
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigations into the N-dealkylation reaction of protected chelating agents†
Jonathan
Martinelli
 abc,
Enrico
Martorana
a,
Angelo
Marcotrigiano
a and
Lorenzo
Tei
*a
abc,
Enrico
Martorana
a,
Angelo
Marcotrigiano
a and
Lorenzo
Tei
*a
aDepartment of Science and Technological Innovation (DISIT), Università del Piemonte Orientale, Alessandria, Italy. E-mail: lorenzo.tei@uniupo.it
bDepartment of Health Sciences (DISSAL), University of Genova, Genova, Italy
cNuclear Medicine Unit, IRCCS Ospedale Policlinico San Martino, Genova, Italy
First published on 20th March 2025
Abstract
To comply with the specific requirements for the coordination of a certain metal ion, it is often necessary to decrease the substitution degree on the nitrogen atoms of well-known poly(aminocarboxylate) ligands used as chelators for the preparation of diagnostic or therapeutic probes. The procedures used so far to prepare such partially-alkylated compounds involve steps that suffer from product loss or the need to introduce protection/deprotection reactions, consequently lowering the final yield. The application of an N-dealkylation reaction to an exhaustively-substituted precursor could in principle allow to achieve the same result in fewer steps and therefore with higher yields. Dealkylation reactions have been known since the early 1900s, but they have never been exploited for such a purpose. We investigated the applicability of the simple iron-Polonovski N-dealkylation reaction to obtain a library of useful ligands starting from the tert-butyl-protected derivatives of chelators widely used in the biomedical fields such as CDTA, EDTA, NOTA, AAZTA and PCTA. The preparation of partially-alkylated ligands has already been reported in the literature but with several drawbacks and possible improvements. In most of the examples reported, it was found that the reaction occurred in an easy and straightforward way by only using an excess of oxidizing agent that was sufficient to convert the N-oxide into the N-dealkylated product without the need for a reducing agent.
Introduction
Dealkylation reactions, defined as the detachment of one or more alkyl groups from a certain compound, can be divided into two types, depending on the heteroatom from which the group is removed: O-dealkylations and N-dealkylations. The former take place in human metabolism, for example during drug degradation processes. The latter can instead play an important role in a variety of preparations, for example in the synthesis of opioids with specific pharmacological activities. N-Dealkylations1,2 have been studied since the beginning of the 20th century, when von Braun first investigated the use of cyanogen bromide for the demethylation of alkaloids.3,4 The toxicity of this reactant5 pushed the development of alternative methods, such as the exploitation of different chloroformates6 or azodicarboxylates.7 Besides other more limited electrochemical,8 photochemical9 and enzymatic10 methods, the most successful and flexible applications were achieved via procedures catalyzed by metal ions such as palladium,11 ruthenium,12 rhodium13 and copper.14 Iron has also been used in N-dealkylations, particularly in a variation of the so-called Polonovski reaction,15 which is a procedure where an oxidizing reagent (e.g., a peroxide or a peroxyacid) is used to produce the N-oxide of a tertiary amine, which is then treated with an activating agent able to promote the detachment of an alkyl group as the corresponding aldehyde.16 The most common activating agents reported for the Polonovski reaction are acetyl chloride,16 acetic anhydride16 and sulfurous anhydride.17 In the non-classical Polonovski variation, upon formation of the N-oxide, iron(II) is used to promote the release of the oxygen atom as a water molecule followed by the formation of an iminium species that is finally hydrolyzed to the targeted secondary amine with the release of an aldehyde.16,18,19 Iron(II) is introduced as a suitable salt (e.g., FeCl2 in H2O18 or FeSO4 in MeOH19) and removed later by the addition of chelators such as ethylenediamine or EDTA (ethylenediaminetetraacetic acid).We envisioned that dealkylation reactions and particularly the non-classical Polonovski method would be a convenient way for preparing polyamino–polycarboxylate ligands for diagnostic or theranostic applications where the N-positions are not exhaustively substituted with acetate arms. Diagnostic techniques such as magnetic resonance imaging (MRI), positron emission tomography (PET) and single photon emission computed tomography (SPECT) often require a (radio)metal complex as a contrast agent or a radiotracer to allow tissue visualization. The suitable metal ions (paramagnetic for MRI, radioactive for PET and SPECT) must be chelated by polydentate ligands according to their coordination number combined with the possible need to leave some positions free for other species to bind (e.g., water for MRI, 18F− for aluminum–fluoride applications in PET).20
Alternatively, one N-position may be used for conjugation to biological moieties for targeting and/or molecular imaging applications.21 In general, such ligands consist of linear, macrocyclic or mesocyclic polyamine backbones functionalized with acetate arms on their N-atoms, such as EDTA,22 CDTA23 (trans-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid), NOTA24 (1,4,7-triazacyclononane-1,4,7-triacetic acid), PCTA25 (3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-1(15),11,13-triene-3,6,9-triacetic acid) and AAZTA26 (6-amino-6-methylperhydro-1,4-diazepine-N,N′,N′′,N′′-tetraacetic acid) (Fig. 1).
 | ||
| Fig. 1 Ligands used in diagnostic/therapeutic applications investigated in this work. | ||
These fully-substituted chelators can be modified to fit metal ions with lower coordination numbers by formally removing one or more arms. However, the preparations reported so far for partially-alkylated compounds are often not satisfactory, because they follow one of these two methods: (1) the use of ca. 3 equivalents of an alkylating reactant (e.g., tert-butyl bromoacetate), with consequent formation of a mixture of mono-, di-, tri- and tetrasubstituted products that need to be separated under difficult conditions due to the similarity of the various species; (2) selective protection of one of the N-positions, alkylation of the remaining ones and successive deprotection, meaning at least 2 additional synthetic steps with the consequent consumption of time, materials and effort, as well as lowering of the final yield. Regarding another important ligand, i.e., DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) and its mono-dealkylated analogue (DO3A), effective procedures have already been set up over the past 20 years for their preparation27 and therefore they have not been considered in this work.
