
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in asymmetric synthesis via cyclopropanol intermediates
Marharyta
Laktsevich-Iskryk
 a,
Alaksiej
Hurski
bc,
Maksim
Ošeka
a and
Dzmitry
Kananovich
*a
a,
Alaksiej
Hurski
bc,
Maksim
Ošeka
a and
Dzmitry
Kananovich
*a
aDepartment of Chemistry and Biotechnology, Tallinn University of Technology, Akadeemia tee 15, 12618, Tallinn, Estonia. E-mail: dzmitry.kananovich@taltech.ee
bRepublican Scientific Center of Human Issues, Belarusian State University, Minsk 220064, Belarus
cScientific Testing Center Campilab Ltd., Dynaraŭka 222202, Belarus
First published on 4th December 2024
Abstract
Cyclopropanols have attracted significant attention in organic synthesis as versatile three-carbon synthons, as this readily available class of donor-activated cyclopropanes undergoes miscellaneous transformations, either via ring-opening or with retention of the cyclopropane ring. This review summarizes stereoselective and stereoretentive transformations suitable for asymmetric synthesis. The utility of cyclopropanols is discussed for two main strategies: (i) substrate-controlled transformations using enantiomerically enriched cyclopropanol intermediates through a traditional approach, and (ii) the use of nonchiral or racemic cyclopropanols, where asymmetric induction is achieved through a chiral catalyst, representing a direction that has recently emerged.
 Marharyta Laktsevich-Iskryk | Marharyta Laktsevich-Iskryk earned her bachelor's degree from Belarusian State University in 2016. During her undergraduate studies, she joined Dr Alaksiej Hurski's group in the Laboratory of Steroidal Chemistry at the Institute of Bioorganic Chemistry (Minsk, Belarus), where she conducted research on transition metal catalysis and photoredox-catalysed transformations of cyclopropanols. After obtaining her master's degree, she visited the Tallinn University of Technology (TalTech), where she explored the enantioselective synthesis of cyclopropanols with Dr Dzmitry Kananovich. In 2023, Marharyta began her PhD studies at TalTech under the supervision of Professor Maksim Ošeka, focusing on enantioselective electrochemical synthesis. |
 Alaksiej Hurski | Alaksiej Hurski graduated from Belarusian State University (Minsk, Belarus) in 2005. In 2011, he received his Ph.D. under the supervision of Professor Oleg Kulinkovich. His current research focuses on the photochemical reactions of titanium alkoxides, the reactions of cyclopropanols, and the synthesis of natural products. |
 Maksim Ošeka | Maksim Ošeka is an Assistant Professor at the Tallinn University of Technology (TalTech), Estonia, where he focuses on advancing organic synthesis, including photochemistry, electrochemistry, asymmetric organocatalysis, and flow chemistry. He earned his PhD from TalTech in 2017 under the supervision of Professor Tõnis Kanger, specializing in asymmetric organocatalysis. As a visiting PhD student with the Melchiorre group at ICIQ in Spain, he contributed to developing a new photocatalytic enantioselective alkylation reaction. Maksim then pursued postdoctoral research under Professor Timothy Noël at the Eindhoven University of Technology, the Netherlands, working on electrochemical flow methods, before returning to TalTech in 2021 to launch his independent career. |
 Dzmitry Kananovich | Dzmitry Kananovich obtained his doctoral degree in 2008 under the guidance of Professor Oleg Kulinkovich at Belarusian State University (BSU), Minsk, Belarus. He continued working with Professor Kulinkovich on the asymmetric synthesis of cyclopropanols, first at BSU and later at the Tallinn University of Technology (TalTech) from 2011, in collaboration with Professor Margus Lopp. In 2015, he began his independent career, focusing on cyclopropanol and cyclopropylamine chemistry. His current research interests include organometallic chemistry, cyclopropane chemistry, and the use of mechanochemistry as a green technique in organic synthesis, developed within the Supramolecular Chemistry group at TalTech (with Professor Riina Aav). |
1. Introduction
Cyclopropanols and related oxocyclopropanes are versatile intermediates and valuable C3 building blocks in organic synthesis, attracting significant interest in recent decades.1–8 The popularity of these cyclopropane derivatives stems from ease of preparation1,9,10 and diverse reactivity, primarily in ring cleavage reactions. The latter occur under mild conditions, driven by the release of the cyclopropane ring strain (28 kcal mol−1) and facilitated by virtue of an electron-donating oxy-substituent (Scheme 1a).11 This unique combination of structural features makes cyclopropanols 1 (Y = H) and their derivatives (Y ≠ H) particularly sensitive to electrophilic reagents and single electron oxidants, which induce either heterolytic or homolytic cleavage of the cyclopropane C–C bonds adjacent to oxygen and produce metal homoenolates A (path a) or β-keto radicals B (path b) as transient species, respectively.1–8 Additional ring-opening opportunities are offered by cationic cyclopropyl–allyl rearrangement (path c), which delivers transient allylic cations C, and by ring expansion via semipinacol rearrangement, resulting in cyclobutanones 2 (path d). Beyond these mainstream ring cleavage processes, cyclopropanols can serve as precursors to other cyclopropane derivatives, notably through the formal substitution of an oxy substituent (path e) and very recently emerging examples of C–H functionalization methodologies (path f). | ||
| Scheme 1 (a) Overview of cyclopropanol chemistry. (b) Cyclopropanols as intermediates in asymmetric synthesis. | ||
The aim of this review is to illuminate the diversity of stereoselective and stereoretentive transformations of cyclopropanols, suitable for asymmetric synthesis. We focused primarily, but not exclusively, on developments from the past decade, especially those not covered in previous reviews.1–8,12–14 The reported transformations have been categorized into two main strategies (Scheme 1b). In the first part (section 2), we review transformations in which the stereochemical integrity of the original stereocenters in the starting cyclopropanols is preserved in the products. This group of traditional methods was the first to emerge historically and it employs enantiomerically enriched cyclopropanol intermediates, available from the chiral pool or through asymmetric synthesis. The second strategy (section 3) appeared very recently and it uses readily available nonchiral or racemic cyclopropanols to generate prochiral intermediates, with asymmetric induction provided by a chiral catalyst.
2. Enantioenriched cyclopropanol intermediates: substrate-controlled transformations
2.1. Enantioselective synthesis of cyclopropanols
Within this strategy, the stereochemical outcome is governed by the cyclopropanol substrate itself, necessitating the preservation of the original stereocenters and enantiomeric purity. Consequently, the synthesis of enantiomerically pure cyclopropanols is essential for this approach and represents the main challenge. Here, we only briefly outline the methods for preparing enantiomerically enriched cyclopropanols and discuss current limitations. A comprehensive description is outside the scope of this review. Recent focused review articles can be recommended for a more detailed overview of the topic.10,15The primary strategies for the preparation of enantioenriched cyclopropanols are presented in Scheme 2. These include functional group interconversion in cyclopropane precursors (method a), cyclopropanation of esters with alkoxytitanacyclopropane reagents (the Kulinkovich reaction, method b),16 cyclopropanation of alkenes with carbenes and carbenoids (method c; e.g., the Simmons–Smith reaction),17 addition reactions to cyclopropenes (method d),18,19 and intramolecular 1,3-cyclization of homoenolates20 and similar 1,3-dipolar synthons (method e).21,22 Finally, given the easy access to racemic cyclopropanols, kinetic resolution (method f) is a compelling addition to synthetic methods a–e.
 | ||
| Scheme 2 Strategies (a–f) for the preparation of enantiomerically enriched cyclopropanols. Approaches b–d and f for which asymmetric catalytic versions are known, are marked with a green highlight. | ||
As a rule, the preparation of enantioenriched cyclopropanols from the chiral pool by means of substrate-controlled asymmetric induction is a relatively straightforward task by using all these approaches and has been extensively applied in the synthesis of natural and bioactive products via cyclopropanol intermediates.5,21,23
In the functional group interconversion methods, Baeyer–Villiger oxidation of cyclopropyl ketones 2 is historically the first method24 used to prepare enantiopure cyclopropanols (Scheme 3a). This method preserves the configuration and enantiomeric purity of the starting cyclopropyl ketone. Recently, this transformation was applied to the synthesis of cyclopropoxy building block 4 for the preparation of the antiviral drug grazoprevir.25
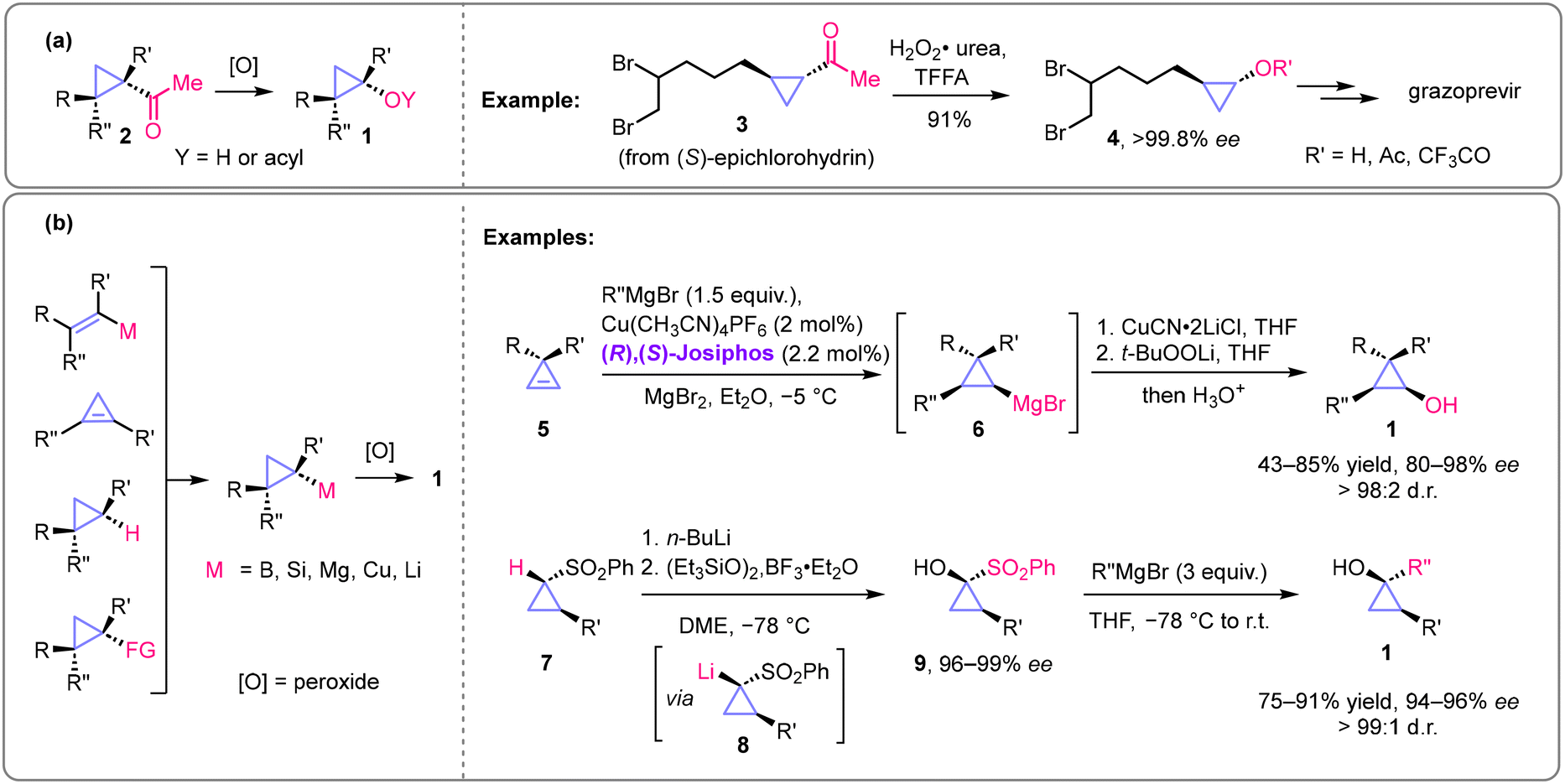 | ||
| Scheme 3 Preparation of enantiomerically pure cyclopropanols via functional group interconversion and through addition to cyclopropenes. General methods and examples of recent developments. TFFA = trifluoroacetic anhydride. | ||
Another general and widely applied method following the same strategy is based on the oxidation of readily available organoelement derivatives of cyclopropane (M = B, Si, Cu, Mg, Li) with peroxide-type oxidants (Scheme 3b). This oxidation generally occurs under mild conditions with retention of configuration and enantiomeric purity. Most commonly, enantiopure organoboron derivatives of cyclopropane are used as precursors.26 Marek and co-workers showed that organocopper and organomagnesium derivatives (e.g., 6), which are available via chiral phosphine ligand-controlled carbocupration27 or carbomagnesiation19 of cyclopropenes (5), could be used similarly. Notably, the carbomagnesiation method19 can also operate as a catalytic asymmetric reaction, affording the respective cyclopropanol products with 80–98% ee and excellent diastereoselectivity. Another notable general methodology was developed recently by Lindsay and co-workers.28 They demonstrated that diverse enantioenriched (94–96% ee) 1,2-disubstituted cyclopropanols 1 could be obtained from optically active sulfonylcyclopropanes 7 by oxidation of cyclopropyllithium derivatives 8 followed by treatment of intermediate 1-sulfonylcyclopropanols 9 with Grignard reagents (Scheme 3b).
By contrast to the chiral pool approaches, asymmetric synthesis enabled by a chiral catalyst can be challenging due to a lack of enantioselectivity, substrate scope limitations, or the total absence of an asymmetric variant. For example, the Kulinkovich reaction is predominantly used in the synthesis of nonchiral or racemic cyclopropanols, and can be equally successful for substrate-controlled cyclopropanation of chiral esters and/or alkenes.5,29,30 However, its asymmetric variant, based on the use of titanium(IV) TADDOLate complexes,31 is much less frequently utilized. This is because it may deliver variable yields and enantioselectivities depending on the cyclopropanol product, and achieving high 84–87% ee values requires stoichiometric amounts (1–2 equiv.) of chiral titanium alkoxide (Scheme 4).32,33 The catalytic version (10–20 mol% of the titanium catalyst) results in decreased ee values,34 likely due to the degradation of pentacoordinated titanium “ate-type” intermediates into less sterically hindered tetrahedral species.33 The mechanism-driven design of chiral ligands that prevent this degradation and stabilize or mimic the spatial structure of the ate complex intermediates may be crucial for developing a robust and highly enantioselective catalytic method.
 | ||
| Scheme 4 Recent examples of the asymmetric construction of cyclopropanols via enantioselective Kulinkovich reaction. | ||
Kinetic resolution of cyclopropyl acetate esters via enzymatic hydrolysis with Candida antarctica B lipase (CAL-B) was first demonstrated by Pietruszka and co-workers in the late 1990s.35 Following this seminal work, non-enzymatic methods remained unexploited until very recently, when Yoshikai and co-workers reported the first examples.36,37 Thus, enantiomerically enriched cyclopropanol (S,S)-1a was obtained in 32% yield and 84% ee via kinetic resolution through a ring-opening reaction using diethyl zinc and amino alcohol (1R,2S)-14 as the catalytic system (Scheme 5).37 However, the theoretical maximum yield of 50% represents a significant limitation of the kinetic resolution approach. Consequently, the development of dynamic kinetic resolution methods is highly appealing. Observations that cyclopropanols can undergo reversible ring-opening and ring-closure through the respective metal homoenolates,38 the configurational lability of stereocenters in organometallic compounds,39 and the generation of a single cyclopropanol diastereomer by intramolecular cyclization of diastereomeric β-metalloketone intermediates20 suggest the feasibility of such methods, which are currently lacking.
 | ||
| Scheme 5 Kinetic resolution of cyclopropanol 1a by Yoshikai and co-workers. | ||
2.2. Stereo-retentive ring opening into β-functionalized carbonyl compounds
The ring-opening of enantioenriched cyclopropanols is the most commonly applied method. This approach dates back to the 1960s with the works of DePuy and co-workers,24 who performed stereochemical studies on the ring-opening reactions of an optically active cyclopropanol 1b to elucidate the mechanism of ring cleavage (Scheme 6). They demonstrated that the ring opening of 1b occurred at the most substituted C1–C2 bond with inversion and retention of configuration at the C-2 stereocenter in aqueous base and acid, respectively.24 Subsequently, the same group provided evidence for the inversion of configuration at the point of electrophilic attack upon the action of electrophilic halogens40 and mercury(II) acetate.41 Importantly, the reaction of 1b with the latter predominantly occurred via cleavage of the C1–C3 bond without affecting the stereocenter at C-2. These seminal results have paved the way for creating stereocenters in acyclic systems via the ring-opening reactions of chiral cyclopropanol intermediates, particularly in the synthesis of chiral α-alkyl ketones. It is crucial to note that carbonyl compounds with an α-stereocenter are prone to racemization under acidic or basic conditions. Hence, the reaction conditions become essential for preserving the C-2 stereocenter intact. Known transformations that meet this criterion are detailed in this section below. Unfortunately, despite the relatively large number of known ring-opening transformations of cyclopropanols into functionalized carbonyl compounds, data on product epimerization and the behaviour of 1,2-disubstituted cyclopropanols are still lacking for many newly discovered reactions. We believe that the stereochemical aspects of such newly discovered transformations should be rigorously investigated whenever possible. It is essential to include 1,2-disubstituted cyclopropanols, preferably in enantiomerically enriched form, in the substrate scope. | ||
| Scheme 6 (a) Early results of DePuy and co-workers on the stereochemistry of ring-opening reactions of optically active cyclopropanol 1b. (b) Current approaches (i)–(iii) for the synthesis of α-alkyl ketones by regioselective C1–C3 ring opening of cyclopropanols. | ||
C1–C3 regioselectivity of the ring-opening is usually observed in the catalytic isomerization of 1,2-dialkylsubstituted cyclopropanols to α-methyl ketones 19 (Scheme 6b, path i), reactions with halogen or carbon-centered electrophiles (path ii), and transition metal-promoted generation of homoenolates A (path iii). 2-Aryl- or heteroaryl-substituted cyclopropanols (e.g.1b) may not follow the same regioselectivity rule favouring C1–C2 cleavage due to electrophilic attack at the activated benzylic C-2 carbon.
Isomerization to α-methyl ketones can be easily performed by heating 1,2-disubstituted cyclopropanols in the presence of potassium hydroxide.42 However, strongly basic conditions are detrimental to the epimerization-sensitive α-stereocenter. Therefore, alternative catalytic systems, including [Pt(C2H4)Cl2]2 (Zeise's dimer),43,44 Pd/C,45,46 EtZnCH2I47 and Mg(OMe)2,29 are shown to be suitable for the task (Scheme 7). Mechanistically, these reactions likely involve the generation of homoenolate (β-metalloketone) intermediates, followed by a protodemetallation step. For example, the application of a platinum catalyst was first reported by Hoberg and Jennings,43 followed by Funk and co-workers44 who additionally demonstrated similar catalytic properties of a dimeric iridium complex, [Cp*IrCl2]2. The reaction with the Pt complex was proposed to occur via the insertion of Pt into the C1–C3 bond of the cyclopropane ring following by rearrangement of the metallacyclobutane intermediate A into the corresponding Pt homoenolate B (Scheme 7a).43 Although the original articles described the preparation of racemic ketones (e.g., 19a and 19b), Cha and co-workers demonstrated stereo-retentive isomerization using a platinum catalyst to prepare α-methyl ketone 19c, a key intermediate in the synthesis of the ingenol core.48 Recently, Newhouse and co-workers successfully employed similar conditions for the isomerization of a chiral cyclopropanol intermediate in the final stage of the total synthesis of (−)-xylogranatopyridine B.49
 | ||
| Scheme 7 Stereo-retentive isomerization of cyclopropanols into α-methyl ketones (cp = cyclopropanol). | ||
Hurski and co-workers discovered magnesium methylate, Mg(OMe)2, as a non-precious metal catalyst for the stereo-retentive isomerization of a broad range of 1,2-disubstituted cyclopropanols,29 including enantiomerically pure steroid-derived substrates.50 The respective α-methyl ketone products (e.g., 19d and 19e, Scheme 7b) were obtained in good yields with negligible epimerization. The reagent acts as a mild base, and the magnesium cation is likely coordinating to the cyclopropanol hydroxyl group during the reaction. However, notable epimerization (degradation of ee up to 20–40%) was noted for ketone (R)-19f with a free hydroxy group in the side chain.33 By contrast, epimerization was absent in structurally similar silyl-protected products (e.g., 19d). These results suggest that the use hydroxyl-protecting groups for the side-chain hydroxyls might be crucial for preserving enantiomeric purity in this method.
Ring-opening with electrophilic halogenating reagents (e.g., Br2, NBS, or t-BuOBr as synthetic equivalents of Br+)40 also proceeds in a stereo-retentive fashion if it occurs along the C1–C3 bond. The halogenating agent can be conveniently generated in situ by oxidizing a halide anion with CAN51 or through electrolysis.52 β-Halogenated ketones, especially bromine and iodine derivatives with α-hydrogens, are prone to dehydrohalogenation to form α-methylene ketones. Nevertheless, β-halogenated ketones, for which the elimination process is impossible or sluggish due to the absence of α-hydrogens or conformational restrictions, can be isolated and used in various C–C bond-forming reactions. Among recent examples, ring-opening bromination with NBS was applied by Abad and co-workers in the synthesis of atisene diterpene for the construction of its C4 quaternary stereocenter (Scheme 8a).53 Hurski and co-workers developed an electrochemical protocol for the ring-opening bromination and iodination of 1,2-disubstituted cyclopropanols, demonstrating that diastereomerically pure β-bromoketone 22b can be used in nickel-catalysed cross-electrophile coupling with 2-bromopropene to produce an α-substituted ketone 23.52 No epimerization was observed during the ring-opening and cross-coupling steps. Moreover, a number of other copper- and nickel-catalysed C–C coupling methods behaved similarly.
 | ||
| Scheme 8 Stereo-retentive ring-opening of cyclopropanols into β-bromo ketones. | ||
For C–C bond forming reactions, regioselective C1–C3 ring-opening of trimethylsiloxycyclopropanes through intramolecular alkylation with a pendant carbon-centered electrophile (oxocarbenium ion) was reported by Cha and co-workers (Scheme 9).54 The reaction enabled the stereoselective preparation of all-cis-2,3,6-trisubstituted tetrahydropyranes (e.g., 25) and occurred without epimerization of the α-stereocenter adjacent to the carbonyl group. This work represents a rare example of stereoselective metal-free ring-opening transformation of cyclopropanols yielding a C–C bond. However, the majority of known methods rely on the generation of metal homoenolate (β-metalloketone) intermediates.3,7
 | ||
| Scheme 9 Stereoselective synthesis of 2,4,6-trisubstituted tetrahydropyrans reported by Cha and co-workers. | ||
The stereoselective metal-mediated processes usually proceed via heterolytic C1–C3 bond cleavage that retains the C2 stereocenter, followed by carbon–carbon or carbon–heteroatom cross-coupling reactions. Typically, this mode of ring opening occurs with electrophilic metal species that do not exhibit single-electron oxidant properties. Examples include transition metals with a single preferred oxidation state (e.g., HgII, ZnII) or low-valent derivatives of noble metals (e.g., PdII, RuII) that react in a two-electron transfer mode. In contrast, this mode of reactivity is less common for the first-row transition metals, which tend to react via single-electron transfer (SET). Conversely, single-electron oxidation of cyclopropanols triggers homolytic C1–C2 bond cleavage, leading to racemization at C2 due to the formation of secondary β-keto radicals, a reaction typically observed with FeIII or MnIII.55,56 Nevertheless, pro-chiral β-keto radicals can still be employed in catalyst- or substrate-controlled enantioselective transformations (as described in section 3). For certain first-row transition metals (e.g., Ni, Cu), the preferred mechanism of ring-opening can be influenced by factors such as the oxidation state of the metal, ligand properties, substrate structure, solvent, and temperature. These factors can result in the generation of either C1–C3 or C1–C2 ring opening products or their mixtures.57
Stereoselective copper-catalysed ring-opening cross-coupling of enantiomerically enriched zinc cyclopropoxides 1-Zn (Scheme 10) with allylic and propargylic bromides was demonstrated by Nomura and Matsubara,22 who generated these species by the reactions of optically active α,β-epoxy ketones or α-sulfonyloxy ketones with bis(iodozincio)methane. Subsequently, similar reactions were thoroughly investigated by Cha and co-workers58–61 Cyclopropanols were converted into zinc cyclopropoxides by the reaction with diethylzinc and further engaged in copper-catalysed carbon–carbon bond-forming reactions with allyl, propargyl, alkynyl halides, phosphates, sulfonates,58 or allenes.61 The ring-opening cross-coupling with acyl chlorides was achieved under palladium catalysis.59 The reactions were shown to be suitable for the enantioselective preparation of α-substituted ketones. For example, the densely functionalized ketone 28 was obtained using this approach.58
 | ||
| Scheme 10 Stereo-retentive cross-couplings via zinc homoenolates. | ||
Palladium(II) complexes catalyse ring-opening cross-coupling of cyclopropanols with aryl, vinyl, benzyl halides, triflates, arenes, N-acyl-glutarimides, alkynes, and carbon monoxide (Scheme 11).3,4,62 Arylation, alkenylation, and carbonylation occur in a stereo-retentive fashion. Thus, Cheng and Walsh reported ring-opening arylation of enantiomerically enriched cyclopropanol 1c to prepare α-benzyloctanal (S)-29 in 65% yield without noticeable loss of enantiomeric purity.63 More recently, Dai and co-workers described a carbonylative ring-opening lactonization of 1,2-disubstituted cyclopropanols to prepare fused bicyclic lactones.64 The reaction proceeds via diastereoselective generation of an organopalladium hemiacetal B from the respective homoenolate A, followed by carbon monoxide insertion and lactonization. This method was applied in the synthesis of the natural products α-levantanolide65 and paeonilide.64
 | ||
| Scheme 11 Stereo-retentive cross-couplings via palladium(II) homoenolates. | ||
Ru(II)-catalysed ring opening of 1-allenyl- and 1-alkynylcyclopropanols occurred along the C1–C3 bond, followed by cyclization of the respective Ru(II) homoenolates into cyclopentenones and cyclobutanones (Scheme 12a).66 However, since only racemic cyclopropanols have been tested in this reaction, the preservation of stereochemistry remains uncertain.
 | ||
| Scheme 12 (a) Ru-catalysed ring expansion of 1-allenylcyclopropanols into cyclopentenones reported by Cha and co-workers. (b) Rh-catalysed ring-opening amidation of cyclopropanols reported by Chang and co-workers. NFTB = nonafluoro-tert-butyl alcohol. | ||
Chang and co-workers developed a Rh-catalysed amidative ring-opening of 1,2-disubstituted cyclopropanols by reacting them with dioxazolones 32 (Scheme 12b).67 The method delivers β-acylaminoketones (e.g., 33) via stereo-retentive ring opening along the C1–C3 bond, which generates a nitrenoid rhodium homoenolate complex A. This is followed by migratory nitrene insertion to form the carbon–nitrogen bond.
As recently reported by Yoshikai and co-workers, nickel-catalysed ring-opening allylation is notable as a rare example of regioselective C1–C3 bond cleavage reactions mediated by non-precious metals (Scheme 13a).68 The reaction presumably occurs via a Ni(II)–homoenolate intermediate. However, only racemic cyclopropanols were tested and the preservation of stereochemistry at C2 was not confirmed.
 | ||
| Scheme 13 (a) Regioselective Ni-catalysed ring-opening allylation reported by Yoshikai and co-workers. (b) Stereoselective Cu-catalysed ring-opening cyclization to tetrahydrofurans and tetrahydropyrans developed by Dai and co-workers. | ||
Dai and co-workers described a regioselective and stereoselective copper-catalysed ring-opening cyclization of diastereomerically pure cyclopropanols 1, bearing a secondary alcohol side chain (Scheme 13b). The reaction produced tetrahydrofurans 36 in up to 91% yield and tetrahydropyranes 37, albeit with reduced efficiency (up to 41% yield).69 The respective cyclic ethers were produced as single diastereomers, with the stereochemical configuration determined by the configuration of the cyclopropanol precursor. Tetrahydrofuran 36h was also produced in enantioenriched form (92% ee) from the corresponding enantioenriched homoallylic alcohol 1h (92% ee). The stereochemistry of the secondary alcohol side chain was found to affect the yields of the diastereomeric tetrahydrofuran products (e.g., 1hvs. 1i), thus highlighting its crucial role in determining the cyclopropanol ring-opening pathways. These observations were rationalized by assuming the generation of copper(II) chelate complex A from 1h, with the phenyl group residing in the less sterically hindered pseudo-equatorial position and avoiding steric repulsion with the methylene group of the cyclopropane ring that is observed in complex A′, which is generated from 1i. Subsequent regioselective C1–C3 bond cleavage of the cyclopropane affords copper(II) homoenolate B, which in turn produces 36h after oxidation to CuIII followed by reductive elimination of CuI to form the C–O bond. The method was applied to the diastereoselective synthesis of racemic hyperiones A and B.
2.3. Stereospecific ring expansion into cyclobutanones and β-lactams
Lindsay and co-workers established a general methodology to access various enantioenriched 1-substituted cyclopropanol derivatives 38 by reacting 1-sulfonylcyclopropanols 9, acting as cyclopropanone surrogates, with carbon or nitrogen nucleophiles (Scheme 14).28,70,71 Generation of an electron-deficient center in the α-position to the cyclopropane ring by treatment of 38 with an electrophilic reagent or a Lewis acid results in a semipinacol rearrangement, leading to ring expansion into cyclobutanones. Notably, the reaction occurs with preservation of the enantiomeric purity of the starting cyclopropanol 9 or with only negligible erosion. Following this general strategy, enantiomerically pure (>99% ee) β-lactam 40 was prepared by the isomerization of hemiaminal 39 triggered by aluminum triflate as a Lewis acid (Scheme 14a).28 The ring expansion was achieved in two steps without isolating the intermediate 39. Similarly, 1-vinyl-substituted cyclopropanols 41, obtained by the reaction of 9 with vinyl magnesium bromides, undergo ring expansion to enantioenriched (96–99% ee) α-methylenecyclobutanones 42 upon treatment with electrophilic bromine (Scheme 14b).71 Strong protic acid (HCl) or treatment with electrophilic oxygen (mCPBA) resulted in analogous ring expansion reactions of 41, yielding the respective enantioenriched cyclobutanone products 43, 44 or γ-lactone 45. As another example, spiro[3.3]heptan-1-one 46 was obtained in 63% yield by an AlCl3-mediated strain-relocating semipinacol rearrangement of cyclopropanol intermediate 47 (Scheme 14c).70 A high enantiomeric purity of 96% ee was achieved, evidencing only insignificant degradation of the enantiomeric purity of the starting 1-sulfonylcyclopropanol 9a. | ||
| Scheme 14 Stereospecific ring expansion of enantioenriched cyclopropanol derivatives into cyclobutanones, γ-lactones and β-lactams. | ||
2.4. Miscellaneous ring-opening reactions
Only a few examples of unconventional (non-homoenolate-type) transition-metal-mediated stereoselective ring-opening reactions of cyclopropanols to carbonyl compounds have been reported.Stereoselective one-pot generation of enantioenriched copper cyclopropoxides 50 and their transformation into α,α-disubstituted β,γ-unsaturated aldehydes 51 was described by Marek and co-workers (Scheme 15).27 The carbocupration of cyclopropene 48 was completely syn-stereoselective due to the directing properties of the acetoxymethyl substituent. Oxidation of organocopper derivative 49 occurred with retention of configuration, followed by a ring-opening process in 50 facilitated by the presence of the acetoxy leaving group at C2. As a result, aldehydes 51 with a quaternary α-stereocenter were obtained with 86% ee, without degradation of the enantiomeric purity of the starting material.
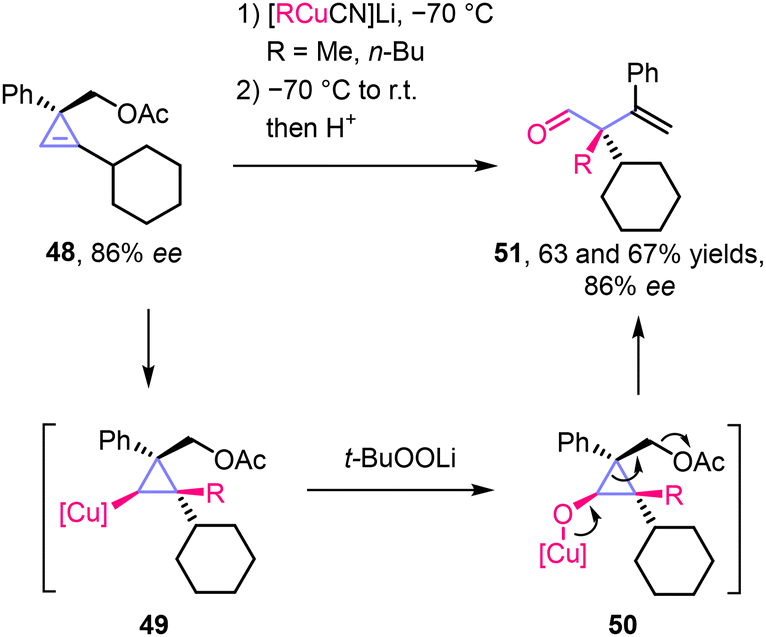 | ||
| Scheme 15 Stereoselective one-pot formation of aldehydes 51 from cyclopropene 48via generation and ring-opening of copper cyclopropoxide 50. | ||
Shortly thereafter, the same group reported the preparation of similar aldehyde products via a palladium-catalysed ring-opening cross-coupling of ω-ene cyclopropanols (e.g., 1j in Scheme 16).72 The reaction mechanism is notably distinct from the mainstream Pd–homoenolate rearrangement and begins with the Heck arylation of the terminal alkene moiety, triggering ‘metal walking’ of the palladium atom along the alkyl side chain. At the terminal point, cyclopropane ring fragmentation and elimination of H[Pd]X result in aldehydes 52 with a quaternary α-stereocenter, obtained with the same enantiomeric ratios as the starting cyclopropanol 1j.
 | ||
| Scheme 16 Pd-catalysed remote ring-opening of ω-ene cyclopropanols reported by Marek and co-workers. | ||
The same group employed a transition metal “chain-walking” strategy to enable iridium-catalysed double bond migration in cis-1,2-disubstituted cyclopropanols 53, leading to the in situ generation of 1,2-divinylcyclopropanes 54 (Scheme 17).73 A subsequent Cope rearrangement of 54 produced bicyclic cycloheptanones 55, a common motif in natural products, in 34–77% yield and with high diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). Although this reaction may be suitable for preparing enantiomerically pure products from non-racemic substrates, enantiomerically enriched cyclopropanols were not validated.
:1 dr). Although this reaction may be suitable for preparing enantiomerically pure products from non-racemic substrates, enantiomerically enriched cyclopropanols were not validated.
 | ||
| Scheme 17 Diastereoselective synthesis of bicyclic cycloheptanones reported by Marek and co-workers. cod = cyclooctadiene, ArF = 3,5-bis(trifluoromethyl)phenyl. | ||
Jeon and co-workers described an unprecedented rhodium-catalysed transformation of cyclopropyl acetates 55 into dioxasilolanes 57 (Scheme 18).74 First, iridium-catalysed hydrosilylation of the acetate group was performed, producing hemiacetals 56 with a Si–H moiety. The subsequent rhodium-catalysed ring-opening reaction led to atypical functionalization at the C1 carbon, rather than at C2 or C3, via highly regioselective cleavage of the less substituted C1–C3 bond, facilitated by the application of a tris(pyrazolyl)borate [Tp(CF3)2Na(THF)] ligand. The reaction of 1,2-disubstituted cyclopropanols proceeded in a highly diastereoselective manner, presumably due to π-coordination between the rhodium atom and the C1 aryl substituent in intermediate 59, which resulted in a tightly organized geometry. Despite the high diastereoselectivity and preservation of the C2 stereocenter, enantioenriched cyclopropanol substrates have not been tested to confirm the preservation of enantiomeric purity in this transformation.
 | ||
| Scheme 18 Ring-opening silylation of cyclopropanols reported by Jeon and co-workers (coe = cyclooctene, nbd = norbornadiene, nbe = norbornene). | ||
Xu, Doyle, and co-workers reported the synthesis of chiral spiroketals 64via a one-pot sequence starting with rhodium-catalysed asymmetric cyclopropanation of 2-aryl- or alkyl-substituted 4,4-dimethyl-5-methylene-4,5-dihydrooxazoles 61 with TBS-protected enoldiazoacetates 62, followed by synchronous ring-opening and intramolecular cyclization of the intermediate donor–acceptor cyclopropanes 63 (Scheme 19).75 The ring-opening process was triggered by tetrabutylammonium fluoride and completed within a few minutes. Spiroketals 64 were obtained in 53–86% yield over two steps with 95–99% ee. The presence of the silylated enol unit in 62 was crucial for achieving high enantioselectivity. When an alkene was reacted with diazoacetoacetate, product 64 formed directly, but its enantiomeric purity was only 69% ee, presumably because the isomerization of the intermediate donor–acceptor cyclopropane proceeded via a stepwise, rather than synchronous, mechanism.
 | ||
| Scheme 19 Enantioselective synthesis of spiroketals 64 reported by Xu, Doyle and co-workers. S-TCPTTL = (S)-4,5,6,7-tetrachloro-alpha-(1,1-dimethylethyl)-1,3-dihydro-1,3-dioxo-2H-isoindole-2-acetato ligand, TBAF = tetrabutylammonium fluoride. | ||
2.5. Transformations with preservation of the cyclopropane ring
Interconversion to other cyclopropane derivatives via displacement of oxy substituents is much less common than ring-opening reactions but is certainly not a less important aspect of cyclopropanol reactivity. The uncommon nature of these transformations stems from the specific bonding properties of cyclopropane and its high ring strain, which result in disrotatory ring opening to an allyl cation after the heterolytic breaking of the carbon–oxygen bond, unless cyclopropyl–allyl rearrangement is prohibited by steric constraints or conjugation with donor substituents.1,76,77 Consequently, the majority of known nucleophilic displacement reactions proceed via alternative mechanistic pathways, such as elimination–addition,8,77,78 including the generation of transition metal π-complexes as intermediates.79 Importantly, the substitution reactions occur in a stereoselective manner, allowing for the asymmetric synthesis of cyclopropane derivatives.78b,79Recently, the Rousseaux group introduced several new opportunities to achieve formal substitution of the hydroxyl group in cyclopropanols while preserving the cyclopropane ring. The conversion of readily available secondary cyclopropanols (e.g., 1k in Scheme 20) into pharmaceutically relevant cyclopropylamines (e.g., 65a) was accomplished by utilizing the electrophilic properties of the carbonyl group in Zn homoenolates (e.g., 66), obtained through the ring-opening reaction.80 The homoenolate intermediates readily reacted with secondary amines, producing the corresponding iminium intermediates (e.g., 67), which underwent facile ring closure resulting in the formation of cyclopropylamines. While the described transformation necessitates the disruption of the three-membered ring, it proceeds in a highly diastereoselective manner, resulting in the almost exclusive formation of trans-cyclopropylamine products, even from cis-cyclopropanol substrates. This phenomenon was ascribed to minimization of 1,3-allylic strain in the iminium intermediate during the ring-closure step. Even more importantly, enantioenriched cyclopropanol 1k (99% ee) yielded cyclopropylamines 65 with a high level of enantiomeric purity (88–92% ee), evidencing only insignificant degradation of the enantiomeric purity of the starting material.
 | ||
| Scheme 20 Enantiospecific conversion of cyclopropanols into cyclopropylamines via a ring-opening and ring-closure sequence. | ||
Homolytic cleavage of the carbon–oxygen bond offers an additional workaround to circumvent ring opening and access substitution products through the functionalization of cyclopropyl radical intermediate A (Scheme 21). This concept was pioneered by Rousseaux and co-workers,81 who achieved Ni-catalysed C(sp3)–O arylation of cyclopropanols via installation of a redox-active thiocarbamate leaving group. Besides the unique reactivity, complete diastereoselectivity was observed, with the relative cis- and trans-configuration of 66 preserved (>20:1 dr) in arylation products 67. Since cyclopropyl radicals may undergo rapid configurational inversion (k ∼ 108 s−1),82 the preservation of stereochemistry evidences fast recombination of radical A with Ni(II) in a solvent cage. The synthetic utility of the approach was further expanded by the same group, demonstrating iridium-photocatalysed rearrangement of 66 into cyclopropanethiol carbamates (e.g., 68).83 Interestingly, the diastereomeric ratio of trans-68 varied based on the diastereomer of the starting material 66 subjected to the reaction conditions, which aligns with the configurational inversion of intermediate cyclopropyl radicals. Although these transformations are only favourable for cyclopropanol substrates capable of producing resonance-stabilized cyclopropyl radicals (e.g., by an α-aryl substituent), the unconventional reactivity marks a significant milestone in cyclopropanol chemistry. The diastereoselective nature of these transformations and intact C2 stereocenter imply that the use of enantiopure cyclopropanol substrates should proceed with the preservation of enantiomeric purity.
 | ||
| Scheme 21 Stereoselective conversion of tertiary cyclopropanol thiocarbamates 66 into aryl-substituted cyclopropanes 67 and cyclopropanethiol carbamates 68. | ||
3. Cyclopropanols as prochiral intermediates: catalyst-controlled asymmetric transformations
3.1. Transition-metal-induced ring-opening reactions
The use of readily available racemic or nonchiral cyclopropanols as prochiral building blocks offers an appealing alternative to the strategy described above, avoiding the necessity to prepare enantiomerically pure cyclopropanol intermediates. However, it introduces another challenge inherent to asymmetric catalysis: the identification of an appropriate chiral catalyst that enables a high level of asymmetric induction. This research direction is conceptually novel to cyclopropanol chemistry, having gained significant attention only in the last decade. This surge of interest is undoubtedly inspired by the tremendous progress and breakthroughs in asymmetric catalysis achieved since 2000. Although the number of known examples of these transformations is currently relatively small, the field holds great promise for further developments.Mechanistically, most of the reported methods involve the generation of metal homoenolates from cyclopropanols by utilizing cobalt, copper, nickel, and zinc complexes. The process can occur directly through a ring-opening reaction with a transition metal complex, or via the generation of β-keto radicals, followed by recombination with a metal. The subsequent coupling reaction forms a carbon–carbon bond with a new stereogenic centre, with asymmetric induction enabled by a chiral ligand coordinated to the metal. Importantly, the process can be performed in a catalytic manner.
In 2018, Yoshikai and co-workers reported the first example of enantioselective cobalt-catalysed ring opening of nonchiral 1-phenylcyclopropanol (1, R = Ph, Scheme 22a).84 The group developed the synthesis of cyclopentenols (e.g., 69) by reacting cyclopropanols with alkynes, catalysed by Co–phosphine complexes. The ring opening of tertiary cyclopropanols was achieved using a catalytic Co(I) species, generated by reducing CoBr2 with Zn in the presence of diphosphine ligands, with DABCO serving as a proton shuttle. It was demonstrated that cyclopentenol 69 could be prepared with 93% ee and 28% yield when the reaction was performed in the presence of the chiral phosphine ligand (R,R)-QuinoxP*.
 | ||
| Scheme 22 Enantioselective cobalt-catalysed ring opening reactions of 1-substituted cyclopropanols via generation of cobalt homoenolates. | ||
Subsequently, the Yoshikai and Meng groups reported that the cyclopropanol-derived Co homoenolates A could also participate in the carbometallation of strained alkenes, with the reactions proceeding enantioselectively in the presence of chiral phosphine ligands. The Yoshikai group further developed a chemodivergent and enantioselective ring-opening addition of cyclopropanols to oxabicyclic alkenes (Scheme 22b).85 Depending on the nature of the Co-precatalyst counterion, the reaction furnishes either ring-opening products 70 (with CoCl2) or hydroalkylation products 71 [with Co(OAc)2]. The reaction proceeded efficiently for a wide range of substrates, delivering the corresponding products in up to 99% yield and up to 94% ee, with excellent enantioselectivity enabled by the (R,R)-bdpp ligand.
Following the same approach, the Meng group developed a diastereo- and enantioselective hydroalkylation of cyclopropenes (Scheme 22c).86 This transformation, promoted by a cobalt complex with the (R,R)-Et-DUPHOS ligand, was effective across a wide range of cyclopropanol and cyclopropene substrates. The acetate counterion acted as a hydrogen-atom abstractor, facilitating the initial coordination of the cyclopropanol to the metal centre, thereby eliminating the need for a stoichiometric amount of base (Scheme 22c, conditions I). An exception was observed with 1-alkoxycarbonyl-substituted cyclopropanols, where the electron-withdrawing nature of the alkoxycarbonyl group reduced the ability to coordinate to the metal centre, resulting in diminished reactivity. In this case, better results were achieved in the presence of DABCO and MeOH as promoters (Scheme 22c, conditions II), though with somewhat lower yields (36–59%) and enantioselectivities (46–90%) compared to those obtained with 1-aryl- and alkenyl-substituted cyclopropanols (59–99% yields, 72–96% ee).
This methodology was further extended to achieve the enantioselective hydroalkylation of 1-alkoxycarbonyl-substituted cyclobutenes 75 (Scheme 22d, conditions I).87 The relatively lower olefinic strain (1.9 kcal mol−1) compared to cyclopropenes (27.7 kcal mol−1) resulted in the reduced reactivity of cyclobutenes and required elevated temperatures for the reaction. The addition of metallic zinc was essential for the reduction of CoII to CoI which possessed higher affinity to the alkene functionality and promoted its coordination. While electron-rich ligands proved to be inefficient, the highest yield of 84% for product 73 (R = 4-MeOC6H4) with 86% ee was achieved using the phosphine–oxazoline ligand L1. Further examples of cyclopropanol substrates provided the corresponding hydroalkylation products 73 in yields of 55–99% and 86–90% ee. In contrast, the enantioselective hydroalkylation of succinimide-fused cyclobutene 76 gave the best outcome (89% yield, 80% ee) in the presence of phosphine ligand L2 and under different reaction conditions (Scheme 22d, conditions II). The hydroalkylation reactions could potentially be accompanied by a competitive process of β-H elimination in A, followed by the conjugate addition of cobalt hydride to enone complex B, forming enolate C. Although this process was completely suppressed in the hydroalkylation of cyclopropenes and cyclobutenes, the Meng group later demonstrated its utility in enantioselective ring-opening coupling of cyclopropanols with Ts-protected aldimines (Scheme 22e).88 The optimal outcome was observed with oxazoline ligand L3, where steric repulsion with the bulky tosyl group of the aldimine substrate directed the formation of the major diastereomers of β-amino ketones 77, obtained in 41–95% yield, 62–96% ee, and up to 92:8 diastereoselectivity.
The enantioselective catalytic conjugate addition of zinc homoenolates, generated from 1-substituted cyclopropanols 1, to α,β-unsaturated ketones 78 was first demonstrated by Yoshikai and co-workers (Scheme 23).36 Importantly, this reaction was promoted by substoichiometric amounts of a zinc complex, formed in situ from diethylzinc and the chiral aminoalcohol 14. The reaction likely proceeds via zinc homoenolate species A, which further coordinates with enone 78 to form a bimetallic associate complex B. The 1,4-addition to the enone in complex B, followed by proton transfer, yields product 79 and regenerates the catalyst. The resulting 1,6-diketones 79 undergo aldol-type cyclization, furnishing cyclopentene products 80 with an asymmetric centre. Aryl-, alkenyl-, and alkyl-substituted cyclopropanol substrates were successfully employed in this reaction, delivering carbocycles 80 in 34–93% yield and 24–94% ee.
 | ||
| Scheme 23 Enantioselective conjugate addition of catalytically generated zinc homoenolates. | ||
Later, the same research group discovered an unprecedented transformation of zinc cyclopropoxides 1-Zn into bimetallic zinc species B, formed as a result of the deprotonation of homoenolate A with diethylzinc (Scheme 24a).89 Bimetallic species B was capable of reacting with electrophiles similarly to allylzinc reagents. The reactions of B with aldehydes 81 proceeded with high γ-regioselectivity, producing 1,2-diols with diastereoselectivities of up to >20:1 in favour of the anti-diol products (Scheme 24b). To rationalize the anti-selectivity, DFT calculations revealed that the transition state leading to the anti-1,2-diol (anti-TS) was 1.0 kcal mol−1 lower in energy than the syn-TS. In the chair-like 6-membered transition state, the coordination of the aldehyde to both the enolate and homoenolate zinc atoms favours the pseudoaxial orientation of the phenyl group in anti-TS over the pseudoequatorial orientation in syn-TS, due to two gauche interactions involving the aldehyde substituent in the latter. Bimetallic zinc species B also showed promise for use in asymmetric synthesis: when cinchonidine (2.2 equiv.) was added as a chiral inducer, the reaction produced 1,2-diol 82a with a moderate 68% ee.
 | ||
| Scheme 24 Dia- and enantioselective reactions of cyclopropanols through enolized zinc homoenolates. | ||
Very recently, Kanemoto and Yoshikai applied the bimetallic zinc species generated from 1-aryl-substituted cyclopropanols 1 to the enantioselective hydroxyallylation of cyclopropenes 83 (Scheme 25).90 In this reaction, cycloalkene 83 underwent carbometallation on the less sterically hindered side. The reaction was promoted by chiral aminoalcohol-derived NHC ligand 84, yielding hydroxyallylation products 85 in 47–76% yield, 82–88% ee, and high diastereomeric purity (>20:1 dr).
 | ||
| Scheme 25 Enantioselective hydroxyallylation of cyclopropenes with cyclopropanols. | ||
Enantioselective copper-catalysed reactions via the generation of copper(I) homoenolates were demonstrated in the ring-opening allylation of tertiary cyclopropanols 1 with allylic phosphates by Yin, Trost and Sawamura groups (Scheme 26).91–93 Yin and co-workers introduced a methodology utilizing a Cu(I) catalyst ligated with chiral NHC ligands derived from imidazolium salts (R,R)-L4 and (R,R)-L5 (Scheme 26a).91 The latter ligand was found to be more suitable for constructing challenging quaternary carbon stereocenters in compounds (S)-87b. Following this methodology, a wide range of allylation products (S)-87a,b were obtained in yields of 63–98% and enantioselectivities of 80–97% ee.
 | ||
| Scheme 26 Enantioselective copper-catalysed ring-opening cross-coupling of cyclopropanols with allylic phosphates via generation of transient copper(I) homoenolates. | ||
Trost and co-workers described a similar methodology utilizing a bimetallic Cu/Zn complex generated in situ from the NHC–ProPhenol ligand L6 (Scheme 26b).92 The reaction of cyclopropanols with ZnEt2 delivered Zn–homoenolates, which in turn produced the corresponding Cu(I) derivatives through transmetalation. The subsequent reaction with allylic phosphates resulted in the predominant formation of S-configured γ-vinyl ketone products 87c, in 51–87% yield and 70–92% ee, including the assembly of quaternary carbon stereocenters. DFT studies supported the preferred nucleophilic attack of the formed Cu(I) homoenolate at the Si face of the allyl phosphate, as the opposite Re-face attack was unfavourable due to significant steric repulsion between the allyl fragment and phenyl groups of the prolinol moiety. The synthetic utility of this method was demonstrated through the asymmetric synthesis of the fish deterrent (+)-sporochnol A.
Sawamura and co-workers explored a methodology based on the use of a chiral NHC ligand for copper(I), derived from the imidazolium salt (S,S)-L7 (Scheme 26c).93 The lithium ion, introduced with t-BuOLi base, was shown to be crucial for achieving a high 80% yield and 89% ee for the allylation product (R)-87d. Cooperative bimetallic Li–Cu activation, assisting in the generation of the homoenolate, was demonstrated through a DFT study. Enantioselectivity in the syn-SN2′ attack of the Cu–homoenolate on the allyl phosphate was governed by C–H/π interactions between the allyl phosphate and the N-naphthoxy group of the chiral catalyst.
Single electron oxidation of cyclopropanols to generate β-keto radicals provides an alternative strategy. Until recently, an asymmetric version of this transformation remained elusive. In 2020, the Liu group reported the first examples, utilizing copper catalysts ligated with chiral bisoxazoline (BOX) ligands L8–L10 (Scheme 27a).94 Enantioselective ring-opening cyanation was achieved with TMSCN by trapping β-keto radicals B with copper dicyanide species, generated through oxidation of a CuI-complex with benzoyl peroxide for cyclopropanols 1, or N-fluorobenzenesulfonimide (NFSI) for cyclopropanone acetals 88. For the ring opening of cyclopropanols 1 (R = Me, Et, n-Pr), the ligand (1R,2S)-L8 provided β-cyanoketones 89 in 40–91% yield and 85–95% ee, with the R-configuration of the stereogenic centre confirmed by comparison of the specific rotation of selected compound with the literature values. Conversely, the ring-opening reaction of cyclopropanone acetals 88 (R = Me, Et, i-Pr, Bn) produced β-cyanoesters 90 as S-enantiomers in 42–75% yield and 87–97% ee in the presence of (1S,2R)-L9. The preparative value was demonstrated through the 2-step synthesis of the GABA receptor agonist (R)-baclofen. However, the substrate scope is limited to 2-aryl-substituted cyclopropanols, which produce resonance-stabilized benzylic-type β-keto radicals.
 | ||
| Scheme 27 Enantioselective copper-catalysed ring-opening cyanation and trifluoromethylation of cyclopropanols. | ||
The same group developed an enantioselective version of copper-catalysed ring-opening trifluoromethylation of cyclopropanols using Togni-I reagent as both an oxidant and a CF3 source (Scheme 27b).95 The non-asymmetric version of this reaction yields a mixture of isomeric β-CF3 ketone products when applied to 1,2-disubstituted cyclopropanols, except for 2-aryl-substituted substrates.96 These 2-aryl-substituted substrates were also employed in the asymmetric version, producing the respective β-CF3 ketones (S)-91 in 42–74% yield and 59–95% ee, with asymmetric induction facilitated by the quinolinyl-containing bisoxazoline ligand (S,S)-L10 for the copper catalyst. The electrophilic nature of the trifluoromethylation reagent played a crucial role in achieving high enantioselectivity. The use of a nucleophilic CF3 source, such as TMSCF3, led to competitive coordination of the CF3 anion with the copper centre, resulting in the decomposition of the chiral copper complex and consequently poor enantioselectivity. The developed protocol was applied in the 3-step synthesis of an (R)-CF3-modified analogue of the calcimimetic agent cinacalcet.
The first example of an asymmetric nickel-catalysed ring-opening cross-coupling reaction of cyclopropanols with bromoarenes was reported by Wang and Li in 2022.97 This process combines photoredox and Ni catalysis to generate secondary β-keto radicals B from cyclopropanols 1l (Scheme 28) via proton-coupled electron transfer (PCET) from a photoexcited iridium complex, with collidine acting as a base. The recombination of the β-keto radical with a chiral Ni complex results in the corresponding Ni homoenolate A, which subsequently participates in cross-coupling with a bromoarene. The substrate scope included 1,2-disubstituted cyclopropanols 1l bearing a silyl-protected hydroxymethyl group, delivering the respective β-arylated ketones (S)-92 in 50–83% yield and 76–90% ee, with asymmetric induction facilitated by the adamantyl-substituted oxazoline ligand (S)-L11. The silyl-protected hydroxymethyl group in 1l was essential for achieving high enantioselectivity, as 2-methyl- or 2-benzyl-substituted cyclopropanols yielded β-arylated products with reduced enantioselectivities of 36–40% ee. This protocol was demonstrated to be suitable for late-stage functionalization of complex bioactive molecules, such as fenofibrate, lithocholic acid, and isoxepac. It was also applied in a concise synthesis of an analogue of the marine natural product ent-calyxolane B.
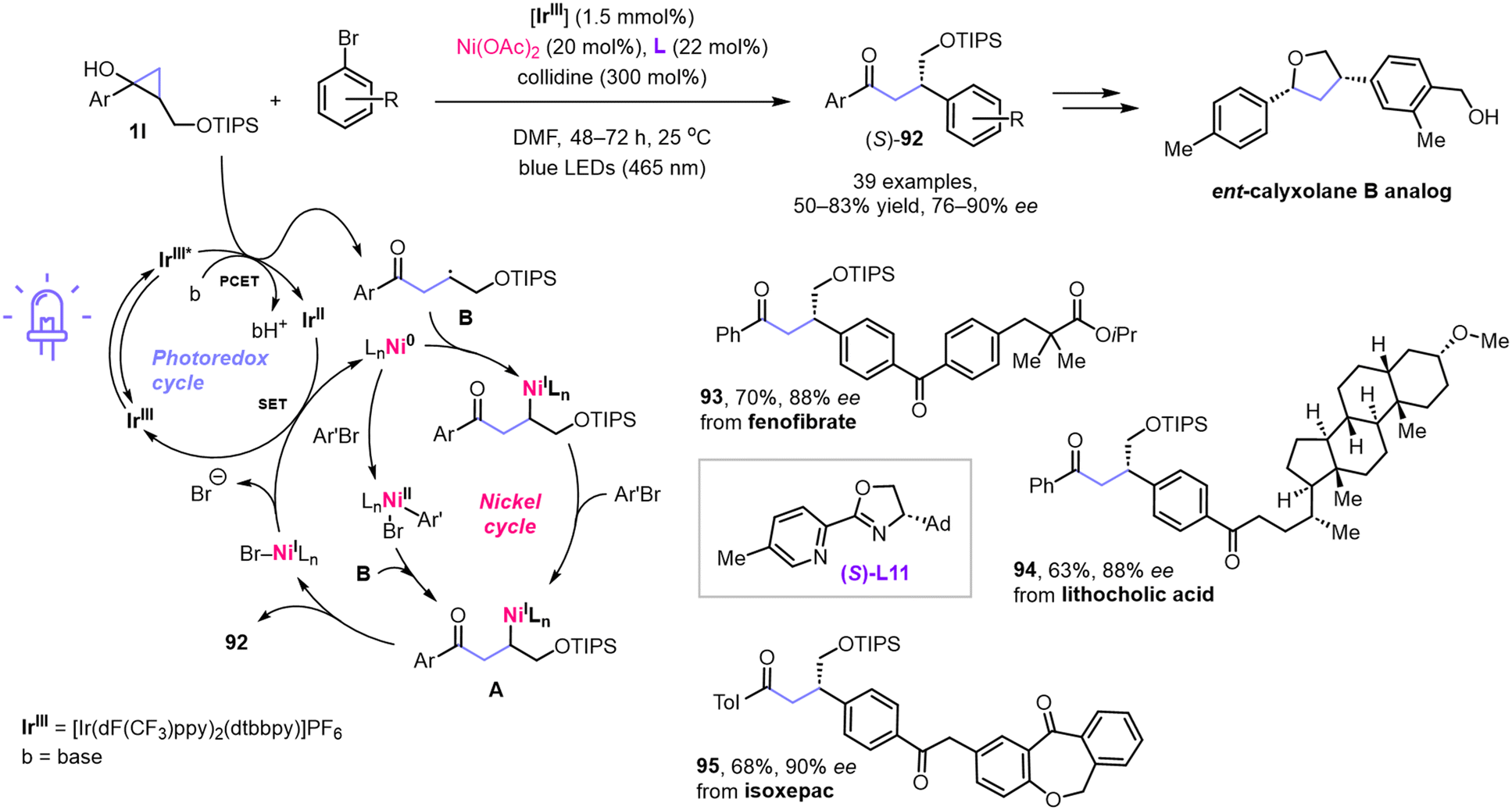 | ||
| Scheme 28 Enantioselective nickel-catalysed ring-opening cross-coupling of cyclopropanols with bromoarenes. | ||
Besides radical and homoenolate chemistry, transition metal catalysis was shown to be effective in enantioselective ring expansion reactions of cyclopropanols. In 2017, Voituriez described a gold-promoted enantioselective transformation of 1-vinyl-1-ethoxycyclopropanes 96 into cyclobutanones 97 (Scheme 29).98 Previously, only a diastereoselective version of the ring-expansion cyclization was reported by Echavarren.99 The transformation was catalysed by the [(R)-MeO–DTBM–BIPHEP–(AuCl)2] complex. The developed protocol was applied for the construction of the tricyclic backbone of the natural product russujaponol D.
 | ||
| Scheme 29 Enantioselective gold-catalysed ring expansion of vinyl-substituted cyclopropanol ethers. | ||
3.2. Asymmetric organocatalytic transformations involving ring opening of cyclopropanols
Over the last decades, organocatalysis has proved to be an efficient tool for the synthesis of enantiomerically enriched compounds as an alternative to metal and enzyme-mediated catalysis.100 Various activation modes and reaction types can be employed and generally the developed methods do not require an inert atmosphere or harsh reaction conditions.The Alexakis group developed a series of enantioselective halogenative semipinacol rearrangements catalysed by chiral phosphoric acids (Scheme 30a).101 While both substrate 99 and halogenating agent 100 are simultaneously activated by chiral catalyst 102, the reaction is induced by stereoselective electrophilic halogenation of the vinylic moiety, triggering a [1,2]-alkyl shift. The reaction is driven by strain-releasing ring-expansion and yields spirocyclobutanones 101 with a high level of stereocontrol (80–94% ee). Recently, an alternative to the halogenation concept was introduced by Jacobsen and co-workers.102 The described protio-semipinacol rearrangement reaction is based on co-catalysis between a strong Brønsted acid and chiral hydrogen-bond donor 104. The method provides access to highly enantioenriched cyclobutanones 105 obtained with 88–94% ee (Scheme 30b). Through a series of mechanistic studies, the authors concluded that the reaction proceeded through a stepwise pathway where protonation of the alkene was the rate-determining step. Nevertheless, the concerted pathway cannot be ruled out completely. Chen, Tu and Bao have also demonstrated an enantioselective sulfenylation/semipinacol rearrangement on the only example of 1-vinyl-substituted cyclopropanol by using a chiral Lewis base and a chiral Brønsted acid as cocatalysts.103
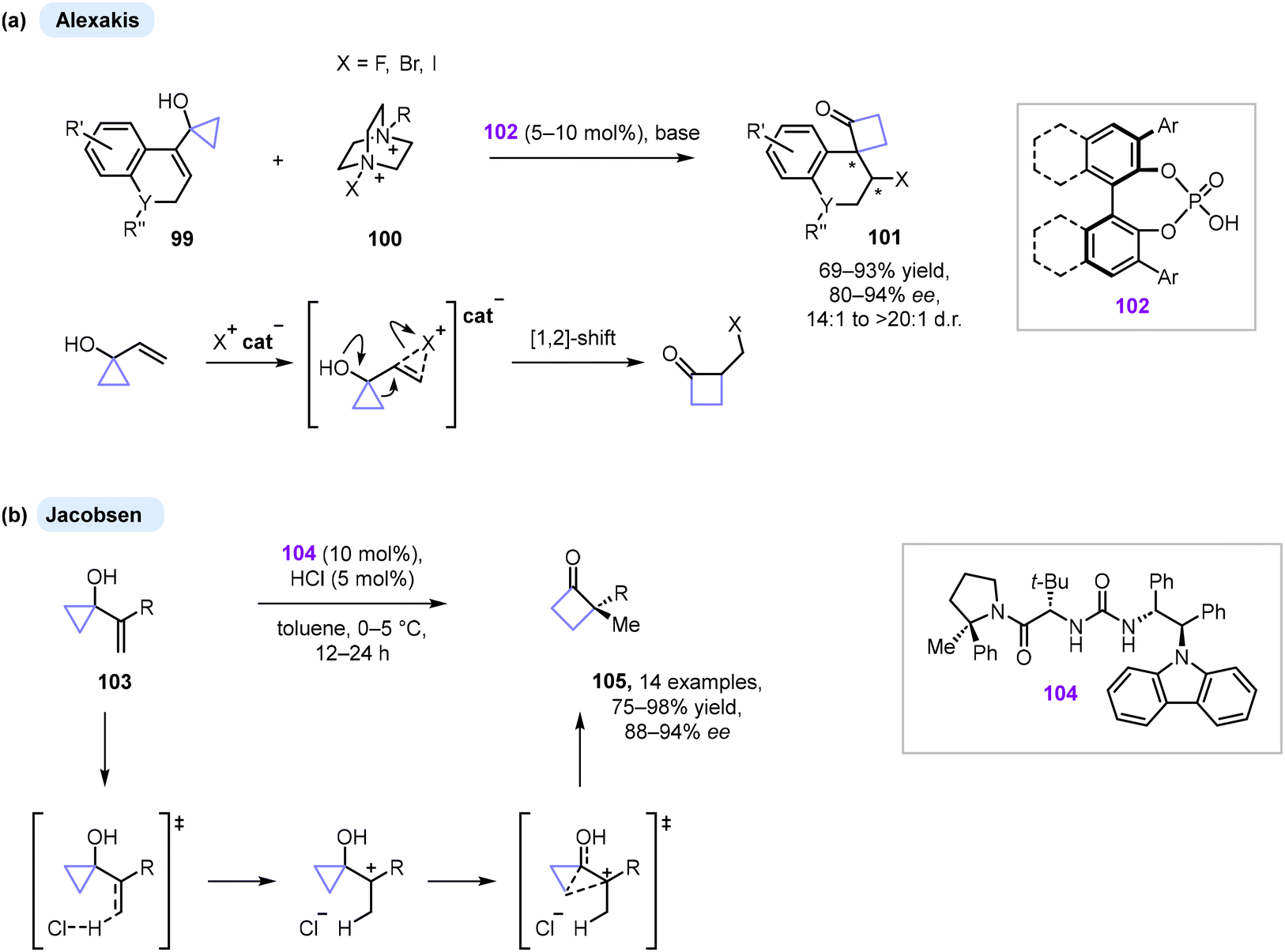 | ||
| Scheme 30 Enantioselective organocatalytic semipinacol rearrangements of cyclopropanols. | ||
An example of enantioselective sulfonylation promoted by quinine-derived squaramide catalyst 107 was presented by Ye and Wu (Scheme 31).104 It was shown previously by the authors that γ-keto sulfinate intermediate A could be generated in situ by the reaction of cyclopropanols with sulfur dioxide and subsequently trapped by a variety of electrophiles.105 Thus, nucleophilic addition of A to allene intermediate B, formed from naphthalene-2-ols in a presence of chiral base 107, produced axially chiral (S)–(E)-1-((1-alkylsulfonyl)-2-arylvinyl)naphtalen-2-ols 108 in 36–87% yield and 80–98% ee.
 | ||
| Scheme 31 Asymmetric sulfonylation of naphthalene-2-ols by cyclopropanol-derived γ-keto sulfinates. | ||
A covalent catalysis strategy was reported by Lupton and co-workers.106 In their study, chiral N-heterocyclic carbene (NHC) organocatalyst 111A was employed to activate acyl fluoride 110 by generating the corresponding acyl azolium intermediate B and releasing a fluorine anion, which was responsible for O-TMS deprotection of donor–acceptor cyclopropane 109 (Scheme 32). TMS-deprotected cyclopropoxide C readily undergoes the ring-opening reaction leading to the formation of enolate D. Nucleophilic 1,2-addition of the enolate to the acyl azolium intermediate was followed by Claisen rearrangement. In the final steps, an intramolecular aldol reaction provides a five-member carbocycle with three adjacent stereogenic centres. The catalytic cycle is closed by β-lactonization of the cyclic intermediate to yield product 112 as a single diastereomer (in most cases) with moderate to excellent enantiomeric purity (55–98% ee).
 | ||
| Scheme 32 Enantioselective [3 + 2] annulation of α,β-unsaturated acyl azoliums and donor–acceptor cyclopropanes. | ||
Merging organocatalysis with other activation modes involving the formation of highly reactive radical species, such as photo- and electrochemistry, provided access to reaction pathways, which were otherwise impossible to reach under classical thermochemical conditions.107 The method reported by the Melchiorre group is a great example of such a synergetic approach, which employs cleavage of the cyclopropanol ring (Scheme 33).108 The described transformation relies on the oxidative properties of the photoexcited iminium ion A. This ion forms through the condensation of cinnamyl aldehyde with the chiral L-proline-derived catalyst 114, followed by activation with visible light. Being a strong oxidant, the excited iminium ion abstracts an electron from electron-rich cyclopropanol leading to cleavage of the cyclopropyl ring and formation of β-keto radical cation C. Subsequent radical–radical coupling is followed by an aldol reaction and hydrolysis provides enantiomerically enriched product 115 and regenerates the catalyst. It is worth noting that the catalyst used in this study is a modified version of the Jørgensen–Hayashi catalyst, with fluorine substituents in the pyrrolidine ring, which enhance its redox stability under the reaction conditions. Under the optimal reaction conditions, formal [3 + 2] cycloaddition products 115 were isolated in 43–89% yield and 90–99% ee.
 | ||
| Scheme 33 Enantioselective photochemical organocascade catalysis to enable formal [3 + 2] cycloaddition of cyclopropanols and cinnamyl aldehydes. | ||
In addition to generating transient prochiral intermediates from cyclopropanols, stable prochiral intermediates can also be formed. This alternative strategy provides a foundation for developing one-pot tandem or cascade enantioselective transformations and presents an opportunity to integrate various catalytic approaches. In 2017, Kananovich and co-workers reported a route to enantiomerically enriched (R)-epoxy ketones 116 through prochiral 1,2-dioxolane intermediates 117a (equilibrating with β-peroxyketones 117b) generated by Mn(III)-catalysed aerobic oxidation of tertiary cyclopropanols (Scheme 34).109 The formation of (R)-116 in 58–84% yield and 80–97% ee was achieved by treatment of 117 with a strong organic base (1,8-diazabicyclo[5.4.0]undec-7-ene, DBU) in the presence of the poly-L-leucine (PLL) catalyst immobilized on silica gel. The method was subsequently applied in a short synthesis of chlamydocin, a natural histone deacetylase inhibitor with an (S)-epoxy ketone warhead, by using the poly-D-leucine (PDL) catalyst.110
 | ||
| Scheme 34 Enantioselective one-pot synthesis of α,β-epoxy ketones via aerobic oxidation of cyclopropanols. | ||
3.3. Asymmetric transformations with preservation of the cyclopropane ring
The unique bonding properties and increased acidity of C–H bonds in cyclopropane (pKa ≈ 46)2 present an opportunity for C–H functionalization. Until very recently, transition-metal-mediated C–H activation methodologies remained elusive for oxo-substituted cyclopropanes, although corresponding stereo- and regioselective reactions are well established for other cyclopropane derivates, including cyclopropylamines.111 This limitation arises from the high susceptibility of electron-rich C–C bonds in cyclopropanol derivatives to cleavage by electron-deficient transition metals. An alternative approach could involve directed metalation with organolithium bases.112 While this method occurs stereoselectively, no asymmetric versions have yet been reported.In 2023, Gao and Xu reported the first and currently only example of an asymmetric transition-metal-mediated C–H functionalization methodology for cyclopropanol derivatives (Scheme 35).113 The authors developed a general protocol for iridium-catalysed site- and stereoselective borylation of cycloalkanols, using carbamates as the directing group. The reaction of 1-substituted cyclopropyl carbamates 117 led to their facile desymmetrization, delivering borylated products 118 with 85–95% ee, with asymmetric induction enabled by chiral bidentate boryl ligand L12 (Scheme 35a). Racemic trans-2-substituted cyclopropyl carbamates (±)-119 underwent kinetic resolution under similar conditions, producing borylated cyclopropanes 120 with high enantiomeric purity (90–95% ee), while the enantiomerically enriched starting cyclopropyl carbamates 119 were recovered with 60–89% ee (Scheme 35b).
 | ||
| Scheme 35 Enantioselective iridium-catalysed C–H borylation of cyclopropyl carbamates. | ||
Another elegant solution to the challenging C–H functionalization problem was reported by Sekiguchi and Yoshikai, who demonstrated a formal C–H allylation of cyclopropanols 1via ring opening to generate enolized homoenolate species A, acting as a latent α,β-bisnucleophile (Scheme 36).37 The addition of Morita–Baylis–Hillman carbonate 121 to the enolate unit in A produces zinc homoenolate B, which undergoes ring closure to restore the cyclopropane ring in cyclopropoxide C, eventually yielding cyclopropyl-fused α-alkylidene-δ-valerolactones 122 in a diastereoselective fashion. Several chiral amino alcohol ligands were screened, with the highest efficiency (up to 75% yield) achieved using the azetidine derivative (S)-L13. However, only minor asymmetric induction (up to 18% ee) was observed with L13 and similar amino alcohol ligands (e.g., L14). Despite the low ee, the process establishes a unique and highly diastereoselective approach to access densely functionalized cyclopropanol derivatives via a ring-opening/ring-closing sequence.
 | ||
| Scheme 36 Zinc-catalysed β-functionalization of cyclopropanols via enolized homoenolate. | ||
4. Conclusions and perspectives
The chemistry of cyclopropanols has proved to be essential in addressing intricate synthetic challenges, offering elegant shortcuts to access a diverse array of valuable compounds. In this review, we presented the current landscape of transformations suitable for asymmetric synthesis via cyclopropanol intermediates, pinpointing areas that warrant further exploration.Enantioenriched cyclopropanols are powerful synthetic intermediates in stereoretentive ring-opening reactions, offering straightforward routes to branched skeletons and quaternary stereocenters that are challenging to construct by other means. However, the limited availability of enantioenriched cyclopropanols (unless derived from a chiral pool) remains a significant bottleneck. To address this challenge, further advances in the catalytic asymmetric synthesis of enantiomerically pure cyclopropanols are required, while currently unknown dynamic kinetic resolution strategies could offer a valuable complementary approach. Additionally, stereochemical outcomes should be systematically studied in new transformations using enantiomerically enriched cyclopropanol substrates.
Enantioselective catalytic transformations of nonchiral cyclopropanols as prochiral substrates or racemic cyclopropanols in stereoablative reactions are conceptually novel synthetic strategies and their emergence marks a significant milestone in the field. This approach, though still in its infancy, offers promising new avenues for asymmetric catalysis, particularly in the formation of challenging carbon–carbon and carbon–heteroatom bonds with high stereocontrol. However, it is crucial to expand it further beyond the mainstream use of cyclopropanols as synthetic equivalents of homoenolates to other reactivity modes, yet unexplored in the asymmetric domain (e.g., cationic cyclopropyl–allyl rearrangement). Mechanistic studies that shed light on the structure of reactive intermediates and the origin of asymmetric induction in these transformations are required as a prerequisite for their rational development.
Due to their challenging nature, asymmetric transformations that preserve the cyclopropane ring remain underexplored. In this area, recent studies have established new stereoselective reaction pathways, such as ring-opening followed by ring closure and the generation of cyclopropyl radicals via homolytic cleavage of the C–O bond.
Continued exploration of both established and novel reactivity patterns, coupled with the development of more accessible and highly enantioselective methods grounded in mechanistic understanding, will be essential to further unlock the synthetic potential of cyclopropanols in asymmetric synthesis. We hope that the overview of methods summarized in this article will serve as a useful guide for asymmetric synthesis based on cyclopropanols, provide a “triangulation strategy”2 for planning the total synthesis of natural products, and promote further developments in the area.
Author contributions
M. L. I.: writing – original draft, editing, literature search and analysis (section 3). A. H.: writing – original draft, writing – review and editing, literature search and analysis (section 2). M. O.: writing – original draft, literature search and analysis (section 3.2). D. K.: conceptualisation, writing – original draft, writing – review and editing, literature search and analysis, project supervision.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research by M. L. I. and M. O. was funded by the Estonian Research Council grant (PSG828). D. K. acknowledges funding from the Centre of Excellence in Molecular Cell Engineering (Grant 2014-2020.4.01.15-0013) and Tallinn University of Technology (Grant No. B58) for supporting the cyclopropane research project in 2016–2020. M. L. I., M. O. and D. K. acknowledge the funding provided by the Ministry of Education and Research of Esronia through the Centre of Excellence in Circular Economy for Strategic Mineral and Carbon Resources (SOURCES, TK228, 01.01.2024–31.12.2030). D. K. would like to thank his colleagues and collaborators at TalTech, especially Prof. Margus Lopp and Prof. Riina Aav, for fostering a friendly and supportive environment within the department, making research both rewarding and enjoyable.References
- O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597–2632 CrossRef CAS.
- O. Kulinkovich, Cyclopropanes in Organic Synthesis, John Wiley & Sons, Inc., 2015 Search PubMed.
- A. Nikolaev and A. Orellana, Synthesis, 2016, 1741–1768 CAS.
- J. Le Bras and J. Muzart, Tetrahedron, 2020, 76, 130879 CrossRef CAS.
- X. Cai, W. Liang and M. Dai, Tetrahedron, 2019, 75, 193–208 CrossRef CAS.
- T. R. McDonald, L. R. Mills, M. S. West and S. A. L. Rousseaux, Chem. Rev., 2021, 121, 3–79 CrossRef CAS PubMed.
- Y. Sekiguchi and N. Yoshikai, Bull. Chem. Soc. Jpn., 2021, 94, 265–280 CrossRef CAS.
- Y. Jang, R. Machín-Rivera and V. N. G. Lindsay, Synthesis, 2021, 3909–3934 CrossRef.
- J. K. Cha and O. G. Kulinkovich, Org. React., 2012, 77, 1–160 CAS.
- Q. Liu, B. You, G. Xie and X. Wang, Org. Biomol. Chem., 2020, 18, 191–204 RSC.
- A. de Meijere, Angew. Chem., Int. Ed. Engl., 1979, 18, 809–826 CrossRef.
- V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed.
- Y. Cohen, A. Cohen and I. Marek, Chem. Rev., 2021, 121, 140–161 CrossRef CAS PubMed.
- H. Yan, G. S. Smith and F.-E. Chen, Green Synth. Catal., 2022, 3, 219–226 CrossRef CAS.
- H. Pellissier, A. Lattanzi and R. Dalpozzo, Asymmetric Cyclopropanation, in Asymmetric Synthesis of Three–Membered Rings, Wiley–VCH, 2017, pp. 1–204 Search PubMed.
- Y. A. Konik and D. G. Kananovich, Tetrahedron Lett., 2020, 61, 152036 CrossRef CAS.
- (a) T. Sugimura, T. Futagawa, M. Yoshikawa, T. Katagiri, R. Miyashige, M. Mizuguchi, S. Nagano, S. Sugimori, A. Tai, T. Tei and T. Okuyama, Tetrahedron, 2001, 57, 7495–7499 CrossRef CAS; (b) H. Du, J. Long and Y. Shi, Org. Lett., 2006, 8, 2827–2829 CrossRef CAS; (c) M. G. Siriboe, D. A. Vargas and R. Fasan, J. Org. Chem., 2023, 88, 7630–7640 CrossRef CAS; (d) M. Puriņš and J. Waser, Angew. Chem., Int. Ed., 2022, 61, e202113925 CrossRef PubMed.
- (a) B. K. Alnasleh, M. Rubina and M. Rubin, Chem. Commun., 2016, 52, 7494–7496 RSC; (b) P. Ryabchuk, J. P. Matheny, M. Rubina and M. Rubin, Org. Lett., 2016, 18, 6272–6275 CrossRef CAS PubMed; (c) P. Yamanushkin, M. Lu-Diaz, A. Edwards, N. A. Aksenov, M. Rubina and M. Rubin, Org. Biomol. Chem., 2017, 15, 8153–8165 CAS; (d) V. A. Maslivetc, D. N. Turner, K. N. McNair, L. Frolova, S. Rogelj, A. A. Maslivetc, N. A. Aksenov, M. Rubina and M. Rubin, J. Org. Chem., 2018, 83, 5650–5664 CAS; (e) H. Straub, P. Ryabchuk, M. Rubina and M. Rubin, Molecules, 2022, 27, 7069 CAS.
- M. Simaan and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 1543–1546 CAS.
- D. M. Zubrytski, D. G. Kananovich and O. G. Kulinkovich, Tetrahedron, 2014, 70, 2944–2950 CrossRef CAS.
- S. Kawamura, H. Chu, J. Felding and P. S. Baran, Nature, 2016, 532, 90–93 CrossRef CAS.
- K. Nomura and S. Matsubara, Chem. – Asian J., 2010, 5, 147–152 CrossRef CAS PubMed.
- (a) A. Watanabe, M. Nagatomo, A. Hirose, Y. Hikone, N. Kishimoto, S. Miura, T. Yasutake, T. Abe, S. Misumi and M. Inoue, J. Am. Chem. Soc., 2024, 146, 8746–8756 CrossRef CAS PubMed; (b) W. Zhang, P.-C. Yu, C.-Y. Feng and C.-C. Li, J. Am. Chem. Soc., 2024, 146, 2928–2932 CrossRef CAS.
- C. H. DePuy, F. W. Breitbeil and K. R. DeBruin, J. Am. Chem. Soc., 1966, 88, 3347–3354 CrossRef CAS.
- F. Xu, Y.-L. Zhong, H. Li, J. Qi, R. Desmond, Z. J. Song, J. Park, T. Wang, M. Truppo, G. R. Humphrey and R. T. Ruck, Org. Lett., 2017, 19, 5880–5883 CrossRef CAS.
- (a) M. Montesinos-Magraner, M. Costantini, R. Ramírez-Contreras, M. E. Muratore, M. J. Johansson and A. Mendoza, Angew. Chem., Int. Ed., 2019, 58, 5930–5935 CrossRef CAS; (b) Á. Gutiérrez-Bonet, S. Popov, M. H. Emmert, J. M. E. Hughes, A. F. Nolting, S. Ruccolo and Y. Wang, Org. Lett., 2022, 24, 3455–3460 CrossRef; (c) J. Altarejos, D. Sucunza, J. J. Vaquero and J. Carreras, Org. Lett., 2021, 23, 6174–6178 CrossRef CAS PubMed; (d) H. Iwamoto, Y. Ozawa, Y. Hayashi, T. Imamoto and H. Ito, J. Am. Chem. Soc., 2022, 144, 10483–10494 CrossRef CAS PubMed; (e) T. Xie, L. Chen, Z. Shen and S. Xu, Angew. Chem., Int. Ed., 2023, 62, e202300199 CrossRef CAS; (f) V. Martín-Heras, A. Parra and M. Tortosa, Synthesis, 2018, 470–484 Search PubMed; (g) Y. Shi, Q. Gao and S. Xu, Synlett, 2019, 2107–2112 CAS.
- M. Simaan, P.-O. Delaye, M. Shi and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 12345–12348 CrossRef CAS PubMed.
- C. M. Poteat, Y. Jang, M. Jung, J. D. Johnson, R. G. Williams and V. N. G. Lindsay, Angew. Chem., Int. Ed., 2020, 59, 18655–18661 CrossRef CAS PubMed.
- M. V. Barysevich, V. V. Kazlova, A. G. Kukel, A. I. Liubina, A. L. Hurski, V. N. Zhabinskii and V. A. Khripach, Chem. Commun., 2018, 54, 2800–2803 RSC.
- D. Astashko, J. K. Cha, N. N. Rao and B. B. Parida, Eur. J. Org. Chem., 2014, 181–187 CrossRef CAS.
- E. J. Corey, S. A. Rao and M. C. Noe, J. Am. Chem. Soc., 1994, 116, 9345–9346 CrossRef CAS.
- O. G. Kulinkovich, D. G. Kananovich, M. Lopp and V. Snieckus, Adv. Synth. Catal., 2014, 356, 3615–3626 CrossRef CAS.
- M. Iskryk, M. Barysevich, M. Ošeka, J. Adamson and D. Kananovich, Synthesis, 2019, 1935–1948 CAS.
- Y. A. Konik, D. G. Kananovich and O. G. Kulinkovich, Tetrahedron, 2013, 69, 6673–6678 CrossRef CAS.
- (a) J. Pietruszka, A. C. M. Rieche, T. Wilhelm and A. Witt, Adv. Synth. Catal., 2003, 345, 1273–1286 CrossRef CAS; (b) J. Pietruszka, T. Wilhelm and A. Witt, Synlett, 1999, 1981–1983 CrossRef CAS.
- Y. Sekiguchi and N. Yoshikai, J. Am. Chem. Soc., 2021, 143, 4775–4781 CrossRef CAS.
- Y. Sekiguchi and N. Yoshikai, J. Am. Chem. Soc., 2021, 143, 18400–18405 CrossRef CAS PubMed.
- C. P. Casey and N. A. Strotman, Can. J. Chem., 2006, 84, 1208–1217 CrossRef CAS.
- (a) R. W. Hoffmann, Chem. Soc. Rev., 2003, 32, 225–230 RSC; (b) A. Guijarro and R. D. Rieke, Angew. Chem., Int. Ed., 2000, 39, 1475–1479 CrossRef CAS.
- C. H. DePuy, W. C. Arney Jr. and D. H. Gibson, J. Am. Chem. Soc., 1968, 90, 1830–1840 CrossRef CAS.
- A. DeBoer and C. H. DePuy, J. Am. Chem. Soc., 1970, 92, 4008–4013 CrossRef CAS.
- O. L. Epstein and O. G. Kulinkovich, Tetrahedron Lett., 2001, 42, 3757–3758 CrossRef CAS.
- J. O. Hoberg and P. W. Jennings, Organometallics, 1996, 15, 3902–3904 CrossRef CAS.
- D. T. Ziegler, A. M. Steffens and T. W. Funk, Tetrahedron Lett., 2010, 51, 6726–6729 CrossRef CAS.
- M. Shan and G. A. O'Doherty, Org. Lett., 2008, 10, 3381–3384 CrossRef CAS PubMed.
- M. Shan and G. A. O'Doherty, Synthesis, 2008, 3171–3179 CAS.
- P. P. Das, B. B. Parida and J. K. Cha, ARKIVOC, 2012, 2012, 74–84 Search PubMed.
- O. L. Epstein and J. K. Cha, Angew. Chem., Int. Ed., 2005, 44, 121–123 CrossRef CAS.
- A. W. Schuppe, D. Huang, Y. Chen and T. R. Newhouse, J. Am. Chem. Soc., 2018, 140, 2062–2066 CrossRef CAS PubMed.
- M. V. Barysevich, M. V. Laktsevich-Iskryk, A. M. Scherbakov, D. I. Salnikova, O. E. Andreeva, D. V. Sorokin, Y. Y. Shchegolev, A. L. Hurski, V. N. Zhabinskii and V. A. Khripach, Steroids, 2022, 188, 109135 CrossRef CAS.
- J. Jiao, L. X. Nguyen, D. R. Patterson and R. A. Flowers, Org. Lett., 2007, 9, 1323–1326 CrossRef CAS.
- M. V. Barysevich, Y. M. Aniskevich and A. L. Hurski, Synlett, 2021, 1934–1938 CAS.
- A. Abad, C. Agulló, A. C. Cuñat, I. de Alfonso Marzal, A. Gris, I. Navarro and C. Ramírez de Arellano, Tetrahedron, 2007, 63, 1664–1679 CrossRef CAS.
- H. G. Lee, I. L. Lysenko and J. K. Cha, Angew. Chem., Int. Ed., 2007, 46, 3326–3328 CrossRef CAS.
- N. Jha, S. Mondal and M. Kapur, Chem. Commun., 2023, 59, 12491–12494 RSC.
- T. Feng, C. Liu, Z. Wu, X. Wu and C. Zhu, Chem. Sci., 2022, 13, 2669–2673 RSC.
- (a) N. Varabyeva, M. Barysevich, Y. Aniskevich and A. Hurski, Org. Lett., 2021, 23, 5452–5456 CrossRef CAS PubMed; (b) E. Hasegawa, K. Nemoto, R. Nagumo, E. Tayama and H. Iwamoto, J. Org. Chem., 2016, 81, 2692–2703 CrossRef CAS PubMed.
- P. P. Das, K. Belmore and J. K. Cha, Angew. Chem., Int. Ed., 2012, 51, 9517–9520 CrossRef CAS PubMed.
- B. B. Parida, P. P. Das, M. Niocel and J. K. Cha, Org. Lett., 2013, 15, 1780–1783 CrossRef CAS.
- R. V. N. S. Murali, N. N. Rao and J. K. Cha, Org. Lett., 2015, 17, 3854–3856 CrossRef CAS PubMed.
- N. Tumma, E. Gyanchander and J. K. Cha, J. Org. Chem., 2017, 82, 4379–4385 CrossRef CAS.
- L. Fang, S. Jia, S. Fan and J. Zhu, Chem. Commun., 2023, 59, 10392–10395 RSC.
- K. Cheng and P. J. Walsh, Org. Lett., 2013, 15, 2298–2301 CrossRef CAS PubMed.
- X. Cai, W. Liang, M. Liu, X. Li and M. Dai, J. Am. Chem. Soc., 2020, 142, 13677–13682 CrossRef CAS PubMed.
- D. C. Davis, K. L. Walker, C. Hu, R. N. Zare, R. M. Waymouth and M. Dai, J. Am. Chem. Soc., 2016, 138, 10693–10699 CrossRef CAS.
- E. Gyanchander, S. Ydhyam, N. Tumma, K. Belmore and J. K. Cha, Org. Lett., 2016, 18, 6098–6101 CrossRef CAS PubMed.
- M. Lee, J. Heo, D. Kim and S. Chang, J. Am. Chem. Soc., 2022, 144, 3667–3675 CrossRef CAS PubMed.
- Y. Sekiguchi, Y. Y. Lee and N. Yoshikai, Org. Lett., 2021, 23, 5993–5997 CrossRef CAS PubMed.
- W. Liang, X. Cai and M. Dai, Chem. Sci., 2021, 12, 1311–1316 RSC.
- M. Jung, J. E. Muir and V. N. G. Lindsay, Tetrahedron, 2023, 134, 133296 CrossRef CAS PubMed.
- C. M. Poteat and V. N. G. Lindsay, Org. Lett., 2021, 23, 6482–6487 CrossRef CAS PubMed.
- S. Singh, M. Simaan and I. Marek, Chem. – Eur. J., 2018, 24, 8553–8557 CrossRef CAS PubMed.
- H. Sommer, T. Weissbrod and I. Marek, ACS Catal., 2019, 9, 2400–2406 CrossRef CAS.
- T. Avullala, P. Asgari, Y. Hua, A. Bokka, S. G. Ridlen, K. Yum, H. V. R. Dias and J. Jeon, ACS Catal., 2019, 9, 402–408 CrossRef CAS PubMed.
- K. Dong, R. Gurung, X. Xu and M. P. Doyle, Org. Lett., 2021, 23, 3955–3959 CrossRef CAS.
- D. H. Gibson and C. H. DePuy, Chem. Rev., 1974, 74, 605–623 CrossRef CAS.
- J. Salaun, Chem. Rev., 1983, 83, 619–632 CrossRef CAS.
- (a) M. V. Raiman, N. A. Il'ina and O. G. Kulinkovich, Synlett, 1999, 1053–1054 CrossRef CAS; (b) A. Fadel and A. Khesrani, Tetrahedron: Asymmetry, 1998, 9, 305–320 CrossRef CAS.
- J. Salaün, Chem. Rev., 2005, 105, 285–312 CrossRef PubMed.
- L. R. Mills, L. M. Barrera Arbelaez and S. A. L. Rousseaux, J. Am. Chem. Soc., 2017, 139, 11357–11360 CrossRef CAS PubMed.
- L. R. Mills, J. J. Monteith, G. dos Passos Gomes, A. Aspuru-Guzik and S. A. L. Rousseaux, J. Am. Chem. Soc., 2020, 142, 13246–13254 CrossRef CAS PubMed.
- H. M. Walborsky, Tetrahedron, 1981, 37, 1625–1651 CrossRef CAS.
- J. J. Monteith, J. W. Pearson and S. A. L. Rousseaux, Angew. Chem., Int. Ed., 2024, 63, e202402912 CrossRef CAS.
- J. Yang, Y. Shen, Y. J. Lim and N. Yoshikai, Chem. Sci., 2018, 9, 6928–6934 RSC.
- J. Yang, Y. Sekiguchi and N. Yoshikai, ACS Catal., 2019, 9, 5638–5644 CrossRef CAS.
- W. Huang and F. Meng, Angew. Chem., Int. Ed., 2021, 60, 2694–2698 CrossRef CAS.
- Z. Liang, L. Wang, Y. Wang, L. Wang, Q. Chong and F. Meng, J. Am. Chem. Soc., 2023, 145, 3588–3598 CrossRef CAS PubMed.
- Z. Liang, Q. Chong and F. Meng, Sci. China: Chem., 2021, 64, 1750–1755 CrossRef CAS.
- Y. Sekiguchi and N. Yoshikai, Org. Lett., 2022, 24, 960–965 CrossRef CAS PubMed.
- K. Tsukiji, A. Matsumoto, K. Kanemoto and N. Yoshikai, Angew. Chem., Int. Ed., 2024, e202412456 CAS.
- Q. Zhang, S.-W. Zhou, C.-Y. Shi and L. Yin, Angew. Chem., Int. Ed., 2021, 60, 26351–26356 CrossRef CAS PubMed.
- B. M. Trost, G. Zhang, M. Xu and X. Qi, Chem. – Eur. J., 2022, 28, e202104268 CrossRef CAS PubMed.
- A. Kitabayashi, S. Mizushima, K. Higashida, Y. Yasuda, Y. Shimizu and M. Sawamura, Adv. Synth. Catal., 2022, 364, 1855–1862 CrossRef CAS.
- L. Wu, L. Wang, P. Chen, Y.-L. Guo and G. Liu, Adv. Synth. Catal., 2020, 362, 2189–2194 CrossRef.
- C. Jiang, L. Wang, H. Zhang, P. Chen, Y.-L. Guo and G. Liu, Chem, 2020, 6, 2407–2419 CAS.
- (a) D. G. Kananovich, Y. A. Konik, D. M. Zubrytski, I. Järving and M. Lopp, Chem. Commun., 2015, 51, 8349–8352 RSC; (b) Y. Li, Z. Ye, T. M. Bellman, T. Chi and M. Dai, Org. Lett., 2015, 17, 2186–2189 CrossRef CAS PubMed.
- J. Wang and X. Li, Chem. Sci., 2022, 13, 3020–3026 RSC.
- Z. Wu, D. Lebœuf, P. Retailleau, V. Gandon, A. Marinetti and A. Voituriez, Chem. Commun., 2017, 53, 7026–7029 RSC.
- E. Jiménez-Núñez, C. K. Claverie, C. Nieto-Oberhuber and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5452–5455 CrossRef PubMed.
- (a) F. Giacalone, M. Gruttadauria, P. Agrigento and R. Noto, Chem. Soc. Rev., 2012, 41, 2406–2447 RSC; (b) B. Han, X.-H. He, Y.-Q. Liu, G. He, C. Peng and J.-L. Li, Chem. Soc. Rev., 2021, 50, 1522–1586 RSC.
- (a) F. Romanov-Michailidis, L. Guénée and A. Alexakis, Angew. Chem., Int. Ed., 2013, 52, 9266–9270 CrossRef CAS PubMed; (b) F. Romanov-Michailidis, L. Guénée and A. Alexakis, Org. Lett., 2013, 15, 5890–5893 CrossRef CAS PubMed; (c) F. Romanov-Michailidis, M. Pupier, L. Guénée and A. Alexakis, Chem. Commun., 2014, 50, 13461–13464 RSC; (d) F. Romanov-Michailidis, M. Romanova-Michaelides, M. Pupier and A. Alexakis, Chem. – Eur. J., 2015, 21, 5561–5583 CrossRef CAS PubMed.
- M. A. S. Blackburn, C. C. Wagen, M. R. Bodrogean, P. M. Tadross, A. J. Bendelsmith, D. A. Kutateladze and E. N. Jacobsen, J. Am. Chem. Soc., 2023, 145, 15036–15042 CrossRef CAS PubMed.
- Y.-Y. Xie, Z.-M. Chen, H.-Y. Luo, H. Shao, Y.-Q. Tu, X. Bao, R.-F. Cao, S.-Y. Zhang and J.-M. Tian, Angew. Chem., Int. Ed., 2019, 58, 12491–12496 CrossRef CAS PubMed.
- C. Zhang, S. Ye and J. Wu, Org. Lett., 2024, 26, 3321–3325 CrossRef CAS PubMed.
- C. Zhang, J. Huang, S. Ye, J. Tang and J. Wu, Chem. Commun., 2020, 56, 13852–13855 RSC.
- L. Candish, C. M. Forsyth and D. W. Lupton, Angew. Chem., Int. Ed., 2013, 52, 9149–9152 CrossRef CAS PubMed.
- S.-H. Xiang and B. Tan, Nat. Commun., 2020, 11, 3786 CrossRef CAS PubMed.
- Ł. Woźniak, G. Magagnano and P. Melchiorre, Angew. Chem., Int. Ed., 2018, 57, 1068–1072 CrossRef PubMed.
- G. Z. Elek, V. Borovkov, M. Lopp and D. G. Kananovich, Org. Lett., 2017, 19, 3544–3547 CrossRef CAS PubMed.
- G. Z. Elek, K. Koppel, D. M. Zubrytski, N. Konrad, I. Järving, M. Lopp and D. G. Kananovich, Org. Lett., 2019, 21, 8473–8478 CrossRef CAS PubMed.
- L. Dian and I. Marek, Chem. Rev., 2018, 118, 8415–8434 CrossRef CAS PubMed.
- V. D. Gvozdev, K. N. Shavrin, A. A. Ageshina and O. M. Nefedov, Russ. Chem. Bull., 2017, 66, 862–866 CrossRef CAS.
- Q. Gao and S. Xu, Angew. Chem., Int. Ed., 2023, 62, e202218025 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |