
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of 6,8-ditrifluoromethyl halogenated phenazine as a potent bacterial biofilm-eradicating agent†
Qiwen
Gao
 ab,
Hongfen
Yang
b,
Jeremy
Sheiber
c,
Priscila Cristina
Bartolomeu Halicki
c,
Ke
Liu
b,
David
Blanco
b,
Sadie
Milhous
b,
Shouguang
Jin
d,
Kyle H.
Rohde
c,
Renee M.
Fleeman
c and
Robert W.
Huigens III
*abef
ab,
Hongfen
Yang
b,
Jeremy
Sheiber
c,
Priscila Cristina
Bartolomeu Halicki
c,
Ke
Liu
b,
David
Blanco
b,
Sadie
Milhous
b,
Shouguang
Jin
d,
Kyle H.
Rohde
c,
Renee M.
Fleeman
c and
Robert W.
Huigens III
*abef
aDepartment of Pharmaceutical & Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, Georgia 30602, USA. E-mail: Robert.Huigens@uga.edu; qiwen.gao@uga.edu; Tel: +706-542-7787
bDepartment of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, USA. E-mail: yanghf88@gmail.com; liu_ke@gzlab.ac.cn; DBlanco5016@gmail.com; smilhous@unc.edu
cDivision of Immunity and Pathogenesis, Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida, Orlando, Florida 32827, USA. E-mail: je503372@ucf.edu; PriscilaCristina.BartolomeuHalicki@ucf.edu; kyle.rohde@ucf.edu; renee.fleeman@ucf.edu
dDepartment of Molecular Genetics & Microbiology, College of Medicine, University of Florida, Gainesville, Florida 32610, USA. E-mail: sjin3499@gmail.com
eDepartment of Chemistry, Franklin College of Arts and Sciences, University of Georgia, Athens, Georgia 30602, USA
fDepartment of Infectious Diseases, College of Veterinary Medicine, University of Georgia, Athens, Georgia 30602, USA
First published on 6th January 2025
Abstract
Bacterial biofilms are surface-attached communities consisting of non-replicating persister cells encased within an extracellular matrix of biomolecules. Unlike bacteria that have acquired resistance to antibiotics, persister cells enable biofilms to demonstrate innate tolerance toward all classes of conventional antibiotic therapies. It is estimated that 50–80% of bacterial infections are biofilm associated, which is considered the underlying cause of chronic and recurring infections. Herein, we report a modular three-step synthetic route to new halogenated phenazine (HP) analogues from diverse aniline and nitroarene building blocks. The HPs were evaluated for antibacterial and biofilm-killing properties against a panel of lab strains and multidrug-resistant clinical isolates. Several HPs demonstrated potent antibacterial (MIC ≤ 0.39 μM) and biofilm-eradicating activities (MBEC < 10 μM) with 6,8-ditrifluoromethyl-HP 15 demonstrated remarkable biofilm-killing potencies (MBEC = 0.15–1.17 μM) against Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus clinical isolates. Confocal microscopy showed HP 15 induced significant losses in the polysaccharide matrix in MRSA biofilms. In addition, HP 15 showed increased antibacterial activities against dormant Mycobacterium tuberculosis (Mtb, MIC = 1.35 μM) when compared to replicating Mtb (MIC = 3.69 μM). Overall, this new modular route has enabled rapid access to an interesting series of potent halogenated phenazine analogues to explore their unique antibacterial and biofilm-killing properties.
Bacterial infections have become increasingly challenging to treat with conventional antibiotics due to acquired resistance and innate tolerance.1–3 Every class of clinically used antibiotic was initially discovered to inhibit rapidly-dividing planktonic bacteria. Unfortunately, pathogenic bacteria have acquired resistance to antibiotics through a multitude of mechanisms, including: target mutation or overexpression, efflux pump action, enzyme-mediated antibiotic inactivation, and altered membrane chemistry to decrease drug penetration.3–6 In addition, individual (free-floating) planktonic bacteria utilize a sophisticated signaling system known as quorum sensing to communicate and coordinate group behaviors, including the attachment to a surface and subsequent biofilm formation (Fig. 1).7 Bacterial biofilm communities consist of enriched populations of non-replicating persister cells encased within an extracellular polymeric matrix of polysaccharides, proteins, and extracellular DNA.3,8,9 Due to their non-replicating phenotype, biofilms demonstrate high levels of innate antibiotic tolerance and are credited as the primary cause of chronic and recurring infections.8,10–12
 | ||
| Fig. 1 Individual, free-floating planktonic bacteria coordinate the simultaneous attachment to a surface where subsequent development leads to a mature biofilm community consisting of enriched populations of non-replicating (metabolically-dormant) persister cells. Established biofilms will disperse planktonic cells back into the surrounding environment and are credited as the underlying cause of chronic and recurring bacterial infection. | ||
Despite an antibiotic pipeline that has struggled to produce clinically useful agents over the last ∼50 years, innovative approaches have delivered exciting advances in recent years.3,13–16 Lewis and colleagues have developed an incredible natural product-focused discovery platform that has resulted in (1) ClpP-activating agent ADEP4 that kills persister cells and eradicates a chronic biofilm infection in mice,17 (2) the development of iChip technology that led to the identification of the lipid II and III binding antibiotic teixobactin from “unculturable” bacteria,18 and (3) the discovery of darobactin, an antibiotic that targets BamA and kills Gram-negative pathogens, isolated from a nematode bacterial symbiont.19 Myers and co-workers have developed robust total synthesis platforms to access new tetracyclines20–24 (including FDA-approved eravacycline), macrolides,25–28 clindamycins (iboxamycin, IBX, is a promising lead),29 and a bridged macrobicyclic antibiotic cresomycin30 designed to overcome multidrug-resistant pathogens. The Boger lab has reported new vancomycin analogues that (1) enable dual binding of transpeptidase's D-Ala–D-Ala (wild type) and D-Ala–D-Lac (vancomycin resistant) moiety, (2) results in transglycosylase inhibition, and (3) cause membrane permeabilization to overcome vancomycin resistance.31–34 In recent years, the Hergenrother team has (1) defined predictive compound accumulation (eNTRy) rules to target Gram-negative pathogens,35–39 (2) reported porin-independent accumulation rules for Pseudomonas aeruginosa,40 and (3) discovered lolamicin, a Gram-negative-specific antibiotic targeting the lipoprotein transport system while sparing the gut microbiota in mice.41 Smith, Heise, and colleagues at Genentech reported G0775, an optimized arylomycin, that inhibits the essential bacterial type I signal peptidase through covalent modification and demonstrates efficacy against Gram-negative pathogens in mouse models of infection.42
Our work to address problems associated with antibiotic-resistant and -tolerant pathogens has been inspired by the action of phenazine antibiotics.43–45 Individuals suffering from cystic fibrosis (CF) are inflicted with chronic lung infections. Oftentimes, young CF patients are initially infected by Staphylococcus aureus and, as they age, Pseudomonas aeruginosa later co-infects their lungs. It is believed that P. aeruginosa secretes the redox-active phenazine pyocyanin in microbial warfare to kill S. aureus and become the primary pathogen infecting the CF patient's lungs.46 As CF-related lung infections are chronic, we hypothesized that pyocyanin must play a role in eradicating S. aureus biofilms and initiated a research program to explore the antibacterial properties of phenazine molecules.