Herein, we report the study and application of a potentially easier way for preparing partially-alkylated derivatives of common ligands for diagnostic or therapeutic purposes, based on the preliminary preparation of the corresponding exhaustively-substituted chelators followed by their N-dealkylation. The non-classical iron-Polonovski reaction was chosen because of the readily available materials and the mild conditions it requires.
Results and discussion
All investigations were carried out starting from the tert-butyl esters of the ligands because (1) they are by far the most common precursors in the preparation of chelators, (2) they are easier to manipulate in terms of solubility and lack of side reactivity, and (3) the presence of an electron-withdrawing group increases the acidity of the alpha-CH2 hydrogens so that the dealkylation should take place exclusively on this site.16,28 Six model compounds were considered in this study (Fig. 1): CDTA(Ot-Bu)4 and its unsaturated and more rigid analogue, compound 2 (Fig. 1); linear EDTA(Ot-Bu)4 and macrocyclic NOTA(Ot-Bu)3, where all the amine groups are equivalent; mesocyclic AAZTA(Ot-Bu)4 and macrocyclic PCTA(Ot-Bu)3, bearing non-equivalent tertiary amines.At first, we studied the N-dealkylation of the CDTA tert-butyl ester to obtain CD3A(Ot-Bu)3. CD3A is the scaffold of bifunctional chelators particularly effective in binding aluminum–fluoride, 68Ga or 111In ions for PET applications, sometimes referred to as RESCA (REStrained Complexing Agent) ligands.29–34 The first reported preparation starts with the protection of one of the amine groups of racemic trans-1,2-cyclohexanediamine, followed by alkylation of the remaining free positions on the N-atoms and final deprotection. An available binding site is thus obtained, which can be exploited to conjugate a targeting vector such as a peptide, an antibody, or a PSMA (prostate-specific membrane antigen) binding motif via a reactive tetrafluorophenyl ester or by click chemistry. Later, the synthesis was modified by Wong and co-workers32 by using methyl 2-(4-formylphenyl)acetate as a protecting group and optimizing the reaction conditions to reduce the chromatographic purifications. According to our strategy, we started with the easy preparation of the CDTA tert-butyl ester from trans-1,2-cyclohexanediamine and a slight excess of tert-butyl bromoacetate (see the ESI†), followed by treatment with meta-chloroperoxybenzoic acid (mCPBA) to form the corresponding N-oxide under Polonovski reaction conditions (Scheme 1). Commercially available mCPBA in 77% purity was first purified following the procedure reported by Traylor et al.,35i.e., by washing with PBS buffer and recrystallizing from petroleum ether. Test reactions were carried out on 10 mg of tetraalkylated CDTA and variable amounts of mCPBA (1–10 equiv.) to investigate the possible formation of different intermediates and products (e.g., bis-N-oxide, bis-dealkylated products, etc.).
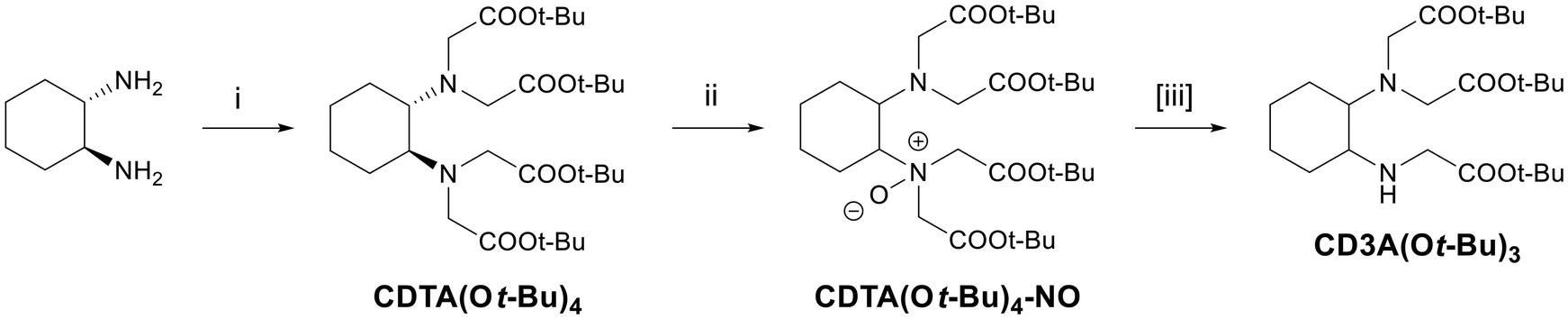 | ||
| Scheme 1 Steps and observed species in the preparation of CD3A(Ot-Bu)3 from CDTA(Ot-Bu)4via the Polonovski reaction. (i) BrCH2COOt-Bu, K2CO3, ACN, 70 °C, overnight; (ii) mCPBA (1–10 equiv.), DCM, rt, 1–24 h; [iii] FeCl2, DCM, rt, 1–15 h. The third step ([iii]) was only necessary when 7 or 10 equiv. of mCPBA were used. | ||
The next step should have been the treatment with iron(II) to promote the dealkylation. However, ESI MS analyses (Fig. S16†) showed that some CD3A(Ot-Bu)3 had already formed, depending on the amount of mCPBA added to the tetraalkylated species. In detail, while with 1 equivalent only a partial conversion into the mono-N-oxide had taken place, it was observed that with 2 equivalents of mCPBA the N-dealkylated product had already formed after 1 hour and was present together with some starting material; when 3 or 5 equivalents of oxidant were used, the main species were the N-oxide and the dealkylated product; with 7 or 10 equivalents of mCPBA, only the intermediate N-oxide could be identified. On the other hand, no other intermediates or by-products were formed, regardless of the reaction stoichiometry. Each reaction mixture was stirred overnight at room temperature and then checked again by ESI MS (Fig. S16†): while for the reaction with 2 equivalents of mCPBA no significant difference was observed, in the presence of 3 and 5 equivalents, all starting materials had converted into the mono-dealkylated product and no N-oxide remained. In the case of the reaction carried out with a 7-fold excess of mCPBA, some N-oxide transformed into the product, but with 10 equivalents, the N-oxide was still the only observable species. Noteworthily, no other species were formed such as the bis-N-oxide or poly-dealkylated products. The composition of the reaction mixtures was confirmed by HPLC-MS analysis (Fig. S17†), where CDTA(Ot-Bu)3, CDTA(Ot-Bu)4–NO and CDTA(Ot-Bu)4 are eluted with retention times of 13.6, 14.9 and 15.2 min, respectively, under the applied conditions. The peak due to mCPBA could be observed in the UV trace at 10.9 min.
