
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the bioorthogonal chemistry toolbox: innovative synthetic strategies for cyclooctynes
Andrea
Diana
a,
Giuseppina I.
Truglio
 b,
Elena
Petricci
b and
Daniela
Lanari
*a
b,
Elena
Petricci
b and
Daniela
Lanari
*a
aDepartment of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, 06123, Perugia, Italy. E-mail: daniela.lanari@unipg.it
bDepartment of Biotechnology, Chemistry and Pharmacy, University of Siena, Via Aldo Moro 2, 53100, Siena, Italy
First published on 21st May 2025
Abstract
Cyclooctyne derivatives represent an important class of compounds that play a key role in bioorthogonal chemistry. The presence of an endocyclic triple bond endows these molecules with the necessary reactivity for strain-promoted azide–alkyne cycloaddition (SPAAC); however, stability issues may hamper their use in biological systems. Many research groups, with the aid of computational studies, are devoting their efforts to finding ideal cyclooctyne candidates that strike a delicate balance between reactivity and stability. In this context, providing reliable and general synthetic procedures for accessing such chemical scaffolds is of critical importance. This review covers the recent synthetic strategies found in the literature to achieve this goal. Specifically, six main methodologies are discussed, highlighting the synthetic pathways, the key precursors for each, the applicability to a wide range of cyclooctyne derivatives and the challenges encountered in fulfilling this target.
1. Introduction
Since their first appearance in a paper by Blomquist1 in 1953, cyclooctynes have long been considered curiosity-driven molecules, with synthetic methodologies for their formation seen merely as exercises to test chemists’ ability to synthesize highly reactive and strained molecular structures. However, at the beginning of the new century, interest in the unique reactivity profile of this class of compounds surged as they were recognized as valuable moieties in different fields of chemistry for a variety of specialized applications. Their most remarkable use is found in the field of bioorthogonal chemistry,2–4 where they participate in the strain-promoted azide–alkyne cycloaddition (SPAAC) reaction.5 This transformation avoids the use of metal catalysts and efficiently occurs under physiological conditions, making cyclooctynes exceptionally suitable for in vivo and in situ applications. Specifically, cyclooctynes enable the precise labelling of biomolecules with fluorophores or other spectroscopic probes, facilitating dynamic studies of biological processes without perturbing cellular environments.6,7 The success of cyclooctynes is strictly related to their reactivity, which directly depends on the ideal ring size that provides the necessary balance between stability and reactivity. Smaller cycloalkynes usually exhibit high ring strain due to a strong deviation of the triple bond angle from linearity (180°), making them challenging to synthesize and store. In contrast, larger ones, in which the triple bond angle resembles that of the linear alkynes, lack sufficient reactivity under these conditions.8,9 Thus, cyclooctynes, featuring a triple bond angle of 155°,10 just slightly deviating from linearity (180°), stand out as the optimal choice for these important chemical transformations.Early methodologies for the synthesis of cycloalkynes date back to the 1970s, including the classical protocol for introducing a triple bond, i.e. the two-step procedure involving bromination of an alkene followed by elimination, and also the electrocyclic ring opening of 1,1-dibromo derivatives followed by the capture of the corresponding trans-allylic cation to yield the vinyl bromide intermediate, which is finally converted to the desired cyclooctyne under basic conditions.
Such methodologies were applied to the synthesis of several cyclooctynes;11–16 in particular, the bromination/elimination strategy proved to be very efficient for the synthesis of dibenzoazacyclooctyne (DBCO) on a gram scale, without the aid of column chromatography for the purification steps.17 Even though this methodology allowed access to the cyclooctyne moiety, it appeared to be inadequate for exploiting a wider variety of such strained molecules. Computational studies in fact, along with the first data on kinetic studies, called for the synthesis of new and improved cyclooctynes with the right lipophilicity profile (as these molecules are primarily intended for in vivo use) and the introduction of different heteroatoms, which could be useful either for electronic effects or for further functionalization. It was clear that the synthetic aspect could not dictate a limitation for the construction of novel and promising cyclooctynes. Therefore, a series of new methodologies have been developed over time with the aim of effectively tackling the most diverse classes of cyclooctynes. This review will illustrate such synthetic procedures, highlighting the scope, the key steps and the strengths and weaknesses. Bromination/elimination reactions and ring opening reactions of 1,1-dibromo derivatives will not be discussed here, as these methodologies were already known before the advent of SPAAC in chemical biology. The main procedures discussed in this review for the synthesis of cyclooctyne derivatives are reported in Fig. 1 in the order of chronological appearance of the first publication.
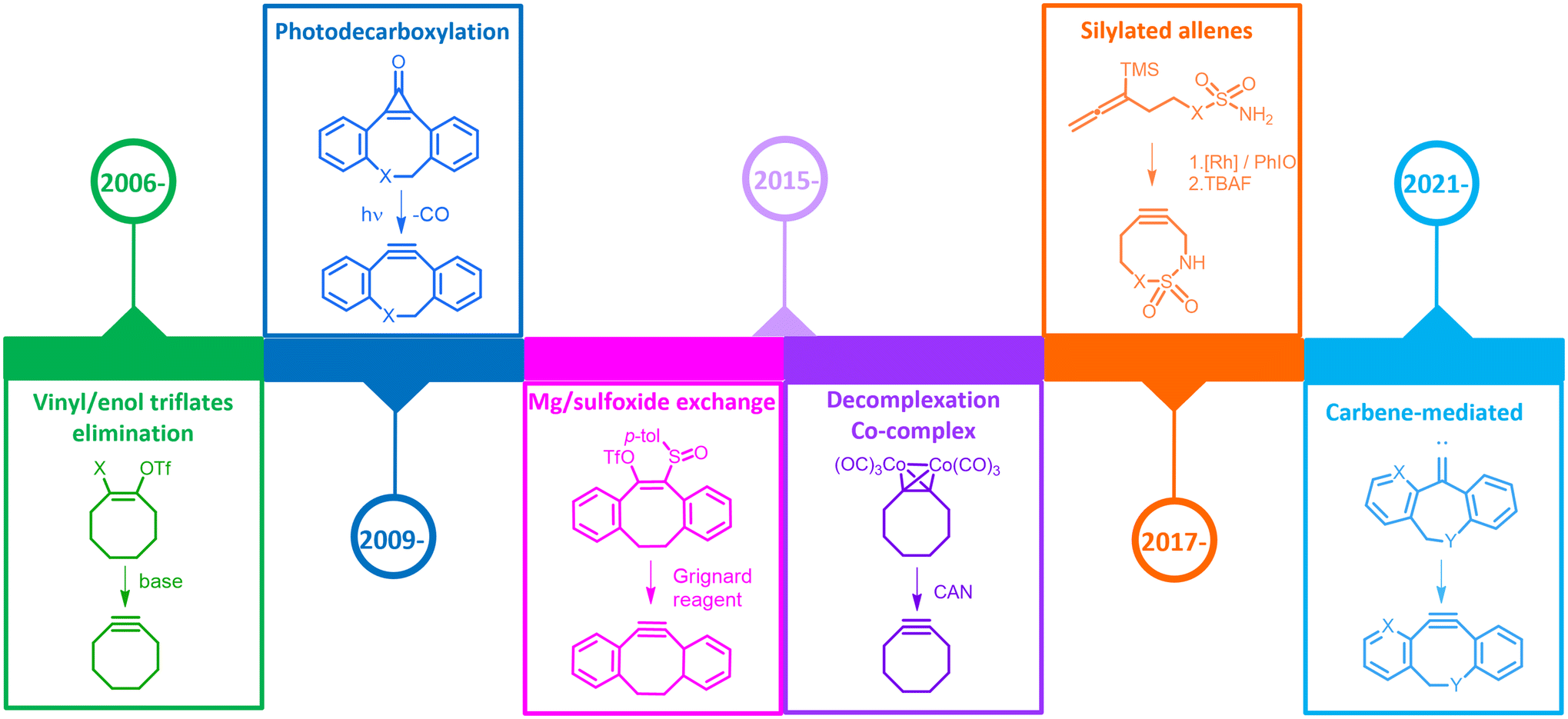 | ||
| Fig. 1 Timeline of synthetic methodologies applied to cyclooctyne synthesis. | ||
2. Synthetic methodologies
2.1. Vinyl/enol triflate elimination
Over the years, Bertozzi and co-workers have made a fundamental contribution to the development of bioorthogonal chemistry. For the synthesis of the cycloalkyne moiety, they initially employed the 1,1-dibromo derivative strategy.15,16 However, this approach was not adequate for the synthesis of peculiar cyclooctynes, such as dibenzo-substituted substrates or cyclooctynes bearing an electron-withdrawing moiety at the propargylic position. The urgent need for such molecules arises from their outstanding reactivities toward bioorthogonal reactions in various biological applications.Therefore, the introduction of a synthetic methodology that was feasible, scalable and modular was urgent. In order to access fluorine-substituted cyclooctynes with promising electronic and reactivity features, the authors explored the chemistry of vinyl triflates. Stang and co-workers were the first to synthesize 3,3-dimethyl-1-buten-2-yl triflate to yield, in the presence of pyridine, the corresponding tert-butylacetylene.18 Later, other examples on the use of vinyl triflates appeared in the literature.19–21 This methodology was then applied to the synthesis of cyclooctynes by Bertozzi. Specifically, the key steps in this protocol involved the formation of vinyl triflates from the corresponding substituted cyclooctanones in the presence of potassium bis(trimethylsilyl)amide (KHMDS) and a triflate source, and the final step was a base-mediated elimination reaction (Scheme 1).
 | ||
| Scheme 1 Synthesis of MOFO using the vinyl triflate elimination strategy. | ||
The first cyclooctyne synthesized using this strategy was a monofluorinated cyclooctyne (MOFO),16 in which a fluorine atom was introduced at the propargylic position. This substitution was performed with the aim of lowering the LUMO level and enhancing the interaction with the HOMO of the azide partner,22 as recently elucidated by density functional theory calculations.23 Deprotonation at −78 °C with lithium diisopropylamide (LDA) at the 2-position of 2-fluorocyclooctan-1-one24 (1), followed by alkylation with methyl 4-(bromomethyl)benzoate, resulted in an overall yield of 43% and yielded the intermediate fluoroketone 2. The corresponding vinyl triflate 3 was obtained (73% yield) after deprotonation mediated by KHMDS and triflation with N-phenyl-bis(trifluoromethanesulfonimide) (Tf2NPh). The final step was performed using LDA as a base at 0 °C in THF, followed by hydrolysis with LiOH in dioxane/water at 50 °C to yield the desired cyclooctyne MOFO in 58% yield.
A structural evolution of MOFO is the difluorinated cyclooctyne named DIFO,25 in which an electron-withdrawing moiety, i.e., the difluoride unit, is introduced at the propargylic position (Scheme 2). This moiety, as similarly described for MOFO, increases the reactivity toward the azide in SPAAC reactions; furthermore, it is easy to install and has the advantage of being inert toward biological systems. The synthesis employed cis-1,5-cyclooctanediol (4) as the starting material, which was first O-monoalkylated with allyl bromide, and then the remaining hydroxyl group was oxidized with pyridinium chlorochromate (PCC). Subsequent treatment with lithium bis(trimethylsilyl)amide (LiHMDS) and chlorotriethylsilane (Et3SiCl) afforded the key intermediate Et3Si-enolate 5. Selectfluor™ was used to introduce the first fluorine unit to afford the corresponding diastereoisomeric mixture of fluoroketones 6a and 6b in a 2.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Epimerization in the presence of catalytic LiHMDS increased the amount of the desired stereoisomer 6b, which was transformed into the corresponding α-difluoroketone intermediate after treatment with KHMDS and Et3SiCl, followed by Selectfluor™. The terminal alkene was then converted into a carboxylic moiety with RuCl3 and NaIO4 to yield the desired intermediate 7. Treatment of 7 with KHMDS and Tf2NPh yielded the desired vinyl triflate 8 in 47% yield. Finally, the cyclooctyne DIFO was obtained by LDA-mediated elimination of intermediate 8.
:1 ratio. Epimerization in the presence of catalytic LiHMDS increased the amount of the desired stereoisomer 6b, which was transformed into the corresponding α-difluoroketone intermediate after treatment with KHMDS and Et3SiCl, followed by Selectfluor™. The terminal alkene was then converted into a carboxylic moiety with RuCl3 and NaIO4 to yield the desired intermediate 7. Treatment of 7 with KHMDS and Tf2NPh yielded the desired vinyl triflate 8 in 47% yield. Finally, the cyclooctyne DIFO was obtained by LDA-mediated elimination of intermediate 8.
 | ||
| Scheme 2 Synthesis of DIFO employing the vinyl triflate elimination strategy. | ||
DIFO cyclooctyne displays outstanding reactivity, labelling azides linked to biomolecules within minutes through the Cu-free [3 + 2] dipolar cycloaddition; however, the synthetic process to obtain this molecule is quite lengthy, with a very low overall yield of 1.4% from commercially available 4.
In search of alternative DIFO-like cyclooctynes with comparable reactivity but with a simpler synthetic pathway, Bertozzi and co-workers proposed a second generation of DIFO, namely DIFO2,26 which retains the same difluorinated moiety but features a benzyl chain instead of an alkoxy handle. This structural modification allowed for a more straightforward synthesis (Scheme 3).
 | ||
| Scheme 3 Synthesis of DIFO2. | ||
1,3-Cyclooctanedione (9) was fluorinated with Selectfluor™ and cesium carbonate (Cs2CO3) to give the intermediate 2,2-difluoro-1,3-cyclooctanedione 10 in 73% yield. To install the handle for further functionalization, a Wittig reaction between 10 and phosphonium salt 11 in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was performed, followed by double bond reduction with H2 over Pd/C to yield the corresponding intermediate 12. The usual step for vinyl triflate formation (i.e. treatment with KHMDS and Tf2NPh) was performed to obtain the desired compound 13 in 88% yield. The step to afford the triple bond was run using LDA as a base, and final hydrolysis of the ester moiety yielded the terminal carboxylic acid. The overall yield of DIFO2 was 36%, approximately 25 times higher than that obtained for DIFO, even though the starting material for this synthesis, the 1,3-cyclooctanedione, was not commercially available but obtained in 67% yield in two steps from diethyl pimelate.27 Furthermore, a similar synthetic procedure that employed intermediate 10 was also used to synthesize an analogue of DIFO2 without a phenyl moiety in the handle, in order to decrease the hydrophobicity that could lead to undesired nonspecific interactions.26
A successful alternative to the use of vinyl triflates was a strategy inspired by the synthesis of cyclohexyne reported by Guitan and co-workers, which envisioned the formation of the parent enol triflate compounds.28 Such intermediates were obtained from the corresponding α-silyl ketones, which were in turn converted to cyclooctynes by treatment with cesium fluoride (CsF).
Bertozzi and co-workers employed the latter strategy for their research, as they were interested in establishing a modular protocol for the synthesis of a biarylazacyclooctynone (BARAC)29 (Scheme 4). The synthesis of this new cyclooctyne was inspired by the 4-dibenzocyclooctynol (DIBO)30 introduced by Boons, which exhibits enhanced reactivity due to the increased strain energy of the ring. In the synthesis of BARAC, the authors aimed to enhance the reactivity of the cyclooctyne skeleton by introducing an additional degree of unsaturation through the amide moiety, which could also serve as a handle for further functionalization. The synthetic strategies introduced so far were not feasible for such a substrate as they were too long and low-yielding,31,32 so the authors developed a new procedure that involved the formation of an α-silyl ketone as a key intermediate, which was converted into the corresponding enol triflate and eventually transformed into the desired strained cyclooctyne.28
 | ||
| Scheme 4 BARAC synthesis using the enol triflate elimination strategy. | ||
Commercially available indole 14 was used as the starting material for this synthesis. This precursor is ideal, as a large variety of derivatives can also be synthesized using the Fisher indole synthesis, eventually leading to different BARAC substrates. N-Alkylation with allyl bromide was performed under phase transfer conditions using tributylammonium bromide (TBAB). Then the trimethylsilyl group was installed using trimethylsilylchloride (TMSCl) after deprotonation with n-BuLi to yield intermediate 15, with an overall yield of 81% from 14. Oxidation of indole 15 in the presence of meta-chloroperbenzoic acid (m-CPBA) led to ring opening and the formation of keto-amide 16. Enolate formation using KHMDS followed by Tf2O yielded the key intermediate enol triflate 17. Notably, the early formation of the carbon–silicon bond is critical for a positive outcome of this reaction, as the formation of α-silyl ketones under enolate forming conditions is very difficult due to the high affinity of the silicon atom for oxygen.33 The final functionalization of the terminal double bond was achieved using a nitrile oxide, which was prepared in situ from chlorooxime 18 upon treatment with triethylamine (TEA), affording intermediate 19 decorated with the desired handle for further conjugation reaction. The formation of the triple bond to yield BARAC (85% yield) was successfully achieved in only 30 min at room temperature using CsF. This synthetic pathway affords an overall yield of 36%, and BARAC is also stable during chromatographic purification as well as on a benchtop. Such features make BARAC an ideal candidate for bioconjugation reactions.
A similar strategy was employed by the same authors for the synthesis of dibenzofluorocyclooctyne (DIFBO),34 aiming to combine the beneficial effects of the electron-withdrawing moiety of the difluoro group (DIFO) on reactivity with the presence of the aromatic units (DIBO). Studies by Goddard's group, in fact, highlighted that, unlike DIBO, the presence of just one aryl moiety could enhance cycloaddition reactivity with benzyl azide, favoring the formation of at least one regioisomer.35 The synthetic pathway employed, as a starting material, the commercially available 1-benzosuberone 20, which, following a procedure described by Stavber and colleagues,36 was allowed to react with hexylamine and then fluorinated using Selectfluor™ to afford α-difluoroketone 21 in 60% yield. A Lewis acid-mediated homologation reaction using (trimethylsilyl)diazomethane (TMSCHN2) was used for the first time in this work on fluoroketones and successfully yielded the desired intermediate 22 in 95% yield. Treatment with KHMDS followed by Tf2O afforded enol triflate 23, which, according to the previous synthesis, easily yields the desired cyclooctyne DIFBO (Scheme 5). Unfortunately, this product was highly unstable and difficult to isolate, as it underwent spontaneous homotrimerization. However, its structure was confirmed through in situ trapping with benzyl azide, and stabilization was also achieved by complexation with β-cyclodextrins.
 | ||
| Scheme 5 DIFBO synthesis. | ||
As previously mentioned, DIFBO is a promising substrate for bioorthogonal reactions, but unfortunately stability issues hindered its use. Bertozzi and co-workers suggested that a promising structural modification to release part of the strain in the cyclooctyne moiety while maintaining high reactivity could be the insertion of a bulkier heteroatom into the 8-membered skeleton. Specifically, they investigated the installation of a sulfur atom in the cyclooctyne ring. The replacement of a carbon atom with sulfur resulted in a longer atom-to-atom length (C–C length is equal to 1.54 A, whilst that of C–S is 1.84 A), leading to a small but significant ring expansion. In order to test the effect of the sulfur atom, the authors synthesized two different molecules, namely thia-cyclooctyne (thia-OCT) and thia-dibenzofluorocyclooctyne (thia-DIFBO).37 The synthetic strategy that involved the formation of a vinyl triflate intermediate was also applied in this work (Scheme 6). Specifically, for the synthesis of thia-OCT, 1,6-dienyl-4-oxysilane 24 was monoepoxidized with dimethyldioxirane (DMDO), and the unreacted double bond underwent a thiol–ene reaction with thioacetic acid to yield intermediate 25. Intermolecular cyclization was induced through deprotection with NaH to give intermediate 26 in 72% yield. A two-step procedure involving the protection of the free hydroxyl group with pivaloyl chloride (PivCl) and the removal of the triisopropylsilyl ether group (TIPS) with tetrabutylammonium fluoride (TBAF) yielded the corresponding alcohol 27, which was oxidized using Dess–Martin periodinane to afford ketone 28. Finally, intermediate 28 yielded vinyl triflate 29 after treatment with sodium bis(trimethylsilyl)amide (NaHMDS) and Tf2O. LDA-mediated syn-elimination afforded the desired cyclooctyne thia-OCT in 53% yield.
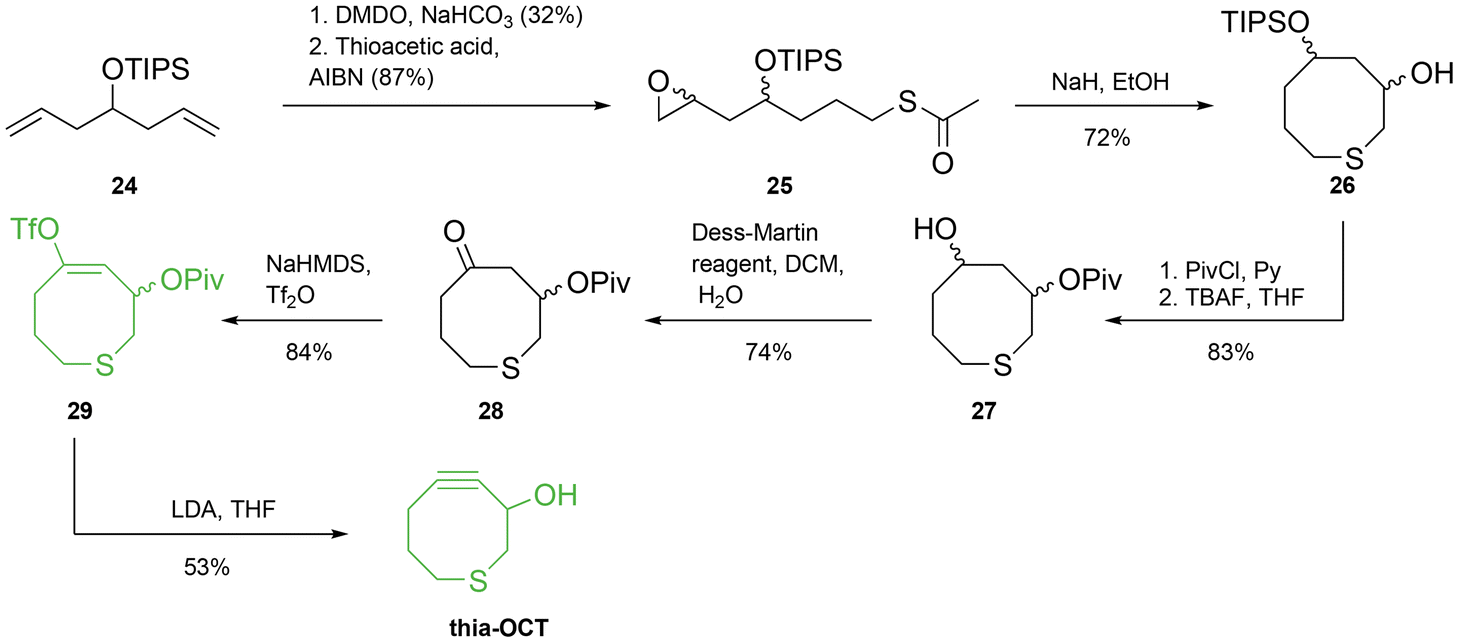 | ||
| Scheme 6 Thia-OCT synthesis employing the vinyl triflate elimination strategy. | ||
The second-order rate constant of the reaction between thia-OCT and benzyl azide was found to be one order of magnitude lower than that of OCT, indicating that the sulfur atom had indeed rendered the cycloalkyne more stable to be isolated, but at the same time it had also considerably reduced the reactivity of the new cyclooctyne. Next, the authors sought the right structural balance by synthesizing the sulfur analogue of DIFBO, i.e.thia-DIFBO, that featured a propargylic gem-difluoro group and a fused aryl ring, but in this case also, the rate constant for the cycloaddition reaction with benzyl azide was not satisfactory, resulting in a value twenty times lower than that obtained using DIFBO. Notably, the authors were able to find a balance between stability and reactivity only by employing a sulfur-containing seven-membered ring, namely 3,3,6,6,-tetramethylthiacycloheptyne (TMTH), previously synthesized by Krebs and co-workers.38 When TMTH was allowed to react with benzyl azide in CD3CN, a second-order rate constant of 4 M−1 s−1 was observed.