Initial efforts from our lab focused on the synthesis and antibacterial assessment (in minimum inhibitory concentration, or MIC assays) of 13 diverse phenazine antibiotics and synthetic phenazines.47 We were encouraged to discover marine Streptomyces derived 2-bromo-1-hydroxyphenazine (MIC = 6.25 μM = 1.72 μg mL−1) demonstrates significantly more potent antibacterial activities than pyocyanin (MIC = 50 μM = 10.5 μg mL−1) against S. aureus and S. epidermidis.47 In addition, we found synthetic analogue 2,4-dibromo-1-hydroxyphenazine (halogenated phenazine analogue 1, or HP-1; Fig. 2) to demonstrate potent planktonic growth inhibition (MIC = 1.56 μM = 0.55 μg mL−1).47 In other work, HP-1 was shown to eradicate S. aureus biofilms at a minimum biofilm eradication concentration, or MBEC, value of 100 μM (35.4 μg mL−1).48
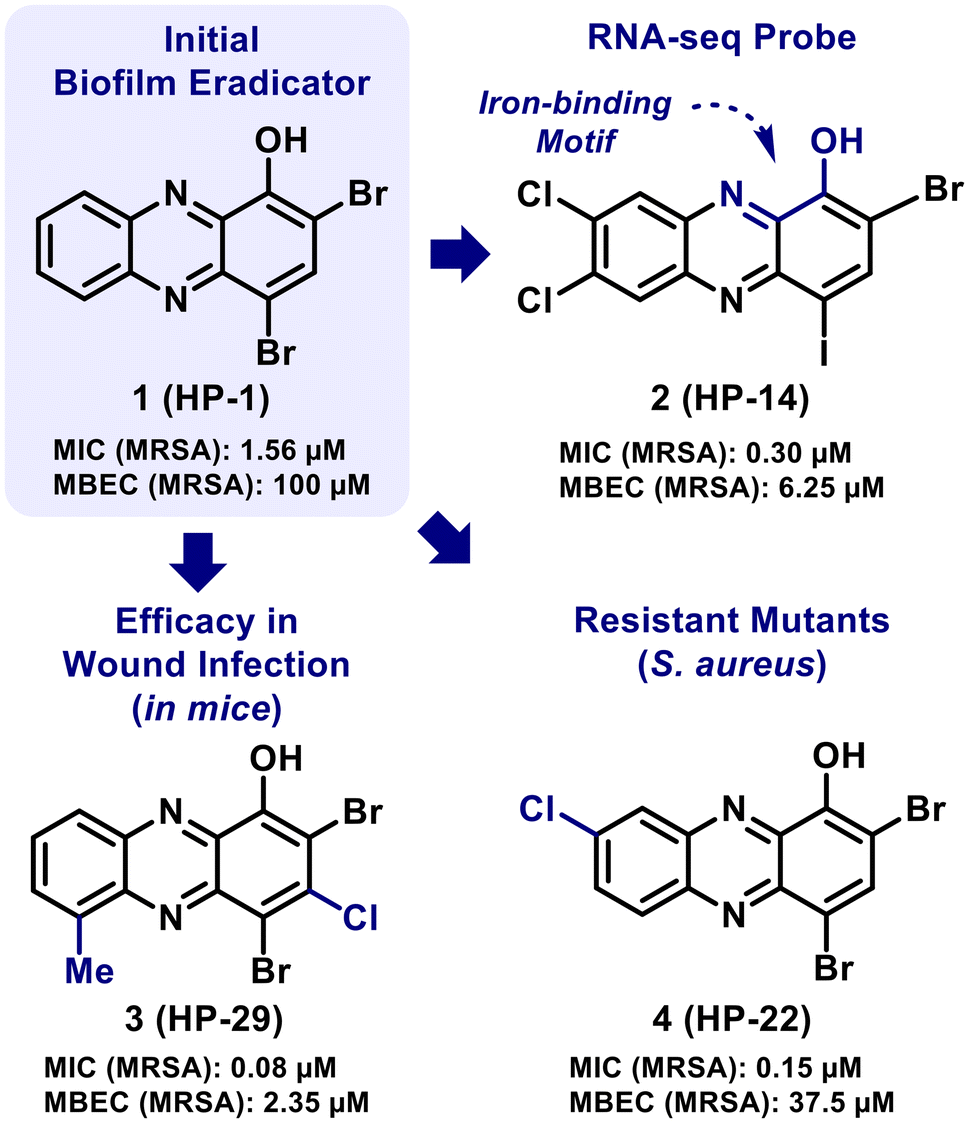 | ||
| Fig. 2 Halogenated phenazine (HP) analogues that have demonstrated potent biofilm eradication activities and/or have been used as probes to explore mechanistic insights. | ||
Our more recent efforts related to HPs have focused on exploring structure–activity relationships (SAR), using select analogues as probes in mode of action studies, and treating wound infections in mice (Fig. 2).49–54 These collective efforts have led to a detailed SAR of more than 150 HPs with the identification of several potent HP analogues that eradicate (kill) biofilms of Gram-positive pathogens (e.g., S. aureus, S. epidermidis; MBEC ≤ 10 μM). In addition, key mechanistic insights were obtained from transcript profiling (RNA-seq) studies that revealed HP-14 induces rapid iron starvation in MRSA biofilms through the direct binding of iron.51 Additional work has shown potent HP analogues to induce rapid starvation in Staphylococcal biofilms using RT-qPCR of select iron uptake biomarkers (e.g., isdB, sbnC, sfaA, MW0695).53–55 In a separate study, an HP-resistant mutant was generated in S. aureus using HP-22 that had a single amino acid change in transcriptional regulator TetR21 (Arg116Cys).56 RNA-seq analysis showed the TetR21R116C mutant to have up-regulated the transcription of hprS (halogenated phenazine resistant protein of S. aureus) and HprS was discovered as an efflux pump for phenazines.56 In addition, HP-29 demonstrated good in vivo efficacy against S. aureus UAMS-1 and E. faecalis OG1RF in wound infection models in mice.53
In the present study, we developed a modular route to several new halogenated phenazines from diverse nitroarene and aniline building blocks. This focused collection of HP small molecules was evaluated against a panel of pathogens in standard antibacterial assays and Calgary Biofilm Device assays to determine biofilm eradication activities against lab strains and clinical isolates. From these studies, we identified 6,8-ditrifluoromethyl-HP 15 to be the most potent biofilm-eradicating HP small molecule to date. To determine its ability to kill other drug-tolerant bacterial pathogens, HP 15 was also evaluated against both replicating and dormant Mycobacterium tuberculosis (Mtb) cultures. Using confocal microscopy experiments, HP 15 showed significant impacts on the extracellular polysaccharide matrix of established MRSA biofilms.
Chemical synthesis of diverse halogenated phenazines
We synthesized nine halogenated phenazines using a three-step route from simple aniline and nitroarene building blocks (Scheme 1). To access the phenazine core, an aniline and a nitroarene were reacted in the presence of potassium tert-butoxide to join these materials through a regioselective C–N bond formation that results in a nitroso intermediate (select nitroso compounds were characterized in the ESI;† however, most were taken directly to the next synthetic step without purification), which was directly subjected to N,O-bis(trimethylsilyl)acetamide (BSA) to yield diverse 1-methoxyphenazines in 27–48% yield (36% average yield). This modular phenazine synthesis was inspired by work done in the Wróbel lab,57–60 which we previously reported to access HP 15.61 With the phenazine nucleus intact, 1-methoxyphenazines were then transformed into halogenated phenazines via a demethylation step using boron tribromide (BBr3: 53–100% yield, 9 examples) or aluminum(III) chloride (AlCl3: 91–95% yield, 2 examples), and a final bromination reaction with N-bromosuccinimide (NBS: 28–99% yield). Three halogenated phenazines in Scheme 1 have been reported by our lab (7-Cl-HP 10, 7-CF3-HP 14, 6,8-CF3-HP 15)52,54,61 and were included to evaluate the efficiency of this synthetic route while expanding our understanding of their biological activities. | ||
| Scheme 1 Modular synthesis of halogenated phenazines from diverse nitroarene and aniline materials. Notes. (a) Demethylation or bromination yields from previous studies. (b) Previously reported halogenated phenazines. | ||
Antibacterial evaluation of halogenated phenazines
Following synthesis, we evaluated the HP analogues to determine their ability to inhibit planktonic growth in minimum inhibition concentration (MIC) assays. MIC values were determined for HPs against a panel of Gram-positive pathogens (lab strains and drug-resistant clinical isolates; see Table 1 and ESI Table 1†), replicating and dormant Mycobacterium tuberculosis cultures, and Gram-negative pathogens (ESI Table 2†). Although this collection of HPs demonstrated impressive antibacterial activities against the Gram-positive pathogens, the MIC profiles of 10, 14, and 15 have been reported52,54,61 and served as comparators to benchmark activity for new HPs described during these studies. All 7-position substituted HPs reported herein demonstrated moderate to significant improvements in antibacterial activities (up to 30-fold more potent MIC values; Table 1) when compared to parent HP-1 against methicillin-resistant Staphylococcus aureus (MRSA), methicillin-resistant S. epidermidis (MRSE), methicillin-sensitive S. epidermidis (MSSE), vancomycin-resistant Enterococcus faecium (VRE), multidrug-resistant Enterococcus faecalis (strain OG1RF), and Streptococcus pneumoniae strains.| Compound | MRSA 1707 | MRSA 44 | MRSE 35984 | S. epi 12228 | VRE 700221 | E. faecalis OG1RF | S. pneumoniae 6303 | Mtb (rep) | Mtb (dorm) | HeLa cytotox. (IC50) |
|---|---|---|---|---|---|---|---|---|---|---|
| All MIC values against bacterial pathogens and cytotoxicity against HeLa cells are reported in micromolar (μM) concentrations. All biological results were acquired from three or more independent experiments.a Midpoint value for 2-fold range in MIC values observed.b Lowest concentration tested. “Mtb (rep)” refers to replicating Mtb CDC1551 bacteria; “Mtb (dorm)” refers to dormant Mtb CDC1551 bacteria. | ||||||||||
| 1 (HP-1) | 1.17a | 1.56 | 1.17a | 1.17a | 4.69a | 18.8a | 1.56 | 15.8 | 200 | >100 |
| 9 | 0.30a | 0.39 | 0.39 | — | 2.35a | 6.25 | 0.39 | — | — | >100 |
| 10 | 0.08a | 0.10 | 0.15a | 0.10 | 1.56 | 3.13 | 0.20 | — | — | >100 |
| 11 | 0.05 | 0.10 | 0.30a | — | 1.17a | 3.13 | 0.39 | — | — | >100 |
| 12 | 0.08a | 0.05b | 0.15a | 0.08a | 0.78 | — | 0.20 | — | — | >100 |
| 13 | 0.04a | 0.05b | 0.10 | 0.10 | 0.39 | 0.78 | 0.08a | — | — | >100 |
| 14 | 0.05 | 0.05b | 0.10 | — | 0.39 | 0.78 | 0.08a | — | — | >100 |
| 15 | 0.05b | 0.05b | 0.08a | 0.15a | 0.05b | 0.20 | 0.08a | 3.69 | 1.35 | >100 |
| 16 | 1.17a | 0.78 | 0.20 | 1.56 | 3.13 | 4.69a | 1.56 | — | — | — |
| 17 | 0.30a | 0.20 | 0.15a | 0.10 | 0.59a | 4.69a | 0.78 | — | — | — |
| Vancomycin | 0.59a | 0.59a | 1.17a | 1.56 | >100 | 0.78 | 0.39 | — | — | — |
| Isoniazid | — | — | — | — | — | — | — | 0.1 | 114.7 | — |
| Rifamycin | — | — | — | — | — | — | — | 0.001 | 1.43 | — |
Here, we report the activity profiles of all halogens substituted at the 7-position of the HP scaffold, which demonstrated potent antibacterial activities (MIC = 0.04–1.56 μM against most Gram-positive strains; Table 1; Fig. 3A and B). In most instances, 7-bromo-HP 11 and 7-iodo-HP 12 analogues report equipotent MIC values compared to 7-chloro-HP 10; however, 7-fluoro-HP 9 typically reports MIC values that are up to 4-fold less potent when compared to the other halogens at the 7-position of the HP scaffold. HP analogue 13 bears a –SCF3 group at the 7-position of the HP core and demonstrated highly potent activities against the Gram-positive strains (MIC = 0.04–0.78 μM). HP 17 contains a benzyl group at the 7-position of the HP scaffold and demonstrated potent antibacterial activities (MIC = 0.10–0.78 μM) against nearly all Gram-positive strains in the Table 1 panel. To probe the SAR of the highly potent 6,8-ditrifluoromethyl-HP agent 15 (MIC = 0.05–0.20 μM against Gram-positive pathogens), we synthesized HP analogue 16 bearing a 3-chlorine atom as the lone difference between analogues; however, 16 (MIC = 0.20–4.69 μM) lost considerable activities compared to HP 15 and we believe this trend could correlate with losses in water solubility.
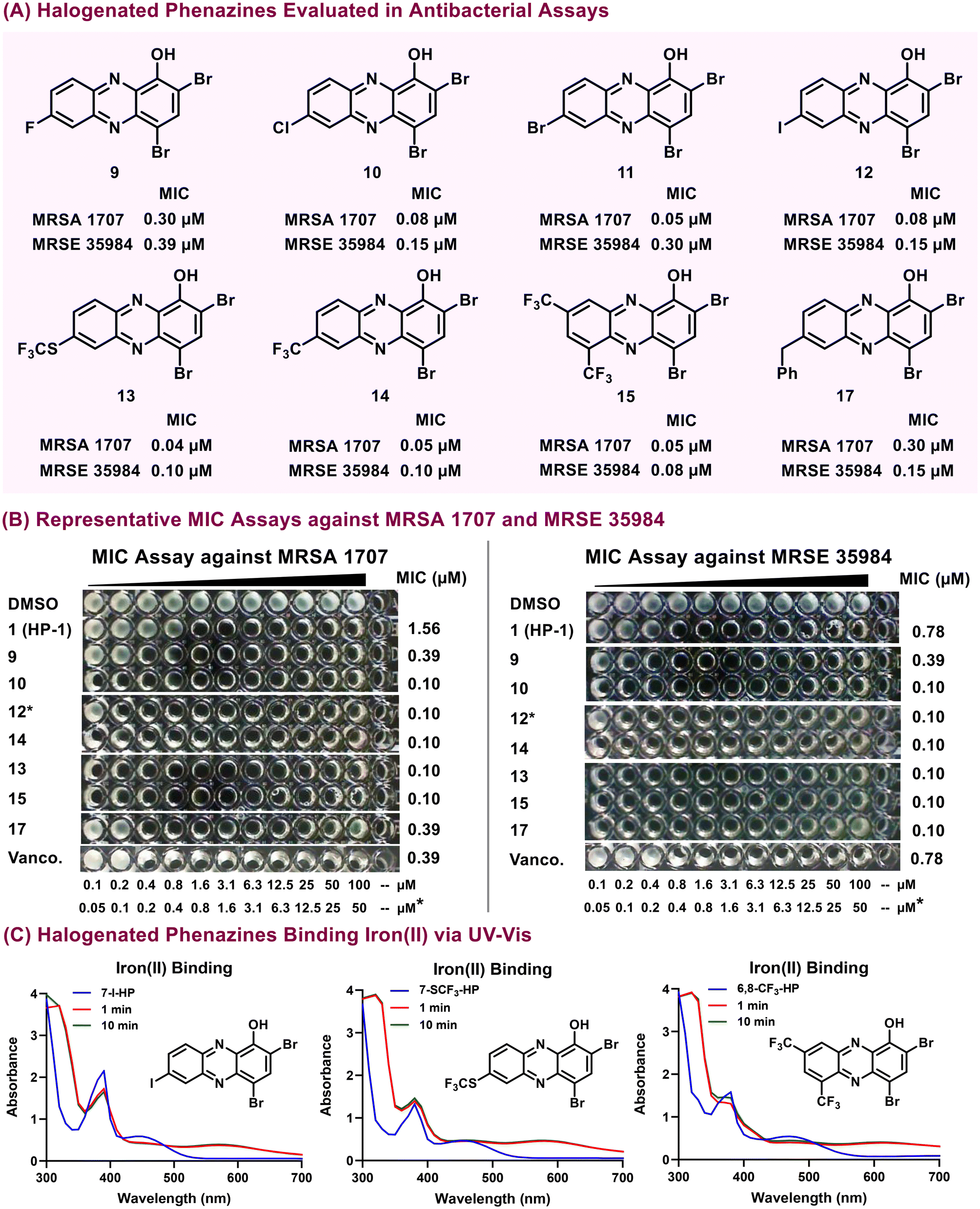 | ||
| Fig. 3 (A) Chemical structures and focused antibacterial profiles (against MRSA 1707 & MRSE 35984) for potent halogenated phenazines investigated during these studies. (B) Images of representative MIC assays against MRSA 1707 and MRSE 35984. (C) UV-vis experiments to show select HPs directly binding iron(II). | ||
When evaluated against Gram-negative pathogens, new HPs reported minimal antibacterial activities. With that, HP 15 was found to report moderate to weak antibacterial activities against A. baumannii strains (MIC = 12.5–75 μM; ESI Table 2†); however, 15 was inactive against P. aeruginosa or E. coli strains (MIC > 100 μM).
Following initial MIC assessment, we performed UV-vis spectroscopy experiments to evaluate iron(II)-binding properties of synthesized HP analogues (Fig. 3C; ESI†). In these UV-vis experiments, we observed an elevated absorbance between 550 nm to 650 nm of HP samples following the addition of iron(II) due to the rapid binding between the HP analogues and iron(II) after 1 and 10 minutes. Our findings that HP analogues directly bind iron(II) support the notion that these HPs are able to induce iron starvation in bacteria as their primary mode of action and aligns with previous studies.53–55,61
In addition, we have found several HPs to demonstrate good antibacterial activities against Mycobacterium tuberculosis (MIC < 2 μM) in previous work.53 With the antibacterial activity profiles in hand, we were encouraged to investigate HP 15 against both replicating and dormant Mtb cultures (Table 1). Although HP-1 was able to inhibit replicating Mtb with an MIC = 15.8 μM, only weak activity was observed against dormant bacilli (MIC = 200 μM). A similar pattern of drug tolerance was reported for the first-line anti-TB drugs isoniazid (INH) and rifampin (RIF), with dormant Mtb becoming more than 1000 times more tolerant to these drugs than the replicating bacteria. In contrast, HP 15 exhibited significantly improved potency against replicating Mtb (MIC = 3.69 μM) than HP-1 and, unexpectedly, even better activity against dormant Mtb (MIC = 1.35 μM). Additional work is needed to better understand the unique activity HP 15 demonstrates against dormant Mtb.
Mammalian cytotoxicity
Following MIC assessment, eight HPs were evaluated against HeLa cells to determine bacteria-to-mammalian selectivity using 24 hours lactate dehydrogenase assays at 25, 50, and 100 μM test concentrations (Table 1). Similar to previous studies,52,53 these HPs demonstrated minimal to no cytotoxicity against HeLa cells (IC50 > 100 μM). Selectivity indexes (SI) were determined based on relative IC50 values against HeLa cells compared to MIC values against S. aureus (HeLa [IC50]/S. aureus [MIC]) to show most HPs demonstrate impressive SI > 300 during these studies.Biofilm eradication studies with halogenated phenazines
After determining initial antibacterial activities from MIC and HeLa cell cytotoxicity assays, we evaluated HPs against established MRSA, MRSE, MSSE (methicillin-sensitive S. epidermidis), and VRE biofilms using Calgary Biofilm Device (CBD) assays62,63 (Fig. 4; Table 2). CBD assays are performed in 96-well plates and utilize specialized lids with pegs that are submerged into media (1 peg per microtiter well; see ESI†) to provide a surface for bacteria to attach and form biofilms.62,63 Biofilm eradication assays have three distinct phases, which include: (1) establishing biofilms on CBD peg surfaces (bacteria in media alone), (2) compound challenge (biofilm-associated CBD pegs are submerged into a 96-well plate containing test compounds in 2-fold serial dilution), and (3) biofilm dispersion and growth (this final phase includes CBD pegs incubated in media alone to facilitate the dispersion of viable planktonic bacteria from biofilms followed by growth to give a final turbid readout; note: biofilms that have been eradicated result in a complete lack of turbidity at the assay end point). | ||
| Fig. 4 (A) Select halogenated phenazine biofilm eradication activities. (B) Representative MBEC images of halogenated phenazines evaluated in Calgary Biofilm Device (CBD) assays. (C) Biofilm eradication profile for HP 15. Note: *Compounds were tested from 0.2 μM to 200 μM (10 mM DMSO stock solution). **Compounds were tested from 0.1 μM to 100 μM (5 mM DMSO stock solution). ***Compounds were tested from 2 μM to 2000 μM (100 mM DMSO stock solution). | ||
| Compound | MRSA 1707 | MRSA 44 | MRSE 35984 | S. epidermidis 12228 | VRE 700221 | % Hemolysis at 200 μM |
|---|---|---|---|---|---|---|
| MBC/MBEC | MBC/MBEC | MBC/MBEC | MBC/MBEC | MBC/MBEC | ||
| All biological results in this table are reported in micromolar (μM) concentrations and were acquired from three or more independent experiments.a Midpoint value for experiments that yielded a 2-fold range from CBD assays.b Midpoint value for experiments that yielded a 4-fold range from CBD assays.c Lowest concentration tested.d For HP compound 14, we previously reported CBD assay values of MBC = 3.13 μM & MBEC = 4.69 μM against MRSE 35984 (see compound 5h in ref. 54). nd = not determined due to an uncharacteristically large range of planktonic killing activities in these assays against VRE 700221. | ||||||
| 1 (HP-1) | 50b/100b | 100/100b | 50b/100 | 25b/50b | 18.8a/9.38a | ≤1 |
| 9 | 37.5a/37.5a | 50/12.5 | 50/75a | 3.13/37.5a | nd/3.13b | 1.6 |
| 10 | 1.17a/4.69a | 12.5/4.69a | 25/50 | — | 1.56b/0.59a | ≤1 |
| 11 | 4.69a/9.38a | 9.38a/18.8a | 12.5/12.5 | — | 18.8a/25 | 1.6 |
| 12 | 4.69a/4.69a | 37.5a/18.8a | 25/37.5a | 1.56b/4.69a | — | ≤1 |
| 13 | 2.35a/1.56 | 3.13b/3.13b | 3.13/2.35a | 1.17a/1.17a | 0.15a/0.10c | ≤1 |
| 14 | 4.69a/4.69a | 4.69a/9.38a | 12.5/12.5 | 3.75a/3.75a | 9.38a/0.39 | 7.2 |
| 15 | 1.17a/1.17a | 0.30a/0.78b | 0.20c/0.39b | 0.10c/0.15a | 0.10c/0.15a | 7.8 |
| 16 | 25b/12.5 | — | 1.17a/1.17a | — | — | — |
| 17 | 9.38a/6.25 | — | 12.5/75a | 25b/18.8a | — | ≤1 |
| QAC-10 | 93.8a/93.8a | — | 3.13/3.13 | 4.69a/6.25 | 3.0a/3.0a | >99 |
| EDTA | >2000/>2000 | — | 1000/>2000 | — | — | ≤1 |
| TPEN | 375a/>2000 | — | 250/>2000 | — | — | ≤1 |
| Vancomycin | 3.9/>2000 | 7.8/>2000 | 3.0b/>2000 | — | >200/150 | ≤1 |
Each of the three phases of our CBD assays were performed under static conditions for 24 hours at 37 °C (experiments require 72-hours total to complete). Biofilm-attached pegs were gently washed with PBS to remove planktonic cells between each of the three phases of the MBEC assay (lid transfer steps). Upon completion of CBD assays, turbidity measurements were taken using a spectrophotometer (OD600) to quantify (1) viable biofilms that undergo planktonic dispersion and subsequent bacterial growth (visible turbidity), or (2) eradicated biofilms (no turbidity results from completely eradicated biofilms unable to disperse planktonic bacteria; see Fig. 4B for representative CBD assays against MRSA lab strain & clinical isolate). Following the completion of CBD assays, the minimum biofilm eradication concentration (MBEC) for test compounds is determined as the lowest concentration resulting in complete biofilm killing (no turbidity in microtiter wells). In addition to biofilm killing, CBD assays also provide direct planktonic eradication assessment of test compounds following dilution from the test plate (phase 2) into fresh media and subsequent incubation for 24 hours to determine minimum bactericidal concentrations (CBD). This assay allows for the determination of biofilm and planktonic killing dynamics of various small molecules from a single experiment.
During these studies, we evaluated all HPs in CBD assays against MRSA 1707 and MRSE 35984 biofilms to determine MBEC values against these major human pathogens (Table 2). Select HP analogues were also evaluated against MRSA 44, S. epidermidis 12228, and VRE 700221 biofilms. From our CBD experiments, we found biofilm-killing profiles analogous to the antibacterial properties as each 6-, 7-, or 8-substituted HP demonstrated significantly improved biofilm eradication activities compared to HP-1. 7-Fluoro-HP 9 (MBEC = 37.5 μM) demonstrated improved biofilm eradication against MRSA 1707 biofilms when compared to parent HP-1; however, HP 9 was found to be 4- to 8-fold less potent than 7-chloro-HP 10, 7-bromo-HP 11, 7-iodo-HP 12 (Fig. 4), or 7-trifluoromethyl-HP 14 as these HPs demonstrated potent activities (MBEC = 4.69–9.38 μM) in Calgary Biofilm Device assays. HP 17 (7-benzy-HP; MBEC = 6.25 μM) also demonstrated potent biofilm eradication activities against MRSA 1707 biofilms. Impressively, 7-SCF3-HP 13 demonstrated highly potent biofilm-killing activities in CBD assays against MRSA 1707 (MBEC = 1.56 μM, Fig. 4), MRSA 44 (MBEC = 3.13 μM), MRSE 35984 (MBEC = 2.35 μM), S. epidermidis 12228 (MBEC = 1.17 μM), and VRE 70021 (MBEC = 0.10 μM).
We were very encouraged to find 6,8-ditrifluoromethyl-HP 15 eradicates bacterial biofilms with incredible potency against the Gram-positive pathogens in our panel (MRSA 1707, MBEC = 1.17 μM; MRSA 44, MBEC = 0.78 μM; MRSE 35984, MBEC = 0.39 μM; S. epidermidis 12228, MBEC = 0.15 μM; VRE 700221, MBEC = 0.15 μM; Fig. 4 and Table 2). HP 15 is the most potent biofilm eradicating agent we have reported to date. 3-Chloro-6,8-ditrifluoromethyl-HP (16; MBEC = 12.5 μM) showed a 4- to 11-fold reduction in biofilm-killing properties against MRSE 35984 and MRSA 1707 biofilms, respectively, when compared to 6,8-ditrifluoromethyl-HP 15.
In addition to examining HPs for their biofilm eradication properties, we evaluated a focused series of comparator agents, including vancomycin – a frontline antibiotic used to treat MRSA infections (Table 2). We evaluated QAC-10 which is a known quaternary ammonium cation agent that has demonstrated biofilm killing properties through a membrane-lysis mode of action.64 When tested alongside 6,8-ditrifluoromethyl-HP 15, QAC-10 demonstrated an 8- to 80-fold reduction in biofilm-killing potency reporting MBEC values of 93.8 μM against MRSA 1707, 3.13 μM against MRSE 35984, 6.25 μM against S. epidermidis 12228, and 3.0 μM against VRE 700221 biofilms. Since HP biofilm-eradicating agents operate through an iron starvation mode of action, we evaluated metal-chelating agents EDTA (general chelator) and TPEN (cell-permeable chelator) as comparators; however, neither of these compounds were able to eradicate MRSA or MRSE biofilms (MBEC > 2000 μM). Vancomycin was evaluated in CBD assays against MRSA, MRSE and VRE biofilms. Against MRSA and MRSE, we found vancomycin to report potent planktonic killing activities (MBC = 3.0–7.8 μM) while their biofilm counterparts were completely tolerant to this antibiotic (MBEC > 2000 μM). Interestingly, we did observe vancomycin to eradicate VRE 700221 biofilms at high concentrations (MBEC = 150 μM), which is attributed to a less robust biofilm under these assay conditions.
We then performed hemolysis assays to determine if this series of HPs demonstrates membrane-lysis activities following initial biofilm eradication studies. Hemolysis assays are an important secondary experiment when evaluating biofilm-killing profiles of small molecules as general membrane lysis is an effective way to eradicate established biofilms (i.e., quaternary ammonium cations such as QAC-10 induce >99% hemolysis of red blood cells at 200 μM and kill biofilms; Table 2). During these studies, eight HPs were evaluated against red blood cells at 200 μM to give <8% hemolysis activity at this high test concentration demonstrating that these HPs do not operate through a membrane lysis mechanism.
Following initial biofilm eradication assessment, HP-1, 6,8-ditrifluoromethyl-HP 15, 7-SCF3-HP 13, 7-iodo-HP 12, and vancomycin were evaluated against a panel of six MRSA clinical isolates in CBD assays (Table 3). Against MRSA isolates, HP-1 demonstrated moderate to low biofilm-killing activities with MBECs = 150–>200 μM while 7-SCF3-HP 13 and 7-iodo-HP 12 reported good to excellent activities (MBEC values = 4.69–25 μM). 6,8-ditrifluoromethyl-HP 15 demonstrated incredible biofilm-killing properties against MRSA clinical isolates with MBEC values = 0.59–1.17 μM. In addition, vancomycin was used as a comparator against this panel of MRSA clinical isolates. Despite vancomycin's potent activity against planktonic cells (MBC = 3.0–7.8 μM; Table 3) in CBD assays, the drug of last resort for MRSA infections was unable to eradicate MRSA biofilms at the highest concentrations tested (MBEC values > 2000 μM against all isolates).
| Compound | MRSA 1 | MRSA 2 | S. aureus 129 | S. aureus 138 | S. aureus 147 | S. aureus 156 |
|---|---|---|---|---|---|---|
| MBC/MBEC | MBC/MBEC | MBC/MBEC | MBC/MBEC | MBC/MBEC | MBC/MBEC | |
| All biological results are reported in micromolar (μM) concentrations and were acquired from three or more independent experiments.a Midpoint value for experiments that yielded a 2-fold range from CBD assays.b Midpoint value for experiments that yielded a 4-fold range from CBD assays. | ||||||
| 1 (HP-1) | 37.5a/150a | 37.5a/≥200 | 75a/>200 | 50/200 | 50/≥200 | 50/≥200 |
| 12 | 6.25/9.38a | 6.25/18.8a | 12.5b/18.8a | 12.5/18.8a | 9.38a/9.38a | 9.38a/12.5 |
| 13 | 2.35a/4.69a | 4.69a/18.8a | 2.35a/9.38a | 12.5/25 | 6.25/9.38a | 3.13/6.25 |
| 15 | 0.59a/0.78b | 0.59a/0.78b | 0.59a/0.78 | 1.56/1.17a | 0.78b/0.59a | 0.78/0.78b |
| Vancomycin | 3.0a/>2000 | 5.9a/>2000 | 5.9a/>2000 | 7.8/>2000 | 5.9a/>2000 | 7.8/>2000 |
Confocal microscopy reveals matrix loss with HP treatment
To visualize the MRSA biofilm matrix modulation of 6,8-ditrifluoromethyl-HP 15 we utilized confocal z-stack imaging with USA300 S. aureus carrying pCM29_GFP. This plasmid allows for the constitutive expression of green fluorescent protein (GFP) and imaging of the biofilm cell population. To visualize the associated biofilm matrix, we stained the biofilms before imaging with concanavalin A lectin conjugated with Texas Red. This combination allows for visualization of both the bacteria and the matrix polysaccharides of the S. aureus biofilms.We found that treatment of a pre-formed MRSA biofilm with both parent HP-1 (1) and 6,8-ditrifluoromethyl-HP 15 at 25 μM could reduce the biofilm height, although HP 15 was far superior at this concentration (Fig. 5). Specifically, the untreated biofilms had a height of ∼30 μm with a dense layer of polysaccharide encasing the bacterial cells. With HP-1 treatment, this height decreased to ∼20 μm and the polysaccharide matrix was much less dense than the untreated biofilms. Following treatment with 6,8-ditrifluoromethyl-HP 15, there was a drastic reduction in biofilm height (∼10 μm height remained) and the polysaccharide matrix on top of the biofilm was largely removed. The images in Fig. 5 validate the increased potency of the optimized 6,8-ditrifluoromethyl-HP 15 compound over parent HP-1.
 | ||
| Fig. 5 Confocal microscopy images of MRSA biofilms untreated or treated with select HP analogues. The figures show the 3-D rendering of confocal z-stack imaging of biofilms grown with GFP expressing S. aureus and stained with Texas Red conjugated concanavalin A. The top three images show MRSA biofilms with no compound (DMSO Control), the middle and bottom images are those treated with 25 μM of HP-1 and 6,8-ditrifluoromethyl-HP 15, respectively. All imaging was done with n = 3 replicates with representative images shown. | ||
Conclusions
In conclusion, we have developed a modular synthetic route to access HPs utilizing a reaction sequence that starts from nitroarene and aniline materials to yield diverse 1-methoxyphenazines. Several new HPs demonstrated highly potent antibacterial (MIC ≤ 0.39 μM) and biofilm-eradicating properties (MBEC < 10 μM) with 6,8-ditrifluoromethyl-HP 15 demonstrating the most potent biofilm-killing properties (MBEC = 0.15–1.17 μM) against Gram-positive pathogens, including MRSA clinical isolates. Using confocal microscopy, 6,8-ditrifluoromethyl-HP 15 was shown to induce significant polysaccharide loss of MRSA biofilms. In addition, HP 15 demonstrated increased antibacterial activity against dormant Mtb cultures (MIC = 1.35 μM) when compared to replicating Mtb (MIC = 3.69 μM). Overall, this modular synthesis enabled rapid access to new halogenated phenazines to further explore their unique antibacterial and biofilm-killing properties.Experimental
General information
All synthetic reactions were carried out under an inert atmosphere of argon unless otherwise specified. All reagents for chemical synthesis were purchased from commercial sources and used without further purification. Reagents were purchased at ≥95% purity and commercially available controls were used in our biological investigations without further purification. Analytical thin layer chromatography (TLC) was performed using 250 μm Silica Gel 60 F254 pre-coated plates (EMD Chemicals Inc.). Flash column chromatography was performed using 230–400 Mesh 60 Å Silica Gel from Sorbent Technologies. All melting points were obtained, uncorrected, using a Mel-Temp capillary melting point apparatus from Laboratory Services, Inc.NMR experiments were recorded on the following instruments: Bruker Avance III HD and Avance Neo spectrometers (600 MHz and 400 MHz for 1H NMR; 151 MHz and 101 MHz for 13C NMR), and Agilent Systems VNMRS spectrometer (500 MHz for 1H NMR; 126 MHz for 13C NMR). All spectra are presented using MestReNova (Mnova) software and are displayed without the use of the signal suppression function. Spectra were obtained in the following solvents (reference peaks also included for 1H and 13C NMRs): CDCl3 (1H NMR, 7.26 ppm; 13C NMR, 77.23 ppm) and DMSO-d6 (1H NMR: 2.50 ppm, 13C NMR, 39.52 ppm). All NMR experiments were performed at room temperature. Chemical shift values (δ) are reported in parts per million (ppm) for all 1H NMR and 13C NMR spectra. 1H NMR multiplicities are reported as: s = singlet, br. s = broad singlet, d = doublet, q = quartet, m = multiplet. High-Resolution Mass Spectrometry (HRMS) were obtained for new compounds from the Chemistry Department at the University of Florida.
Bacterial strains used during these investigations include: methicillin-resistant Staphylococcus aureus (ATCC BAA-1707 and ATCC BAA-44; Clinical Isolates from Shands Hospital in Gainesville, FL: MRSA 1, MRSA 2, S. aureus 129, S. aureus 138, S. aureus 147, & S. aureus 156), methicillin-sensitive Staphylococcus epidermidis (ATCC 12228), methicillin-resistant Staphylococcus epidermidis (MRSE, ATCC 35984), Enterococcus faecalis (ATCC OG1RF), vancomycin-resistant Enterococcus (VRE, ATCC 700221), Streptococcus pneumonia (ATCC 6303), Mtb-lux (strain CDC1551 transformed with pMV306hsp+LuxG13), multidrug-resistant (MDR) Acinetobacter baumannii (ATCC 1794, ATCC 19606, & ATCC 17978), Pseudomonas aeruginosa (ATCC PA01), and Escherichia coli (Clinical Isolate UAEC-1). S. aureus strain AH3669 constitutively expressing GFP was used for confocal microscopy experiments.
All compounds were stored as dimethyl sulfoxide (DMSO) stocks at room temperature in the absence of light. To ensure compound integrity of DMSO stock solutions of our test compounds, we did not subject them to freeze–thaw cycles.
Minimum inhibitory concentration (MIC) susceptibility assay against ESKAPE pathogens
The minimum inhibitory concentration (MIC) for each test compound was determined by the broth microdilution method as recommended by the Clinical and Laboratory Standards Institute (CLSI).65 In a 96-well plate, two-fold serial dilutions of each test compound were made in a final volume of 100 μL of broth (Lysogeny Broth or LB for S. aureus, S. epidermidis MRSE 35984, S. pneumonia, A. baumannii, P. aeruginosa and E. coli; TSBG was required for S. epidermidis 12228 as this strain for optimal growth for MIC assays; Brain Heart Infusion for E. faecium and E. faecalis). Each microtiter well was inoculated with ∼105 bacterial cells at the initial time of incubation, prepared from a fresh log phase culture (OD600 0.5–1.0). The MIC was defined as the lowest concentration of a compound that completely inhibits bacterial growth after incubating for 16 hours at 37 °C (MIC values were determined by visual inspection of turbidity and spectrophotometric readings at OD600 to show ≥90% bacterial growth inhibition compared to DMSO vehicle control). The concentration range tested during this study was 0.1 to 100 μM. DMSO served as our vehicle and negative control in each microdilution MIC assay. DMSO was serially diluted with a top concentration of 1% v/v. All compounds were tested in three or more independent experiments.Minimum inhibitory concentrations against replicating Mtb
Mtb-lux (strain CDC1551 transformed with pMV306hsp+LuxG13) were grown aerobically in Middlebrook 7H9 (supplemented with glycerol, 0.05% Tween 80, 10% oleic acid-albumin-dextrose-catalase (OADC), 50 μg mL−1 Kanamycin) until log phase (OD600 0.8–1.0). After diluting to OD600 0.02, 15 μL of Mtb-lux was added into each well of 384-well plates containing the drugs serially diluted (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) (final volume per well = 30 μL; final Mtb OD600 per well = 0.01). Luminescence readouts were taken using the Synergy H4 plate reader (BioTek) after 4–5 days of incubation at 37 °C, 5% CO2.66 Isoniazid and rifampicin curves were included against replicating Mtb in complete Dubos medium for comparison of MIC values.66 MIC values against Mtb were defined as the concentration required to give 99% growth inhibition from on dose–response curves generated in GraphPad Prism 10 using the Gompertz model based on positive (rifamycin at 15 μM served as 100% growth inhibition) and negative (1% DMSO) controls. Experimental results were obtained from duplicates in at least two independent assays.
:2) (final volume per well = 30 μL; final Mtb OD600 per well = 0.01). Luminescence readouts were taken using the Synergy H4 plate reader (BioTek) after 4–5 days of incubation at 37 °C, 5% CO2.66 Isoniazid and rifampicin curves were included against replicating Mtb in complete Dubos medium for comparison of MIC values.66 MIC values against Mtb were defined as the concentration required to give 99% growth inhibition from on dose–response curves generated in GraphPad Prism 10 using the Gompertz model based on positive (rifamycin at 15 μM served as 100% growth inhibition) and negative (1% DMSO) controls. Experimental results were obtained from duplicates in at least two independent assays.
Minimum inhibitory concentrations against dormant Mtb
To determine MICs against drug-tolerant non-replicating dormant Mtb, we utilized our previously reported multi-stress dormancy model.66,67 Briefly, replicating Mtb-lux were grown aerobically in Middlebrook 7H9 as described above. After reaching log phase, the culture was pre-adapted in Complete Dubos Medium (CDM) supplemented with 10% ADS for 48 hours. Then, the cells were resuspended in Multiple Stress Dormancy Media (pH = 5.0) and kept for 9 days under hypoxia (37 °C, 5% O2, 10% CO2). At the day 9, the dormant Mtb was exposed to serially diluted test compounds and after 48 hours of treatment, the luminescence readouts were taken using the Synergy H4 plate reader (BioTek). Isoniazid and rifampicin curves were included against dormant Mtb in complete Dubos medium for comparison of MIC values.66 MIC values against Mtb were defined as the concentration required to give 99% growth inhibition from on dose–response curves generated in GraphPad Prism 10 using the Gompertz model based on positive (rifamycin at 15 μM served as 100% growth inhibition) and negative (1% DMSO) controls. Experimental results were obtained from duplicates in at least two independent assays.LDH release assay for HeLa cytotoxicity assessment
HeLa cytotoxicity was assessed using the LDH release assay described by CytoTox96 (Promega G1780). HeLa cells were grown in Dulbecco's Modified Eagle Medium (DMEM; Gibco) supplemented with 10% Fetal Bovine Serum (FBS) at 37 °C with 5% CO2. When the HeLa cultures exhibited 70–80% confluence, halogenated phenazines were then diluted by DMEM (10% FBS) at concentrations of 25, 50 and 100 μM and added to HeLa cells. Triton X-100 (at 2% v/v) was used as the positive control for maximum lactate dehydrogenate (LDH) activity in this assay (i.e., complete cell death) while “medium only” lanes served as negative control lanes (i.e., no cell death). DMSO was used as our vehicle control. HeLa cells were treated with compounds for 24 hours and then 50 μL of the supernatant was transferred into a fresh 96-well plate where 50 μL of the reaction mixture was added to the 96-well plate and incubated at room temperature for 30 minutes. Finally, Stop Solution (50 μL) was added to the incubating plates and the absorbance was measured at 490 nm. Results are from three independent experiments.Calgary biofilm device (CBD) assays
Biofilm eradication experiments were performed using the Calgary Biofilm Device to determine MBC/MBEC values for various compounds of interest (Innovotech, product code: 19111). The Calgary device (96-well plate with lid containing pegs to establish biofilms) was inoculated with 125 μL of a mid-log phase culture diluted 1000-fold in tryptic soy broth with 0.5% glucose (TSBG) to establish bacterial biofilms after incubation at 37 °C for 24 hours. The lid of the Calgary device was then removed, gently washed and transferred to a new 96-well plate containing 2-fold serial dilutions of the test compounds in media (the “challenge plate”). The total volume of media with compound in each well in the challenge plate is 150 μL. The Calgary device was then incubated at 37 °C for 24 hours. The lid was then removed from the challenge plate and MBC/MBEC values were determined using different experimental pathways. To determine MBC values, 20 μL of the challenge plate was transferred into a fresh 96-well plate containing 180 μL TSBG and incubated overnight at 37 °C. The MBC values were determined as the test concentration of compound that resulted in a complete lack of visible bacterial growth (i.e., turbidity). For determination of MBEC values, the Calgary device lid (with attached pegs/treated biofilms) was transferred to a new 96-well plate containing 150 μL of fresh TSBG media in each well and incubated for 24 hours at 37 °C to allow viable biofilms to grow and disperse resulting in turbidity after the incubation period. MBEC values were determined as the lowest test concentration that resulted in microtiter wells with no turbidity as a result of complete biofilm eradication. Notes: All data were obtained from a minimum of three independent experiments. MBC & MBEC values were determined as the lowest test concentration required to demonstrate ≥90% reduction in turbidity compared to the DMSO control as determined with the use of a spectrophotometer based on OD600 values (with background subtracted; see ESI†).Hemolysis assay with red blood cells
Freshly drawn human red blood cells (hRBC with ethylenediaminetetraacetic acid, EDTA, as an anticoagulant) were washed with Tris-buffered saline (0.01 M Tris-base, 0.155 M sodium chloride, pH 7.2) and centrifuged for 5 minutes at 3500 rpm. The washing was repeated three times with the buffer. In a 96-well plate, test compounds were added to the buffer from DMSO stocks. Then 2% hRBCs (50 μL) in buffer were added to test compounds to give a final concentration of 200 μM. The plate was then incubated for 1 hour at 37 °C. After incubation, the plate was then centrifuged for 5 minutes at 3500 rpm. Then, 80 μL of the supernatant was transferred to another 96-well plate and the optical density (OD) was read at 405 nm. DMSO served as our negative control (0% hemolysis) while Triton X served as our positive control (100% hemolysis). The percent hemolysis was calculated as (OD405 of the compound − OD405 DMSO)/(OD405 Triton X − OD405 buffer) from three independent experiments.Confocal microscope images with Staphylococcus aureus biofilms
Staphylococcus aureus strain AH3669 (USA300-0114 MRSA; pCM29_GFP)68 constitutively expressing GFP were grown overnight at 37 °C at 220 rpm in tryptic soy broth (TSB) with 10 μg mL−1 of chloramphenicol. The resulting cultures were used to seed biofilms at an OD600 = 0.5 in Biofilm Media (TSB, 0.5% Glucose) at a volume of 1 mL. Biofilms were seeded in 35 mm Matsunami glass bottom culture dishes (VWR) with glass thickness of #1.5 (0.16–0.19 mm). The edges of the dish were sealed with parafilm, and the biofilms were grown for 24 hours 37 °C under static conditions. After 24 hours of growth, the supernatant was carefully aspirated so as not to disturb bacteria adhered to the dish. The halogenated phenazine compounds were diluted in Mueller Hinton 2 broth (MHBII) from the stock concentration (10 mM) to the appropriate concentration in triplicate. A DMSO control was prepared in triplicate containing an equivalent volume to that of the highest concentration of halogenated phenazine. The dilutions and controls were added to the biofilms, the edges of the dishes were sealed with parafilm, and the biofilms were returned to the 37 °C static bacterial incubator for 24-hour incubation with the treatment. Following treatment, the supernatant was removed, and the biofilms were washed with 1 mL of phosphate buffered saline (PBS). The biofilm polysaccharide matrix material was stained with 1 mL of the 100 μg mL−1 Texas Red conjugated Concanavalin A (Invitrogen) in 0.1 M Sodium Bicarbonate for 5 minutes in the dark on a gel rocker. The dye was aspirated, and the samples were washed with 1 mL of PBS to remove residual dye. Z-stack images were taken with Zeiss LSM 710 confocal microscope using the 63× oil objective with 488 nm and 543 nm laser channels.UV-vis spectroscopy experiments
In a 1.5 mL cuvette were added 970 μL of DMSO and 30 μL of test compound (as a 10 mM DMSO stock solution). In a separate cuvette, 955 μL of DMSO, 30 μL of test compound (10 mM DMSO stock), and 15 μL of ammonium iron(II) sulfate hexahydrate were added (10 mM water solution) and thoroughly mixed. Then, spectral scanning was performed from 300 to 700 nm in 10 nm increments after 1 and 10 minutes. Note: All iron salt solutions were freshly made and added to the cuvette immediately.Abbreviations
| AlCl3 | Aluminium(III) chloride |
| BBr3 | Boron tribromide |
| BHI | Brain Heart Infusion (broth) |
| Bn | Benzyl |
| BSA | N,O-Bis(trimethylsilyl)acetamide |
| t BuOK | Potassium tert-butoxide |
| °C | Degrees celsius |
| CBD | Calgary Biofilm Device |
| CDM | Complete Dubos Medium |
| CF | Cystic fibrosis |
| cytotox. | Cytotoxicity |
| DMEM | Dulbecco's Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| EDTA | Ethylenediaminetetraacetic acid |
| FBS | Fetal Bovine Serum |
| GFP | Green fluorescent protein |
| equiv. | Equivalents |
| HP | Halogenated phenazine |
| HRMS | High resolution mass spectroscopy |
| HSQC | Heteronuclear Single Quantum Coherence |
| INH | Isoniazid |
| LB | Luria–Bertani (broth) |
| LDH | Lactate dehydrogenase |
| M | Molar |
| MBC | Minimum bactericidal concentration |
| MBEC | Minimum biofilm eradication concentration |
| MDR | Multidrug-resistant |
| Me | Methyl |
| MeOH | Methanol |
| mg | Milligram(s) |
| MH | Mueller Hinton (broth) |
| mL | Milliliters |
| mM | Millimolar |
| MIC | Minimum inhibitory concentration |
| μm | Micrometer |
| μM | Micromolar |
| μg | Microgram |
| min | Minute(s) |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| MRSE | Methicillin-resistant Staphylococcus epidermidis |
| MSSE | Methicillin-sensitive Staphylococcus epidermidis |
| Mtb | Mycobacterium tuberculosis |
| NaCl | Sodium chloride |
| NBS | N-Bromosuccinimide |
| nM | Nanomolar |
| nm | Nanometer |
| NMR | Nuclear magnetic resonance (spectroscopy) |
| OADC | Oleic acid-albumin-dextrose-catalase |
| OD | Optical density |
| PBS | Phosphate-buffered silane |
| QAC-10 | Quaternary ammonium cation-10 |
| RBCs | (Human) red blood cells |
| RIF | Rifampicin |
| rpm | Revolutions per minute |
| rt | Room temperature |
| S. epi | Staphylococcus epidermidis |
| SAR | Structure–activity relationship |
| SI | Selectivity index |
| Supp. | Supplemental |
| TLC | Thin-layer chromatography |
| TPEN | N,N,N′,N′-Tetrakis(2-pyridinylmethyl)-1,2-ethanediamine |
| TSB | Tryptic soy broth |
| TSBG | Tryptic soy broth with 0.5% glucose |
| μL | Microliters |
| TxRed | Texas Red (fluorescent dye) |
| VRE | Vancomycin-resistant Enterococcus faecium |
| v/v | Volume per volume |
Author contributions
Q. G. synthesized HPs, evaluated HPs (MIC assays, MBEC assays, iron(II)-binding experiments using UV-vis), and wrote the initial draft of this manuscript. H. Y. synthesized HPs and evaluated HPs (MIC and MBEC assays). J. S. performed confocal imaging experiments with S. aureus biofilms. P. C. B. H. evaluated HPs to determine activity profiles against replicating and dormant Mtb. K. L. evaluated HPs (MIC and MBEC assays). D. B. synthesized a few HPs and evaluated HPs (MIC and MBEC assays). S. M. evaluated HPs in MIC and MBEC assays. S. J. performed cytotoxicity assessments of HPs against HeLa cells. K. R. directed Mtb studies and drafted text describing these experiments. R. M. F. directed confocal imaging experiments and drafted manuscript text pertaining to these experiments. R. H. directed chemical synthesis, evaluation of HPs, coordinated research activities, and finalized writing this manuscript with the input of select authors. All authors have reviewed the data, agree with the findings, and approve the final manuscript.Data availability
The data underlying this study are available in the published article and its ESI.† The data contained in the manuscript include: synthesis results (Scheme 1), MIC & MBEC results (Tables 1–3 & Fig. 3–4), confocal biofilm images (Fig. 5). The contents contained in the ESI† include: supporting figures, characterization data for newly synthesized (with tabulated 1H & 13C NMR data, HRMS, melting points for solids, and NMR spectra), UV-vis spectroscopy results for iron(II) binding, microbiological assay results, cytotoxicity assay results, and HPLC traces with purity analysis for HPs synthesized and evaluated during these studies.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank the University of Georgia, the University of Florida, and the National Institute of General Medical Sciences of the National Institutes of Health (R35GM153272 to R. W. H.). This work was partially supported by the National Institutes of Health R00AI163295 to R. M. F. D.B. was supported by the NIH/NIGMS T32GM136583 “Chemistry-Biology Interface Training Program” at the University of Florida. We are very grateful for Dr Alexander Horswill from University of Colorado providing us with the S. aureus strain AH3669 constitutively expressing GFP. High resolution mass spectra were obtained for novel synthesized compounds from the University of Florida's Mass Spectrometry Research and Education Center and supported by NIH S10 OD021758-01A1. We thank Eli Levit for helpful discussions related to NMR data during the preparation of this manuscript.References
- W. R. Miller and C. A. Arias, ESKAPE Pathogens: Antimicrobial Resistance, Epidemiology, Clinical Impact and Therapeutics, Nat. Rev. Microbiol., 2024, 22, 598–616 CrossRef CAS PubMed.
- O. Ciofu, C. Moser, P. Ø. Jensen and N. Høiby, Tolerance and Resistance of Microbial Biofilms, Nat. Rev. Microbiol., 2022, 20, 621–635 CrossRef CAS PubMed.
- Y. Abouelhassan, A. T. Garrison, H. Yang, A. Chávez-Riveros, G. M. Burch and R. W. Huigens III, Recent Progress in Natural-Product-Inspired Programs Aimed to Address Antibiotic Resistance and Tolerance, J. Med. Chem., 2019, 62, 7618–7642 CrossRef CAS PubMed.
- E. M. Darby, E. Trampari, P. Siasat, M. S. Gaya, I. Alav, M. A. Webber and J. M. A. Blair, Molecular Mechanisms of Antibiotic Resistance Revisited, Nat. Rev. Microbiol., 2023, 21, 280–295 CrossRef CAS PubMed.
- J. M. Munita and C. A. Arias, Mechanisms of Antibiotic Resistance, Microbiol. Spectrum, 2016, 4, VMBF-0016-2015 Search PubMed.
- G. D. Wright, Molecular Mechanisms of Antibiotic Resistance, Chem. Commun., 2011, 47, 4055–4061 RSC.
- A. A. Bridges, J. A. Prentice, N. S. Wingreen and B. L. Bassler, Signal Transduction Network Principles Underlying Bacterial Collective Behaviors, Annu. Rev. Microbiol., 2022, 76, 235–257 CrossRef CAS PubMed.
- K. Lewis, Persister Cells: Molecular Mechanism Related to Antibiotic Tolerance, Handb. Exp. Pharmacol., 2012, 211, 121–133 CAS.
- K. Schilcher and A. R. Horswill, Staphylococcal Biofilm Development: Structure, Regulation, and Treatment Strategies, Microbiol. Mol. Biol. Rev., 2020, 84, e00026–e00019 CrossRef PubMed.
- H. Niu, J. Gu and Y. Zhang, Bacterial Persisters: Molecular Mechanisms and Therapeutic Development, Signal Transduction Targeted Ther., 2024, 9, 174 CrossRef PubMed.
- J. Yan and B. L. Bassler, Surviving as a Community: Antibiotic Tolerance and Persistence in Bacterial Biofilms, Cell Host Microbe, 2019, 26, 15–21 CrossRef CAS PubMed.
- Y. K. Wu, N. C. Cheng and C. M. Cheng, Biofilms in Chronic Wounds: Pathogenesis and Diagnosis, Trends Biotechnol., 2019, 37, 505–517 CrossRef CAS PubMed.
- K. Lewis, The Science of Antibiotic Discovery, Cell, 2020, 181, 29–45 CrossRef CAS PubMed.
- K. Lewis, R. E. Lee, H. Brötz-Oesterhelt, S. Hiller, M. V. Rodnina, T. Schneider, M. Weingarth and I. Wohlgemuth, Sophisticated Natural Products as Antibiotics, Nature, 2024, 632, 39–49 CrossRef CAS PubMed.
- T. Lluka and J. M. Stokes, Antibiotic Discovery in the Artificial Intelligence Era, Ann. N. Y. Acad. Sci., 2023, 1519, 74–93 CrossRef CAS PubMed.
- U. Theuretzbacher, B. Blasco, M. Duffey and L. J. V. Piddock, Unrealized Targets in the Discovery of Antibiotics for Gram-negative Bacterial Infections, Nat. Rev. Drug Discovery, 2023, 22, 957–975 CrossRef CAS PubMed.
- B. P. Conlon, E. S. Nakayasu, L. E. Fleck, M. D. LaFleur, V. M. Isabella, K. Coleman, S. N. Leonard, R. D. Smith, J. N. Adkins and K. Lewis, Activated ClpP Kills Persisters and Eradicates a Chronic Biofilm Infection, Nature, 2013, 503, 365–370 CrossRef CAS PubMed.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, A New Antibiotic Kills Pathogens without Detectable Resistance, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D'Onofrio and K. Lewis, A New Antibiotic Selectively Kills Gram-negative Pathogens, Nature, 2019, 576, 459–464 CrossRef CAS PubMed.
- M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, A Convergent Enantioselective Route to Structurally Diverse 6-Deoxytetracycline Antibiotics, Science, 2005, 308, 395–398 CrossRef CAS PubMed.
- J. D. Brubaker and A. G. Myers, A Practical, Enantioselective Synthetic Route to a Key Precursor to the Tetracycline Antibiotics, Org. Lett., 2007, 9, 3523–3525 CrossRef CAS PubMed.
- C. Sun, Q. Wang, J. D. Brubaker, P. M. Wright, C. D. Lerner, K. Noson, M. Charest, D. R. Siegel, Y. M. Wang and A. G. Myers, A Robust Platform for the Synthesis of New Tetracycline Antibiotics, J. Am. Chem. Soc., 2008, 130, 17913–17927 CrossRef CAS PubMed.
- P. M. Wright, I. B. Seiple and A. G. Myers, The Evolving Role of Chemical Synthesis in Antibacterial Drug Discovery, Angew. Chem., Int. Ed., 2014, 53, 8840–8869 CrossRef CAS PubMed.
- F. Liu and A. G. Myers, Development of a Platform for the Discovery and Practical Synthesis of New Tetracycline Antibiotics, Curr. Opin. Chem. Biol., 2016, 32, 48–57 CrossRef CAS PubMed.
- I. Seiple, D. Hog and A. Myers, Practical Protocols for the Preparation of Highly Enantioenriched Silyl Ethers of (R,)-3-Hydroxypentan-2-one, Building Blocks for the Synthesis of Macrolide Antibiotics, Synlett, 2015, 57–60 Search PubMed.
- Z. Zhang, T. Fukuzaki and A. G. Myers, Synthesis of D-Desosamine and Analogs by Rapid Assembly of 3-Amino Sugars, Angew. Chem., Int. Ed., 2016, 55, 523–527 CrossRef CAS PubMed.
- I. B. Seiple, Z. Zhang, P. Jakubec, A. Langlois-Mercier, P. M. Wright, D. T. Hog, K. Yabu, S. R. Allu, T. Fukuzaki, P. N. Carlsen, Y. Kitamura, X. Zhou, M. L. Condakes, F. T. Szczypiński, W. D. Green and A. G. Myers, A Platform for the Discovery of New Macrolide Antibiotics, Nature, 2016, 533, 338–345 CrossRef CAS PubMed.
- P. C. Hogan, C. L. Chen, K. M. Mulvihill, J. F. Lawrence, E. Moorhead, J. Rickmeier and A. G. Myers, Large-Scale Preparation of Key Building Blocks for the Manufacture of Fully Synthetic Macrolide Antibiotics, J. Antibiot., 2018, 71, 318–325 CrossRef CAS PubMed.
- M. J. Mitcheltree, A. Pisipati, E. A. Syroegin, K. J. Silvestre, D. Klepacki, J. D. Mason, D. W. Terwilliger, G. Testolin, A. R. Pote, K. J. Y. Wu, R. P. Ladley, K. Chatman, A. S. Mankin, Y. S. Polikanov and A. G. Myers, A Synthetic Antibiotic Class Overcoming Bacterial Multidrug Resistance, Nature, 2021, 599, 507–512 CrossRef CAS PubMed.
- J. Xie, J. G. Pierce, R. C. James, A. Okano and D. L. Boger, A Redesigned Vancomycin Engineered for Dual D-Ala-D-ala and D-Ala-D-Lac Binding Exhibits Potent Antimicrobial Activity Against Vancomycin-Resistant Bacteria, J. Am. Chem. Soc., 2011, 133, 13946–13949 CrossRef CAS PubMed.
- J. Xie, A. Okano, J. G. Pierce, R. C. James, S. Stamm, C. M. Crane and D. L. Boger, Total Synthesis of [Ψ[C(
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)NH]Tpg4]Vancomycin Aglycon, [Ψ[C(NH)NH]Tpg4]Vancomycin Aglycon, and Related Key Compounds: Reengineering Vancomycin for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding, J. Am. Chem. Soc., 2012, 134, 1284–1297 CrossRef CAS PubMed.
S)NH]Tpg4]Vancomycin Aglycon, [Ψ[C(NH)NH]Tpg4]Vancomycin Aglycon, and Related Key Compounds: Reengineering Vancomycin for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding, J. Am. Chem. Soc., 2012, 134, 1284–1297 CrossRef CAS PubMed. - A. Okano, A. Nakayama, A. W. Schammel and D. L. Boger, Total Synthesis of [Ψ[C(NH)NH]Tpg4]Vancomycin and its (4-Chlorobiphenyl)methyl Derivative: Impact of Peripheral Modifications on Vancomycin Analogues Redesigned for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding, J. Am. Chem. Soc., 2014, 136, 13522–13525 CrossRef CAS PubMed.
- A. Okano, A. Nakayama, K. Wu, E. A. Lindsey, A. W. Schammel, Y. Feng, K. C. Collins and D. L. Boger, Total Synthesis and Initial Evaluation of [Ψ[C(S)NH]Tpg4]Vancomycin, [Ψ[C(NH)NH]Tpg4]Vancomycin, [Ψ[CH2NH]Tpg4]Vancomycin, and Their (4-Chlorobiphenyl)methyl Derivatives: Synergistic Binding Pocket and Peripheral Modifications for the Glycopeptide Antibiotics, J. Am. Chem. Soc., 2015, 137, 3693–3704 CrossRef CAS PubMed.
- A. Okano, N. A. Isley and D. L. Boger, Peripheral Modifications of [Ψ[CH2NH]Tpg4]Vancomycin with Added Synergistic Mechanisms of Action Provide Durable and Potent Antibiotics, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5052–E5061 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Predictive Compound Accumulation Rules Yield a Broad-Spectrum Antibiotic, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- M. F. Richter and P. J. Hergenrother, The Challenge of Converting Gram-positive-Only Compounds into Broad-Spectrum Antibiotics, Ann. N. Y. Acad. Sci., 2019, 1435, 18–38 CrossRef CAS PubMed.
- S. E. Motika, R. J. Ulrich, E. J. Geddes, H. Y. Lee, G. W. Lau and P. J. Hergenrother, A Gram-negative Antibiotic Active Through Inhibition of an Essential Riboswitch, J. Am. Chem. Soc., 2020, 142, 10856–10862 CrossRef CAS PubMed.
- E. N. Parker, B. S. Drown, E. J. Geddes, H. Y. Lee, N. Ismail, G. W. Lau and P. J. Hergenrother, Implementation of Permeation Rules Leads to a FabI Inhibitor with Activity Against Gram-negative Pathogens, Nat. Microbiol., 2020, 5, 67–75 CrossRef CAS PubMed.
- K. A. Munoz and P. J. Hergenrother, Facilitating Compound Entry as a Means to Discover Antibiotics for Gram-negative Bacteria, Acc. Chem. Res., 2021, 54, 1322–1333 CrossRef CAS PubMed.
- E. J. Geddes, M. K. Gugger, A. Garcia, M. G. Chavez, M. R. Lee, S. J. Perlmutter, C. Bieniossek, L. Guasch and P. J. Hergenrother, Porin-Independent Accumulation in Pseudomonas Enables Antibiotic Discovery, Nature, 2023, 624, 145–153 CrossRef CAS PubMed.
- K. A. Muñoz, R. J. Ulrich, A. K. Vasan, M. Sinclair, P. C. Wen, J. R. Holmes, H. Y. Lee, C. C. Hung, C. J. Fields, E. Tajkhorshid, G. W. Lau and P. J. Hergenrother, A Gram-negative-Selective Antibiotic that Spares the Gut Microbiome, Nature, 2024, 630, 429–436 CrossRef PubMed.
- P. A. Smith, M. F. T. Koehler, H. S. Girgis, D. Yan, Y. Chen, Y. Chen, J. J. Crawford, M. R. Durk, R. I. Higuchi, J. Kang, J. Murray, P. Paraselli, S. Park, W. Phung, J. G. Quinn, T. C. Roberts, L. Rougé, J. B. Schwarz, E. Skippington, J. Wai, M. Xu, Z. Yu, H. Zhang, M. W. Tan and C. E. Heise, Optimized Arylomycins are a New Class of Gram-negative Antibiotics, Nature, 2018, 561, 189–194 CrossRef CAS PubMed.
- J. B. Laursen and J. Nielsen, Phenazine Natural Products: Biosynthesis, Synthetic Analogues, and Biological Activity, Chem. Rev., 2004, 104, 1663–1685 CrossRef CAS PubMed.
- A. Price-Whelan, L. E. P. Dietrich and D. K. Newman, Rethinking “Secondary” Metabolism: Physiological Roles for Phenazine Antibiotics, Nat. Chem. Biol., 2006, 2, 71–78 CrossRef CAS PubMed.
- N. Guttenberger, W. Blankenfeldt and R. Breinbauer, Recent Developments in the Isolation, Biological Function, Biosynthesis, and Synthesis of Phenazine Natural Products, Bioorg. Med. Chem., 2017, 25, 6149–6166 CrossRef CAS PubMed.
- Z. A. Machan, T. L. Pitt, W. White, D. Watson, G. W. Taylor, P. J. Cole and R. Wilson, Interaction Between Pseudomonas aeruginosa and Staphylococcus aureus: Description of an Antistaphylococcal Substance, J. Med. Microbiol., 1991, 34, 213–217 CrossRef CAS PubMed.
- N. V. Borrero, F. Bai, C. Perez, B. Q. Duong, J. R. Rocca, S. Jin and R. W. Huigens III, Phenazine Antibiotic Inspired Discovery of Potent Bromophenazine Antibacterial Agents against Staphylococcus aureus and Staphylococcus epidermidis, Org. Biomol. Chem., 2014, 12, 881–886 RSC.
- A. T. Garrison, Y. Abouelhassan, D. Kallifidas, F. Bai, M. Ukhanova, V. Mai, S. Jin, H. Luesch and R. W. Huigens, Halogenated Phenazines That Potently Eradicate Biofilms, MRSA Persister Cells in Non–Biofilm Cultures, and Mycobacterium tuberculosis, Angew. Chem., Int. Ed., 2015, 54, 14819–14823 CrossRef CAS PubMed.
- A. T. Garrison, Y. Abouelhassan, V. M. Norwood, D. Kallifidas, F. Bai, M. T. Nguyen, M. Rolfe, G. M. Burch, S. Jin, H. Luesch and R. W. Huigens, Structure–Activity Relationships of a Diverse Class of Halogenated Phenazines that Targets Persistent, Antibiotic-Tolerant Bacterial Biofilms and Mycobacterium tuberculosis, J. Med. Chem., 2016, 59, 3808–3825 CrossRef CAS PubMed.
- H. Yang, Y. Abouelhassan, G. M. Burch, D. Kallifidas, G. Huang, H. Yousaf, S. Jin, H. Luesch and R. W. Huigens, A Highly Potent Class of Halogenated Phenazine Antibacterial and Biofilm-Eradicating Agents Accessed Through a Modular Wohl-Aue Synthesis, Sci. Rep., 2017, 7, 2003 CrossRef PubMed.
- Y. Abouelhassan, Y. Zhang, S. Jin and R. W. Huigens III, Transcript Profiling of MRSA Biofilms Treated with a Halogenated Phenazine Eradicating Agent: A Platform for Defining Cellular Targets and Pathways Critical to Biofilm Survival, Angew. Chem., Int. Ed., 2018, 57, 15523–15528 CrossRef CAS PubMed.
- A. T. Garrison, Y. Abouelhassan, D. Kallifidas, H. Tan, Y. S. Kim, S. Jin, H. Luesch and R. W. Huigens, An Efficient Buchwald–Hartwig/Reductive Cyclization for the Scaffold Diversification of Halogenated Phenazines: Potent Antibacterial Targeting, Biofilm Eradication, and Prodrug Exploration, J. Med. Chem., 2018, 61, 3962–3983 CrossRef CAS PubMed.
- H. Yang, S. Kundra, M. Chojnacki, K. Liu, M. A. Fuse, Y. Abouelhassan, D. Kallifidas, P. Zhang, G. Huang, S. Jin, Y. Ding, H. Luesch, K. H. Rohde, P. M. Dunman, J. A. Lemos and R. W. Huigens, A Modular Synthetic Route Involving N-Aryl-2-Nitrosoaniline Intermediates Leads to a New Series of 3-Substituted Halogenated Phenazine Antibacterial Agents, J. Med. Chem., 2021, 64, 7275–7295 CrossRef CAS PubMed.
- K. Liu, M. Brivio, T. Xiao, V. M. Norwood, Y. S. Kim, S. Jin, A. Papagni, L. Vaghi and R. W. Huigens, Modular Synthetic Routes to Fluorine-Containing Halogenated Phenazine and Acridine Agents That Induce Rapid Iron Starvation in Methicillin-Resistant Staphylococcus aureus Biofilms, ACS Infect. Dis., 2022, 8, 280–295 CrossRef CAS PubMed.
- T. Xiao, K. Liu, Q. Gao, M. Chen, Y. S. Kim, S. Jin, Y. Ding and R. W. Huigens III, Design, Synthesis and Evaluation of Carbonate-Linked Halogenated Phenazine-Quinone Prodrugs with Improved Water-Solubility and Potent Antibacterial Profiles, ACS Infect. Dis., 2023, 9, 899–915 CrossRef CAS PubMed.
- T. Fu, Z. Cai, Z. Yue, H. Yang, B. Fang, X. Zhang, Z. Fan, X. Pan, F. Yang, Y. Jin, Z. Cheng, W. Wu, B. Sun, R. W. Huigens, L. Yang and F. Bai, Evolution of Resistance to Phenazine Antibiotics in Staphylococcus aureus and Its Role During Coinfection with Pseudomonas aeruginosa, ACS Infect. Dis., 2021, 7, 636–649 CrossRef CAS PubMed.
- Z. Wróbel and A. Kwast, 2-Nitroso-N-arylanilines: Products of Acid-Promoted Transformation of σH Adducts of Arylamines and Nitroarenes, Synlett, 2007, 1525–1528 CrossRef.
- Z. Wróbel and A. Kwast, Simple Synthesis of N-Aryl-2-nitrosoanilines in the Reaction of Nitroarenes with Aniline Anion Derivatives, Synthesis, 2010, 3865–3872 CrossRef.
- A. Kwast, K. Stachowska, A. Trawczyński and Z. Wróbel, N-Aryl-2-Nitrosoanilines as Intermediates in the Synthesis of Substituted Phenazines from Nitroarenes, Tetrahedron Lett., 2011, 52, 6484–6488 CrossRef CAS.
- Z. Wróbel, K. Plichta and A. Kwast, Reactivity and Substituent Effects in the Cyclization of N-Aryl-2-nitrosoanilines to Phenazines, Tetrahedron, 2017, 73, 3147–3152 CrossRef.
- K. Liu, T. Xiao, H. Yang, M. Chen, Q. Gao, B. R. Brummel, Y. Ding and R. W. Huigens III, Design, Synthesis and Evaluation of Halogenated Phenazine Antibacterial Prodrugs Targeting Nitroreductase Enzymes for Activation, RSC Med. Chem., 2023, 14, 1472–1481 RSC.
- H. Ceri, M. E. Olson, C. Stremick, R. R. Read, D. Morck and A. Buret, The Calgary Biofilm Device: New Technology for Rapid Determination of Antibiotic Susceptibilities of Bacterial Biofilms, J. Clin. Microbiol., 1999, 37, 1771–1776 CrossRef CAS PubMed.
- J. J. Harrison, C. A. Stremick, R. J. Turner, N. D. Allan, M. E. Olson and H. Ceri, Microtiter Susceptibility Testing of Microbes Growing on Peg Lids: A Miniaturized Biofilm Model for High-Throughput Screening, Nat. Protoc., 2010, 5, 1236–1254 CrossRef CAS PubMed.
- M. C. Jennings, L. E. Ator, T. J. Paniak, K. P. C. Minbiole and W. M. Wuest, Biofilm-Eradicating Properties of Quaternary Ammonium Amphiphiles: Simple Mimetics of Antimicrobial Peptides, ChemBioChem, 2014, 15, 2211–2215 CrossRef CAS PubMed.
- Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standards, Clinical and Laboratory Standards Institute, Wayne, PA, 8th edn (M7–M8), 2009 Search PubMed.
- F. C. Rodrigues, R. Gupta, S. Geden, J. Roberts, P. Winder, S. A. Pomponi, M. C. Diaz, J. K. Reed, A. E. Wright and K. H. Rohde, Selective Killing of Dormant Mycobacterium tuberculosis by Marine Natural Products, Antimicrob. Agents Chemother., 2017, 61, e00743–e00717 Search PubMed.
- C. Deb, C. M. Lee, V. S. Dubey, J. Daniel, B. Abomoelak, T. D. Sirakova, S. Pawar, L. Rogers and P. E. Kolattukudy, A Novel In Vitro Multiple-Stress Dormancy Model for Mycobacterium tuberculosis Generates a Lipid-Loaded, Drug-Tolerant, Dormant Pathogen, PLoS One, 2009, 4, e6077 CrossRef PubMed.
- Y. Y. Pang, J. Schwartz, M. Thoendel, L. W. Ackermann, A. R. Horswill and W. M. Nauseef, agr-Dependent Interactions of Staphylococcus aureus USA300 with Human Polymorphonuclear Neutrophils, J. Innate Immun., 2010, 2, 546–559 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures, characterization data for newly synthesized (with tabulated 1H & 13C NMR data, HRMS, melting points for solids, and NMR spectra), UV-vis spectroscopy results for iron(II) binding, microbiological assay results, cytotoxicity assay results, and HPLC traces with purity analysis for HPs synthesized and evaluated during these studies. See DOI: https://doi.org/10.1039/d4ob02011a |
| This journal is © The Royal Society of Chemistry 2025 |