The progress of the conversion was also studied via NMR spectroscopy with 10 mg (0.017 mmol) of tetra-ester and 3 equivalents of mCPBA, by carrying out the reaction in 0.5 mL of CDCl3 in a 5 mm tube and acquiring 1H spectra at different times (Fig. 2). The NMR spectra show that the N-oxide forms quantitatively within 5 min, and then it starts to convert into CDTA(Ot-Bu)3. After 2 days, the conversion is complete. According to the generally accepted mechanism, the alkyl group should leave the substrate as an aldehyde, and in this case this would be even favored by the conjugation with the ester group; however, no evidence of an aldehyde group was observed in the 1H and 13C NMR spectra of the reaction mixtures: the distinctive signals expected for the CHO moiety (ca. 10 and 190 ppm for the proton and carbon resonances, respectively) are completely absent.
 | ||
| Fig. 2 1H NMR spectra (CDCl3, 25 °C) of the reaction between CDTA(Ot-Bu)4 and mCPBA (3 equiv.) in the time range of 0–48 h. | ||
Where the N-oxide persisted (i.e., when 7 and 10 equivalents of mCPBA were used), the intermediate was characterized by NMR (Fig. S3 and S4†) and it was treated with a catalytic amount of iron(II), according to the conditions of the abovementioned non-classical Polonovski reaction. In particular, 10 mol% of FeCl2 tetrahydrate were added directly to each reaction mixture, which was then stirred at room temperature and checked by ESI-MS (NMR spectra cannot be acquired because iron is paramagnetic): in both cases, complete conversion to CDTA(Ot-Bu)3 was achieved after 15 hours (overnight) (Fig. S18†).
The novel and simplified dealkylation procedure was then applied on a larger scale, by reacting 0.5 g of CDTA(Ot-Bu)4 with 3 equivalents of mCPBA in DCM at room temperature overnight. The excess mCPBA was then quenched by adding a saturated aqueous solution of Na2SO3, and the product was obtained after washing with H2O and brine and purification by column chromatography. If FeCl2 is used, additional washings with 0.1 M EDTA, H2O and sat. NaHCO3 have to be carried out.
The overall yield (from cyclohexanediamine to CD3A(Ot-Bu)3) was 70%, higher than those reported in the literature for the longer procedures (55–59%).32,36–38 Even the procedure optimized by Wong and co-workers suffered from several disadvantages, such as the formation of over-benzylated impurities with substantial product loss following the necessary chromatographic purifications at each step, or the need to stabilize the protected intermediate by forming an orotate salt with an inconvenient atom economy.32 The yields of the alkylated derivative of CD3A(Ot-Bu)3 for the two reported procedures are 39% (2 steps) and 58% (3 steps), respectively.
Prompted by the unexpected results obtained from the dealkylation of CDTA(Ot-Bu)4, more investigations on other exhaustively substituted tertiary amines were carried out. A bicyclic protected chelator (prepared from trans-bicyclo[2.2.2]oct-5-ene-2,3-diamine, 1),39 similar to CDTA(Ot-Bu)4 and with a double bond on the opposite side with respect to the amine groups (Scheme 2, 2), was tested for the dealkylation reaction with mCPBA. We were interested in checking whether the dealkylation would take place also on such a more rigid and hindered structure and if there would be regioselectivity between the dealkylation and the epoxidation on the alkene. Thus, 2 was prepared (see the ESI†) and reacted in DCM with 2 equivalents of mCPBA at room temperature overnight.
 | ||
| Scheme 2 Reaction between bicyclic unsaturated compound 2 and mCPBA. (i) BrCH2COOt-Bu, K2CO3, ACN, 70 °C, overnight; (ii) mCPBA, DCM, rt, 15 h. | ||
When the reaction mixture was checked by mass spectrometry and NMR spectroscopy (see the ESI†), only the mono-dealkylated product 3 was observed, while the epoxide was absent, confirming the results obtained with CDTA(Ot-Bu)4. Also in this case, no other intermediates (e.g., bis-N-oxide) or poly-dealkylated products were formed. The mono-dealkylation only with mCPBA proved therefore to be reliable for cyclic trans-1,2-diaminoderivatives.