In conclusion, the vinyl enol/triflate strategy has been applied to the synthesis of a wide variety of cyclooctynes, ranging from dibenzosubstituted scaffolds like BARAC, mono- and difluorinated derivatives like MOFO and DIFO and heterocycle-containing targets such as thia-DIFBO. Some procedures rely on a lengthy synthetic pathway, and their optimization should be considered.
2.2. Photodecarboxylation of cyclopropenones
Popik and co-workers39 proposed an alternative strategy to synthesize dibenzocyclooctyne derivatives named DIBOs.30 This strategy involves their photochemical generation from the corresponding cyclopropenone derivatives. Previous studies by the same authors40,41 had already demonstrated that the photodecarboxylation of diaryl-substituted cyclopropenones was a very efficient process that yielded the corresponding alkynes quantitatively. Inspired by these results, they extended this strategy to the synthesis of strained cyclooctynes. In this context, photoactivation was particularly convenient as the formation of the triple bond could be rapidly achieved and therefore could trigger for instance a SPAAC reaction in the desired biological environment.As reported in Scheme 7, DIBO dibenzylcyclopropenone intermediates 31a and 31b were obtained via a Friedel–Crafts reaction between 3,3′-bisbutoxybibenzyl 30 and tetrachlorocyclopropene in the presence of AlCl3, followed by acidic hydrolysis, with a yield of 23% and 12%, respectively. Cyclopropenone 31a was further functionalized to yield, after 7 steps, the biotinylated intermediate 31c to be used for labelling living cells through photo-triggered click reactions. The final dibenzocyclooctynes 32a–c were obtained after decarboxylation induced by efficient irradiation with 350 nm light (Φ355 = 0.33); the transformation was monitored by UV-vis spectroscopy.
 | ||
| Scheme 7 Synthesis of DIBOs and ODIBOs using the photodecarboxylation strategy. | ||
A few years later, Popik42 and co-workers applied the same synthetic strategy to the synthesis of the oxygenated analogues of DIBO, i.e. the oxadibenzocyclooctynes, referred to as ODIBOs. Cyclopropenone derivatives 34a–c were obtained in moderate yields (29% to 12%) starting from the substituted phenyl benzyl ethers 33a–c, which allowed the quantitative synthesis of the corresponding ODIBOs35a–c (Scheme 7).
Later, such cyclopropenone intermediates were employed through multiphoton activation in an AAC reaction for in situ labeling.43 Furthermore the same synthetic strategy was also applied to the synthesis of a 9-membered sulfate-linked dibenzocycloalkyne, the bis-methoxydiphenylacetylene diol cyclic sulfate (DPAD-CS).44
The photodecarboxylation strategy proved to be a valuable methodology for yielding dibenzocyclooctyne derivatives in a rapid and efficient way. Cyclopropenone intermediates have a long shelf life if protected from light. However this methodology is so far restricted to the synthesis of only dibenzocycloalkynes, and key cyclopropenone intermediates are often synthesized with long and complex synthetic procedures.
2.3. Sulfoxide–magnesium exchange
Hosoya45 and co-workers described a new procedure for the synthesis of cycloheptynes and cyclooctynes employing a sulfoxide–magnesium exchange reaction that involved 2-sulfinylcycloalkenyl triflates. Inspired by their own studies on the chemistry of arynes,46 the authors established an efficient three-step procedure, i.e. α-thiolation, triflylation and oxidation, to obtain the desired 2-sulfinyl derivatives from the corresponding cyclic ketones. Reaction with a Grignard reagent triggered a sulfur–magnesium substitution and subsequent β-elimination of the triflyloxy group afforded the desired cycloalkyne (Scheme 8). Phenylmagnesium bromide (PhMgBr) was identified as the optimal Grignard reagent after thorough screening using a cycloheptyne precursor and benzyl azide as the ynophile. | ||
| Scheme 8 General procedure for sulfoxide–magnesium exchange. | ||
The described general procedure was suitable for the synthesis of unsubstituted cyclooctyne, which was not isolated but readily reacted with benzyl azide. DIBO, instead, was synthesized as described and isolated with 18% overall yield (Scheme 9). 11,12-Dihydrodibenzo[a,e]cycloocten-5(6H)-one 36, obtained as reported in the paper from the catalytic hydrogenation of commercially available dibenzo[a,e]cycloocten-5(6H)-one, was deprotonated employing LDA and thiolated at the α-position with S-p-tolyl p-toluenethiosulfonate at −78 °C to afford intermediate 37 in 34% yield. Addition of KHMDS and Tf2NPh at −78 °C, followed by final oxidation of the sulfur atom with m-CPBA, yielded enol triflate 38 in 58% overall yield. Finally, sulfoxide–magnesium exchange in the presence of PhMgBr afforded the desired DIBO in 92% yield. The described procedure for the synthesis of 38 could also be applied to the formation of different 2-sulfinylcycloalkenyl triflates with six- or seven-membered rings, but the isolation of the corresponding cyclohexyne and cycloheptyne derivatives was not possible, and an alternative procedure involving in situ formation of the alkyne species followed by reaction with benzyl azide was proposed.
 | ||
| Scheme 9 DIBO synthesis applying the sulfur–magnesium exchange strategy. | ||
This synthetic strategy proposed by Hosoya is a short and simple procedure for the synthesis of a variety of cycloheptynes and two cyclooctynes. However, there might be some challenges in applying this strategy to a broader set of triflate precursors, as the exchange reaction with the Grignard reagent may not be suitable for base-sensitive substrates.
