
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An investigation into catalysed xanthene-based dye oxidation by a family of coordination cages†
James R.
Williams
and
Michael D.
Ward
 *
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: m.d.ward@warwick.ac.uk
First published on 7th July 2025
Abstract
The ability of a family of M4, M8 and M12 coordination cages to effect catalytic oxidative degradation of a family of xanthene-based dyes using peroxymonosulfate (PMS) has been investigated in water. The M12 cages bind one dye molecule inside the central cavity; the M8 cages bind multiple anionic dye molecules around the external cage surface; the smallest M4 cages do not interact strongly with the dyes. Three separate sets of experiments showed that octanuclear Co8 was the most effective catalyst due to a combination of (i) its ability to bind multiple dye molecules around its surface in solution, and (ii) the Co(II)/Co(III) redox couple which activates the PMS anion by reducing it to the reactive species SO4˙− close to the cage-bound substrates. Control experiments showed that replacing Co(II) by Fe(II), Ni(II) or Zn(II) in isostructural M8 cages removed catalytic activity, which specifically requires the Co(II)/Co(III) couple; and the effectiveness of the catalysis is guest-dependent according to parameters such as charge, hydrophobicity and inductive effect of substituents on the xanthene core. Overall the Co8 cage fulfils three functions of (i) binding the guest, (ii) activating the PMS using the Co(II)/Co(III) couple, and (iii) accumulating the SO4˙− anions around the cationic cage surface close to bound guests.
Introduction
A particularly appealing application of coordination cage hosts is the catalysis of reactions of bound guests, a subject which has been extensively reviewed in recent years.1 There are many mechanisms by which such catalysis can occur, including (but not limited to) (i) co-location of >1 reaction partners, meaning that each reactant experiences a high local concentration of the other; (ii) electrostatic factors whereby the high charge on a host cage can modify the affinity of a bound guest for protons, leading to substantial changes in acid- or base-catalysed reaction rates; (iii) constrictive binding, whereby folding a flexible guest in a confined space perturbs it towards the geometry of a transition state, thereby making it more accessible; and (iv) light-induced processes in which the host cage surrounding a guest incorporates light-harvesting chromophores or redox-active quenching groups in close proximity to the guest, facilitating reactions based on photoinduced electron transfer.1Our recent work in this area, based on members of our family of coordinated cages that contain ditopic or tritopic ligands with pyrazolyl-pyridine chelating termini,2 has made two distinct contributions. The first is that a range of catalysed reactions between hydrophobic organic guests and anions (which include hydroxide,3a enolates3b and phenolates3c) requires recognition of the anionic reaction partner at binding sites on the cationic cage surface: and these reactions can occur not just on cavity-bound substrates,3 but also on substrates that are too large for the cavity but are nonetheless associated with the cage exterior surface via hydrophobic interactions.4
Our second recent contribution is that Co(II)-containing cages can act as redox partners for cage-catalysed oxidation reactions of substrates.2b,5 Oxidants such as H2O2 and peroxymonosulfate, which are thermodynamically powerful but kinetically slow oxidants, may be activated by conversion to reactive oxygen species (ROS) using a low-potential Mn+/M(n+1)+ redox couple in which the Mn+ ion acts as a one-electron reducing agent.6,7 If this happens using the Co(II) ions in the cage, then hydrophobic organic substrates that interact with the cage surface (exterior or interior) can be oxidised by the resulting high local concentration of reactive oxygen species, which will be further facilitated by the fact that anionic ROS will accumulate around the 16+ cage for electrostatic reasons. In these cases the cage therefore acts not only to co-locate substrate (organic substrate) and reaction partner (oxidant), but also [via the Co(II)/Co(III) couple] to activate the oxidant, thereby participating directly in the reaction cycle and not just acting as a container.5
A specific example of this type of reactivity that we reported recently was the oxidation of xanthene-based dyes such as fluorescein with peroxymonosulfate (PMS), catalysed by the Co12 cage with a cuboctahedral geometry (Fig. 1);5a this cage contains four tritopic (face-capping) ligands and twelve ditopic (edge-bridging) ligands, and has an internal cavity volume of ca. 1100 Å3.8 We described the catalytic process as occurring through the encapsulation of a dye molecule inside the Co12 internal cavity, and generation of the reactive radical anion SO4˙− by the redox reaction of PMS with Co(II) ions in the cage (eqn (1)).‡ The strong tendency of anionic species to accumulate round the cage surface2–4 (the Co12 cage carries a 24+ charge) results in co-location of substrate and oxidant which is the source of the catalysis.
| Co2+ + HSO5 → Co3+ + SO4˙− + HO− | (1) |
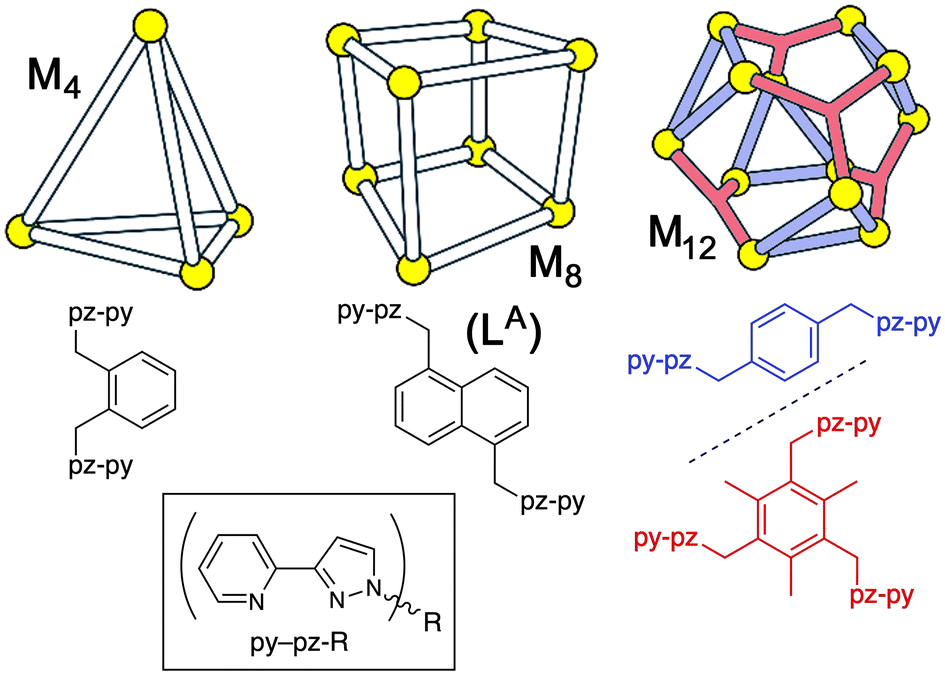 | ||
| Fig. 1 Geometries of the M4, M8 and M12 cages, and the structural formulae of the associated ligands, used in this work. | ||
Significantly, control experiments conformed that both (i) binding of substrate by cage, and (ii) redox activity of the Co(II) ions, were essential. Replacing Co(II) ions with Zn(II) ions in an isostructural cage completely removed the catalytic effect. An equal number of mononuclear Co(II) complexes of similar redox potential – i.e. removing the possibility of a complete cage encapsulating the substrate – likewise removed most of the catalysis.5a
In this paper we report a more extensive study into the PMS-based oxidation of xanthene dyes catalysed by our octanuclear cubic M8 cages.2c The smaller size of these compared to the M12 cages means that interaction of the cages with the dyes is necessarily at the external surface as the dyes cannot fit inside the cavities. The Co8 cage has been shown to bind multiple equivalents of xanthene dyes such as fluorescein (FLU, Fig. 2) at the anion binding sites on each face exterior surface,9 but cannot accommodate guests of that size inside the cavity. As part of this work we have also varied the metal ions used to include Fe(II) and Ni(II) to see the effect of having different metal ions (with different redox properties) on the catalysis which relies on initial activation of the PMS using the M(II)/M(III) couple.
 | ||
| Fig. 2 Structures of the xanthene-based dyes used in this work. | ||
Importantly, the use of dye molecules – of this class and many others – in industrial and large-scale processes is widespread. In many cases, these dyes are acutely toxic to aquatic life, meaning that waste water treatment for large scale processes is an ongoing problem.10 Environmentally friendly oxidants used in advanced oxidation processes, whose activity is based on formation of reactive oxygen species, can result in complete destruction of the dye with only water and carbon dioxide produced from hydrocarbon dyes.11 PMS has recently emerged as a more potent oxidant than hydrogen peroxide: like H2O2, it requires an initial redox-based activation step – often with a redox-active metal ion – to afford the reactive radical anion SO4˙− as discussed above.6,7 Accordingly this is a type of cage-based catalysis of potentially significant value and it may be extendable to other substrates that are susceptible to reaction with H2O2 or PMS and which bind to (or within) the host cage.
Results and discussion
Range of compounds used: cages, control complexes, and substrates
The cage complexes used in this investigation are presented in Fig. 1: these are cuboctahedral Co12,8b cubic M8 (M = Fe, Co, Ni, Zn)2–4 and tetrahedral Co4 cages.12 Of these, full synthesis/characterisation of Fe8 has not been reported before: it is prepared in the same way as the regularly-studied Co8 cage2c and relevant details are included in the ESI,† with the crystal structure of the new Fe8 cage being essentially identical to the structure of those with M = Co, Ni and Zn.In addition simple mononuclear complexes [M(pypz-Me)3]2+ (denoted M1; M = Fe, Co, Ni, Zn, and based on a known ligand13) have been prepared (details in ESI†) to be used for control experiments and electrochemical studies, based on the fact that the metal ions are in the same coordination environment as those in the larger cages: accordingly they should mimic the redox activity of metal ions in the cages, but without any ability to encapsulate guests. Given the inequivalence of the coordinating N atoms in the bidentate ligand pypz-Me, these mononuclear complexes are formed as a statistical (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) mixture of fac and mer isomers,14 as clearly evident in some of the NMR spectra, and which precisely matches what happens during assembly of the M8 cubic cages which contain two fac tris-chelate and six mer tris-chelate vertices.2a,14b Consequently these mononuclear complexes are reasonable mimics of the metal vertices in the larger cages. Crystals of [Co(pypz-Me)3](BF4)2 incorporate only the mer isomer: although one ligand is disordered over two orientations, both components are meridional (see ESI†). In crystals of [Zn(pypz-Me)3](BF4)2 the positional disorder of ligands is more severe so this is not reported in full, we just note that it is isostructural with the Co(II) analogue and is clearly mononuclear [Zn(pypz-Me)3](BF4)2.
:3) mixture of fac and mer isomers,14 as clearly evident in some of the NMR spectra, and which precisely matches what happens during assembly of the M8 cubic cages which contain two fac tris-chelate and six mer tris-chelate vertices.2a,14b Consequently these mononuclear complexes are reasonable mimics of the metal vertices in the larger cages. Crystals of [Co(pypz-Me)3](BF4)2 incorporate only the mer isomer: although one ligand is disordered over two orientations, both components are meridional (see ESI†). In crystals of [Zn(pypz-Me)3](BF4)2 the positional disorder of ligands is more severe so this is not reported in full, we just note that it is isostructural with the Co(II) analogue and is clearly mononuclear [Zn(pypz-Me)3](BF4)2.
The xanthene-based dyes used in this study (Fig. 2) are fluorescein (FLU), 6-carboxyfluorescein (CFLU), eosin-Y (EY), rhodamine-B (RB) and sulforhodamine-B (SRB). Both FLU and EY exist as dianions in neutral aqueous solution; CFLU with its extra carboxylic acid group can be a trianion. RB and SRB both contain an iminium group rendering RB neutral and zwitterionic in neutral solution; and SRB is anionic overall but with charge imbalance over the dye (a cationic iminium group, and two anionic sulfonate units). FLU and EY have both been studied in the investigation on the catalytic activity of the Co12 cage,5a and will therefore serve as points of comparison.
The range of cages, substrates and metal ions used allows for investigation of three variables. In this work we have:
• used a single dye substrate (FLU) but a range of different catalyst types containing 1, 4, 8 or 12 metal ions: what is the effect of catalyst structure/nuclearity on oxidative dye degradation?
• used a single cage (Co8) as catalyst to examine the effectiveness of the catalysed oxidations with different xanthene dye substrates: how do substrate properties such as charge and hydrophobicity affect catalysis?
• used a single cage type (M8) and single dye substrate (FLU) but varied the nature of the metal ion in the cage [Fe(II), Co(II), Ni(II), Zn(II)] to examine effect of metal ion redox properties on the cage-based catalysis.
Crystal structures of M8 cage/dye assemblies
To illustrate how the dye molecules can interact with the cage exterior surface, we crystallised samples of the Ni8 cage in the presence of EY2− and SRB− (as their sodium salts) and performed crystallographic analyses of the single crystals obtained in which the dye anions had replaced some of the tetrafluoroborate anions. Of course crystal structures may not replicate solution speciation but they are usefully illustrative.The structure of Ni8·EY contains EY2− anions in the lattice in spaces between cage complex cations, as these anions are too large to occupy the cage cavity. The EY2− anions occupy two crystallographically different sites: this is illustrated in Fig. 3 in which the EY2− anions are coloured red or purple to indicate the different crystal sites.