In order to evaluate the importance of the rigidity of the 1,2-diaminotetraacetate moiety on the dealkylation reaction, the more flexible and linear EDTA(Ot-Bu)4, prepared as described in the literature,40 was reacted with a 3-fold excess of mCPBA (Scheme 3) according to the conditions optimized for CDTA derivatives as described above. Differently from what was observed for cyclohexanediamine derivatives, where only the mono-N-oxide formed and successively underwent dealkylation (depending on the amount of mCPBA), 10 min after the addition of the oxidant, the bis-N-oxide intermediate had formed quantitatively and remained unaltered over time (1H NMR and HPLC-MS analyses in Fig. S19†). Because of the electrostatic repulsion between the charges on the N- and O-atoms, the structure is conformationally restrained and the acetic CH2 protons become magnetically non-equivalent, therefore generating a couple of doublets in the 1H NMR spectrum (Fig. S19-A†).
 | ||
| Scheme 3 Dealkylation reaction on EDTA(Ot-Bu)4. (i) mCPBA, DCM, rt, 1–5 h; (ii) FeCl2, DCM, rt, 1 h. | ||
As the reaction did not proceed with any dealkylation, catalytic FeCl2 was added to initiate a non-classical Polonovski transformation. HPLC-MS analysis (Fig. S20†) confirmed the progressive formation of ED3A(Ot-Bu)3 and a small amount of ED2A(Ot-Bu)2, with complete consumption of EDTA(Ot-Bu)4–bisNO within ca. 24 hours. Because of the very small quantity of the bis-dealkylated product, it was not possible to isolate it by semi-preparative HPLC and thus determine whether it consisted of N,N- or N,N′-ED2A(Ot-Bu)2. The information obtained from the analysis of the dealkylation data for EDTA(Ot-Bu)4 is important for understanding the mechanism and the dynamics that lead to the formation of the N-oxide intermediate(s) and, in turn, the mono- or poly-dealkylated products. However, for this compound, the literature procedures for the preparation of ED3A(Ot-Bu)3 and ED2A(Ot-Bu)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41 remain preferred.
41 remain preferred.
The macrocyclic ligand NOTA(Ot-Bu)342 was then investigated, as the mono-dealkylated NODA(Ot-Bu)2 ligand is useful for the synthesis of chelators43 for [Al18F]2+ or other radiometals such as 68Ga3+ or 61/67Cu2+.44 Thus, NOTA(Ot-Bu)3 was reacted with 2, 3, 5 or 7 equivalents of mCPBA, and the reaction was monitored by ESI-MS to evaluate the formation of dealkylation products39 (Scheme 4).
 | ||
| Scheme 4 N-Dealkylation reaction on NOTA(Ot-Bu)3. (i) mCPBA, DCM, rt, 1–5 h; [ii] FeCl2, DCM, rt, 1 h. | ||
After 1 hour (Fig. S21†), a certain amount of the mono-dealkylated product had formed only with the lowest amount of mCPBA (2 eq.), whereas with a 3-fold excess of the oxidizing agent, only the mono-N-oxide intermediate was present. With a larger excess of mCPBA, both NOTA(Ot-Bu)3–NO and NOTA(Ot-Bu)3–bisNO were detected. By increasing the reaction time to 5 hours, more NODA(Ot-Bu)2 was obtained in the reaction with 2 equivalents of mCPBA, while the other mixtures remained basically unchanged (Fig. S22†). In order to try and push the conversion, a catalytic amount of FeCl2 was added and stirring was continued for 1 hour. It was then observed that with 2 equivalents of the oxidizing agent, a substantially complete conversion into the mono-dealkylated product had taken place (Fig. S23†), while the other reactions showed a much more complex composition, with unidentified byproducts formed besides the expected NODA(Ot-Bu)2 and NO1A(Ot-Bu) (Fig. S23†). Under these conditions, the preparation of NOTA(Ot-Bu)2via the protocol reported herein is less convenient than other procedures reported in the literature (e.g., 66% yield after a simple acid–base work-up according to Shetty et al.45).
Finally, the dealkylation procedure was investigated on two compounds bearing non-equivalent tertiary amines: pseudo-macrocyclic AAZTA(Ot-Bu)426 and cyclic PCTA(Ot-Bu)3.46 While the mono-dealkylated hexadentate derivative of AAZTA has been investigated as a chelator for paramagnetic Mn(II) ions47 and for 68Ga labelling,48 the mono-dealkylation of the pyridine-based macrocycle PCTA leads to a ligand that has been used to prepare efficient and stable Mn(II) complexes for MRI applications.49
The investigation on AAZTA(Ot-Bu)4, which bears one exocyclic and two endocyclic amino groups, was carried out with mCPBA in the range of 2–10 equivalents (Scheme 5). After 1 hour at room temperature, according to ESI-MS analysis (Fig. S24†), the reaction with two equivalents of oxidant still showed some unreacted starting material together with mono-N-oxide and traces of mono- and bis-dealkylated products. When 3 equivalents were used, the initial ester reacted completely forming basically mono-N-oxide and small amounts of mono- and bis-dealkylated products. In the mixture with 5 equivalents of mCPBA, two new species were also observed: AAZTA(Ot-Bu)4–bisNO and the N-oxide derivative of the mono-dealkylated product. These two intermediates represented the main components upon reaction with 7 and 10 equivalents of peroxyacid. Remarkably, also in this case, it was not necessary to add any catalyst to promote the removal of an acetate arm from the amines. Moreover, it is to be noted that the N-oxidation(s) and the dealkylation(s) can take place on various sites, and the signals detected by ESI MS can refer to different isomers. Leaving the reaction overnight only led to a more complicated mixture. Instead, when a catalytic amount of FeCl2 was added to the first two samples 1 hour after the addition of mCPBA, a mono-dealkylated product seemed to have formed as the main component: in particular, the HPLC ionic trace corresponding to AAZ3A(Ot-Bu)3 (472 m/z) showed one peak, suggesting that only one regioisomer had selectively formed (Fig. S25†).
 | ||
| Scheme 5 Species observed by ESI+ MS in the reaction mixture between AAZTA(Ot-Bu)4 and mCPBA (2–10 equiv.). (i) mCPBA, DCM, rt, 1 h; (ii) FeCl2, rt, 1 h. The ligand in the box, AAZ3A(Ot-Bu)3, is the product isolated after purification. | ||
The reaction was then carried out on a larger scale (100 mg of AAZTA(Ot-Bu)4) with 3 equivalents of mCPBA, and the mono-dealkylated product was purified by column chromatography and characterized by NMR spectroscopy (Fig. S11 and S12†): the obtained set of signals in both the 1H and 13C spectra showed a limited number of resonances, suggesting a symmetrical structure. Therefore, the nature of the compound was ascribed to the product derived from the detachment of an acetate arm from the exocyclic nitrogen, in agreement with the data reported in the literature for such species.47 However, the final yield (42%) was still sub-optimal with respect to the more convenient one-step procedure (55%) previously reported by our group, i.e., the reaction between 6-amino-6-methylperhydro-1,4-diazepine and ca. 3 equivalents of tert-butyl bromoacetate, followed by chromatographic purification.47
The last polyaminoester whose dealkylation was investigated is PCTA(Ot-Bu)3.46 This species bears a pyridine ring, whose N-atom is known to form a stable N-oxide when reacted with oxidizing agents such as peroxyacids. Unlike the N-oxides that are produced from the tertiary amines described so far, such a moiety does not undergo further transformations and needs to be reduced again to obtain the original aromatic group. Considering the several possible nucleophilic sites, the reaction was then carried out with 3, 4 and 6 equivalents of mCPBA (Scheme 6). After 1 hour the composition of the three samples was the same: the starting material seemed to have completely converted mainly into its tris-N-oxide or into the two possible isomers of the bis-N-oxide of the mono-dealkylated species (Scheme 6 and Fig. S26-A†). After 24 hours, no change in the mixture composition was observed; thus, the mixtures were treated with a 5-fold excess of Na2SO3 to quench the remaining mCPBA and reduce the N-oxides back to amines. ESI and HPLC-MS analyses revealed that the main observable species were the initial PCTA(Ot-Bu)3 and the two isomers of the mono-dealkylated product: 1,4- (asymmetrical) and 1,7-PC2A(Ot-Bu)2 (symmetrical) (Fig. S26-B†). By semi-preparative HPLC, it was possible to isolate (16% yield) and characterize one of the two mono-dealkylated products, which resulted in the asymmetric 1,4-PC2A(Ot-Bu)2 by considering the differentiation of all hydrogen atoms in the 1H NMR spectrum and by comparison with the data reported in the literature.49 An even better assignment of the NMR resonances was possible upon conversion of the obtained free ligand into the Na+ complex (Fig. S13†). Besides these qualitative determinations, it should be highlighted that this N-dealkylation procedure still lacks reliability and convenience for the preparation of either isomer of PC2A(Ot-Bu)2.
 | ||
| Scheme 6 Observed products upon reaction between PCTA(Ot-Bu)3 and mCPBA and successive treatment with Na2SO3. (i) mCPBA (3–6 equiv.), DCM, rt, 1 h; (ii) Na2SO3 (5 equiv.), H2O, rt, overnight. | ||
The results described above (summarized in Table 1) allow us to draw some conclusions: (i) only in the case of EDTA(Ot-Bu)4 and in the presence of a large excess of mCPBA, it was necessary to add the iron catalyst to achieve the dealkylation of a tertiary amine; (ii) in the other cases, it was sufficient to use an excess of peroxyacid to induce the formation of the N-oxide that successively converted into the targeted product.
| CDTA(Ot-Bu)4 | 2 | EDTA(Ot-Bu)4 | NOTA(Ot-Bu)3 | AAZTA(Ot-Bu)4 | PCTA(Ot-Bu)3 | |
|---|---|---|---|---|---|---|
| a Y = observed; N = not observed. In parentheses: equivalents of mCPBA required. | ||||||
| Mono-NO | Y | Y | N | Y (3) | Y (2,3,5) | N |
| Poly-NO | N | N | Y (3) | Y (5,7) | Y (5,7,10) | Y (3,4,6) |
| Mono-dealk without Fe-cat | Y | Y | N | Y (2) | Y (2,3) | Y (3,4,6) |
| Poly-dealk without Fe-cat | N | N | N | N | Y (2,3) | N |
| Mono-dealk with Fe-cat | Y | Y | Y (3) | Y (2) | Y (2,3) | n/d |
| Poly-dealk with Fe-cat | N | N | Y (3) | Y (3,5,7) | n/d | n/d |
On this basis, it was hypothesized that a [1,2]-Meisenheimer rearrangement50 takes place with the formation of a tert-butyl 2-((dialkylamino)oxy)acetate, followed by protonation of the amino group by the 3-chlorobenzoic acid derived from the first oxidation step and elimination of tert-butyl 2-oxoacetate (tert-butyl glyoxylate) that probably is readily oxidized to mono-tert-butyl oxalate by the Bayer–Villiger reaction51 with a second equivalent of mCPBA (Scheme 7). This hypothesis can be supported by the absence of the aldehyde resonances in the NMR spectra of the reaction between CDTA(Ot-Bu)4 and mCPBA (Fig. 2).
 | ||
| Scheme 7 Proposed mechanism for the iron-free dealkylation based on a [1,2]-Meisenheimer rearrangement followed by a Baeyer–Villiger oxidation. | ||
Another interesting observation concerns the regiochemistry of the substrates. In the case of forcibly oriented amines, two N-oxide moieties likely cannot form when the dihedral angle constrains them to stay too close in space. This is the case for trans-1,2-cyclohexanediamine derivatives, where the 60° dihedral angle would imply that the positive nitrogen ions bear a strong Coulomb repulsion. Therefore, only one N-oxide can form and consequently undergo dealkylation as experimentally observed for CDTA(Ot-Bu)4 and compound 2. On the other hand, the rotational freedom of EDTA derivatives allows the placement of the cations in anti-periplanar positions, thus minimizing the electrostatic forces. When the nitrogen atoms are [also] part of a ring structure, like for NOTA(Ot-Bu)3, AAZTA(Ot-Bu)4 and PCTA(Ot-Bu)3, the situation becomes more complicated and can lead to multiple sites of oxidation with different possible dealkylation pathways. In the case of the AAZTA derivative, the exocyclic nitrogen is a preferential site for the formation of the N-oxide and successive detachment of the alkyl group probably for steric reasons; the main/only regioisomer that forms is the 1,4,N-trisubstituted isomer, while the 1,N,N-isomer was not observed. With PCTA(Ot-Bu)3, a situation similar to that for EDTA probably occurs because of the alternate direction of the nitrogens’ lone pairs, and even the tris-NO intermediate can form. Dealkylation can therefore take place leading to, for example, the symmetrical and asymmetrical isomers of PC2A(Ot-Bu)2.
Conclusions
An investigation was carried out on the dealkylation of exhaustively-substituted polyamino–polyesters, commonly used as precursors for important chelating ligands for metal ions of diagnostic and therapeutic interest. The non-classical Polonovski reaction was chosen, which reportedly includes two steps: the reaction with meta-chloroperoxybenzoic acid to form an N-oxide moiety, followed by the addition of an iron(II)-based catalyst to promote the detachment of the alkyl chain. However, we demonstrated that in most of the investigated cases, dealkylation takes place without the need for the metal catalyst when an excess of mCPBA is used. A mechanism based on the [1,2]-Meisenheimer rearrangement was proposed for this reaction. The N-dealkylation reaction was particularly successful for some constrained substrates resulting in higher yields than those reported in the literature for the synthesis of analogous systems but via longer pathways. For example, CD3A(Ot-Bu)3 was obtained in 70% yield from trans-1,2-cyclohexanediamine in 2 steps, compared to a maximum of 58% in the previously reported procedures. On the other hand, this procedure is not advantageous with respect to the literature ones for some of the considered model compounds, at least at this stage of development, due to the formation of byproducts and lower yields. In any case, the N-dealkylation reaction was confirmed on macro- or mesocyclic ligands such as NOTA, PCTA and AAZTA, whose mono-dealkylated derivatives have already been used for MRI or nuclear medicine applications, and therefore it is of high interest to optimize it in terms of oxidant and other reaction conditions (time, temperature, solvents, etc.), as it definitely represents an important improvement for the synthesis of already widely used partially-alkylated ligands, as well as an opportunity for the development of new ones.Author contributions
J. M: conceptualization, data curation, formal analysis, investigation, methodology, project administration, supervision, validation, visualization, and writing – original draft and review & editing. E. M: data curation, formal analysis, investigation, methodology, visualization, and writing – review & editing. A. M: data curation, investigation, and writing – review & editing. L. T: conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, visualization, and writing – original draft and review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
L.T. and A.M. acknowledge the EU under the Next Generation program for support of the PRIN 2022 project (ID 20224B4285).References
- A. A. Najmi, R. Bischoff and H. P. Permentier, N-Dealkylation of Amines, Molecules, 2022, 27, 3293–3326 CAS.
- S. Thavaneswaran, K. McCamley and P. J. Scammells, N-Demethylation of Alkaloids, Nat. Prod. Commun., 2006, 1, 885–897 CAS.
- J. V. Braun, Untersuchungen Über Morphium-Alkaloide, Ber. Dtsch. Chem. Ges., 1914, 47, 2312–2330 Search PubMed.
- H. A. Hageman, The von Braun Cyanogen Bromide Reaction, in Organic Reactions, 2011, pp. 198–262 Search PubMed.
- J. Morris, L. Kovács and K. Ohe, Cyanogen Bromide, Encyclopedia of Reagents for Organic Synthesis (EROS), 2015, pp. 1–8 Search PubMed.
- J. Cooley and E. Evain, Amine Dealkylations with Acyl Chlorides, Synthesis, 1989, 1–7 CrossRef CAS.
- J. Marton, C. Simon, S. Hosztafi, Z. Szabó, Á. Márki, A. Borsodi and S. Makleit, New Nepenthone and Thevinone Derivatives, Bioorg. Med. Chem., 1997, 5, 369–382 CrossRef CAS.
- A. Just Pedersen, L. Ambach, S. König and W. Weinmann, Electrochemical Simulation of Phase I Metabolism for 21 Drugs Using Four Different Working Electrodes in an Automated Screening Setup with MS Detection, Bioanalysis, 2014, 6, 2607–2621 CrossRef CAS PubMed.
- J. Santamaria, M. T. Kaddachi and J. Rigaudy, Electron-Transfer Activation. Photocyanation of Tertiary Amines, Tetrahedron Lett., 1990, 31, 4735–4738 CrossRef CAS.
- C. Shen, H. Liu, W. Dai, X. Liu, J. Liu and B. Yu, Specific N-Demethylation of Verapamil by Cytochrome P450 from Streptomyces Griseus ATCC 13273, Eng. Life Sci., 2019, 19, 292–301 CrossRef CAS.
- B. Gutmann, U. Weigl, D. P. Cox and C. O. Kappe, Batch- and Continuous-Flow Aerobic Oxidation of 14-Hydroxy Opioids to 1,3-Oxazolidines—A Concise Synthesis of Noroxymorphone, Chem.- Eur. J., 2016, 22, 10393–10398 CrossRef CAS PubMed.
- D. Hollmann, S. Bähn, A. Tillack and M. Beller, N-Dealkylation of Aliphatic Amines and Selective Synthesis of Monoalkylated Aryl Amines, Chem. Commun., 2008, 3199–3201 RSC.
- Z. Ling, L. Yun, L. Liu, B. Wu and X. Fu, Aerobic Oxidative N-Dealkylation of Tertiary Amines in Aqueous Solution Catalyzed by Rhodium Porphyrins, Chem. Commun., 2013, 49, 4214–4216 CAS.
- J. Genovino, D. Sames and B. B. Touré, Access to Drug metabolites via C–H functionalization: Copper-catalyzed aerobic oxidation of N,N-Dimethylalkylamines in complex pharmacauticals, Tetrahedron Lett., 2015, 56, 3066–3069 CAS.
- M. Polonovski, Bull. Soc. Chim. Fr., 1929, 41, 1190–1208 Search PubMed.
- D. Grierson, The Polonovski Reaction, in Organic Reactions, 2004, pp. 85–295 Search PubMed.
- G. J. Kubas, A. C. Larson and R. R. Ryan, Reactions of Quinuclidine N-Oxide and Other Amine Oxides with Sulfur Dioxide. Structure of Quinuclidine Sulfur Trioxide, J. Org. Chem., 1979, 44, 3867–3871 CAS.
- I. Monković, H. Wong and C. Bachand, Secondary amines from the Iron(II) Ion-Catalyzed Reaction of Amine Oxides: A General Method for the Dealkylation of Tertiary Amines, Synthesis, 1985, 770–773 Search PubMed.
- A. Mary, D. Z. Renko, C. Guillou and C. Thal, Selective N-Demethylation of Galanthamine to Norgalanthamine via a Non-Classical Polonovski Reaction, Tetrahedron Lett., 1997, 38, 5151–5152 CAS.
- E. W. Price and C. Orvig, Matching Chelators to Radiometals for Radiopharmaceuticals, Chem. Soc. Rev., 2014, 43, 260–290 CAS.
- L. Lattuada, A. Barge, G. Cravotto, G. B. Giovenzana and L. Tei, The Synthesis and Application of Polyamino Polycarboxylic Bifunctional Chelating Agents, Chem. Soc. Rev., 2011, 40, 3019–3049 CAS.
- F. K. Kalman and G. Tircso, Kinetic Inertness of the Mn2+ Complexes Formed with AAZTA and Some Open-Chain EDTA Derivatives, Inorg. Chem., 2012, 51, 10065–10067 CrossRef CAS.
- E. Szilagyi and E. Brucher, Kinetics and mechanisms of formation of the Lanthanide(III)-trans-1,2-Diaminocyclohexane-N,N,N′N′-Tetraacetate Complexes, J. Chem. Soc., Dalton Trans., 2000, 2229–2233 RSC.
- E. T. Clarke and A. E. Martell, Stabilities of Trivalent Metal-Ion Complexes of the Tetraacetate Derivatives of 12-Membered, 13-Membered and 14-Membered Tetraazamacrocycles, Inorg. Chim. Acta, 1991, 190, 37–46 CAS.
- S. Aime, M. Botta, S. G. Crich, G. B. Giovenzana, G. Jommi, R. Pagliarin and M. Sisti, Synthesis and NMR Studies of Three Pyridine-Containing Triaza Macrocyclic Triacetate Ligands and Their Complexes with Lanthanide Ions, Inorg. Chem., 1997, 36, 2992–3000 CAS.
- S. Aime, L. Calabi, C. Cavallotti, E. Gianolio, G. B. Giovenzana, P. Losi, A. Maiocchi, G. Palmisano and M. Sisti, [Gd-AAZTA]-: A New Structural Entry for an Improved Generation of MRI Contrast Agents, Inorg. Chem., 2004, 43, 7588–7590 CAS.
- M. Botta, F. Carniato, D. Esteban-Gomez, C. Platas-Iglesias and L. Tei, Mn(II) Compounds as an Alternative to Gd-Based MRI Probes, Future Med. Chem., 2019, 11, 1461–1483 CAS.
- R. A. Jessop and J. R. L. Smith, Amine Oxidation. Part XII. Reactions of Some N,N-Dimethylbenzylamine N-Oxides with Acetic Anhydride and of Some N-Acetoxy-N,N-Dimethylbenzylammonium Perchlorates with Acetate Ion. The Polonovski Reaction, J. Chem. Soc., Perkin Trans. 1, 1976, 1801–1805 CAS.
- F. Cleeren, J. Lecina, J. Bridoux, N. Devoogdt, T. Tshibangu, C. Xavier and G. Bormans, Direct fluorine-18 labeling of heat-sensitive biomolecules for Positron Emission Tomography imaging using the Al18F-RESCA method, Nat. Protoc., 2018, 13, 2330–2347 CrossRef CAS PubMed.
- F. Cleeren, J. Lecina, E. M. F. Billaud, M. Ahamed, A. Verbruggen and G. M. Bormans, New Chelators for Low Temperature Al18F-Labeling of Biomolecules, Bioconjugate Chem., 2016, 27, 790–798 CrossRef CAS PubMed.
- F. Cleeren, J. Lecina, M. Ahamed, G. Raes, N. Devoogdt, V. Caveliers, P. McQuade, D. J. Rubins, W. Li, A. Verbruggen, C. Xavier and G. Bormans, Al18F-Labeling of Heat-Sensitive Biomolecules for Positron Emission Tomography Imaging, Theranostics, 2017, 7, 2924–2939 Search PubMed.
- N. Wong, E. Zhao, J. Bao, S. G. Koenig, C. Molinaro, J. C. Timmerman, H. Zhang, C. G. Sowell and F. Gosselin, Practical Synthesis of Reactive Immuno-PET Linker-Chelator (1R,2R)-RESCA-TFP, Org. Process Res. Dev., 2023, 27, 1400–1405 CrossRef CAS.
- W. Sihver, M. Walther, M. Ullrich, A.-K. Nitt-Weber, J. Böhme, F. Reissig, M. Saager, K. Zarschler, C. Neuber, J. Steinbach, K. Kopka, H.-J. Pietzsch, R. Wodtke and J. Pietzsch, Cyclohexanediamine Triazole (CHDT) Functionalization Enables Labeling of Target Molecules with Al18F/68Ga/111In, Bioconjugate Chem., 2024, 35, 1402–1416 Search PubMed.
- M. N. Iannone, S. Valtorta, S. Stucchi, S. Altomonte, E. A. Turolla, E. Vino, P. Rainone, V. Zecca, A. Lo Dico, M. Maspero, M. Figini, M. Bellone, S. Ciceri, D. Colombo, C. Chinello, L. Pagani, R. M. Moresco, S. Todde and P. Ferraboschi, Automated Radiosynthesis and Preclinical Evaluation of Two New PSMA-617 Derivatives Radiolabelled via [18F]AlF2+ Method, EJNMMI Radiopharm. Chem., 2024, 9, 50 Search PubMed.
- T. G. Traylor, T. Nakano, B. E. Dunlap, P. S. Traylor and D. Dolphin, Mechanisms of Hemin-Catalyzed alkene epoxidation. The effect of catalyst on the regiochemistry of epoxidation, J. Am. Chem. Soc., 1986, 108, 2782–2784 Search PubMed.
- A. Mohamadi and L. W. Miller, Efficient Route to Pre-Organized and Linear Polyaminopolycarboxylates: Cy-TTHA, Cy-DTPA and Mono/Di- Reactive, Tert-Butyl Protected ITHA/Cy-TTHA, Tetrahedron Lett., 2017, 58, 1441–1444 CAS.
- E. M. Gale, I. P. Atanasova, F. Blasi, I. Ay and P. Caravan, A Manganese Alternative to Gadolinium for MRI Contrast, J. Am. Chem. Soc., 2015, 137, 15548–15557 CAS.
- A. M. S. Musthakahmed, E. Billaud, G. Bormans, F. Cleeren, J. Lecina and A. Verbruggen, Methods for Low Temperature Fluorine-18 Radiolabeling of Biomolecules, WO2016065435A2, 2016.
- S. A. Moteki, S. M. Xu, S. Arimitsu and K. Maruoka, Design of structurally rigid trans-Diamine-based Tf-Amide organocatalysts with a dihydroanthracene framework for asymmetric conjugate additions of heterosubstituted aldehydes to vinyl sulfones, J. Am. Chem. Soc., 2010, 132, 17074–17076 CrossRef CAS PubMed.
- G. B. Giovenzana, D. Imperio, L. Lattuada and F. Uggeri, Stevens Rearrangement as a Tool for the Structural Modification of Polyaminopolycarboxylic Ligands, Org. Biomol. Chem., 2011, 9, 679–681 RSC.
- G. Bechara, N. Leygue, C. Galaup, B. Mestre-Voegtlé and C. Picard, Polyazamacrocycles based on a Tetraaminoacetate moiety and a (Poly)Pyridine intracyclic unit: direct synthesis and application to the photosensitization of Eu(III) and Tb(III) ions in aqueous solutions, Tetrahedron, 2010, 66, 8594–8604 CrossRef CAS.
- Y. Huang, Y. Liu, S. Liu, R. Wu and Z. Wu, An Efficient Synthesis of N,N,N-Substituted 1,4,7-Triazacyclononane, Eur. J. Org. Chem., 2018, 2018, 1546–1551 Search PubMed.
- C. A. D'Souza, W. J. McBride, R. M. Sharkey, L. J. Todaro and D. M. Goldenberg, High-Yielding Aqueous F-18-Labeling of Peptides via (AlF)-F-18 Chelation, Bioconjugate Chem., 2011, 22, 1793–1803 Search PubMed.
- T. Joshi, M. Kubeil, A. Nsubuga, G. Singh, G. Gasser and H. Stephan, Harnessing the Coordination Chemistry of 1,4,7-Triazacyclononane for Biomimicry and Radiopharmaceutical Applications, ChemPlusChem, 2018, 83, 554–564 Search PubMed.
- D. Shetty, S. Y. Choi, J. M. Jeong, J. Y. Lee, L. Hoigebazar, Y.-S. Lee, D. S. Lee, J.-K. Chung, M. C. Lee and Y. K. Chung, Stable Aluminium Fluoride Chelates with Triazacyclononane Derivatives Proved by X-Ray Crystallography and 18F-Labeling Study, Chem. Commun., 2011, 47, 9732–9734 CAS.
- M. Enel, N. Leygue, N. Saffon, C. Galaup and C. Picard, Facile Access to the 12-Membered Macrocyclic Ligand PCTA and Its Derivatives with Carboxylate, Amide, and Phosphinate Ligating Functionalities, Eur. J. Org. Chem., 2018, 1765–1773 CAS.
- L. Tei, G. Gugliotta, M. Fekete, F. K. Kalman and M. Botta, Mn(II) Complexes of Novel Hexadentate AAZTA-like Chelators: A Solution Thermodynamics and Relaxometric Study, Dalton Trans., 2011, 40, 2025–2032 CAS.
- J. Seemann, B. P. Waldron, F. Roesch and D. Parker, Approaching ‘Kit-Type’ Labelling with 68Ga: The DATA Chelators, ChemMedChem, 2015, 10, 1019–1026 Search PubMed.
- Z. Garda, E. Molnár, N. Hamon, J. L. Barriada, D. Esteban-Gómez, B. Váradi, V. Nagy, K. Pota, F. K. Kálmán, I. Tóth, N. Lihi, C. Platas-Iglesias, É. Tóth, R. Tripier and G. Tircsó, Complexation of Mn(II) by Rigid Pyclen Diacetates: Equilibrium, Kinetic, Relaxometric, Density Functional Theory, and Superoxide Dismutase Activity Studies, Inorg. Chem., 2021, 60, 1133–1148 CAS.
- Z. Wang, Meisenheimer Rearrangement, in Comprehensive Organic Name Reactions and Reagents, 2010, pp. 1889–1892 Search PubMed.
- Z. Wang, Baeyer–Villiger Oxidation, in Comprehensive Organic Name Reactions and Reagents, 2010, pp. 150–155 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; characterization data; 1H and 13C NMR spectra; MS spectra. See DOI: https://doi.org/10.1039/d5ob00114e |
| This journal is © The Royal Society of Chemistry 2025 |