
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomodimerization of 3-substituted-2-oxindoles for the construction of vicinal all-carbon quaternary centers: chemical, photochemical and electrochemical approaches
Sulekha
Sharma
,
Harapriya
Behera
,
Shivani
Ahlawat
 and
Amit
Paul
*
and
Amit
Paul
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER) Bhopal, MP- 462 066, India. E-mail: apaul@iiserb.ac.in
First published on 4th March 2025
Abstract
Advancements in organic synthesis are revolutionizing the synthesis of complex natural products, which are essential in biomedical research and drug discovery due to their intricate structures. Natural products such as chimonanthine, folicanthine, calycanthine, psychotriadine, etc., with vicinal all-carbon quaternary stereocenters, are particularly significant for their strong binding properties and biological activities. One common feature of these natural products is the presence of dimeric 3-substituted-2-oxindoles having vicinal all-carbon quaternary stereocenters. This review focuses on the chemical, photochemical, and electrochemical approaches for the homodimerization of 3-substituted-2-oxindoles employed by different researchers, with a strong focus on the mechanistic details of proton-coupled electron transfer (PCET). The article also demonstrates that PCET facilitates the reduction of kinetic barriers through the formation of low-energy intermediates and the expansion of synthetic possibilities. Furthermore, natural product syntheses (folicanthine and chimonanthine) from dimeric 3-substituted-2-oxindoles are discussed. Chemical syntheses are time-consuming and, even more importantly, generate significant waste due to the use of metal-based oxidants and catalysts. In this regard, electrochemical synthesis methods offer promising solutions by avoiding the use of chemical oxidants and metal catalysts, thus minimizing environmental impact. The article also outlines the advantages and disadvantages of different synthesis methods and proposes a new direction for future research in this field.
 Sulekha Sharma | Sulekha Sharma is currently working as a postdoctoral research associate in the Institute of Chemistry at the University of Graz, Austria. She earned her PhD from IISER Bhopal in 2023 under the mentorship of Prof. Amit Paul. Her doctoral research focused on developing green electrochemical methodologies for synthesizing dimeric 3-substituted-2-oxindoles, which are directly applicable to the synthesis of various biologically important molecules and natural products. In addition to this work, she conducted detailed mechanistic investigations of these electrochemical reactions. |
 Harapriya Behera | Harapriya Behera is currently pursuing her PhD in the Department of Chemistry at the Indian Institute of Science Education and Research (IISER), Bhopal, under the supervision of Prof. Amit Paul. In 2022, she graduated with a master's degree from the National Institute of Technology (NIT), Rourkela. Her research interest is centered on electro-organic synthesis and the mechanistic understanding of such syntheses. |
 Shivani Ahlawat | Shivani Ahlawat is currently pursuing her PhD in the Department of Chemistry, IISER Bhopal, under the guidance of Prof. Amit Paul. She completed her master's degree at Jamia Millia Islamia, New Delhi, in 2020. Her research work is focused on electro-organic synthesis and proton-coupled electron transfer (PCET). |
 Amit Paul | Amit Paul received his B.Sc. from Jadavpur University, M.Sc. from IIT Bombay, and PhD from the University of Pittsburgh under the supervision of Prof. David H. Waldeck. He then worked as a postdoctoral research associate as a part of the Energy Frontier Research Center (EFRC) at the University of North Carolina at Chapel Hill under Prof. Thomas. J. Meyer. In October 2011, he joined the Indian Institute of Science Education and Research (IISER) Bhopal as an Assistant Professor. Since 2023, he has been working as a Professor in the same department. He was awarded the DAE Young Scientist Award in 2013 and the Chirantan Rasayan Sanstha (CRS) Bronze Medal in 2024. His research interests include electro-organic synthesis, electrochemical supercapacitors, heterogeneous water oxidation, solid-state proton conduction, etc. |
Introduction
The field of organic synthesis, focused on producing naturally occurring organic molecules, is undergoing substantial development due to emerging synthesis methodologies. Natural products are pivotal in biomedical research and drug discovery due to their intricate and diverse structures, which have evolved over a very long period of time, optimizing their functions.1–3 Through the application of innovative synthesis methodologies, scientists have been able to replicate some of nature's most fascinating molecules in laboratory environments. In this regard, natural products containing vicinal all-carbon quaternary stereocenters hold particular significance due to their distinctive structural characteristics and their propensity for strong binding to target molecules.1The synthesis of sterically constrained vicinal stereogenic centers has gained remarkable attention from synthetic chemists due to their pervasive structural motifs within complex natural products and pharmaceuticals.4–6 The dimeric hexahydropyrrolo[2,3-b]indole alkaloids, also known as pyrroloindolines (Scheme 1), are particularly fascinating targets as they share contiguous stereogenic centres and exhibit diverse biological properties, including antibacterial and anticancer effects.7–9 In recent years, synthetic chemists have made significant efforts in the synthesis of their representative alkaloids, such as (+)-chimonanthine (I), (+)-folicanthine (II), (−)-calycanthine (III), and psychotriadine (IV) (Scheme 1a).10–19
 | ||
| Scheme 1 (a) Naturally occurring hexahydropyrroloindole-derived family of alkaloids and (b) general structure of 2-oxindole and 3,3′-bisoxindoles. | ||
Specifically, nature-inspired strategies such as C–H oxidative methods have gained significant attention as they eliminate the need to protect and deprotect functional groups.6,20–23 In this regard, 3,3′-bisoxindoles (2) (Scheme 1b) are a class of molecules that play a crucial role as intermediates in the total synthesis of natural products such as chimonanthine and folicanthine.24
However, sterically hindered vicinal all-carbon quaternary stereocenters pose a formidable challenge in synthetic organic chemistry due to the constraints faced by chemical oxidants in approaching these reaction centres.3 Reported methodologies for the synthesis of these types of molecules can be classified into three categories: chemical, photochemical, and electrochemical methods. Despite the advancements that have been made in chemical and photochemical methodologies for the formation of all-carbon quaternary stereocenters, the chemical oxidant-, organocatalyst- or metal-catalyst-free construction of vicinal all-carbon quaternary stereocenters remains a considerable challenge in contemporary organic synthesis for making the synthesis process greener. Research in the development of efficient methods has shown that electrochemical methodologies are valuable as they avoid the use of chemical oxidants or metal catalysts in reactions and are, therefore, atom-economical and environment-friendly.25–29 These methodologies are considered green since they utilize the electrode potential to drive oxidation and reduction reactions, minimizing the need for traditional chemical oxidants or reductants.30 Furthermore, harnessing the electrode potential to match the specific potential required for substrate oxidation/reduction allows precise control of redox reactions.30,31 Moreover, this approach is considered inherently sustainable since electricity can be generated from renewable energy resources such as solar, wind, geothermal, etc.25
On the other hand, the interaction between electron and proton transfers has been a significant focus in both experimental and theoretical studies in chemistry and biochemistry. A key concept in this area is that electron acceptance often requires the addition of an acid, while electron donation requires a base. In this regard, proton-coupled electron transfer (PCET)32–34 represents redox mechanisms wherein electron and proton transfers occur via stepwise or concerted pathways, enabling energy-efficient bond cleavage or formation35 by avoiding the formation of high-energy intermediates.35 PCET mechanisms have gained remarkable attention in various domains of chemistry/catalysis,36 with broad applications in numerous biological redox catalysis such as photosynthesis,37,38 enzymatic C–H oxidation,39,40 energy conversion, etc.35,41–43 Despite these advancements, the utilization of PCET in electro-organic synthesis has not received great attention and requires further exploration due to its role in lowering the kinetic barrier.33
This review article discusses the construction of structurally diverse homo-dimeric vicinal all-carbon quaternary centers according to reaction types in a systematic manner (chemical, photochemical and electrochemical) and the most probable reaction mechanisms that the reactions follow in terms of electron and proton transfer. The primary focus of this article is on the formation of sterically constrained 3,3′-C(sp3)–C(sp3) bonds (shown in red in Scheme 1), which, despite being longer than the typical C(sp3)–C(sp3) single bond (1.54 Å), are still commonly found in nature.