2.4. Decomplexation of Co-complexes
Tomooka47 and co-workers reported the synthesis of heteroatom-embedded cycloalkynes with medium-sized rings. These compounds are amenable to further functionalization through the insertion of lateral chains attached to the heteroatom. The formation of the triple bond relies on the use of alkyne-dicobalt hexacarbonyl complexes. These intermediates, known from the pioneering work of Nicholas,48 give, in the presence of a Lewis acid, a dicobalt hexacarbonyl-stabilized propargylic cation. Subsequent addition of a nucleophile and application of mild oxidative conditions yield the desired alkyne. The authors applied this approach to a variety of cycloalkynes with rings containing 8 to 11 atoms and embedded with N, O, or S atoms. Regarding cyclooctyne synthesis, they reported a protocol for the formation of a N,S-containing cyclooctyne. In the first step, the reaction of a cobalt complex 39, decorated with two hydroxyl moieties as leaving groups,49 and N-(2-mercaptoethyl)-4-methylbenzenesulfonamide yielded, through a double Nicholas reaction promoted by BF3Et2O, the corresponding complex 40 (76% isolated yield). The next step was performed using the optimized conditions for cobalt removal, i.e. the use of ammonium cerium(IV) nitrate (CAN) with silica gel, which afforded the desired alkyne 41 (Scheme 10). Unfortunately, the yield of the decomplexation step was only modest (35%), especially when compared to those of larger cycloalkyne rings (from 64% to 99%), due to the instability of the cyclooctyne ring under the oxidative reaction conditions. The positive outcome of the Nicholas reaction was ascribed to the use of a sulfonamide group on the heteroatom, which acted as the nucleophile in the first step of the transformation. Kinetic studies on the influence of the heteroatom in the cycloalkyne rings highlighted an increased reactivity in the cycloaddition reaction with benzyl azide. Specifically, cyclooctyne 41 showed higher reactivity than its carbocyclic analogue OCT. | ||
| Scheme 10 Cyclooctyne 41 synthesis using the Co-complex decomplexation strategy. | ||
Workentin50 and co-workers demonstrated for the first time that a strained alkyne, specifically the endo/exo bicycle[6.1.0]nonyne-methanol (BCN), can be efficiently complexed and de-complexed with dicobalt octacarbonyl (Co2 (CO)8), (Scheme 11). This strategy allowed the construction of more complex cyclooctynes, particularly in cases where the highly reactive triple bond of the cyclooctyne moiety needed to be protected to introduce different functional groups in the structure. A similar approach was also studied by Yoshida51 and co-workers, who temporarily protected the triple bond moiety of a strained alkyne with a Cu-complex to further functionalize the molecule with a linear azide to perform the SPAAC reaction and finally deprotected the product using an excess of NH3. Co-complexes of alkynes might be considered preferable due to their high tolerance to a variety of reaction conditions, allowing the introduction of different functional groups to perform bioorthogonal chemistry reactions. The authors synthesized the BCN alkyne as an endo/exo mixture following a literature procedure.52 This mixture was quantitatively converted to the corresponding Co-complex exo-42 and endo-42 by reaction with Co2(CO)8 after 60 min at r.t. The de-complexation reaction could be performed under mild reaction conditions employing trimethyl N-oxide (Me3NO) to afford the original strained alkynes exo-BNC and endo-BCN in 85% yield. As proof of concept, a chemical modification was performed on the protected alkyne, which would have been incompatible with the unprotected BCN.
 | ||
| Scheme 11 Use of Co-complex decomplexation for BCN functionalization. | ||
Specifically, the endo and exo isomers of 42 were separated, and the most abundant exo component was further functionalized through the reaction of its alcoholic moiety with 4-nitrophenyl chloroformate to yield the corresponding carbonate, which was further functionalized with 2-azidoethanamine to yield the linear azide derivative 43 in 74% yield. The synthesis of this compound would not have been possible without the Co-complexation of the highly reactive triple bond. After the introduction of this new functional group, the highly functionalized intermediate 43 reacted smoothly with phenylacetylene to yield the corresponding triazole in a click reaction catalyzed by CuI. The most critical step, i.e. decomplexation, which can often be very difficult, was successfully accomplished, releasing the desired cyclooctyne 44.
Balova53 and co-workers attempted the synthesis of a new class of cycloalkynes, where a heterocyclic core is fused with a heterocycloalkyne moiety, such as benzothiophene and isocoumarin, with the goal of achieving a balance between stability and reactivity, thereby enabling their use in SPAAC reactions (Scheme 12). In order to synthesize the 8-membered benzothiophene (BT8O) and isocoumarin (IC8O) cyclooctynes, the alkyne intermediates 45a and 45b, obtained from a Sonogashira reaction described in the paper, were allowed to react with Co2(CO)8 to form the Co-complexes 46a and 46b, which were then employed in the following Nicholas reaction mediated by BF3Et2O to yield cyclooctyne precursors 47a and 47b. Unfortunately, all the attempts to decomplex intermediates 47a and 47b with common reagents (e.g., CAN, NMO, TBAF, EDA) failed. The authors pointed out that the structure of this new oxygen-containing analogue of monobenzocyclooctyne (MOBO) was not stable enough. On the other hand, the corresponding cyclononynes were successfully synthesized using the described strategy.
 | ||
| Scheme 12 Synthesis attempts of BT80 and IC80. | ||
Rothman described an attempt to synthesize a cyclooctyne endocyclic amino acid (CEAA).54 The general aim of this work was to create a novel class of cycloalkynes that resemble a proline analogue and, therefore, more similar to natural amino acids, to be employed in bioorthogonal reactions and likely presenting fewer issues of perturbation of the native structural configuration (Scheme 13). Moreover, the inclusion of endocyclic heteroatoms in the ring increased the reactivity of the triple bond compared to that of a carbocyclic ring, because of the hyperconjugation effect as a sigma-acceptor in an antiperiplanar position. The synthetic strategy proposed by the author envisioned a mild Mitsonobu reaction using triphenylphosphine (PPh3) and diisopropyl azodicarboxylate (DIAD) as the key step for the cyclization of a dicobalt adduct 48 (its synthesis is reported in the paper), yielding the intermediate 49, which is then converted to the free cycloalkyne 50 through a Nicholas-related procedure. Unfortunately, the decomplexation process of cyclooctyne 49 proved very challenging, and unlike 9–11-membered cycloalkynes, its isolation was difficult, and the complete synthetic strategy could not be accomplished. Hosoya55 and co-workers applied a synthetic strategy that involved the use of a cobalt complex triple bond intermediate for the synthesis of dibenzoazacyclooctyne (DIBAC, also referred to as DBCO17) derivatives (Scheme 14). These derivatives take advantage of the ring strain of an sp2 center, such as the aromatic ring, fused to the cyclooctyne to increase the reactivity. Furthermore, the endocyclic nitrogen increases hydrophilicity, and it can be used as a functionalization handle. Compared with the classic strategy reported for this moiety, i.e. bromination of the double bond/elimination,13 this procedure was more group-tolerant and enabled the formation of a series of functionalized DIBACs. Specifically, amide 51, obtained in 2 steps in 91% yield from o-iodoaniline, was allowed to react through a Sonogashira coupling with phenylacetylene to form the alkyne precursor 52, which was subsequently complexed with Co2(CO)8 to afford intermediate 53 in 97% yield. The key step in performing the cyclization of the azacyclooctyne moiety was accomplished using the Pictet–Spegler reaction,50 where trimethylsilyl triflate (TMSOTf) activated the Co-complex, with the formation of an iminium salt that finally afforded the desired complexed-dibenzocyclooctyne 54. For the final decomplexation step, classical literature procedures, e.g. Me3NO in CH2Cl2 at r.t. or CAN in silica gel/Et2O at 0 °C, gave only poor yields; therefore, a screening for this step was performed using the Co-complex of dibenzocyclooctynol as a test substrate. Optimal reaction conditions were achieved, in terms of yield, reaction time and amount of additive, when 1 equivalent of Me3NO was employed in pyridine without any protective atmosphere at r.t. for 3 hours. Such conditions were successfully applied to Co-complex 54, yielding the desired DIBAC in 96% yield. This strategy was effectively applied to the other DIBAC derivatives functionalized with the Halotag ligand, coumarin and biotin. The bisDIBAC synthesized through this procedure is a bis-reactive molecule with important applications in azido-installed protein modification through a strain-promoted double click reaction (SPDC).56
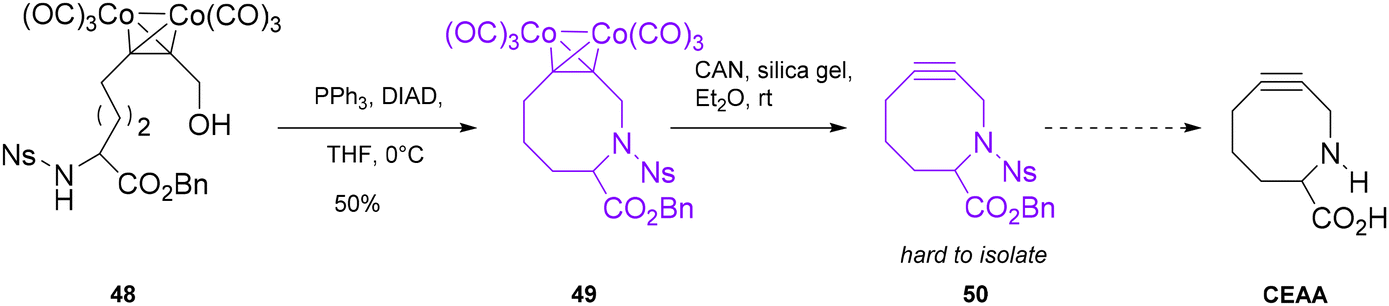 | ||
| Scheme 13 Synthesis attempt of CEAA. | ||
 | ||
| Scheme 14 DIBAC synthesis using the Co-complex decomplexation methodology. | ||
The introduction of a Co-masked triple bond using the Nicholas reaction for the synthesis of cyclooctynes showed good efficacy for producing dibenzocyclooctyne derivatives but presented some limitations due to the difficulties in the decomplexation step for other substrates. On the other hand, this methodology is a valid strategy for temporarily protecting a strained triple bond to insert other functionalities on the alkyne moiety.