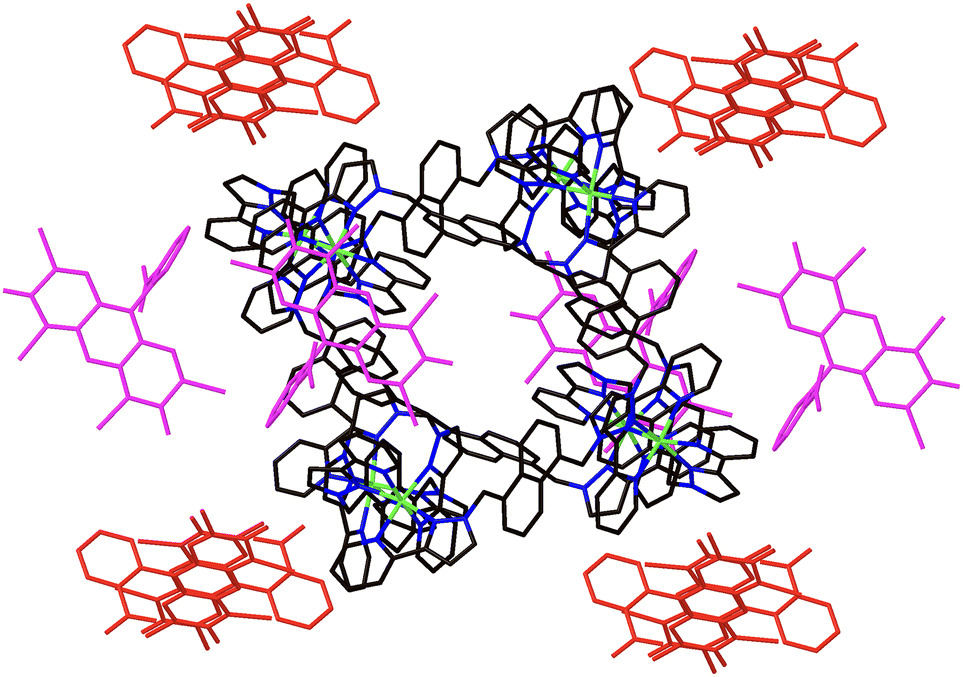 | ||
| Fig. 3 View of the crystal structure of Ni8·EY with the two crystallographically inequivalent EY2− molecules shown in different colours. | ||
There are 1.7 EY2− anions per Ni8 cage cation (which carries a charge of 16+), with the anions coloured red having a site occupancy of 1.0, and those in the alternate site (coloured purple) having a site occupancy of 0.7. This leaves 12.6 tetrafluoroborate anions required per cage to balance the charge, of which 10.45 could be accounted for during the refinement, with the balance likely being a casualty of the extensive disorder of anions/solvent molecules which required use of the solvent mask function to remove diffuse electron density that could not be modelled. As usual, some of the BF4− anions are associated with the windows in the centre of the cage faces where they are anchored by multiple CH⋯F hydrogen-bonding interactions (see ESI†). The EY2− anions are positioned so as to lie close to and ‘embracing’ parts of the cage surface close to the Ni2+ ions, in the regions of highest positive electrostatic potential around the cage vertices, where there are multiple electrostatically-assisted supramolecular interactions (CH⋯π, π⋯π and CH⋯O) between cage surface and anions (Fig. 4).
 | ||
| Fig. 4 Partial views of the structure of Ni8·EY showing the interactions of the anionic dye guests with the external surface of the cage around the vertices (orange spheres are the Br atoms). | ||
Similar behaviour was observed in the Ni8·SRB complex (Fig. 5 and 6). The SRB− anions are all crystallographically equivalent, with a site occupancy of 0.75; the asymmetric unit of the structure contains half of the cage (which lies across an inversion centre) as well as 0.75 SRB− anions, such that the balance is 1.5 SRB− anions per complete Ni8 cage. This means that there must be 14.5 fluoroborate anions of which 7.8 could be crystallographically located, with the balance again being a casualty of disorder/removal of diffuse electron density using a ‘solvent mask’ function.
 | ||
| Fig. 5 View of the crystal structure of Ni8·SRB with the SRB− anions shown in darker colours than the cage for emphasis (S atoms coloured yellow; O atoms coloured red). | ||
 | ||
| Fig. 6 Partial views of the structure of Ni8·SRB showing the interaction of the SRB− dye guest with the external surface of the cage and a fluoroborate anion in one of the surface portals. View (a) is looking down onto one of the cage faces showing all of the SRB− guest; (b) is edge-on to the SRB− guest showing the contact between the sulfonate groups and the cationic cage surface. | ||
As with the Ni8·EY structure, the conformation of the anion (substantial twist between the phenyl ring and the xanthene core) facilitates a variety of close contacts between SRB− and the cage exterior surface, with the negatively-charged sulfonate groups in particular participating in multiple CH⋯O hydrogen-bonding interactions close to a cationic cage vertex, of which the shortest, between a methylene proton H(46F) and sulfonate O atom O(43G), is 2.17 Å. Conversely the cationic NEt2+ terminus of SRB− lies over one the portals in the face centres, close to one of the surface-bound tetrafluoroborate anions, such that there is evidence for weak CH⋯F interactions between an NEt2+ terminus of the dye and BF4− [separations of C(27G) from F(13X) and F(15X) are 3.52 and 3.49 Å respectively]. This combination of interactions between SRB−, the cage vertices, and the portal-bound anion results in the SRB− anion lying relatively flat to the cage surface, see Fig. 6. We have shown in recent work how such close cage/dye interactions can facilitate fast photoinduced electron transfer between a dye donor (EY) that is bound to a M8 cage exterior surface, and a naphthoquinone acceptor that is bound in the cage interior cavity.15
Effect of cage catalyst: catalysis of fluorescein oxidation using different Con-based cage sizes (n = 0, 1, 4, 8, 12)
In our earlier initial work on redox-based catalysis, we established that FLU2− underwent complete oxidative decomposition in minutes using the Co12 cage as catalyst in aqueous solution with PMS as oxidant, which was facilitated by strong (logK = 6.7) 1:1 cage:guest binding; a stoichiometry which suggests that the FLU2− substrate could bind inside the Co12 cage cavity.5a Accordingly we were interested to see how this compared with catalysis by the smaller Co8, Co4 and Co1 complexes under the same conditions. We also know from earlier work that the Co8 cage has the capacity to bind multiple anionic dye molecules of this general type around its external surface:9 this binding is strong due to a combination of the negative charge on the dye molecules, and their large hydrophobic surface area, with (for example) a 1:1 binding constant between FLU and Co8 of 1 × 105 M−1.§ As FLU is too large to bind as a guest inside the smaller cavity of Co8 (compared to Co12) this binding has to be association with the external surface, in a way that we routinely see with smaller anions that occupy the windows in the M8 cage surfaces.2a,16 Further support for this came from a Job plot experiment which showed that several FLU anions could bind to the surface of each Co8 cage (ca. 5:1, with similarly large values for related dye molecules) when the molar ratio of components was optimised for this: a stoichiometry completely incompatible with cavity binding, which suggests that in solution that the anionic dye molecules can associate with each face of the cubic cage.9¶
In contrast we found no such strong interaction between FLU and the smaller Co4 cage: a fluorescence-based titration of Co4 into a solution of FLU, under the same conditions as used for Co12 and Co8 (10−5 M concentration domain in 2% dmso/98% aqueous buffer) showed very little quenching of FLU by Co4, indicative of a much weaker Co4/FLU interaction. We suggest on the basis of this that strong binding of FLU to Co12 is driven by 1:1 cavity binding; strong binding of FLU to Co8 is driven by association with the external surfaces where the portals provide good H-bond donor sites to anionic guests; but as Co4 possesses neither of the necessary structural features to bind FLU strongly (neither a significantly-sized central cavity to encapsulate the guest, nor surface windows to provide a convergent H-bond donor site) the Co4/FLU association is insignificant in this solvent at the concentrations used.
Armed with this knowledge of relative affinities we can understand the data in Fig. 7, which shows graphs plotting disappearance of the absorption maximum of FLU at 489 nm as a function of time, at concentrations of Co12, Co8, Co4 and Co1 catalysts as required to give the same total concentration of Co(II) ions for the redox activation of PMS which is the ultimate oxidant. There are some obvious conclusions that we can draw from this based on knowledge of cage/guest binding behaviour. Firstly, in the absence of any Co(II) complex as catalyst, oxidation of FLU by PMS is very slow due to the lack of redox activation (dotted line). The mononuclear complex Co1 provides some redox activation: the catalysis associated with this (green line) cannot involve any kind of guest encapsulation in a cage, but it is likely that some modest degree of FLU2−/Co1 association can occur for electrostatic reasons to bring the substrate close to where the SO4˙− ions are generated. The Co4 cage performs similarly to Co1 as a catalyst [for the same overall concentration of Co(II) ions]: the reaction rate is slightly faster at early times (steeper gradient of the purple line compared to green in Fig. 7) and tails off later. Given the lack of a strong interaction between Co4 and FLU this general lack of any improved catalysis by Co4 compared to Co1 for the same concentration of Co(II) ions makes sense. Then we see better catalysis and faster oxidative destruction of FLU2− by Co12, as we reported earlier, based on strong 1:1 cage:guest binding involving cavity encapsulation.5a Finally – the most interesting observation – we see that the Co8 cage is a substantially more effective catalyst than Co12 with an initial reaction rate that is ≈3 times higher for the same overall Co(II) ion concentration. We attribute this to the ability of the Co8 cage to bind multiple FLU2− guests around the exterior surface, at the anion binding sites on the faces, as the Job plot experiment demonstrate (ref. 9 and ESI†): this provides the possibility for each Co8 cage to assemble multiple substrates in the vicinity of the Co(II) ions where redox activation of PMS is occurring and the reactive SO4˙− species are generated. In the early stages of these reactions, plots of ln[FLU] vs. time show a linear decrease indicating a reaction that is first-order in substrate (see ESI†); the derived reaction rate constants are in Table 1, and it is clear that Co8 stands out having both the fastest initial rate as well as the reaction proceeding furthest in the time window studied (Fig. 7) and is therefore the most effective catalyst for redox-based oxidative degradation of dyes of this type.
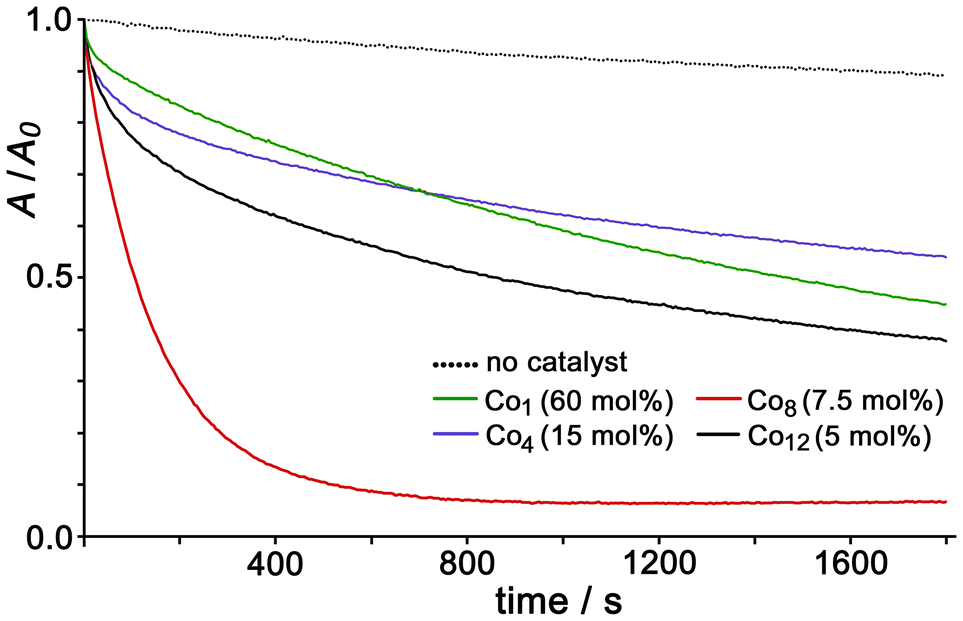 | ||
| Fig. 7 Normalised UV/vis absorption data showing destruction of FLU (7.5 μM, λmax = 489 nm) by oxidation with PMS (45 eq.) over time with no catalyst, Co12 (5 mol%), Co8 (7.5 mol%), Co4 (15 mol%) and Co1 (60 mol%), with cage concentrations adjusted to give the same concentration of Co(II) centres in each case. Solvent was aqueous buffer containing TWEEN-20 surfactant (see ref. 5a) at pH 7. | ||
| Catalyst | Substrate | Background,c 104k/s−1 | Catalysed,d 104k/s−1 | Relative increase |
|---|---|---|---|---|
| a Conditions are given in captions to Fig. 7 and 8 [7.5 μM substrate, 45 eq. PMS, catalyst loadings varied as indicated to give same concentration of Co(II) units]. b Rate constants can be converted to second-order on dividing by catalyst concentration, but to facilitate comparison for the same Co(II) ion concentration they have been left as first order. c Rate for dye degradation in presence of PMS but with no metal complex catalyst present. d Background reaction rate subtracted in each case. | ||||