Chemical approaches for dimerization of 2-oxindole
Chemical strategies for the dimerization of 2-oxindole are particularly valuable as they enable the synthesis of intricate dimeric products with control over stereochemistry and regiochemistry, which are crucial for tailoring biological activity. Advances in catalytic systems, reaction conditions, and mechanistic understanding have expanded the scope of 2-oxindole dimerization, facilitating the synthesis of diverse dimeric frameworks. This reaction is of considerable interest not only for its synthetic utility but also for its potential applications in drug discovery, wherein dimeric 2-oxindole derivatives are explored for their therapeutic potential.3 In this context, the dimerization of 2-oxindole through chemical synthesis is a topic of ongoing research, with a significant focus on developing efficient, selective, and sustainable methodologies that contribute to the broader research area of complex molecule synthesis. In the following section, the dimerization of 2-oxindole through chemical routes is discussed, focusing on the synthesis methodologies, challenges, and significance of this reaction in the broader context of organic synthesis.The dimerization of 2-oxindole at the C-3 position to form an all-carbon quaternary center was first described by Rodrigo et al. in 1994.11 They performed the reaction of 3-alkylcarboxylate-2-oxindole (3) with 1.05 M sodium hydride in tetrahydrofuran (THF), followed by addition of 0.48 mol of carbon tetraiodide (CI4) in tetrahydrofuran (THF) at −65 °C, which resulted in a mixture from which 53% of the desired dimer (4) was isolated (Scheme 2). The mechanism of the reaction involved a radical anion chain reaction, which involved an initial electron transfer (ET) from the oxindole enolate (5) (R−) formed after the removal of a proton by a base (NaH) to 3-(triiodomethyl)oxindole (6). This step was followed by the chain propagation steps, which finally led to the dimerized product (4) (Scheme 2).
 | ||
| Scheme 2 Dimerization of 3-alkylcarboxylate-2-oxindole and the radical anion chain mechanism proposed by Rodrigo et al.11 | ||
In 2013, Kim and coworkers developed a method for the direct dimerization of 3-substituted-2-oxindole derivatives using manganese(III) acetate or copper acetate/silver acetate as catalysts.44 In their initial attempts, employing palladium acetate as the catalyst and 3-substituted-2-oxindole (7) as the substrate, they achieved a 74% yield of the oxindole dimer (8), along with the minor byproduct 3-hydroxy oxindole (9) (Scheme 3). The use of Cu(OAc)2 and AgOAc under an O2 atmosphere resulted in a moderate 78% yield of 3-hydroxy oxindole (9) in a relatively short reaction time of 4 h. However, when AgOAc was used alone under the same conditions, 3-hydroxy-2-oxindole (9) was not detected. Similarly, Cu(OAc)2 alone under O2 was largely ineffective, yielding less than 5% of the oxindole dimer products. Increasing the amount of Cu(OAc)2 improved the yield of the oxindole dimer products to 63% but required a prolonged reaction time of 72 h. Interestingly, switching the reaction to a N2 atmosphere with Cu(OAc)2 and AgOAc further improved the yields to 82%, though the reaction time had to be extended to 36 h. The use of Mn(OAc)3 under a N2 atmosphere provided the best results, affording a 90% yield in just 4 h with no byproducts (Scheme 3).44 The proposed reaction mechanism followed a radical generation pathway, which involved an oxidative H-atom loss to generate a neutral radical species (10) catalyzed by Mn(OAc)3, wherein Mn was likely simultaneously reduced from the +3 to the +2 oxidation state (Scheme 3). The details regarding the involvement of the catalyst in the oxidation step have not been discussed. Thereafter, homocoupling of the radical species (10) resulted in the dimeric oxindole (8). The formation of the neutral radical intermediate was supported by the increased formation of the side product 3-hydroxy oxindole (9) under an O2 atmosphere.
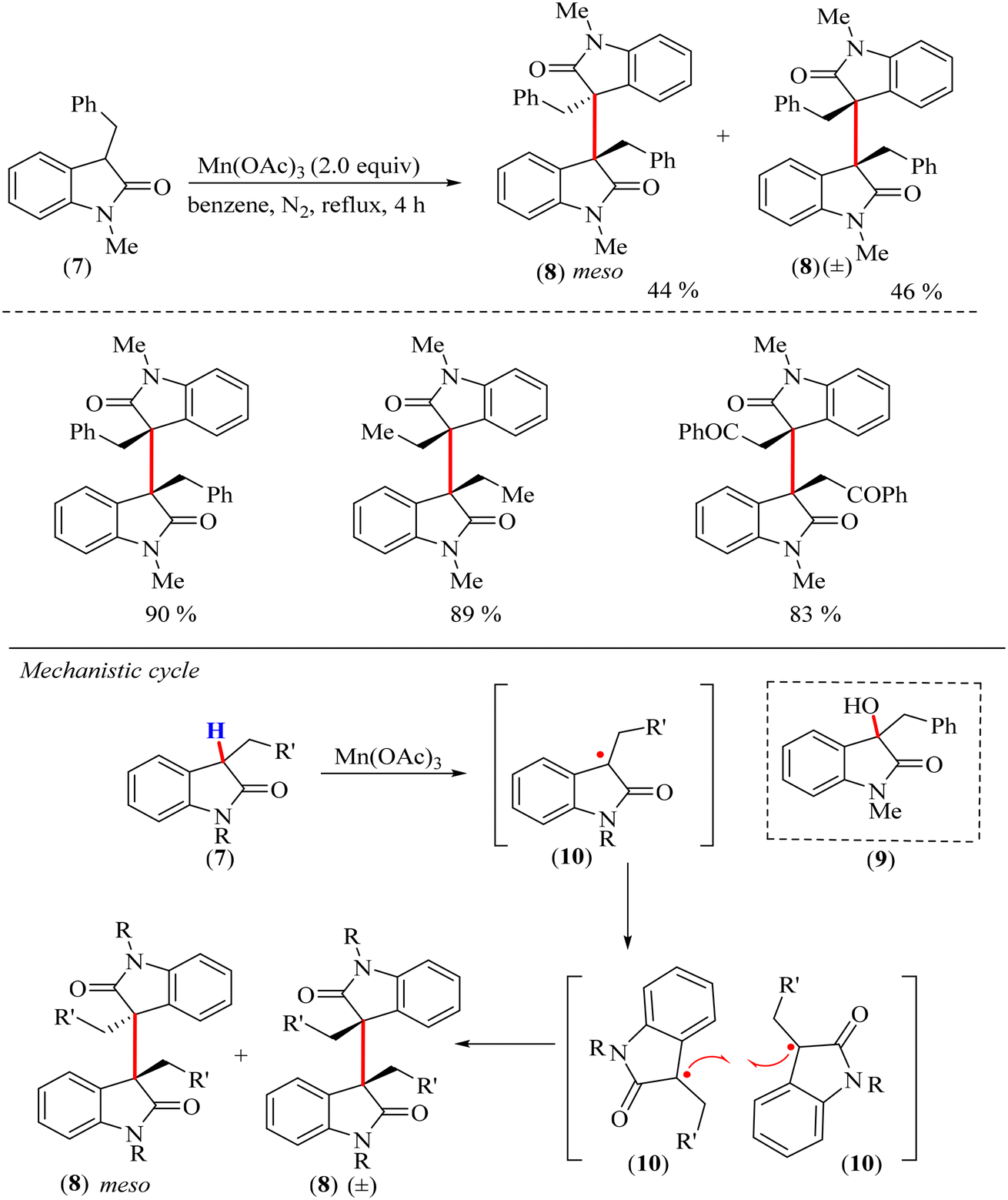 | ||
| Scheme 3 Dimerization of 3-substituted-2-oxindole (7) derivatives and the mechanism for the reaction utilizing manganese(III) acetate as a catalyst. | ||
In 2015, Bisai et al. developed a metal-free methodology for the oxidative coupling of 3-carboxylate-2-oxindoles (11) (Scheme 4). They used 1.2 equiv. of potassium tert-butoxide as a base and 1.2 equiv. of iodine as an oxidant and achieved yields in the range of 70–84% for the dimeric products (12) with a diastereomeric ratio of up to 3.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.24 The reaction mechanism involved a radical-mediated oxidative coupling wherein the base abstracted a proton (PT) from the enol form of the substrate (13) to generate an enolate anion (14). Thereafter, an electron transfer (ET) from the enolate (14) to the oxidant led to the formation of a 3° radical intermediate (15), which was stabilized by resonance. The reaction between two neutral radicals (15) led to the formation of the dimeric oxindole product (12) (Scheme 4). This work represents a significant milestone in the homodimerization of 3-substituted-2-oxindoles by developing a “transition-metal-free reaction” methodology.