2.5. Silylated allene rearrangement
Schomaker57–59 and co-workers extensively studied a class of sulfamate/amide-based cyclooctynes bearing heteroatoms at the homopropargylic position, namely sulfur, nitrogen, and oxygen-containing cyclooctynes (SNO-OCTs). The idea behind the synthesis of molecules with this structure, confirmed by computational studies, lies in the beneficial effect of the heteroatoms on stability and reactivity. In addition, they display multiple sites that are amenable to functionalization. The general synthetic strategy involved a two-step one-pot procedure using α-silyl allene sulfamate/sulfamide, which underwent Rh-catalyzed aziridination,60 followed by ring expansion triggered by TBAF, promoting TMS elimination and ring-opening of the aziridine moiety (scheme of Table 1). The presence of the trimethylsilyl group was of critical importance, as it regiochemically directed the formation of the aziridine to the distal allene carbon for steric reasons, as well as for stabilization of the incipient positive charge through hyperconjugation.61 The procedure had the advantage of being mild and rapid as the aziridine intermediate was not isolated.|
|
|||
|---|---|---|---|
| SNO-OCT generation | Synthesis of homoallenic precursor | Homoallenic reactant | SNO-OCT (yield%) |
| Part A | Acid-catalyzed Claisen rearrangement |

|

|
| First generation: alkyl group at C1-carbon |

|

|
|

|

|
||
| Part B | Zn-mediated Reformatsky–Claisen rearrangement |

|

|
| Second generation: alkyl group at C1-carbon and gem-difluoro at C4 | |||
| Part C | Au(I)-catalyzed Claisen rearrangement |

|

|
| Third generation: functional handle at C4 or C1–C4 |
|

|
|

|
|
||

The first generation of SNO-OCTs57 (56a–c), featuring alkyl functionalization at the C1 position (Table 1, part A), was obtained in excellent to good yields (89% to 66%) from homoallenic sulfamates 55a and 55b and sulfamide 55c under the reaction conditions described in Scheme 15. The carbon skeleton of precursors 55a–c was derived from the β-allenic esters 61a and 61b, which were synthesized via a propargylic Claisen rearrangement of a TMS-propargylic alcohol with an orthoester.62 These intermediates were subjected to reduction of the ester moiety with LiAlH4 to yield homoallenic alcohols 62a and 62b in 81% and 99% yields, respectively. Final functionalization of 62a and 62b employing chlorosulfonyl isocyanate (CSI) and formic acid yielded sulfamates 55a and TBS-55b. The latter was completely deprotected during the ring expansion stage using an excess of TBAF. On the other hand, 62a was converted into precursor 55c using tert-butyl aminosulfonylcarbamate (Boc-sulfamide) in the presence of diisopropyldicarboxylate (DIAD) with 76% yield.
 | ||
| Scheme 15 Synthesis of homoallenic sulfamates 55a and 55b and sulfamide 55c. | ||
Cyclooctyne 56a proved to be very stable under both basic and acidic conditions. Furthermore, the 1,3-dipolar cycloaddition with benzyl azide delivered very high rate constants, greater than those obtained with fluorinated cyclooctynes like DIFO. Bioconjugation studies were performed to assess the viability of using SNO-OCTs in a biological context and for the functionalization of polymers.63
The second generation of SNO-OCT (58)58 (Table 1, part B) consisted of a difluorinated derivative that displayed improved rate constants in the cycloaddition reaction with diazoacetamides, greater than 5.13 M−1 s−1, and in general with Type I–II dipoles, whilst showing a diminished preference for engaging in inverse electron demand Diels–Alder (IEDDA) reactions. Additionally, such a substrate could be further manipulated to afford a triple ligation system for bioorthogonal reactions. The synthetic strategy envisioned the use of a chlorodifluoro ester precursor 63, synthesized according to the literature,58 for the introduction of a gem-difluoro moiety at C4 and an ethyl group at C1 (Scheme 16). This precursor underwent a Zn-mediated Reformatsky–Claisen rearrangement64 to yield the corresponding allenic difluoroacid, which was promptly methylated with trimethylsilyldiazomethane (TMSCHN2), giving the desired homoallenic methyl ester intermediate 64 in 78% overall yield. Finally, the homoallenic sulfamate 57 was obtained after reduction of the ester moiety with sodium borohydride (NaBH4), followed by the introduction of the sulfamate group using CSI and formic acid.
 | ||
| Scheme 16 Synthesis of homoallenic sulfamate 57. | ||
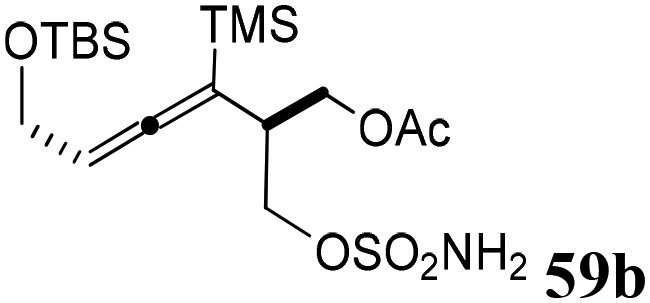
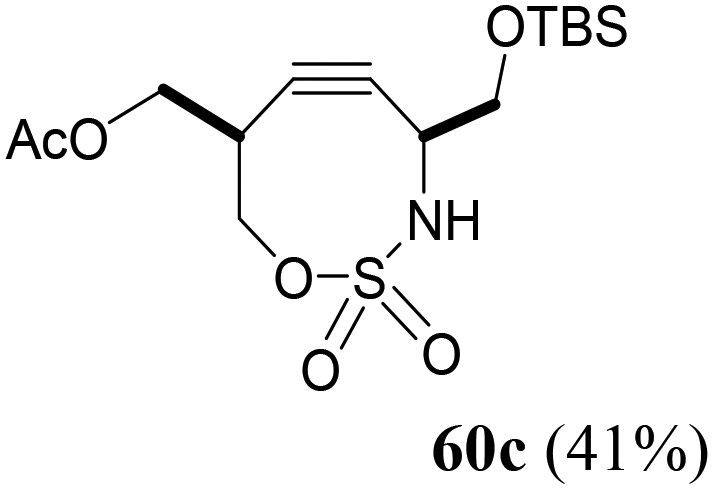
The third generation of SNO-OCTs59 (60a–c) is represented by highly functionalized cyclooctynes at C4 or C4–C1, designed to be employed as labelling reagents (Table 1, part C). The synthesis of the first SNO-OCT 60a with only a C4 handle allowed the authors to optimize a new strategy for this class of compounds (Scheme 17). Specifically, the first step involved an Au(I)-catalyzed Claisen rearrangement65 of intermediate 65, which was synthesized as described in the paper from a TMS-protected propargylic alcohol. Careful screening of the catalytic conditions for the first step revealed that gold(I) chloride (AuCl, 5 mol%) in the presence of sodium tetrafluoroborate (NaBF4, 10 mol%) as an additive provided the best experimental conditions to be employed. Compound 65, which contained both a propargyl unit and an allylic acetate, yielded, after Claisen rearrangement followed by reduction with NaBH4, the corresponding alcohol intermediate 66 with a tertiary allenic center. Intermediate 66 was then converted into homoallenic precursor 59a with CSI and formic acid with a 68% yield.
 | ||
| Scheme 17 Synthesis of homoallenic precursor 59a. | ||
Under these optimized conditions, intermediate 67 (obtained from the reaction of a propargylic alcohol previously synthesized by the authors57 and methyl propiolate) was subjected to Au(I)-catalyzed rearrangement, yielding the products 68a and 68b as a diastereoisomeric mixture, which was separated by column chromatography (Scheme 18). Finally, treatment with hexafluoroisopropyl sulfamate (HFIPS) yielded the desired homoallenic sulfamides 59b (66% yield) and 59c (72% yield), which were employed for the synthesis of third-generation SNO-OCTs (60b and 60c).
 | ||
| Scheme 18 Synthesis of homoallenic sulfamides 59b and 59c. | ||
The strategy proposed by Schomaker and co-workers, i.e. the synthesis of diverse SNO-OCTs via a rearrangement of silylated allenes, proved to be very versatile, as the cyclooctyne moiety can be decorated with different handles at different positions, allowing the formation of highly dense functionalized molecules. However, the drawback of this procedure is that it is limited to the synthesis of heteroatom-containing cyclooctynes.
2.6. Carbene-mediated strategy
Raines and co-workers66 sought the synthesis and application of a new kind of heterocycle-containing cyclooctyne moiety, namely 2-azadibenzocyclooctyne (ABC). The structure of this molecular scaffold was proposed based on computational studies, which identified the ABC moiety as offering an optimal combination of increased strain and electronic tuning to achieve better rate constants in 1,3-dipolar cycloaddition reactions. The synthetic strategy toward ABC involved, as a key reaction step, a ring-expansion through a 1,2 rearrangement of an alkylidene carbene. These intermediates have a very short lifetime, as they immediately undergo a 1,2 migration of a β-substituent upon formation, generating an alkyne.67–69The starting material of the synthetic pathway was the commercially available ketone 69, which reacted with N-morpholinomethyl-5-lithiotetrazole (formed in situ from 4-((N-tetrazolyl)methyl)morpholine and a 1 M solution of LiHMDS) to yield the corresponding 5-hydroxyalkyl-1H-tetrazole 70 in 52% yield after acidic hydrolysis.70 Tetrazole 70 was treated with 1-ethyl-3-(3-dimethyl-aminopropyl)carbodiimide (EDC) to afford, through a dehydrative elimination, the intermediate tetraazafulvene 71, which rapidly expelled two nitrogen molecules to yield the unstable alkylidene carbene 72 that, in turn, after a [1,2] rearrangement, afforded the final cyclooctyne product ABC in 40% yield (Scheme 19).69,71
 | ||
| Scheme 19 Carbene-mediated synthesis of ABC. | ||
The same synthetic strategy has been applied in this paper to the synthesis of the known DIBO30 employing the commercially available dibenzosuberone as the starting material, which yielded the corresponding cyclooctyne in 51% yield.
Later, Gold72 and co-workers attempted to employ this approach for the synthesis of the oxygen-containing analogues oxa-azabenzocyclooctynes (O-ABCs), as reported in Scheme 20. The starting materials employed were ketones 73 and 74 synthesized using a literature procedure that envisioned the use of a two-step substitution–Parham-type cyclization.73
 | ||
| Scheme 20 Carbene-mediated synthesis of O-ABCs. | ||
The cyclooctyne O-ABC-77, derived from 73, showed stability issues upon solvent concentration after flash chromatography or when exposed to air; therefore, it was isolated only in 6% yield. However, it was possible to prove its formation by trapping with benzyl azide. O-ABC-78, derived from ketone 74, was too unstable to be isolated, but the in situ reaction with benzyl azide afforded the corresponding cycloadduct in 53% yield from the tetrazole intermediate.
In conclusion, Raines and Gold proposed a convenient strategy for the synthesis of ABC cyclooctyne employing the commercially available starting material 69. However, extending this strategy to the corresponding oxygenated derivatives O-ABCs presented issues in terms of low yield and stability. Additionally, in the latter case, starting compounds need to be synthesized, as they are not readily available.
3. Conclusions
Cyclooctyne derivatives are scaffolds of strategic importance in bioorthogonal chemistry. Researchers in this field face a stimulating challenge in synthesizing these derivatives, as success relies on the essential balance between reactivity and stability. In addition, the high selectivity these substrates must exhibit in biological environments adds an additional layer of complexity. Many efforts have been made in recent years to develop efficient synthetic protocols applicable to a wide range of substrates. While much progress has been attained over the years, there is still room for improvement. All the procedures reported so far, despite their differences, share a common feature, i.e. the formation or release of the endocyclic triple bond at a late stage in the synthetic protocol, due to its inherent reactivity or instability. The vinyl enol/triflate strategy has been successfully applied to the synthesis of a wide range of cyclooctynes such as MOFO, DIFOs and BARAC. This method can be considered one of the most flexible strategies, even though it does present challenges in achieving good yields in certain cases. On the other hand, the Co-complex decomplexation strategy has proven effective for the synthesis of various cyclooctyne derivatives, with some ongoing studies still needed to complete the final step of decomplexation in some cases. Other procedures, although more specific, still contribute to expanding and diversifying the pool of available cyclooctynes. As an example, the silylated allene rearrangement strategy for the synthesis of SNO-OCTs, developed by the research group of Schomaker, effectively yielded a wide variety of heteroatom-containing cyclooctynes. Future research in this field will hopefully lead to the development of new synthetic protocols that combine wide applicability for the target molecules with a reasonable synthetic pathway and an adequate overall yield. These advancements will undoubtedly provide a winning combination for the effective use of cyclooctyne derivatives in bioorthogonal reactions within biological environments.Data availability
No primary research results, software or code have been included. Multistep and overall yield data were calculated from the individual yields reported in the literature sources cited in this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. D. gratefully acknowledges the Ministero dell'Università e della Ricerca (MUR) for the PON PhD fellowship DOT1423134-3 and E. P. and D. L. acknowledge MUR for PRIN project no. 20207CNBE4 “FAITH”.References
- A. T. Blomquist and L. H. Liu, J. Am. Chem. Soc., 1953, 75, 2153–2154 CrossRef CAS.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 Search PubMed.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Nat. Rev. Methods Primers, 2021, 1, 1–23 CrossRef.
- C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 651–653 CrossRef CAS.
- C. J. Pickens, S. N. Johnson, M. M. Pressnall, M. A. Leon and C. J. Berkland, Bioconjugate Chem., 2018, 29, 686–701 CrossRef CAS.
- J. Kaur, M. Saxena and N. Rishi, Bioconjugate Chem., 2021, 32, 1455–1471 CrossRef CAS PubMed.
- Q. Zhang, G. Kuang, L. Wang, P. Duan, W. Sun and F. Ye, Research, 2023, 6, 0251 CrossRef CAS PubMed.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. (Ton) J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS.
- T. Harris and I. V. Alabugin, Mendeleev Commun., 2019, 29, 237–248 CrossRef CAS.
- M. Trætteberg, W. Lüttke, R. Machinek, A. Krebs and H. J. Hohlt, J. Mol. Struct., 1985, 128, 217–232 CrossRef.
- C. Gröst and T. Berg, Org. Biomol. Chem., 2015, 13, 3866–3870 RSC.
- C. Lis and T. Berg, Synlett, 2019, 30, 939–942 CrossRef CAS.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- B. R. Varga, M. Kállay, K. Hegyi, S. Béni and P. Kele, Chem. – Eur. J., 2012, 18, 822–828 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644–648 CrossRef CAS PubMed.
- S. A. McNelles, J. L. Pantaleo and A. Adronov, Org. Process Res. Dev., 2019, 23, 2740–2745 CrossRef CAS.
- R. J. Hargrove and P. J. Stang, J. Org. Chem., 1974, 39, 581–582 CrossRef CAS.
- W. Bleckmann and M. Hanack, Chem. Ber., 1984, 117, 3021–3033 CrossRef CAS.
- I. Fleming and J. M. Mwaniki, J. Chem. Soc., Perkin Trans. 1, 1998, 1237–1248 RSC.
- X. Pérez-García, A. Rumbo, M. J. Larriba, P. Ordóñez, A. Muñoz and A. Mouriño, Org. Lett., 2003, 5, 4033–4036 CrossRef PubMed.
- L. T. Nguyen, F. D. Proft, V. L. Dao, M. T. Nguyen and P. Geerlings, J. Phys. Org. Chem., 2003, 16, 615–625 CrossRef CAS.
- D. H. Ess, G. O. Jones and K. N. Houk, Org. Lett., 2008, 10, 1633–1636 CrossRef CAS PubMed.
- P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS PubMed.
- M. C. Pirrung and N. J. G. Webster, J. Org. Chem., 1987, 52, 3603–3613 CrossRef CAS.
- N. Atanes, S. Escudero, D. Pérez, E. Guitián and L. Castedo, Tetrahedron Lett., 1998, 39, 3039–3040 CrossRef CAS.
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS.
- X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Org. Lett., 2008, 10, 3097–3099 CrossRef CAS.
- P. Sampson and D. F. Wiemer, J. Chem. Soc., Chem. Commun., 1985, 1746–1747 RSC.
- E. M. Sletten, H. Nakamura, J. C. Jewett and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 11799–11805 CrossRef CAS.
- K. Chenoweth, D. Chenoweth and W. A. G. Iii, Org. Biomol. Chem., 2009, 7, 5255–5258 RSC.
- I. Pravst, M. Zupan and S. Stavber, Synthesis, 2005, 3140–3146 CAS.
- G. de Almeida, E. M. Sletten, H. Nakamura, K. K. Palaniappan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2012, 51, 2443–2447 CrossRef CAS PubMed.
- A. Krebs and H. Kimling, Tetrahedron Lett., 1970, 11, 761–764 CrossRef.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS.
- A. Poloukhtine and V. V. Popik, J. Org. Chem., 2003, 68, 7833–7840 CrossRef CAS PubMed.
- N. K. Urdabayev, A. Poloukhtine and V. V. Popik, Chem. Commun., 2006, 454–456 RSC.
- C. D. McNitt and V. V. Popik, Org. Biomol. Chem., 2012, 10, 8200–8202 RSC.
- C. D. McNitt, H. Cheng, S. Ullrich, V. V. Popik and M. Bjerknes, J. Am. Chem. Soc., 2017, 139, 14029–14032 CrossRef CAS PubMed.
- M. J. Holzmann, N. Khanal, P. Yamanushkin and B. Gold, Org. Lett., 2023, 25, 309–313 CrossRef CAS PubMed.
- S. Yoshida, F. Karaki, K. Uchida and T. Hosoya, Chem. Commun., 2015, 51, 8745–8748 RSC.
- S. Yoshida, K. Uchida and T. Hosoya, Chem. Lett., 2014, 43, 116–118 CrossRef CAS.
- R. Ni, N. Mitsuda, T. Kashiwagi, K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2015, 54, 1190–1194 CrossRef CAS PubMed.
- K. M. Nicholas, Acc. Chem. Res., 1987, 20, 207–214 Search PubMed.
- M. Gruselle, B. Malézieux, J. Vaissermann and H. Amouri, Organometallics, 1998, 17, 2337–2343 Search PubMed.
- P. Gobbo, T. Romagnoli, S. M. Barbon, J. T. Price, J. Keir, J. B. Gilroy and M. S. Workentin, Chem. Commun., 2015, 51, 6647–6650 RSC.
- S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, J. Am. Chem. Soc., 2014, 136, 13590–13593 CrossRef CAS.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 Search PubMed.
- N. A. Danilkina, A. I. Govdi, A. F. Khlebnikov, A. O. Tikhomirov, V. V. Sharoyko, A. A. Shtyrov, M. N. Ryazantsev, S. Bräse and I. A. Balova, J. Am. Chem. Soc., 2021, 143, 16519–16537 CrossRef CAS PubMed.
- J. H. Rothman, ACS Omega, 2022, 7, 9053–9060 CrossRef CAS PubMed.
- Y. Sakata, R. Nabekura, Y. Hazama, M. Hanya, T. Nishiyama, I. Kii and T. Hosoya, Org. Lett., 2023, 25, 1051–1055 Search PubMed.
- A. R. Popchock, S. Jana, R. A. Mehl and W. Qiu, ACS Chem. Biol., 2018, 13, 2229–2236 CrossRef CAS PubMed.
- E. G. Burke, B. Gold, T. T. Hoang, R. T. Raines and J. M. Schomaker, J. Am. Chem. Soc., 2017, 139, 8029–8037 CrossRef CAS PubMed.
- Y. Hu, J. M. Roberts, H. R. Kilgore, A. S. Mat Lani, R. T. Raines and J. M. Schomaker, J. Am. Chem. Soc., 2020, 142, 18826–18835 Search PubMed.
- Y. Hu, R. Spiegelhoff, K. S. Lee, K. M. Sanders and J. M. Schomaker, J. Org. Chem., 2024, 89, 4512–4522 CrossRef CAS.
- E. G. Burke and J. M. Schomaker, Angew. Chem., Int. Ed., 2015, 54, 12097–12101 CrossRef CAS PubMed.
- T. M. Gregg, M. K. Farrugia and J. R. Frost, Org. Lett., 2009, 11, 4434–4436 Search PubMed.
- D. Tejedor, G. Méndez-Abt, L. Cotos and F. García-Tellado, Chem. Soc. Rev., 2012, 42, 458–471 RSC.
- E. G. Burke and J. M. Schomaker, J. Org. Chem., 2017, 82, 9038–9046 CrossRef CAS.
- Y.-Y. Yang, W.-D. Meng and F.-L. Qing, Org. Lett., 2004, 6, 4257–4259 CrossRef CAS.
- B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 15978–15979 CrossRef CAS.
- J. M. Dones, N. S. Abularrage, N. Khanal, B. Gold and R. T. Raines, J. Am. Chem. Soc., 2021, 143, 9489–9497 CrossRef CAS PubMed.
- E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 13, 3769–3772 CrossRef.
- W. Kirmse, Angew. Chem., Int. Ed. Engl., 1997, 36, 1164–1170 CrossRef CAS.
- H. J. A. Dale, C. Nottingham, C. Poree and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2021, 143, 2097–2107 CrossRef CAS PubMed.
- P. D. Alexakos and D. J. Wardrop, J. Org. Chem., 2019, 84, 12430–12436 CrossRef CAS PubMed.
- D. J. Wardrop and J. P. Komenda, Org. Lett., 2012, 14, 1548–1551 CrossRef CAS.
- E. Das, M. A. M. Feliciano, P. Yamanushkin, X. Lin and B. Gold, Org. Biomol. Chem., 2023, 21, 8857–8862 RSC.
- J. Farrokh, C. Campos and D. A. Hunt, Tetrahedron Lett., 2015, 56, 5245–5247 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |