetrasaccharide related to the exocellular polysaccharide from Rhodococcus sp. RHA1, Carbohydr. Res., 2014, 394, 26–31 CrossRef CAS PubMed.
- A. Kafle, J. Liu and L. Cui, Controlling the stereoselectivity of glycosylation via solvent effects, Can. J. Chem., 2016, 94, 894–901 CrossRef CAS.
- S.-R. Lu, Y.-H. Lai, J.-H. Chen, C.-Y. Liu and K.-K. T. Mong, Dimethyl formamide: an unusual glycosylation modulator, Angew. Chem., Int. Ed., 2011, 50, 7315–7320 CrossRef CAS PubMed.
- K. Boubbou, D. C. Zhu, C. Vasileiou, B. Borhan, D. Prosperi, W. Li and X. Huang, Magnetic Glyco-Nanoparticles: A tool to Detect, differentiate, and unlock the Glyco-Codes of cancer via magnetic resonance imaging, J. Am. Chem. Soc., 2010, 132, 4490–4499 CrossRef PubMed.
- B. Thollas and C. Biosset, Total Synthesis of Floridoside, Synlett, 2007, 1736–1738 CrossRef CAS.
- P. A. Gent and R. Gigg, The allyl ether as a protecting group in chemistry. Part V. Preparation of benzyl ethers of carbohydrates for use in oligosaccharide synthesis, J. Chem. Soc., Perkin Trans. 1, 1974, 1446–1455 RSC.
- Y. Demizu, Y. Kubo, H. Miyoshi, T. Maki, Y. Matsumura, N. Moriyama and O. Onomura, Regioselective protection of sugars catalyzed by Dimethyltin Dichloride, Org. Lett., 2008, 10, 5075–5077 Search PubMed.
- A. K. Mishra, B. K. Ghotekar, C. K. Puley and S. S. Kulkarni, Expanding the scope of a One-pot double displacement protocol to access the all-rare-sugar-containing trisaccharide unit of Pseudomonas stutzeri OX1, Org. Lett., 2024, 26, 9955–9960 Search PubMed.
- B. Mukhopadhyay, K. P. R. Kartha, D. A. Russell and R. A. Field, Streamlined synthesis of per-O-acetylated sugars, glycosyl iodides, or thioglycosides from unprotected reducing sugars, J. Org. Chem., 2004, 69, 7758–7760 CrossRef CAS PubMed.
- G. H. Veeneman, S. H. van Leeuwen and J. H. van Boom, Iodonium ion promoted reactions at the anomeric centre. II An efficient thioglycoside mediated approach toward the formation of 1,2-trans linked glycosides and glycosidic esters, Tetrahedron Lett., 1990, 31, 1331–1334 Search PubMed.
- G. Gu, Y. Du and R. J. Linhardt, Facile synthesis of saponins containing 2,3-branched oligosaccharides by using partially protected glycosyl donors, J. Org. Chem., 2004, 69, 5497–5500 CrossRef CAS PubMed.
- X. Zhang, G. Gu and Z. Guo, Synthesis of a trisaccharide repeating unit of the O-antigen from Burkholderia multivorans and Its oligomers, Eur. J. Org. Chem., 2015, 7075–7085 CrossRef CAS.
- B. Mukhopadhyay and N. Roy, Synthesis of the pentasaccharide related to the repeating unit of the antigen from Shigella dysenteriae, type-4 In the form of Its methyl ester 2-(trimethylsilyl)ethyl glycoside, Carbohydr. Res., 2003, 338, 589–596 CrossRef CAS PubMed.
- G. Zemplén and A. Kunz, Studien über Amygdalin, IV: Synthese des natürlichen l–Amygdalins, Ber. Deutsch. Chem. Ges., 1924, 57, 1357–1359 CrossRef.
- V. S. Rao and A. S. Perlin, Removal of O-benzyl protecting-groups of carbohydrate derivatives by catalytic, transfer hydrogenation, Carbohydr. Res., 1980, 83, 175–177 CrossRef CAS.
- D. D. Perrin, W. L. Amarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon, London, 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the NMR spectra of all new compounds. See DOI: https://doi.org/10.1039/d5ob00730e |
| This journal is © The Royal Society of Chemistry 2025 |