| Co1 | FLU | 1.05 | 11.1 | 10.6 |
| Co4 | FLU | 1.05 | 16.6 | 15.8 |
| Co8 | FLU | 1.05 | 64.8 | 61.7 |
| Co12 | FLU | 1.05 | 23.9 | 22.8 |
| Co8 | CFLU | 6.03 | 83.4 | 13.8 |
| Co8 | EY | 2.09 | 26.2 | 12.5 |
| Co8 | RB | 12.2 | 59.6 | 4.9 |
| Co8 | SRB | 20.4 | 121 | 5.9 |
Effect of substrate: catalysis of oxidation of different dyes using the same Co8 cage
Given that the cubic Co8 cage is clearly the best catalyst for this reaction type from the series Co1/Co4/Co8/Co12 (Fig. 7), we next extended the study on oxidative dye degradation to a broader set of substrates shown above but using the same Co8 catalyst in each case, with the aim now of looking at effects of differences in substrate structure. The binding behaviour of FLU, CFLU and EY with a closely-related Co8 cage was reported in detail earlier,9¶ but RB and SRB have not been investigated before in this context. Fluorescence quenching titrations performed by adding portions of Co8 to samples of these dyes afforded binding constants (using Bindfit)17 of 5 × 104 M−1 for RB and 7 × 104 M−1 for SRB, both slightly smaller than what we measured with FLU, possibly for simple electrostatic reasons given their smaller negative charges (0 and −1 respectively). The hydrophobicity of the xanthene dye is a significant additional factor in driving association with Co8,9 as we have reported before with a range of different guest types,2–4 including diacetyl-fluorescein which bound strongly to the external surface of a Co8 cage despite being neutral.4b Job plots to investigate the stoichiometry of possible aggregates between Co8 and the dyes RB and SRB− confirm that, as observed earlier with FLU, CFLU and EY, multiple dye units can accumulate around the Co8 cage surface under different mole fraction conditions (see ESI for examples†).Fig. 8 shows the time-dependent oxidative destruction of the five different dye substrates using PMS as oxidant with Co8 as catalyst. In all cases there is an obvious and substantial increase in reaction rate in the presence of Co8 compared to the much slower reactions in its absence (see for example Fig. 7, dotted line). Again the early-time kinetic data is first order in substrate (see ESI†): after subtraction of the uncatalysed background reaction rates (absence of Co8) in each case we find initial rate accelerations compared to background spanning the range of factors from 5 (for RB and SRB) to 62 (for FLU), see Table 1. Notably from Fig. 8 we can see that destruction of FLU, CFLU and SRB approaches completion fairly quickly, within ca. 600 seconds, whereas destruction of EY and RB tails off more slowly with substantial amounts of unreacted substrate remaining even after 2000 seconds.
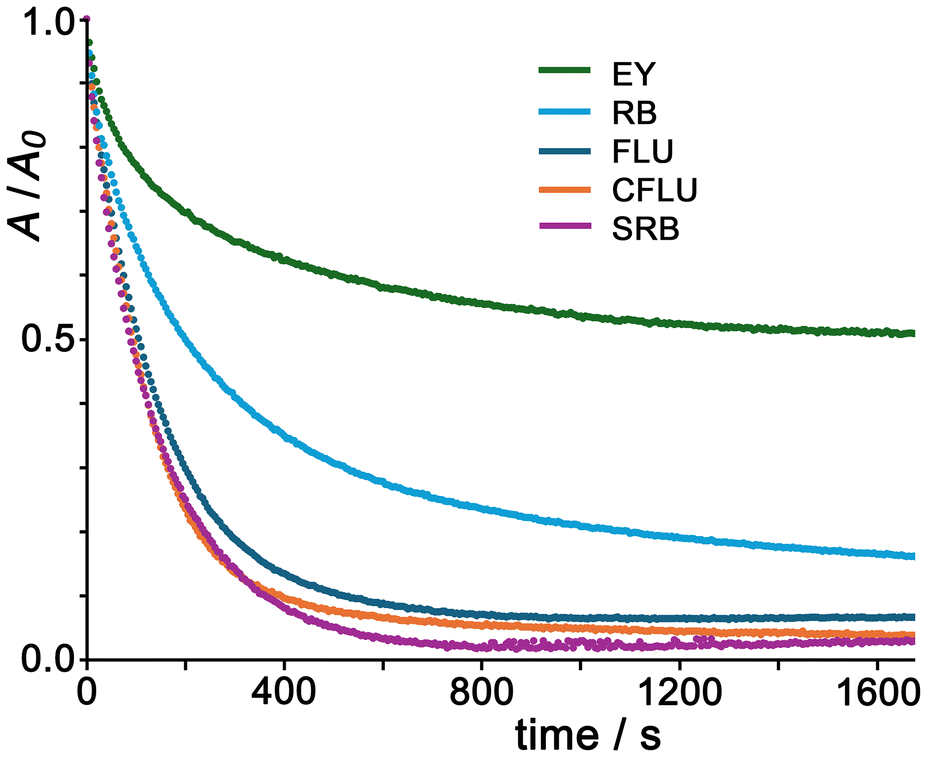 | ||
| Fig. 8 Normalised UV/vis absorption data for the relevant dye maxima showing the destruction of FLU, CFLU, EY, RB and SRB (7.5 μM in each case) with PMS (45 eq.) over time with Co8 as catalyst, at 7.5 mol% catalyst loading. Solvent was aqueous buffer containing TWEEN-20 surfactant (see ref. 5a) at pH 7. | ||
This variation in extent and rate of reaction with different substrates is interesting as it does not simply relate to any one obvious parameter like guest charge. The least effective catalysis, in terms of the extent of reaction which is <50% complete after 30 minutes, is with the substrate EY: which might suggest that electrostatic repulsion between the substrate (2− charge) and the reactive species SO4˙− slows down the reaction. However the other dianionic substrate FLU reacts significantly faster, and also shows the greatest increase relative to the background reaction, which undermines that suggestion.
A factor here could be that as the reaction is an exhaustive oxidation of the dye, the electron-withdrawing Br atom substituents on EY make the xanthene core more resistant to oxidation. Whilst the ground-state redox potentials for the first oxidation of FLU and EY are similar to one another,18 this is because they are localised in each case on the benzoate unit which is pendant from the xanthene core and orthogonal to it, such that the Br substituents have little electronic effect.18b However, subsequent oxidations of the xanthene unit will necessarily be affected by the electron-withdrawing effects of the Br substituents which are well known to make redox potentials more positive, cf. the simple example of the series ferrocene, bromoferrocene and 1,1′-dibromoferrocene whose Fe(II)/Fe(III) couples occur at +0.45, +0.63 and +0.76 V respectively in MeCN vs. Ag/AgCl.19 This effect plausibly accounts for EY being the least reactive substrate in this set of experiments.
SRB is the substrate which shows the most complete reaction, with >95% dye destruction after <10 minutes, possibly because the presence of only a single negative charge results in less electrostatic repulsion with SO4˙− than was the case with the dianionic substrates, though SRB also had the highest uncatalysed reaction rate. If we believe that SO4˙−/substrate electrostatic repulsion has a significant effect, then by that logic neutral RB should show the best catalysis: but this is clearly not the case, and we note that RB has the weakest binding to Co8 of the whole set, so will have the smallest proportion of any of the substrates brought into the reaction zone around the cage surface of any of the substrates under the conditions used.
Thus, inductive effects of substituents, and binding constant of the dye to the catalyst (which in turn relates to charge and hydrophobicity),9 all play a role in determining the relative reactivity of the different substrates with a given catalyst: with the substrate that stands out most being EY with its resistance to oxidation arising from the electron-withdrawing Br substituents.
Effect of different metal ions in isostructural M8 cage catalysts
Given the obvious effectiveness of Co8 as a catalyst for this oxidative degradation reaction using PMS, and the fact that it requires the Co(II)/Co(III) redox activity of the metal ions in the cage to convert PMS to SO4˙−, the last variable we considered is the nature of the metal ion. Accordingly, we prepared the isostructural Fe8/Ni8/Zn8 cages to accompany the Co8 cage [Cu(II) does not form a similar cage structure]. Of these Fe8 is new but isostructural with the others (see ESI†). The effectiveness of the catalysis as the metal ion is varied across the isostructural cage series is shown in Fig. 9. | ||
| Fig. 9 Normalised UV/vis absorption data showing the destruction of FLU (7.5 uM, λmax = 489 nm) with PMS (45 eq.) over time with no catalyst, Co8, Fe8, Ni8 and Zn8, all at 7.5 mol% catalyst loading. Solvent was aqueous buffer containing TWEEN-20 surfactant (see ref. 5a) at pH 7. | ||
The results are striking. We would not expect any catalytic activity from Zn8 compared to the absence of cage, given the impossibility of Zn(II)/Zn(III) redox activity, and indeed there is none: but Fe8 behaves similarly. Ni8 affords very slightly higher reactivity after a long period than background but the effect is tiny. Co8 stands out amongst this isostructural series as being a uniquely effective catalyst for this reaction, and that must be because (i) the Co(II)/Co(III) redox process occurs at a potential which aligns with what is required to activate PMS via a one-electron reduction (eqn (1)), and (ii) it is reversible, allowing for multiple redox cycles.
To check this we measured cyclic voltammograms of the mononuclear model complexes Fe1, Co1, Ni1 and Zn1 in MeCN: this is a convenient solvent for simple electrochemical studies given its high potential window, and the redox potentials of metal-centred redox processes are not expected to be significantly solvent dependent when the metal ions are coordinatively saturated. As these mononuclear complexes contain the metal ions in the same coordination environment that exists at the M8 cage vertices, down to the 3:1 mix of fac and mer isomers,14 this experiment provides a convenient basis for comparison between this set of metal ions. The CVs are shown in Fig. 10.
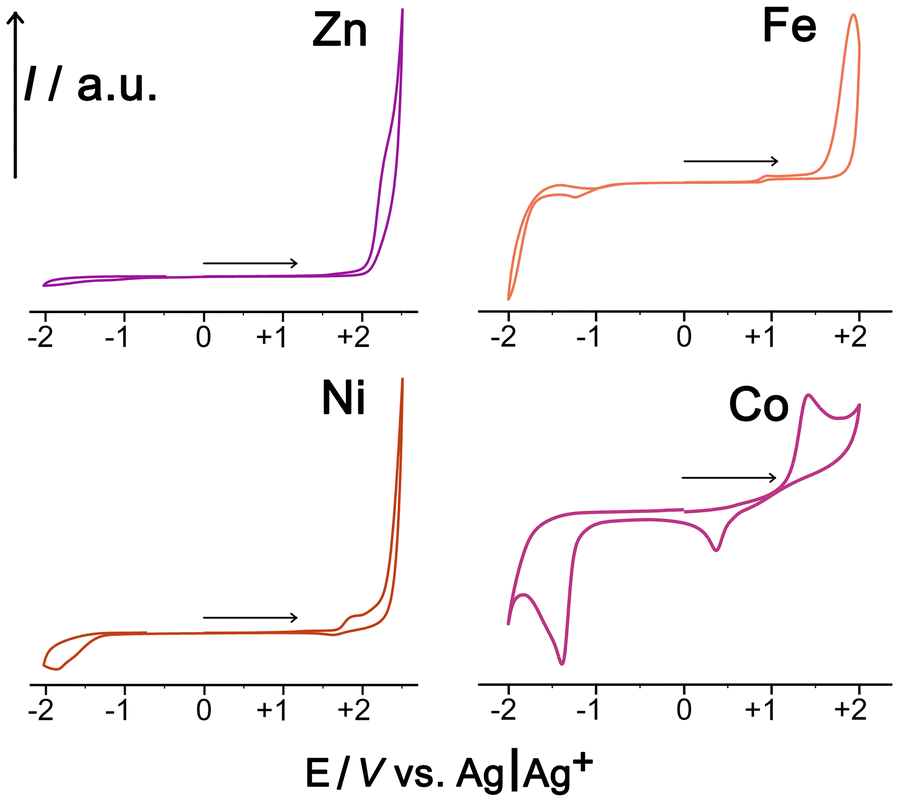 | ||
| Fig. 10 Cyclic voltammograms of the M1 complexes (M = Fe, Co, Ni, Zn) in MeCN/0.1M TBAPF6 at a scan rate of 0.1 V sec−1 using a boron-doped diamond working electrode, Pt wire counter-electrode, and an Ag/Ag+ reference electrode. The horizontal arrows show the initial potential sweep directions. | ||
The Zn(II) complex, as expected, shows no redox activity at modest potentials and can therefore be used as a baseline for comparison purposes. The Ni(II) complex shows an additional non-reversible wave at ca. +1.8 V vs. Ag/Ag+ which means that Ni(II) in this complex is not an effective reductant. The Fe(II) complex shows a very low-intensity wave around +1 V which can be tentatively ascribed to an impurity given how weak it is: the obvious main oxidation process, which is fully irreversible due to the absence of any return wave, occurs at ca. +1.8 V vs. Ag/Ag+. The Co(II) complex behaves distinctly differently with an oxidation wave at ca. +1.5 V that is accompanied by a return wave at ca. 0.5 V; we ascribe this pair to a chemically reversible but electrochemically irreversible Co(II)/Co(III) process, with the large additional wave at −1.4 V vs. Ag/Ag+ possibly being an irreversible Co(II)/Co(I) process. Thus Co1 differs from the other complexes in having a lower M(II)/M(III) redox potential (i.e. the M2+ ion is a better reductant for activation of PMS according to eqn (1)), and it also has the chemical reversibility which is essential for multiple redox cycles and catalytic turnover.
Finally, the obvious question arises as to how the catalytic cycle is completed by reduction of Co(III) back to Co(II). PMS oxidation is mechanistically complex and can generate multiple by-products including peroxide and superoxide,21 both of which are capable of acting as reducing agents (generating O2), which could plausibly provide a basis for completing the catalytic cycle.5b
Conclusions
This study has demonstrated the factors behind the effectiveness of members of our Co(II)-based coordination cage family at acting as catalysts for the peroxy-monosulfate based oxidative degradation of a range of xanthene dyes. These factors are (i) association of the dye ‘guests’ with the cage, either internally (Co12 cage) or at the exterior surface (Co8 cage); (ii) the ability of the Co(II)/Co(III) couple provided by the metal ions in the cage superstructure to activate PMS by converting it to the reactive species SO4˙−; and (iii) the fact that the SO4˙− anions, which are not only generated at the cage surface but also held close to it by the high positive charge of the cage, will therefore be clustered around the bound dye molecule guests. The cage therefore performs the multiple functions of attracting substrates, generating the anionic reaction partner using a reversible cage-based redox process, and holding substrate and reaction partner in close proximity to effect the catalysis.It is worth emphasising that this aligns with conceptually related examples of the use of coordination cages in photo-redox catalysis which combine guest binding by a cage; incorporation of a photosensitising unit into the cage/guest assembly; and (as here) redox activity of components of the cage superstructure, often metal ions.20 The Co8 cage is the most effective catalyst of this cage family for the PMS-catalysed oxidative degradation reactions under investigation, due in particular to its ability to bind multiple anions around the exterior surface. These results suggest multiple new avenues of study for use of coordination cages in supramolecular catalysis.
Author contributions
J. R. W. performed all experimental work (synthesis, catalysis, crystallography). M. D. W. conceived and supervised the project. Both authors contributed to the data analysis and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been uploaded as part of the ESI.†Acknowledgements
We thank the University of Warwick for financial support, and Ms. Anjali John and Prof. Julie MacPherson for assistance with recording cyclic voltammograms.References
- (a) H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171 CrossRef CAS; (b) E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480 CrossRef PubMed; (c) Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707 RSC; (d) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed; (e) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS; (f) M. Otte, ACS Catal., 2016, 6, 6491 CrossRef CAS; (g) C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447 CrossRef CAS PubMed; (h) W.-X. Gao, H.-N. Zhang and G.-X. Jin, Coord. Chem. Rev., 2019, 386, 69 CrossRef CAS; (i) L. Zhao, X. Jing, X. Li, X. Guo, L. Zeng, C. He and C. Duan, Coord. Chem. Rev., 2019, 378, 151 CrossRef CAS; (j) M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969 CrossRef CAS; (k) T. Piskorz, V. Martí-Centelles, R. L. Spicer, F. Duarte and P. J. Lusby, Chem. Sci., 2023, 14, 11300 RSC; (l) Y. Jin, Q. Zhang, Y. Zhang and C. Duan, Chem. Soc. Rev., 2020, 49, 5561 RSC.
- (a) M. D. Ward, C. A. Hunter and N. H. Williams, Acc. Chem. Res., 2018, 51, 2073 CrossRef CAS PubMed; (b) M. D. Ward, Chem. Commun., 2024, 60, 10464 RSC; (c) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS.
- (a) W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231 CrossRef CAS PubMed; (b) C. Mozaceanu, C. G. P. Taylor, J. R. Piper, S. P. Argent and M. D. Ward, Chemistry, 2020, 2, 22 CrossRef CAS; (c) W. Cullen, A. J. Metherell, A. B. Wragg, C. G. P. Taylor, N. H. Williams and M. D. Ward, J. Am. Chem. Soc., 2018, 140, 2821 CrossRef CAS PubMed; (d) J. C. Doratt, C. G. P. Taylor, R. J. Young, A. B. Solea, D. R. Turner, G. H. Dennison, M. D. Ward and K. L. Tuck, Chem. – Eur. J., 2024, 30, e202400501 CrossRef PubMed; (e) M. D. Ludden, C. G. P. Taylor, M. B. Tipping, J. S. Train, N. H. Williams, J. C. Dorratt, K. L. Tuck and M. D. Ward, Chem. Sci., 2021, 12, 14781 RSC.
- (a) C. G. P. Taylor, A. J. Metherell, S. P. Argent, F. M. Ashour, N. H. Williams and M. D. Ward, Chem. – Eur. J., 2020, 26, 3065 CrossRef CAS PubMed; (b) A. B. Solea, B. Sudittapong, C. G. P. Taylor and M. D. Ward, Dalton Trans., 2022, 51, 11277 RSC.
- (a) X. Zhang, B. Sudittapong and M. D. Ward, Inorg. Chem. Front., 2023, 10, 1270 RSC; (b) A. B. Solea and M. D. Ward, Dalton Trans., 2023, 52, 4456 RSC.
- (a) A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121 CrossRef CAS PubMed; (b) P. Kumari and A. Kumar, Res. Surf. Interfaces, 2023, 11, 100122 CrossRef; (c) D. Ma, H. Yi, C. Lai, X. Liu, X. Huo, Z. An, L. Li, Y. Fu, B. Li, M. Zhang, L. Qin, S. Liu and L. Yang, Chemosphere, 2021, 275, 130104 CrossRef CAS PubMed; (d) Y. R. Wang and W. Chu, J. Hazard. Mater., 2011, 186, 1455 CrossRef CAS PubMed.
- (a) J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502 CrossRef CAS; (b) F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41 CrossRef CAS; (c) X. Zheng, X. Niu, D. Zhang, M. Lv, X. Ye, J. Ma, Z. Lin and M. Fu, Chem. Eng. J., 2022, 429, 132323 CrossRef CAS; (d) J. Lee, U. von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 3064 CrossRef CAS PubMed.
- (a) S. P. Argent, F. C. Jackson, H. M. Chan, S. Meyrick, C. G. P. Taylor, T. K. Ronson, J. P. Rourke and M. D. Ward, Chem. Sci., 2020, 11, 10167 RSC; (b) N. K. Al-Rasbi, I. S. Tidmarsh, S. P. Argent, H. Adams, L. P. Harding and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 11641 CrossRef CAS PubMed.
- M. D. Ludden and M. D. Ward, Dalton Trans., 2021, 50, 2782 RSC.
- (a) R. Al-Tohamy, S. S. Ali, F. Li, K. M. Okasha, Y. A.-G. Mahmoud, T. Elsamahy, H. Jiao, Y. Fu and J. Sun, Ecotoxicol. Environ. Saf., 2022, 231, 113160 CrossRef CAS PubMed; (b) L. D. Ardila-Leal, R. A. Poutou-Piñales, A. M. Pedroza-Rodríguez and B. E. Quevedo-Hidalgo, Molecules, 2021, 26, 3813 CrossRef CAS PubMed; (c) P. Verma and S. K. Samanta, Environ. Chem. Lett., 2018, 16, 969 CrossRef CAS.
- (a) A. V. Mohod, M. Momotko, N. S. Shah, M. Marchel, M. Imran, L. Kong and G. Boczkaj, Water Res. Ind., 2023, 30, 100220 CrossRef CAS; (b) P. Zawadki and M. Deska, Catalysts, 2021, 11, 974 CrossRef; (c) A. Iqbal, A. Yusaf, M. Usman, T. H. Bokhari and A. Mansha, Int. J. Environ. Anal. Chem., 2024, 104, 5503 CrossRef CAS.
- (a) J. S. Fleming, K. L. V. Mann, C.-A. Carraz, E. Psillakis, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 1998, 37, 1279 CrossRef CAS; (b) I. S. Tidmarsh, B. F. Taylor, M. J. Hardie, L. Russo, W. Clegg and M. D. Ward, New J. Chem., 2009, 33, 366 RSC.
- (a) M. Dakkach, M. I. Lopez, I. Romero, M. Rodriguez, A. Atlamsani, T. Parella, X. Fontrodona and A. Llobet, Inorg. Chem., 2010, 49, 7072 CrossRef CAS PubMed; (b) D. Sykes, S. C. Parker, I. V. Sazanovich, A. Stephenson, J. A. Weinstein and M. D. Ward, Inorg. Chem., 2013, 52, 10500 CrossRef CAS PubMed.
- (a) A. J. Metherell, W. Cullen, A. Stephenson, C. A. Hunter and M. D. Ward, Dalton Trans., 2014, 43, 71 RSC; (b) A. J. Metherell and M. D. Ward, Dalton Trans., 2016, 45, 16096 RSC.
- M. B. Tipping, J. M. Woolley, J. R. Williams and M. D. Ward, Chem. – Eur. J., 2025, 31, e202404647 CrossRef CAS PubMed.
- M. D. Ludden, C. G. P. Taylor and M. D. Ward, Chem. Sci., 2021, 12, 12640 RSC.
- Bindfit: P. Thordarson, https://supramolecular.org(accessed 20/5/25).
- (a) M. Schmalzbauer, M. Marcon and B. Konig, Angew. Chem., Int. Ed., 2021, 60, 6270 CrossRef CAS PubMed; (b) X.-F. Zhang, I. Zhang and L. Liu, Photochem. Photobiol., 2010, 86, 492 CrossRef CAS PubMed.
- F. S. T. Khan, A. L. Waldbusser, M. C. Carrasco, H. Pourhadi and S. Hematian, Dalton Trans., 2021, 50, 7433 RSC.
- (a) X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100 CrossRef CAS PubMed; (b) R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Chem. Rev., 2023, 123, 5225 CrossRef CAS PubMed; (c) S. Gaikwad, A. Bhattacharjee and E. Elacqua, Chem. – Eur. J., 2025, 31, e202404699 CrossRef CAS PubMed.
- (a) H. J. Lim, D. J. Kim, K. Rigby, W. Chen, H. Xu, X. Wu and J.-H. Kim, Environ. Sci. Technol., 2023, 57, 19054 CrossRef CAS PubMed; (b) X. Xia, F. Zhu, J. Li, H. Yang, L. Wei, Q. Li, J. Jiang, G. Zhang and Q. Zhao, Front. Chem., 2020, 8, 592056 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: All experimental details including synthesis/characterisation data for new compounds; details of spectroscopic and catalytic studies, and X-ray crystallography. CCDC 2455100–2455103. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5nr02293b |
| ‡ Note that SO4˙− does not oxidise the ligands used in the cages. Xanthene dyes are particularly prone to oxidative degradation, hence their popularity for use as substrates to monitor efficacy of advanced oxidation process.11 |
| § We note that the conditions under which the fluorescence quenching titrations are done – with a substantial molar excess of Co8 beyond the early stages of the titration – means that guest molecules will be distributed across an excess of Co8 host molecules, such that fitting the curves to a 1:1 cage:guest stoichiometry is justified, even if aggregates with different ratios are possible early in the titration when the substrate is in excess (cf. Job plots); accordingly we only quote the 1:1 binding constants to one significant figure. |
| ¶ In this new work the conditions for measuring binding constants are slightly different from what was used in ref. 9 (solvent is 2% dmso/98% water rather than pure water, to facilitate solubility of some of the cage catalysts, and there are no substituents on the exterior of this Co8 cage): but we checked that the dye/cage binding is essentially unchanged. We confirmed strong binding for the FLU/Co8 combination (K = 1 × 105 M−1 for the 1:1 binding constant, as before) based on a fluorescence titration: and a Job plot experiment again confirmed the possibility for multiple FLU dye units to aggregate around the cage surface, with a maximum at a FLU:Co8 ratio of ca. 4:1 being possible even at the low concentrations used (see ESI†). |
| This journal is © The Royal Society of Chemistry 2025 |