:1.24 The reaction mechanism involved a radical-mediated oxidative coupling wherein the base abstracted a proton (PT) from the enol form of the substrate (13) to generate an enolate anion (14). Thereafter, an electron transfer (ET) from the enolate (14) to the oxidant led to the formation of a 3° radical intermediate (15), which was stabilized by resonance. The reaction between two neutral radicals (15) led to the formation of the dimeric oxindole product (12) (Scheme 4). This work represents a significant milestone in the homodimerization of 3-substituted-2-oxindoles by developing a “transition-metal-free reaction” methodology.
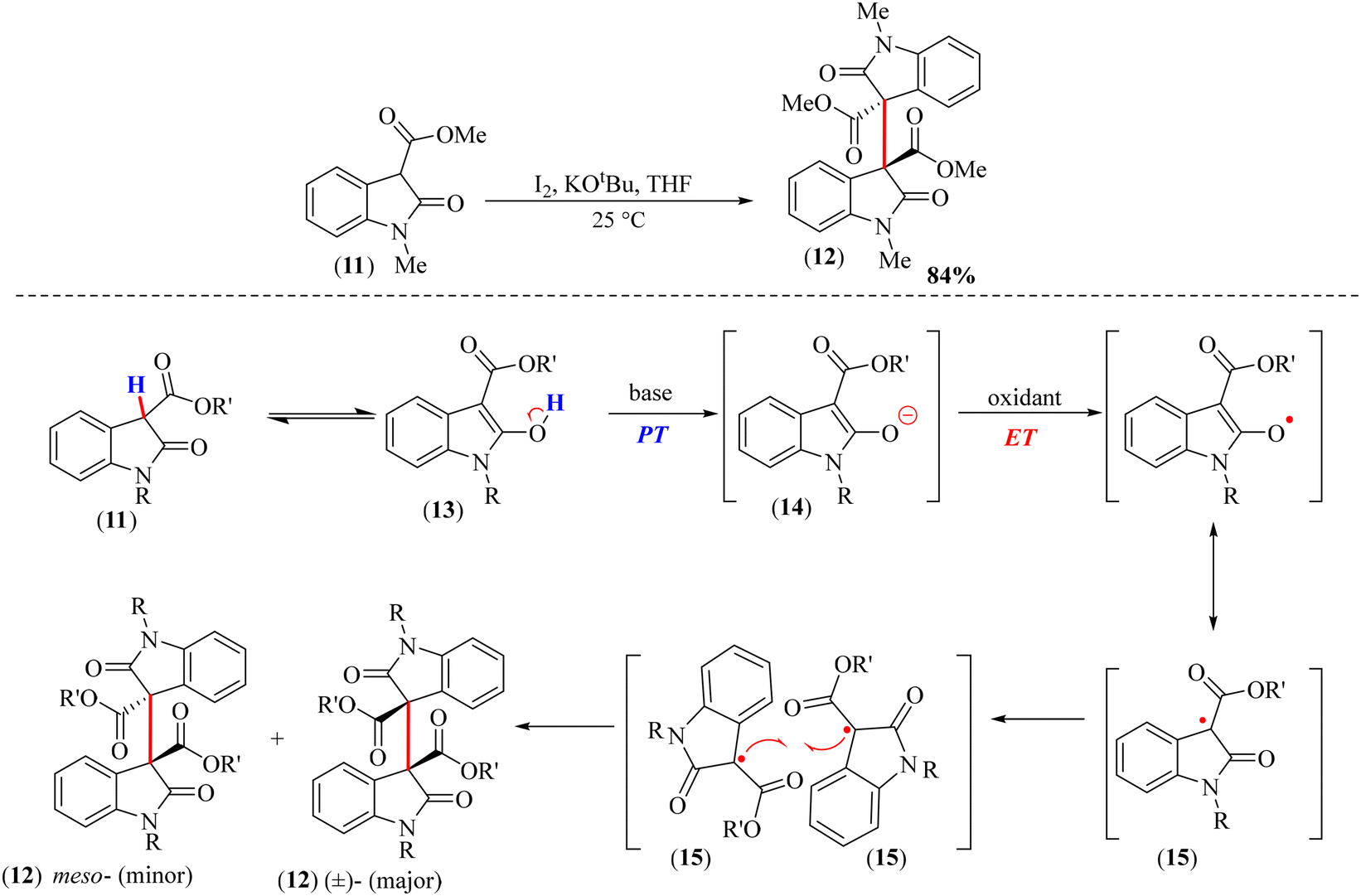 | ||
| Scheme 4 Dimerization of 3-carboxylate-2-oxindole (11) in the presence of potassium tert-butoxide as a base and iodine as an oxidant, with the reaction mechanism. | ||
In 2017, Ooi and co-workers reported an intramolecular ion-pairing electron transfer catalyst, acridinium phenoxide (17), that combines a redox-active component with a basic site within a single molecular framework.43 The effectiveness of acridinium phenoxide as a chemical redox catalyst was demonstrated in the homodimerization of 3-aryl-2-oxindoles (16).43 By employing 2 mol% of the catalyst (17) and 3-aryl-2-oxindoles (16) under an oxygen-free atmosphere at room temperature in toluene with 3 h of stirring, the desired dimeric 3-aryl-2-oxindole (18) was isolated in 82% yield (Scheme 5). The mechanism of this transformation was proposed to follow a PCET pathway. A detailed mechanistic investigation, including kinetic studies, was conducted using in situ infrared (IR) spectroscopy, which revealed that the catalyst (17) significantly accelerated the reaction. The proposed mechanism involved an initial establishment of equilibrium between acridinium phenoxide (17) and 16, followed by sluggish single-electron transfer (ET) from the enolate ion of 16 to the acridinium unit, generating radical pair 19 (Scheme 5). Thereafter, this pair underwent instantaneous single-electron oxidation by O2 to produce a radical enolate of the substrate (19) for subsequent coupling and an ionic radical pair 17·H·[1/2O2]˙. From this ionic radical pair, 17 was regenerated either through the abstraction of the internal phenolic proton by the radical anion of O2 ([1/2O2]˙−) or through the deprotonation of 16 by [1/2O2]˙− to provide 17·16 (Scheme 5). The role of O2 was crucial, as evidenced by the low yield of the dimeric product (18) under argon, and hydrogen peroxide was detected as a byproduct, supporting the PCET mechanism.
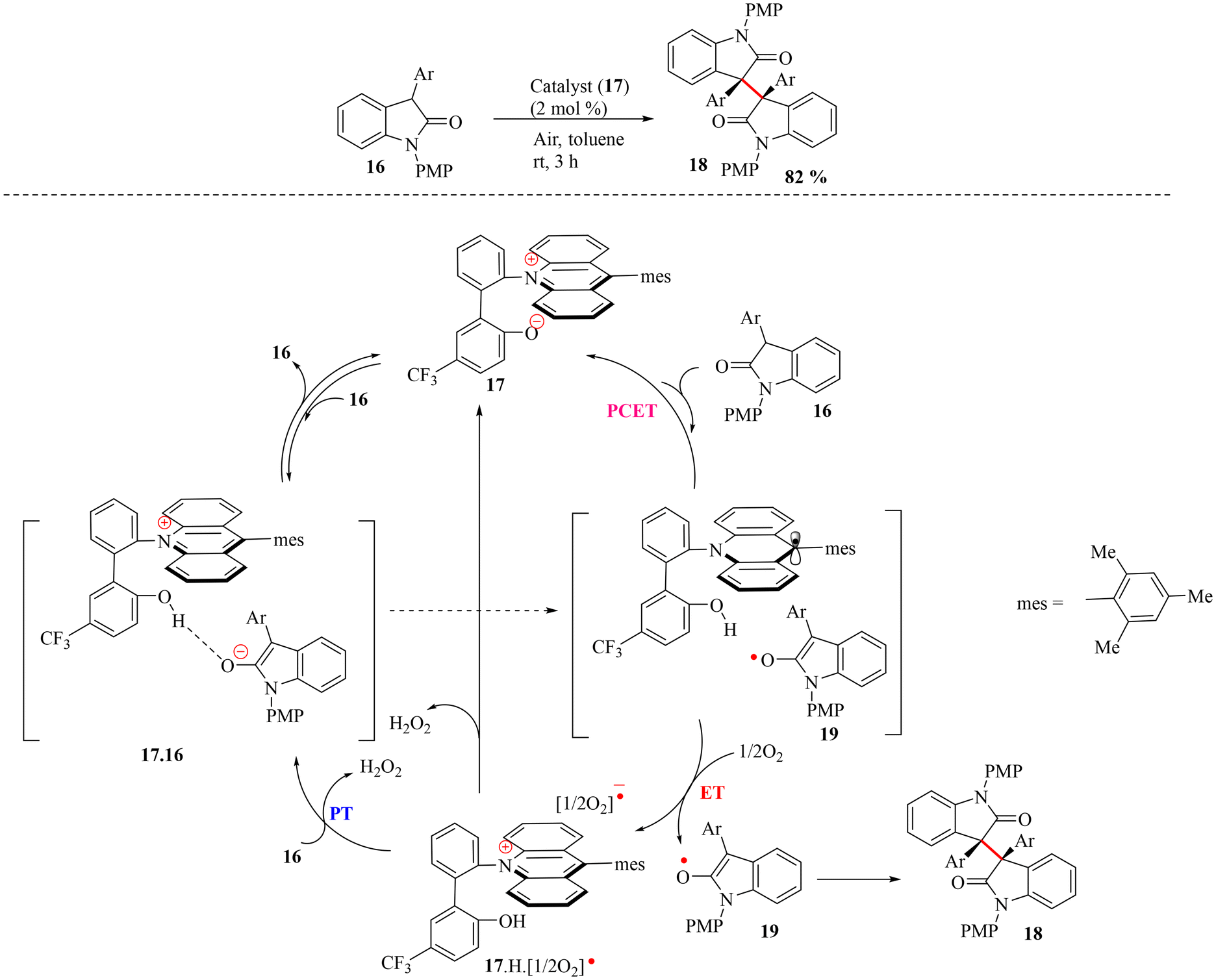 | ||
| Scheme 5 Dimerization of 3-aryl-2-oxindole by an acridinium phenoxide catalyst and the reaction mechanism by Ooi and coworkers.43 | ||
In 2018, Wei et al. developed a copper-catalyzed oxidative dimerization of 3-substituted-2-oxindoles (20) employing di-tert-butyl peroxide (DTBP) (21) as the radical initiator under base-free conditions (Scheme 6).45 For optimization, initial experiments were carried out under base-free conditions with Cu(OAc)2 as the catalyst and DTBP as the oxidant, achieving 80% yield of the dimer product (22) after 4 h at 120 °C. Further optimization revealed that the absence of either Cu(OAc)2 or DTBP resulted in significantly reduced yields. Alternatively, copper catalysts, including CuSO4, CuCl2, CuBr2, CuI, and CuO, as well as different oxidants such as tert-butyl hydroperoxide (TBHP) and K2S2O8, did not surpass the performance of the original conditions. Solvent screening identified 1,4-dioxane as the most effective, providing an 80% yield, whereas solvents such as toluene and DMF resulted in lower yields. This method offered an efficient pathway for synthesizing diverse dimeric oxindoles (2) and was a straightforward strategy for achieving C(sp3)–H functionalization and, subsequently, C(sp3)–C(sp3) bond formation.45 The transformation occurred through a mechanism wherein the hydrogen atom (considered as concerted ET and PT from a single bond) of 20 was abstracted by the t-BuO˙ radical (22), generated from DTBP (21) under heating conditions with the aid of Cu(I) species (Scheme 6). This led to the generation of a key neutral radical intermediate of 3-substituted-2-oxindole (23) and Cu(II)Ot−Bu (24). Subsequently, the enol tautomer of 3-substituted-2-oxindole (20) reacted with the 3-substituted-2-oxindole radical (23) and Cu(II)Ot−Bu (24), which resulted in the formation of intermediate 25 (Scheme 6) and an O to C migration to generate another intermediate (26). The dimerization product of 3-substituted-2-oxindoles (27) was obtained in the final step through a reductive elimination pathway (Scheme 6).
 | ||
| Scheme 6 Copper-catalyzed dimerization of 3-substituted-2-oxindole and the mechanism by Wei and coworkers.45 | ||
In 2021, Petersen and co-workers developed a new method to synthesize dimeric oxindoles from acyclic β-oxoanilides using Mn(OAc)3·2H2O as the catalyst.46 This novel one-step process involved oxidative cross-dehydrogenative cyclization, fragmentation, and dimerization, characterized by C(sp2)–H/C(sp3)–H activation. The reaction yielded dimeric oxindoles from aldehyde and carboxylic acid with 92% and 81% yields, respectively, demonstrating its versatility across a range of substrates and its applicability in the formal synthesis of (±)-folicanthine (Scheme 7).46 The proposed mechanism involved the initial formation of oxindole 30 from 28via two sequential oxidative single electron transfer (ET) processes, followed by proton transfer (PT), i.e. an ET–ET–PT step. Deprotonation of oxindole (30) generated a carboxylate anion, which underwent an oxidative third ET process, forming a methine radical (32) after concomitant loss of CO2. The neutral radical (32) then homocoupled to form dimeric oxindole (33) (Scheme 7). Similarly, 29 underwent two consecutive oxidative ET steps, followed by one PT to form oxindole (31). In the next step, the methine radical (32) was generated from 31 through deformylation processes mediated by polynuclear metal complexes. In the final step, 32 homocoupled to form dimeric oxindole (33). The neutral radical species (32) was successfully trapped by 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as a radical scavenger, aligning with the cascade sequence hypothesis. The detailed involvement of the catalyst (Mn(OAc)3) in the reaction steps was not discussed. This work introduced a new dimension to this field by forming three C–C bonds in a single one-pot reaction, shown in red in Scheme 7.
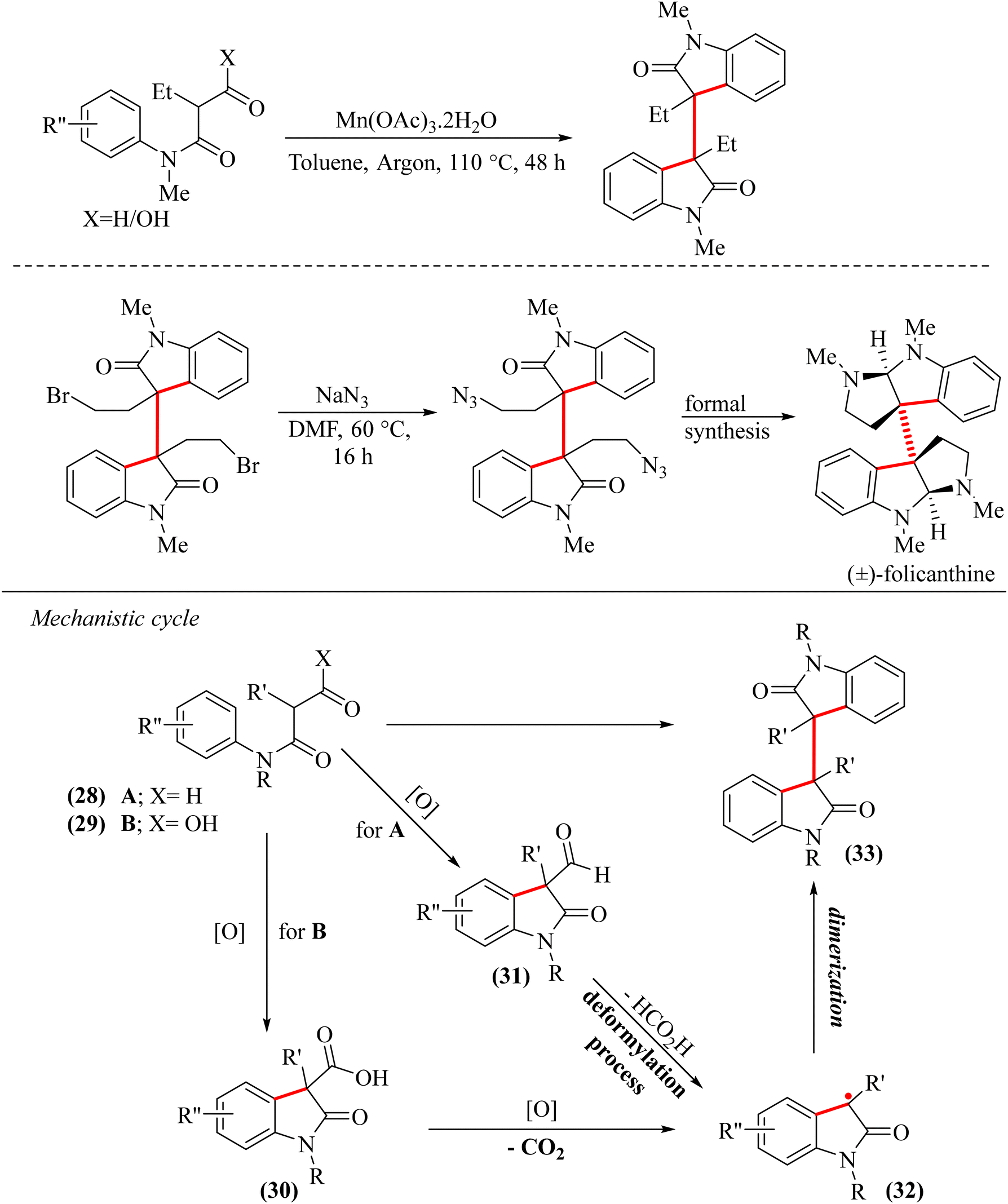 | ||
| Scheme 7 Cross-dehydrogenative cyclization and dimerization for the synthesis of 3,3′-bisoxindole and the PCET mechanism by Peterson and coworkers.46 | ||
In 2023, Nagasawa and co-workers developed an oxidative enolate coupling reaction involving oxindoles that yielded optically active oxindole dimers, which featured consecutive all-carbon quaternary stereogenic centers. This reaction was facilitated by a chiral guanidinium iodide catalyst (34) and cumene hydroperoxide (CHP) as an oxidant, which resulted in the formation of the chiral dimeric oxindole (R,R-(40)) in up to 99% yield with 99% enantioselectivity and a diastereomeric ratio up to 19:1 (dl:meso) (Scheme 8).47 The reaction mechanism was supported by control experiments, indicating that the active catalyst was chiral guanidinium-hypoiodite, generated in situ as an essential step from chiral guanidinium-iodide salt. The radical scavenger TEMPO had no effect, confirming that a two-electron transfer pathway was operational and the absence of a radical intermediate. Kinetic studies showed zeroth-order dependence on the substrate (35) and first-order dependence on both the catalyst (34) and cumene hydroperoxide (36) (CHP), indicating that oxidation of the iodide anion to hypoiodite was the rate-determining step (Scheme 8). The enantioselective step involved hydrogen bonding and ionic interactions between the oxindole enolate and/or iodonium enolate and the catalyst (guanidinium cation), leading to the enantioselective formation of dimeric oxindole (R,R-40) (Scheme 8). The proposed mechanistic cycle involved several key steps. Initially, the oxidation of the chiral guanidinium iodide anion by CHP generated chiral guanidinium-hypoiodite (37) in the rate-determining step. The chiral guanidinium-hypoiodite (37) then reacted with the 2-oxindole substrate (35), which underwent oxidation and deprotonation (PT) to generate intermediate 38. Subsequently, the intermediate (38) reacted with the enolate anion of the substrate attached with the guanidinium cation (39) and led to the formation of the dimeric oxindole product (40) and regenerated the catalyst. This last step of the reaction determined the enantioselectivity of the reaction. The authors proposed that the CH–π and π–π interactions involving the substrate, the aromatic ring of the benzyl (Bn) group of the catalyst and the deep cavity formed by the catalyst in the transition state contributed to enantioselectivity in the reaction. This study introduced a new direction in this field by offering an enantioselective reaction pathway.
 | ||
| Scheme 8 Mechanism of chiral guanidinium hypoiodite-catalyzed enantioselective dimerization of 3-carboxylate-2-oxindole. | ||
Photochemical approaches for dimerization of 2-oxindole
Visible light photoredox chemistry has rapidly advanced as a transformative tool in synthetic organic chemistry, offering a range of advantages such as mild and environmentally friendly reaction conditions, exceptional functional group tolerance, and high reactivity. The ability to harness visible light as a sustainable energy source has enabled chemists to explore new reaction pathways and intermediates. Photocatalysts such as fac-Ir(ppy)3 and Ru(bpy)3Cl2 are popular due to their excellent photophysical properties, such as strong absorption in the visible light range, long-lived excited states, and high redox potentials.48 These properties enable effective single-electron transfer (SET) processes that are crucial for generating radical species. These catalysts are also used to promote the formation of radical intermediates from enols or enolates. In their excited state, photocatalysts can either donate an electron to the enolate, generating a radical anion, or accept an electron from the enol to form a radical cation. A notable example is the work by Xiao and colleagues, wherein a visible-light-induced photocatalytic formyloxylation of 3-alkyl-3-bromooxindole was demonstrated.49 This process involves the generation of a 3-alkyl-2-oxoindolin-3-yl radical via electron transfer from an excited photocatalyst, highlighting the potential of photoredox catalysis in facilitating complex transformations. The photochemical approach for the dimerization of 2-oxindole was first reported by Wu et al. in 2016.50 The group reported the homocoupling of 3-halo-oxindole (44), and the transformation proceeded through visible light irradiation, employing 0.5 mol% of the photocatalyst fac-Ir(ppy)3 (41) and 1,5-diazabicyclo[4.3.0]non-5-ene (DBU) (47) as the electron donor. Initial reactions in dry THF led to the formation of a dimeric product in 57% yield, but further optimization identified acetonitrile as the most effective solvent, significantly improving the yield to 82%. Replacing DBU with other electron donors such as 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) was slightly less efficient, producing the dimer in a 76% yield. Other electron donors, including 1,4-diazabicyclo[2.2.2]octane (DABCO) and triethylamine (Et3N), were less effective. Furthermore, among various photocatalysts, Ru(bpy)3Cl2 and Eosin B were the most ineffective, while Eosin Y and FIrPic produced yields of 51% and 75%, respectively, though both were inferior to fac-Ir(ppy)3.50 This report includes several crucial intermediates that were essential for the total synthesis of natural products.50 The proposed mechanism involved a radical intermediate pathway (Scheme 9). First, [Ir(III)] (41) transformed into excited [Ir(III)]* (42) after the absorption of a photon (Scheme 9). Thereafter, [Ir(III)]* (42), which is a strong reductant, oxidized to [Ir(IV)] (43) via electron transfer (ET) to 3-halo-oxindole (44), leading to the formation of a C-centered radical anion (45) (Scheme 9). A spontaneous loss of a halide anion from this radical anion (45) produced a 3-alkyl-2-oxoindolin-3-yl radical (46) (Scheme 9). Subsequent ET between [Ir(IV)] and DBU (47) regenerated the photocatalyst and formed the DBU radical cation (48), which oxidized to the corresponding iminium ion viaPT. Finally, the homocoupling of the 3-alkyl-2-oxoindolin-3-yl radical (46) led to the desired dimeric oxindole product 49 (Scheme 9). | ||
| Scheme 9 Visible light photocatalytic dimerization of 3-halooxindole by fac-Ir(ppy)3 and the reaction mechanism. | ||
Electrochemical approaches for dimerization of 2-oxindole
In pursuing sustainable development, seeking new reaction pathways that circumvent the need for expensive toxic metals and chemical oxidants is highly advantageous. Electrochemical reactions have emerged as a favourable option due to their environmentally friendly nature, safety, and atom-efficient characteristics, making them a cornerstone of green chemistry.51,52 Electro-organic synthesis, in particular, offers a promising alternative by eliminating harmful oxidants and reductants, leveraging the electrode potential as a reversible electron transfer agent instead.27,53–56 This approach facilitates selective oxidation, allowing for precise targeting of specific bonds while minimizing the generation of undesired by-products.57 Recent advancements in electro-organic synthesis have witnessed a notable rise in applications, specifically in metal-free functionalization, serving as a pivotal step in the total synthesis of natural products.57In 2020, Paul and Bisai et al. introduced an electrochemical dimerization method for 3-carboxylate-2-oxindole under milder reaction conditions, enabling access to a diverse array of dimeric 2-oxindoles featuring vicinal all-carbon quaternary centers with good yields and satisfactory diastereoselectivity.51 Most importantly, this approach obviates the need for toxic chemical oxidants and metal by-products. The study used a simple undivided cell with a two-electrode setup, wherein the electrodes acted as the greener oxidant. Furthermore, the choice of solvent, base, and electrolyte greatly influenced the reaction, and it was found that DMF (solvent), NaH (base), and NH4PF6 (electrolyte) demonstrated notable efficiency. In addition, a wide variety of substrate scopes were demonstrated to depict the versatility of this methodology. Moreover, these dimeric frameworks played a pivotal role in the total synthesis of pyrroloindoline alkaloids. Electrochemical investigations were conducted to elucidate the mechanistic intricacies of a reaction involving the compound 3-carboxylate-2-oxindole (35). Cyclic voltammetry unveiled two oxidation peaks: the first one at 0.17 V vs. Ag/Ag+, indicative of radical cation formation, and the second peak at 1.14 V, suggesting carbocation generation.51 Controlled potential experiments at 0.2 V vs. Ag/Ag+, involving only one electron oxidation, yielded the desired product with low efficiency in 22% yield, supporting the hypothesis of a radical–radical reaction; nevertheless, a low radical concentration near the electrode surface led to a slow reaction. The formation of radicals was verified by performing the reaction between the substrate and DPPH (2,2-diphenyl-1-picrylhydrazyl), a known radical scavenger, under the same electrolytic conditions. The successful identification of the DPPH–substrate complex confirmed the generation of a radical. In the absence of a base, the reaction mechanism proceeded stepwise, starting with single electron transfer (ET) from 3-carboxylate-2-oxindole (35), generating a radical cation (50), followed by deprotonation (PT) yielding a neutral radical (51), and further oxidation (ET) producing a resonance-stabilized carbocation (52/53) (Path A of Scheme 10). Subsequently, the reaction between the carbocation (53) and the substrate's enol form (54) resulted in the desired dimeric oxindole product 40, after losing a proton (Path A of Scheme 10). Therefore, this mechanism suggests an electron transfer–proton transfer–electron transfer (ET–PT–ET) pathway. An alternative pathway was also involved wherein the dimerization of neutral radicals (51) potentially led to the formation of the targeted dimeric product (40), bearing a vicinal all-carbon quaternary center (see Path B in Scheme 10).
 | ||
| Scheme 10 Electrochemical synthesis and the reaction mechanism for C–C coupling of 3-carboxylate-2-oxindole without the addition of a base under constant current conditions. | ||
Additional experiments were conducted to explore the impact of a base (NaH) in the reaction mechanism.51 From the CV experiments in the presence of a base, two peaks at 0.55 and 1.15 V vs. Ag/Ag+ were observed. Therefore, controlled potential electrolysis at 0.55 V vs. Ag/Ag+ was conducted with DPPH, and the formation of a substrate–DPPH complex confirmed that the first CV peak corresponded to the generation of a neutral radical species (51) (Scheme 11) and the second peak presumably corresponded to the generation of a carbocation species (52) (Scheme 11). Based on these observations, the proposed base-assisted mechanism involved proton abstraction (PT) by the base from the substrate (35), forming an enolate ion (55). Oxidation (ET) of this enolate ion (55) generated a neutral radical (51), followed by further 1e− oxidation (ET) to yield a resonance-stabilized carbocation (52). The reaction between the carbocation (52/53) and the substrate after the removal of a proton by the base resulted in the formation of the dimeric oxindole product (40) (Scheme 11). This mechanism suggests a proton transfer–electron transfer–electron transfer (PT–ET–ET) pathway, differing from the pathway observed in the absence of a base. Another pathway was also proposed, which involved the dimerization of the neutral radical (51), leading to the formation of the dimerized product (40) in a low yield (Path B in Scheme 11).
 | ||
| Scheme 11 Electrochemical C–C coupling of 3-carboxylate-2-oxindole with the addition of a base under constant current conditions. | ||
In 2021, the extension of the previous work was carried out, focusing on the effect of the pKa of the C–H bond at the pseudobenzylic position of 2-oxindoles on the reactivity of the substrate and the mechanism of the reaction.52 In this regard, 3-alkyl-2-oxindole (pKa ∼ 21–24), having a higher pKa than 3-carboxylate-2-oxindole (pKa ∼ 17–18), was chosen as the substrate. A comprehensive optimization of electrochemical synthesis conditions was conducted utilizing Pt electrodes in a two-electrode configuration. The experimental findings indicated that acetonitrile, possessing the highest dielectric constant and low viscosity, favoured synthesis over other solvents, while nBu4NPF6 emerged as the optimal electrolyte. Furthermore, a base (NaH, 5 mM) was necessary to enhance the yield.52 The effect of the pKa of the C–H proton at the pseudobenzylic position of 2-oxindole on the reaction mechanism was investigated, and the generation of a neutral radical species was confirmed by cyclic voltammetry experiments in the presence of nitrogen and oxygen and controlled potential experiments with DPPH, a radical scavenger. Based on these findings, the mechanism for the electrochemical oxidative dimerization of 3-alkyl-2-oxindole (7) was proposed, involving a stepwise proton-coupled electron transfer (PCET) pathway comprising an electron transfer (ET) from the substrate 3-alkyl-2-oxindole (7) to generate a radical cation (11), followed by a proton transfer (PT) step to generate a neutral radical species (10) (Scheme 12). Thereafter, two neutral radicals (10) reacted and led to the formation of dimerized 3-alkyl-2-oxindole (8) (Scheme 12). This mechanism remained consistent both in the presence and absence of a base. Unlike prior investigations, the electro-oxidative dimerization of 3-carboxylate-2-oxindole (35) showcased a notable shift in the mechanism. This observation underscores a complete alteration in the reaction mechanism, which correlates with the variation in the pKa of the C–H bond at the pseudobenzylic position.
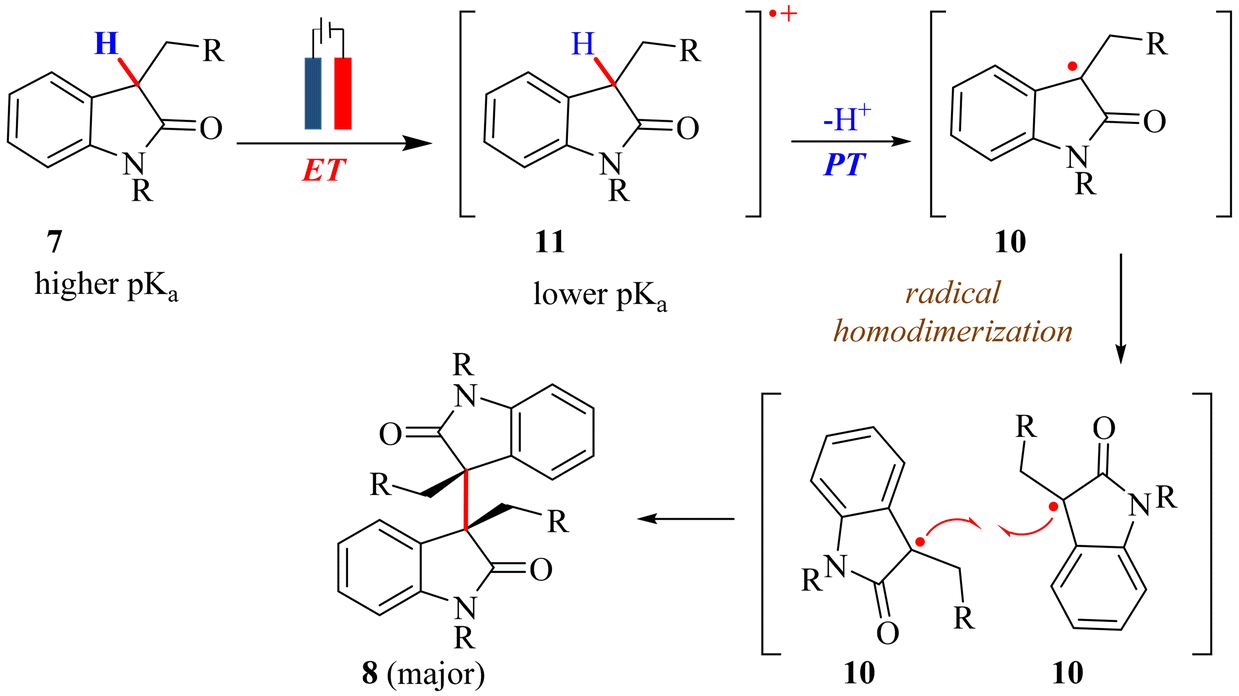 | ||
| Scheme 12 Electro-oxidative dimerization of 3-alkyl-2-oxindole under constant current conditions. | ||
In 2023, Paul and Bisai's research group developed an indirect electrocatalytic dimerization method for 3-substituted-2-oxindoles using TEMPO as a redox mediator.57 This method was able to resolve the electrode passivation problem and lowered the activation barrier for reactions. The reaction was conducted in an undivided cell at only 0.8 V (vs. Ag/Ag+) with a three-electrode setup, and the methodology used greener electrochemical conditions to synthesize various dimeric 2-oxindoles with moderate to good yields. The reaction did not proceed without TEMPO, highlighting its crucial role as a redox mediator/electrocatalyst. Therefore, compared to previous methods, TEMPO-mediated indirect oxidation was milder and more energy-efficient, allowing product formation at lower potentials, thereby reducing energy requirements. Additionally, electrochemical experiments were performed to assess electron transfer rates and diffusion coefficients in different solvents and electrolytes, using TEMPO as the electrocatalyst to streamline optimization. The mechanism of the reaction was found to be significantly different from the previous report (Scheme 13).57 In this case, the proposed mechanism involved a hydride transfer from 3-carboxylate-2-oxindoles (35) to TEMPO+ thus generating TEMPOH. At the working electrode surface, TEMPO underwent oxidation to TEMPO+, which then accepted a hydride from 3-carboxylate-2-oxindoles (35) to form TEMPOH, and a resonance-stabilized carbocation (53) was formed. The carbocation (53) reacted with the enol form of 3-carboxylate-2-oxindoles (54), producing the dimerized product (40) after releasing a proton (Scheme 13). TEMPOH can regenerate TEMPO through either a comproportionation reaction with TEMPO+ or direct oxidation at the electrode surface. The mechanism was supported by spectroelectrochemical experiments, which indicated the formation of TEMPOH during the reaction. This mechanism outlined a TEMPO-catalyzed two-electron transfer pathway for the dimerization of 3-carboxylate-2-oxindoles (35) and similar substrates.
 | ||
| Scheme 13 TEMPO-catalyzed electro-oxidative dimerization of 3-carboxylate-2-oxindole. | ||
Furthermore, for 3-alkyl substituted-2-oxindole (7), the proposed mechanism was different from that of the dimerization of 3-carboxylate-2-oxindoles (35). In this case, the applied potential oxidized TEMPO to TEMPO+, and simultaneously, the base (NaH) abstracted a proton from 3-alkyl-2-oxindole (7), producing the carbanion (56). This carbanion (56) then reacted with TEMPO+ (oxoammonium cation) to form an adduct (57) (Scheme 14), which underwent homolytic cleavage to regenerate TEMPO and produce the neutral radical species (10) (Scheme 14). Subsequently, this radical species (10) dimerized to form the dimeric oxindole product (8) (Scheme 14). This mechanism demonstrated that the formation of a dimeric product (8) from 3-alkyl substituted-2-oxindole (7) involved a one-electron-transfer pathway from 3-alkyl substituted-2-oxindole (7) to TEMPO+, as the two-electron-transfer pathway was energetically unfavorable.
 | ||
| Scheme 14 TEMPO-catalyzed electro-oxidative dimerization of 3-alkyl-2-oxindole. | ||
Syntheses of natural products from dimeric-2-oxindoles
Over the decades, one of the major purposes of synthesizing dimeric 3-substituted-2-oxindoles has been to demonstrate their application towards synthesizing natural products, such as alkaloids. Rodrigo et al.11 performed a reduction reaction on one of the chemically synthesized dimers ((±) 57) using BH3 in THF solvent at room temperature, which resulted in a bis-borane complex ((±) 58) in 71% yield (Scheme 15). Thereafter, (±) 58 was reacted with saturated NH3 in methanol solvent for 2 h under refluxing conditions, which resulted in (±) folicanthine (II) with an excellent yield (75%) (Scheme 15). | ||
| Scheme 15 (±) Folicanthine (II) synthesis by Rodrigo and co-workers from dimerized oxindole.11 | ||
Bisai et al.24 took the chemically synthesized Nphth-protected dimeric oxindole ((±) 59) and reacted it in two steps employing hydrazine and methylchloroformate, which afforded (±) 60 in 70% yield. Thereafter, (±) 60 was reacted with Red-Al at 110 °C for 15 h, resulting in (±) folicanthine (II) in 68% yield (Scheme 16).
 | ||
| Scheme 16 (±) Folicanthine (II) synthesis by Bisai et al. from Nphth-protected dimeric oxindole.24 | ||
Petersen and co-workers46 employed the chemically synthesized (±)-D,L61 to synthesize azide (±)-D,L62 using NaN3 in DMF solvent at 60 °C for 12 h (Scheme 17). Folicanthine (II) can be synthesized via a formal synthesis route previously shown by Bisai and co-workers58 in three steps from (±)-D,L62 (Scheme 17).
 | ||
| Scheme 17 Proposed synthetic route of (±) folicanthine (II) by Peterson and co-workers.46 | ||
Bisai, Paul and co-workers52 exploited electrochemically synthesized dimeric 3-(2-furyl)-2-oxindoles (±) 63 for folicanthine synthesis (Scheme 18). First, oxidative cleavage of the furan ring with the help of a RuCl3 catalyst and NaIO4 resulted in a dicarboxylic acid. Thereafter, the dicarboxylic acid was reacted with dimethyl sulfate and K2CO3 to obtain the ester derivatives ((±) 64). Ester-aminolysis of (±) 64 with methylamine resulted in bis-carboxamides ((±) 65). In the last step, reduction of (±) 65 by Red-Al in toluene solvent in under reflux conditions yielded (±) folicanthine (II) (Scheme 18).
 | ||
| Scheme 18 (±) Folicanthine (II) synthesis by Bisai, Paul and co-workers from electrochemically synthesized dimeric 3-(2-furyl)-2-oxindoles. | ||
In yet another effort, Bisai, Paul and co-workers57 used dimeric 2-oxindole ((±) 66) for the total synthesis of (±) folicanthine (II) and (±) chimonanthine (I) (Scheme 19). Reduction of (±) 66 by LiAlH4 in THF solvent at 0 °C resulted in a diol ((±) 67) in 95% yield. Thereafter, (±) 67 was converted to bis-azide ((±) 68) using DPPA (diphenyl phosphoryl azide) under Mitsunobu conditions. Thereafter, Staudinger reduction of (±) 68 by triphenylphosphine, followed by protection with methyl chloroformate, yielded bis-carbamate ((±) 69) in 86% yield in two steps. Further reduction of (±) 69 by Red-Al resulted in N-benzyl protected chimonanthine ((±) 70) in 89% yield. Hydrogenation of chimonanthine completed the total synthesis of (±) chimonanthine (I) (61% yield). In the final step, reductive amination of (±) chimonanthine with formaldehyde in the presence of sodium triacetoxyborohydride produced (±) folicanthine (II) in 79% yield.
 | ||
| Scheme 19 Total synthesis of (±) folicanthine (II) and (±) chimonanthine (I) by Bisai, Paul and co-workers. | ||
Conclusions and future outlook
In conclusion, dimerization of different 3-substituted-2-oxindoles employing chemical, photochemical and electrochemical methodologies has been discussed. Furthermore, the total syntheses of folicanthine and chimonanthine from chemically and electrochemically synthesized dimeric 3-substituted-2-oxindoles have been discussed. The chemical methodology revealed both diastereoselectivity and excellent enantioselectivity, although the conditions for chemical synthesis were comparatively harsh, since it was difficult for chemical oxidants to approach the sterically crowded 3-position of oxindoles. However, the use of metal catalysts or stoichiometric amounts of oxidants may introduce limitations, such as increased costs and adverse environmental impact, which should be considered when evaluating the overall efficacy and practicality of the method. Furthermore, the chemical reactions cannot proceed through different mechanistic pathways employing the same set of chemicals. Controlling the oxidation process in terms of the number of electron transfers in chemical methodologies is challenging. On the other hand, the photochemical method is a valuable tool due to its unique capability of using light and efficiency in driving specific chemical reactions. However, this method has a notable drawback in that it requires the use of expensive noble metal catalysts based on iridium or ruthenium. This dependency can lead to higher overall costs, and for large-scale industrial applications, the development of low-cost catalysts will be important. In this regard, electrochemical techniques provided an eco-friendly, atom-economical alternative, eliminating the need for chemical oxidants and metal catalysts by utilizing controlled electron transfer. The effectiveness of electrochemical strategies in creating diverse quaternary centers, emphasizing the role of proton-coupled electron transfers (PCETs), gives the methodology an advantage. Compared to chemical methods, electrochemical approaches are more sustainable and efficient, lowering kinetic barriers and expanding synthetic possibilities in natural product synthesis. Additionally, controlling the oxidation processes in terms of electron transfer is straightforward. This precise control over electron transfer allows the reactions to follow diverse mechanistic pathways, providing flexibility in achieving different reaction outcomes and optimizing the process for specific desired products. The ability to navigate through multiple pathways enhances the versatility and effectiveness of the electrochemical methodology. It is worth noting that from Rodrigo et al.'s11 research in 1994 to our recent work in 2023,57 the homodimerization of 3-substituted-2-oxindoles has seen a remarkable journey. Rodrigo and co-workers employed a chemical methodology that involved harsh reaction conditions, including the use of an oxidant (CI4), a strong base (NaH), and extremely low temperature (−65 °C), with a total reaction time exceeding 75 h.11 In contrast, our recent study introduced a TEMPO-catalyzed electrochemical dimerization of 3-substituted-2-oxindoles that takes only 15 min for synthesis at a very low applied potential (0.8 V vs. Ag/Ag+) using a solvent and electrolyte.57 This development suggests that the formation of the unique 3,3′-C(sp3)–C(sp3) sterically crowded vicinal all-carbon quaternary stereocenters found in natural products can be achieved under much milder conditions, potentially replicating nature's ability to form this bond without the need for harsh chemicals.Finally, it is important to highlight that although significant progress has been made in constructing molecules with quaternary stereogenic centres, more comprehensive investigations are required to fully understand the mechanistic pathways. The development of efficient electrochemical processes in this field is ongoing, with a focus on enhancing reaction efficiency. Identifying and utilizing appropriate catalysts is crucial, as the appropriate catalysts can substantially reduce energy requirements further, making the pathways even more energy efficient. However, the development of enantioselective syntheses is still lacking in electrochemical methodologies. Achieving progress in enantioselective synthesis is vital for producing optically pure compounds. Therefore, future research should prioritize not only the optimization of catalyst selection to improve energy efficiency but also the incorporation of strategies for enantioselective synthesis. This dual focus may broaden the applicability and effectiveness of electrochemical methodologies.
Data availability
This is a review article, and no new results have been presented.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely thank IISER Bhopal and SERB, India [CRG/2020/002493] for financial support. Sulekha Sharma thanks the UGC, New Delhi, for the research fellowship. Harapriya Behera and Shivani Ahlawat sincerely thank IISER Bhopal for providing the fellowship for PhD.References
- P. Arya, R. Joseph and D. T. H. Chou, Chem. Biol., 2002, 9, 145–156 CrossRef CAS PubMed.
- Z. Zuo and D. Ma, Isr. J. Chem., 2011, 51, 434–441 CrossRef CAS.
- R. Long, J. Huang, J. Gong and Z. Yang, Nat. Prod. Rep., 2015, 32, 1584–1601 RSC.
- J. Kim and M. Movassaghi, Chem. Soc. Rev., 2009, 38, 3035–3050 RSC.
- F. Zhou, L. Zhu, B.-W. Pan, Y. Shi, Y.-L. Liu and J. Zhou, Chem. Sci., 2020, 11, 9341–9365 RSC.
- R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557–1569 RSC.
- M. A. Schmidt and M. Movassaghi, Synlett, 2008, 313–324 CAS.
- B. M. Trost and M. Osipov, Angew. Chem., Int. Ed., 2013, 52, 9176–9181 CrossRef CAS PubMed.
- P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Álvarez, Chem. – Eur. J., 2011, 17, 1388–1408 CrossRef CAS PubMed.
- M. Movassaghi and M. A. Schmidt, Angew. Chem., 2007, 119, 3799–3802 CrossRef.
- C.-L. Fang, S. Horne, N. Taylor and R. Rodrigo, J. Am. Chem. Soc., 1994, 116, 9480–9486 CrossRef CAS.
- J. Hendrickson, R. Göschke and R. Rees, Tetrahedron, 1964, 20, 565–579 CrossRef CAS.
- L. E. Overman, D. V. Paone and B. A. Stearns, J. Am. Chem. Soc., 1999, 121, 7702–7703 CrossRef CAS.
- C. Guo, J. Song, J. Z. Huang, P. H. Chen, S. W. Luo and L. Z. Gong, Angew. Chem., Int. Ed., 2012, 4, 1046–1050 CrossRef PubMed.
- R. Liu and J. Zhang, Org. Lett., 2013, 15, 2266–2269 CrossRef CAS PubMed.
- S. Ghosh, S. Chaudhuri and A. Bisai, Org. Lett., 2015, 17, 1373–1376 CrossRef CAS PubMed.
- T. Hino, S. Kodato, K. Takahashi, H. Yamaguchi and M. Nakagawa, Tetrahedron Lett., 1978, 19, 4913–4916 CrossRef.
- L. E. Overman, D. V. Paone and B. A. Stearns, J. Am. Chem. Soc., 1999, 121, 7702–7703 CrossRef CAS.
- M. Movassaghi and M. A. Schmidt, Angew. Chem., Int. Ed., 2007, 46, 3725–3728 CrossRef CAS PubMed.
- G. M. Whitesides, Angew. Chem., Int. Ed., 2015, 54, 3196–3209 CrossRef CAS PubMed.
- K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS PubMed.
- I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193–205 CrossRef CAS PubMed.
- M. Munda, S. Niyogi, K. Shaw, S. Kundu, R. Nandi and A. Bisai, Org. Biomol. Chem., 2022, 20, 727–748 RSC.
- S. Ghosh, S. Chaudhuri and A. Bisai, Org. Lett., 2015, 17, 1373–1376 CrossRef CAS PubMed.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: Fundamentals and Applications, John Wiley & Sons, 2022 Search PubMed.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- D. C. Miller, K. T. Tarantino and R. R. Knowles, Top. Curr. Chem., 2016, 374, 30 CrossRef PubMed.
- J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363–390 CrossRef CAS PubMed.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- O. S. Wenger, Coord. Chem. Rev., 2015, 282–283, 150–158 CrossRef CAS.
- J. D. Megiatto Jr, D. D. Méndez-Hernández, M. E. Tejeda-Ferrari, A.-L. Teillout, M. J. Llansola-Portolés, G. Kodis, O. G. Poluektov, T. Rajh, V. Mujica, T. L. Groy, D. Gust, T. A. Moore and A. L. Moore, Nat. Chem., 2014, 6, 423–428 CrossRef PubMed.
- K. Mittra and M. T. Green, J. Am. Chem. Soc., 2019, 141, 5504–5510 CrossRef CAS PubMed.
- T. H. Yosca, J. Rittle, C. M. Krest, E. L. Onderko, A. Silakov, J. C. Calixto, R. K. Behan and M. T. Green, Science, 2013, 342, 825–829 CrossRef CAS PubMed.
- C. Singh and A. Paul, J. Phys. Chem. C, 2015, 119, 11382–11390 CrossRef CAS.
- Z. Chen, J. J. Concepcion, H. Luo, J. F. Hull, A. Paul and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 17670–17673 CrossRef CAS PubMed.
- D. Uraguchi, M. Torii and T. Ooi, ACS Catal., 2017, 7, 2765–2769 CrossRef CAS.
- H. J. Lee, S. Lee, J. W. Lim and J. N. Kim, Bull. Korean Chem. Soc., 2013, 34, 2446–2450 CrossRef CAS.
- Y.-L. Huang, W.-H. Bao, W.-W. Ying, W.-T. Chen, L.-H. Gao, X.-Y. Wang, G.-P. Chen, G.-P. Ge and W.-T. Wei, Synlett, 2018, 1485–1490 CAS.
- F. Dobah, C. M. Mazodze and W. F. Petersen, Org. Lett., 2021, 23, 5466–5470 CrossRef CAS PubMed.
- M. Odagi, I. Mori, K. Sugimoto and K. Nagasawa, ACS Catal., 2023, 13, 2295–2301 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- Y.-Q. Zou, W. Guo, F.-L. Liu, L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Green Chem., 2014, 16, 3787–3795 RSC.
- W.-L. Jia, J. He, J.-J. Yang, X.-W. Gao, Q. Liu and L.-Z. Wu, J. Org. Chem., 2016, 81, 7172–7181 CrossRef CAS PubMed.
- S. Sharma, A. Roy, K. Shaw, A. Bisai and A. Paul, J. Org. Chem., 2020, 85, 14926–14936 CrossRef CAS PubMed.
- K. Shaw, S. Sharma, A. Khatua, A. Paul and A. Bisai, Org. Biomol. Chem., 2021, 19, 9390–9395 RSC.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS.
- E. C. McKenzie, S. Hosseini, A. G. C. Petro, K. K. Rudman, B. H. Gerroll, M. S. Mubarak, L. A. Baker and R. D. Little, Chem. Rev., 2021, 122, 3292–3335 CrossRef PubMed.
- Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2017, 118, 4485–4540 CrossRef PubMed.
- S. Sharma, S. Shaheeda, K. Shaw, A. Bisai and A. Paul, ACS Catal., 2023, 13, 2118–2134 CrossRef CAS.
- S. Ghosh, S. Chaudhuri and A. Bisai, Chem. – Eur. J., 2015, 21, 17479–17484 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |