
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation and recharge of reactive structural Fe(II) in titanomagnetite (Fe3−xTixO4) nanoparticles†
D. V. Boglaienko a,
J. Liub,
M. P. Prangea,
O. Qafokua,
M. Sassia,
E. Arenholzc,
C. I. Pearce*a and
K. M. Rosso*a
a,
J. Liub,
M. P. Prangea,
O. Qafokua,
M. Sassia,
E. Arenholzc,
C. I. Pearce*a and
K. M. Rosso*a
aPacific Northwest National Laboratory, Richland, WA 99352, USA. E-mail: carolyn.pearce@pnnl.gov; Fax: +1509 376 3650; Tel: +1 509 371 6380
bCollege of Environmental Sciences and Engineering, Peking University, Beijing, 100871, China
cBrookhaven National Laboratory, National Synchrotron Light Source II, Upton, NY 11973, USA
First published on 16th June 2025
Abstract
Mixed-valent iron oxide minerals, such as magnetite (Fe(II)(Fe(III))2O4), are an important source of solid-state ferrous iron (Fe(II)) that can impact the speciation and transport of electron accepting contaminants in the Earth’s subsurface, such as radioactive pertechnetate (99Tc(VII)O4−). However, when oxidizing conditions are encountered, structural Fe(II) at the mineral surface is consumed yielding a maghemite (γ-Fe(III)2O3)-like layer that limits further electron transfer. This oxidized surface layer can be recharged back to the original Fe(II)/(III) ratio by re-exposure to reducing conditions, i.e., aqueous solutions containing Fe2+. However, for substituted magnetite (Fe3−xMxO4, M = transition metal cation), the extent of this redox recyclability is unclear. Here, we examine oxidation and recharge for titanomagnetite (Fe3−xTixO4) nanoparticles, where the Fe(II)/Fe(III) ratio varies by the amount of Fe(II) required to charge balance the titanium (Ti(IV)) substituted into the structure. The nanoparticles were synthesized by aqueous precipitation from a solution containing ferrous, ferric and titanium chloride at room temperature. Transmission electron microscopy combined with electron energy loss spectroscopy revealed that rapid precipitation formed core–shell-like nanoparticles consisting of a hyperstoichiometric magnetite core, with Ti(IV) and charge balancing Fe(II) enriched at the surface. This surface enrichment made Fe(II) more available for electron transfer reactions with redox active solution species. Examination of oxidation by H2O2 followed by recharge with aqueous Fe2+ indicates recyclability of reducing equivalents in the nanoparticles, yielding a core recrystallized to stoichiometric magnetite and a shell bearing excess Fe(II) to charge balance the substituted Ti(IV). The recharged particles are shown to have restored redox reactivity with 99Tc(VII)O4− resulting in reduction to 99Tc(IV)O2 and oxidation of the structural Fe(II) to Fe(III).
1. Introduction
Iron redox cycling, entailing alternation between Fe3+ and Fe2+ valence states, is a common process in natural environments such as rooting zones, soils, and near-shore environments.1 Cycling caused by episodic oxygen (O2) depletion during rainfall, soil inundation and organic matter leaching changes the biogeochemical conditions. Iron (Fe3+, Fe2+) and titanium (Ti4+) belong to major elements of soil originating from terrestrial basalts.2 They are present in a variety of Ti-substituted iron oxides that tend to be chemically resistant to weathering, particularly as their crystallinity increases with multiple redox oscillations.1,3The end member magnetite (Fe3O4) is a common iron mineral in basalts and associated soils. It has an inverse spinel structure (Fe3+)A(Fe2+Fe3+)BO4, where A and B refer to tetrahedral and octahedral sites, respectively. In sediments originating from basalt, transition metals, such as Ti4+, often substitute for Fe in the magnetite structure to form titanomagnetite (Fe3−xTixO4).4 Ti(IV) replaces Fe(III) in octahedral (B) sites and causes reduction of remaining structural Fe(III) to Fe(II) for charge balance, increasing the solid-state Fe(II)/Fe(III) ratio (>0.5) compared to pure magnetite (Fe(II)/Fe(III) = 0.5). Here, the end member is ulvöspinel (Fe3−xTixO4, x = 1), Fe2TiO4, or (Fe(II))A(Fe(II))B (Ti(IV))BO4, where x defines the amount of Ti in the solid solution. At low x, Fe(II) tends to occupy the octahedral sublattice; with increasing x the distribution of Fe(II) and Fe(III) between A and B sites changes, impacting the magnetic, electronic, and structural properties.5–8 The Fe(II)/Fe(III) ratio in titanomagnetites is affected not only by x, but also by the extent of oxidation. The oxidation of titanomagnetite is a topotactic reaction involving loss of iron from the structure to produce titanomaghemite,  , where y is the oxidation parameter that varies from 0, unoxidized, to 1, oxidized; and symbol □ represents cationic site vacancies.9,10 The interfacial reaction of titanomagnetites with redox active contaminants in solution is of interest for predicting the potential of these phases to transform the speciation of key metals or organics in soils and sediments.
, where y is the oxidation parameter that varies from 0, unoxidized, to 1, oxidized; and symbol □ represents cationic site vacancies.9,10 The interfacial reaction of titanomagnetites with redox active contaminants in solution is of interest for predicting the potential of these phases to transform the speciation of key metals or organics in soils and sediments.
The reduction of contaminants by titanomagnetite has been investigated in a limited number of previous studies. Marsac et al. (2017)11 compared the redox activity of titanomagnetite with that of Fe(II)-amended magnetite for the reduction of nitroaromatic compounds. This work confirmed that Fe(II) recharge of magnetite to an equivalent amount of total Fe(II) as in titanomagnetite resulted in higher reduction rate, but did not investigate Fe(II) recharge of titanomagnetite. Latta et al., (2013)12 studied the reaction between aqueous hexavalent uranium (U6+), as uranyl acetate, and titanomagnetite nanoparticles. They demonstrate that titanomagnetites reduced U6+ to sorbed U(IV), in contrast to magnetite that reduced U6+ to uraninite (U(IV)O2). They varied the Fe(II)/Fe(III) ratio by oxidizing the structural Fe(II) with hydrogen peroxide and showed that redox reactivity was controlled by the Fe(II)/Fe(III) ratio. Wylie et al. (2016)13 investigated the effect of titanomagnetite on hexavalent neptunium (Np6+) sorption and speciation and showed that increasing Ti concentration resulted in a greater extent of reduction to sorbed Np(IV). All of these studies report direct relationship between redox reactivity and Fe(II)/Fe(III) ratio controlled by Ti(IV) content. Here, we investigate the reaction of titanomagnetites with technetium-99 as pertechnetate (99Tc(VII)O4−(aq), hereafter referred to as TcO4−), a highly soluble radioactive contaminant present in the subsurface beneath nuclear reprocessing facilities. This reaction has been studied for its prospect of immobilizing TcO4− as the reduced and less soluble Tc(IV)O2·nH2O on the titanomagnetite surface.10 Liu et al. (2012)10 showed that the kinetics of TcO4− reduction by ∼10 nm Fe3−xTixO4 nanoparticles had a positive correlation with Ti content and corresponding solid state Fe(II)/Fe(III) ratio. The mechanism of TcO4− reduction involves the migration of structural Fe(II) to the surface of nanoparticles, where it reduces the TcO4− and is oxidized to Fe(III), a process that continues to maintain Fe(II)/Fe(III) equilibrium at the nanoparticle surface.10 A similar phenomenon has been shown for perrhenate (Re(VII)O4−) reduction by magnetite nanoparticles.14 This continued resupply of reducing equivalents from the interior to the nanoparticle surface differs from the classical understanding of magnetite reactivity established primarily through macroscopic bulk magnetite studies where oxidation through reaction with a redox active aqueous species, e.g., chromate (Cr(VI)O42−), leads to passivation of the magnetite surface.15 Scaria et al. (2025)16 investigated the effect of magnetite stoichiometry (Fe(II)/Fe(III)) on Cr(VI)O42− reduction and Cr(III) surface speciation. The study showed the formation of a spinel-type structure (Fe(II)Cr(III)xFe(III)2−xO4) on stoichiometric magnetite, while the previously characterized tridentate trinuclear surface complex17 was found only on oxidized magnetite, challenging the conventional understanding that Cr(III) primarily exists as precipitated (hydr)oxides.
In the subsurface, fluctuating water levels lead to varying oxygen penetration depths and therefore redox conditions change from oxidizing to reducing at low and high water levels, respectively. Previous work on oxidized magnetite showed that contact with aqueous Fe2+ under reducing conditions can restore stoichiometry, presumably by diffusion of adsorbed Fe2+ into the solid phase (via electron transfer and reduction of octahedral Fe(III) to Fe(II)) and recrystallization at the surface without formation of secondary Fe(II)-bearing precipitates.18,19 Byrne et al. (2015; 2016)20,21 showed that magnetite can also act as a recyclable electron donor and acceptor for phototrophic Fe(II)-oxidizing and Fe(III)-reducing bacteria, respectively, with microbial Fe(II) oxidation occurring at the mineral surface, and microbial Fe(III) reduction proceeding via electron conduction into the interior. The rechargeability of Fe(II) electron equivalents in titanomagnetite upon re-exposure to reducing conditions is an important remaining question. Therefore, the present study examines the rechargeability of titanomagnetite (Fe3−xTixO4) nanoparticles with different initial Fe(II)/Fe(III) ratios controlled by the content of Ti (x = 0.00, 0.15 and 0.38), subjected to peroxide (H2O2) oxidation followed by recharge with Fe2+-containing solutions. The effects of nanoparticle oxidation and recharge on reactivity with respect to TcO4− reduction is also investigated. Multimodal solid characterization techniques including surface sensitive Fe L-edge X-ray magnetic circular dichroism (XMCD), micro-X-ray diffraction (μ-XRD) and transmission electron microscopy (TEM), were used to elucidate the effect of Ti content on the rechargeability of the oxidized nanoparticles and their subsequent reactivity with TcO4−.
2. Materials and methods
2.1 Synthesis
Fe3−xTixO4 nanoparticles were synthesized as described in the literature,8,10 which involves instant precipitation through forced base hydrolysis according to eqn (1):
 | (1) |
The nanoparticles were synthesized in a nitrogen-filled glovebox from a stoichiometric mixture of ferrous chloride (FeCl2), ferric chloride (FeCl3) and titanium chloride (TiCl4), dissolved in 0.3 M hydrochloric acid (HCl, pH < 1), and introduced into a 25% w/v ammonium hydroxide (NH4OH) solution with continuous stirring. The nanoparticles were magnetically separated, washed, and resuspended in deionized water (DIW), where they naturally equilibrated to a solution pH of ∼8.5.
2.2. Wet chemical experiments
The conditions for the wet chemical experiments have been described in Liu et al. (2012).10| 2Fe(II) + H2O2 + 2H+ → 2Fe(III) + 2H2O. | (2) |
The suspensions were left to equilibrate in the H2O2 solution overnight with stirring at 500 rpm. For all batches, the resultant suspension was centrifuged and washed twice with DIW under ultrasonication for 5 min, followed by centrifugation at 3500 rpm for 5 min and resuspension in sufficient DIW to produce the initial suspension volume (50 mL).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The concentration of Fe2+ in the supernatant was analyzed by ferrozine assay (described in section 2.3). A solution of known Fe2+ concentration was prepared as an external standard and analyzed using the same procedure as for the nanoparticle suspensions, to detect any oxidation or adsorption of aqueous Fe2+ during the experiment. All experiments were performed in triplicate.
000 rpm. The concentration of Fe2+ in the supernatant was analyzed by ferrozine assay (described in section 2.3). A solution of known Fe2+ concentration was prepared as an external standard and analyzed using the same procedure as for the nanoparticle suspensions, to detect any oxidation or adsorption of aqueous Fe2+ during the experiment. All experiments were performed in triplicate.TcO4− reduction experiments were carried out in 30 mL glass serum bottles equipped with rubber stoppers and crimp seals. A suspension of solid to solution ratio of 1.3 mg:30 mL was prepared by adding HEPES buffer to nanoparticles. The sealed bottles were shaken for 20 hours. A 5 mM 99Tc stock solution was made by diluting NH4TcO4 (PerkinElmer Life Science Inc., Boston, MA) in DIW. After the 20-hour equilibration period, the suspension of nanoparticles was spiked with TcO4− solution targeting concentration of 10 μM, which was continuously shaken and sampled over time in triplicate. Suspension aliquots (3 mL) were filtered with a 0.1 μm syringe filter, and the 1 mL filtrate was added to 9 mL of scintillation cocktail in the glovebox to be counted utilizing a Packard 2500TR liquid scintillation counter (Packard Instrument Co. Meriden, CT) for 10 min. The HEPES buffer solution without TcO4− was used for background subtraction, and the concentration of 99Tc in solution was calculated in dpm mL−1.
Control experiments were conducted to test for potential TcO4− adsorption onto the glass bottles and the syringe filters. TcO4− solution (1 μM) was spiked into HEPES solution (50 mL) in a glass bottle and shaken for five days. The final TcO4− concentration decreased less than 3%. TcO4− solution (1 μM) was also filtered through the 0.1 μm syringe filters used in the reduction experiments. The TcO4− concentration decreased ∼3.3% after filtration. Thus, the effect of TcO4− adsorption by the glass serum bottles and the syringe filters was minimal.
2.3. Characterization
Chemical analysis was used to determine the total chemical composition of the nanoparticles (Fe(II), total Fe, and Ti content) by digesting samples in N2-sparged 5 M HCl inside the glovebox with shaking overnight. The ferrozine method was used to determine the concentration of Fe2+ in solution.23 Ferrozine reagent (1.8 mL, 1 g L−1 ferrozine in 50 mM HEPES buffer, pH 7.0) was added to the diluted acid digest solution (0.2 mL), and the absorbance at 562 nm was measured using a Shimadzu UV-2501 PC spectrophotometer. A calibration curve of the absorbance at 562 nm for solutions of known Fe2+ concentration was used to determine the Fe2+ concentration in solution. Total Fe and Ti concentrations were determined by adding 0.05 mL of the digested nanoparticles to 4.95 mL 2% HNO3 and analyzing the solutions using inductively coupled plasma – mass spectrometry (ICP-MS) with an Agilent 7500 instrument.A Jeol-JEM 2010 transmission electron microscope (TEM), equipped with a LaB6 filament and a post-column Gatan Image Filter (GIF2000) at an acceleration voltage of 200 kV and point-to-point resolution of 0.2 nm was used to determine particle size and morphology. Scanning TEM (STEM) was combined with electron energy loss spectroscopy (EELS), with energy dispersion of 0.25 eV per pixel, to map the chemistry of the nanoparticles. Images were recorded with a slow-scan charge-coupled device (CCD) camera and processed with Digital Micrograph (Gatan, USA). Samples for TEM analysis were prepared by dipping a 400 mesh copper grid coated with lacey carbon film into a diluted nanoparticle suspension and drying it inside an N2-filled glovebox. Samples were taken out of the glovebox directly before measurement.
For micro-X-ray diffraction (μ-XRD) measurements to characterize nanoparticle suspensions in situ, boron-rich capillary tubes (0.5 mm outer diameter, Charles Supper Company) were loaded with the anaerobic aqueous nanoparticle suspensions, sealed with wax (Charles Supper Company), and kept in the glovebox until immediately prior to measurement. A Rigaku D/Max Rapid II instrument with a 2D image plate detector, equipped with a MicroMax 007HF generator and a rotating chromium anode (λ = 2.2897 Å) was used for μ-XRD measurements. It was focused on a capillary tube using a 30 μm diameter collimator. The diffraction rings were integrated with 2DP, Rigaku 2D Data Processing Software (Ver. 1.0, Rigaku, 2007). JADE 8.5 from Materials Data Inc. and the PDF4+ database from ICSD were used for the analysis of diffraction data. A cubic spline function was used to fit the background, and a pseudo-Voigt profile shape function was applied to the diffraction peaks. The cubic cell parameter and crystallite particle size were determined using a cubic magnetite (Fe3O4) structure (PDF # 00-019-0629) in JADE; and the cell parameter of a Si standard (Silicon powder, NIST 640c) was measured to verify the sample-to-detector distance (deviation of the observed cell parameter from the standard was 0.002 Å).
X-ray absorption (XA) spectra at the Fe L2,3 edges were obtained at the Advanced Light Source (ALS), Berkeley, CA, on the beamline 4.0.2, employing the eight-pole resistive magnet end-station,24 and collected in the total-electron yield (TEY) mode with an effective probing depth of 50 Å.25 Aliquots of the anaerobic nanoparticle suspensions were deposited onto carbon tape attached to the copper sample holder and dried in the N2-filled glovebox. The sample holder was then sealed into an anaerobic container for shipment to ALS. The sample holder was removed from the anaerobic container immediately prior to insertion into the N2-filled end-station at beamline 4.0.2. XA spectra were collected from 609 to 740 eV for two opposite magnetization directions, reversing the applied 0.6 T field at each energy point. The X-ray magnetic circular dichroism (XMCD) spectrum was obtained as the difference between two XA spectra normalized to the incident beam intensity.26 To calculate Fe(II) and Fe(III) distribution over tetrahedral and octahedral sites, the XMCD spectra were fit using a nonlinear least-squares analysis based on ligand field multiplet models,27,28 which simulate the effects of crystal field on the atomic energy levels and take into account the symmetry of the local environment and charge transfer effects. In these calculations, the 10Dq crystal field parameters were taken as 1.2 and 0.6 eV for Fe cations in octahedral and tetrahedral sites. The results were convoluted using a Gaussian of σ = 0.2 eV to account for instrumental broadening and using a Lorentzian of Γ = 0.3 (0.5) eV for the L3 (L2) edge to account for intrinsic core-hole lifetime broadening. The Ti L2,3 XAS data (not sensitive to the magnetic field) were collected on the same beamline 4.0.2 at the ALS. For the ligand field multiplet model fitting, the estimated random error per cation site occupancy value for the XMCD-derived Fe(II)/Fe(III) ratio is ±0.02.29
Fe K-edge and Ti K-edge XANES and EXAFS measurements were made at 20-BM at the Advanced Photon Source (APS), Argonne, IL. A Si (1 1 1) monochromator was used with a 1 mm entrance slit located approximately 50 m from the source. The monochromator allowed an energy resolution of about 1 eV at the Fe edge and 0.7 eV at the Ti edge. Samples of nanoparticle suspension were transferred in the glovebox into Teflon holders with Kapton windows and flash frozen in liquid N2 for shipment. At the beamline, nanoparticle samples were measured at 17 K to avoid oxidation. Data were taken in transmission mode with a Fe foil used for online energy calibration. The XANES data were analyzed using the Athena interface to the IFEFFIT program package.30
2.4. Theoretical modeling of Fe K-edge and Ti K-edge EXAFS
Local structures for Ti atoms were obtained by replacing one or two Fe atoms in the magnetite structure31 with Ti in density functional theory structural relaxations using a PBE+U theory in which the PBE exchange correlation approximation32 is augmented with an on-site Coulomb repulsion of U − J = 4 eV for the Fe d orbitals following Dudarev et al. (1998).33 No Hubbard corrections were used for Ti. The VASP code34 and associated projector-augmented wave datasets35 were used to compute the Kohn–Sham orbitals on a 4 × 4 × 4 grid of k-points in a basis of plane waves defined by an energy cutoff of 400 eV. Although magnetite is ferrimagnetic, spin-polarized simulations were performed in a ferromagnetic configuration with a constrained total moment to prevent the collapse of the high-spin configuration of the Fe(II) sites in the structurally disordered model during the relaxations. Simulations of XA spectra were made using the FEFF code36 to evaluate the path expansion in clusters generated from the relaxed structures. Overlapped atomic potentials, Rmax = 4.36 Å, and otherwise default settings were used. A uniform thermal disorder parameter σ2 = 0.01 Å2 was applied to all paths.3. Results and discussion
3.1 Characterization of as-synthesized titanomagnetite nanoparticles
The as-synthesized titanomagnetite nanoparticles used in this study were synthesized and characterized as described in Pearce et al. (2012)8 and Liu et al. (2012).10 The nanoparticles were characterized by chemical analysis of digested nanoparticles (ferrozine method for UV-vis spectroscopy for Fe2+ and ICP-MS for total Fe and Ti), TEM (particle size), μ-XRD (cell parameter and Fe(II)/Fe(III) ratio), XAS and XMCD (Fe(II)/Fe(III) ratio) and XANES and EXAFS (local coordination environment). The characteristics of the as-synthesized titanomagnetite nanoparticles are summarized in Table 1, as originally reported in Pearce et al. (2012).8 The nanoparticles had near spherical shape with an equivalent diameter of 9.4–10.6 nm (±2 nm) (Fig. 1A). | ||
| Fig. 1 TEM of x = 0.15 (A) before and (B) after oxidation with H2O2; (C) x = 0.15 as-synthesized, euhedral with straight edges, with a diameter of ∼9.5 nm; (D) x = 0.15 oxidized, euhedral with straight edges and a diameter of ∼8.8 nm; (E) x = 0.15 recharged, rounded, rough edges, with a diameter of ∼9.6 nm. | ||
| x | Size (nm) | Cell parameter (Å) | Fe(II) (mmol g−1) | Fe(III) (mmol g−1) | Ti(IV) (mmol g−1) | Composition (experimental) | Composition (theoretical) | Fe(II)/Fe(III) μ-XRD ± 0.05 | Fe(II)/Fe(III) XMCD ± 0.02a |
|---|---|---|---|---|---|---|---|---|---|
| a Estimated according to Coker et al. (2009).29 | |||||||||
| 0.00 | 11.8 | 8.406 | 4.42 | 7.89 | 0.00 | Fe(II)1.08Fe(III)1.92O4 | Fe(II)1.00Fe(III)2.00O4 | 0.556 | 0.604 |
| 0.15 | 9.4 | 8.415 | 4.44 | 6.33 | 0.55 | Fe(II)1.17Fe(III)1.68Ti0.15O4Fe(II)1.27Fe(III)1.35Ti0.38O4 | Fe(II)1.15Fe(III)1.70Ti0.15O4Fe(II)1.38Fe(III)1.24Ti0.38O4 | 0.656 | 0.667 |
| 0.38 | 10.6 | 8.429 | 4.76 | 5.02 | 1.38 | 0.829 | 0.878 | ||
μ-XRD patterns revealed peaks belonging to a single titanomagnetite phase (space group Fd3m). Increasing Ti content (x) caused increasing structural substitution of Ti(IV) for octahedral Fe(III) and corresponding reduction of Fe(III) to a larger Fe(II) cation for the charge balance, which led to the increase of the cubic unit cell edge parameter, as shown in Table 1.7,37,38
Fe L-edge XAS in TEY mode and XMCD analyses provide surface sensitive information, down to 50 Å, and the signal strength decreases exponentially with depth.25 The XMCD signal is sensitive to the oxidation state and local structure of magnetically ordered iron cations at solid surfaces. It is derived from the difference of two XA spectra obtained with circularly polarized light in alternating opposing static applied magnetic fields. Fe(II)/Fe(III) ratios at nanoparticle surfaces were estimated by fitting the Fe L-edge XMCD spectra with weighted sums of the theoretically calculated spectra for Fe octahedral and tetrahedral sites.25,28,39–41 The methods based on multireference configuration interaction (MRCI) calculations and ligand-field multiplet models42 were also used to analyze the XMCD data, and the results were consistent between the models regarding the trends of the Fe(II)/Fe(III) ratios. Data summarized in Table 1 show that the surface Fe(II)/Fe(III) ratio is similar to the bulk Fe(II)/Fe(III) ratio, obtained from the relationship between Fe(II)/Fe(III) ratio and cell parameters from XRD, for the as-synthesized nanoparticles.
3.2 Oxidation of titanomagnetite nanoparticles
The amount of H2O2 required to oxidize the Fe3−xTixO4 nanoparticle suspensions to different extents (25%, 50%, and added in excess for 100%) was calculated based on the extent of Ti substitution (x), i.e. 0.063 mL, 0.162 mL, and 1.62 mL for x = 0.00; 0.0725 mL, 0.145 mL, and 1.45 mL for x = 0.15; 0.085 mL, 0.17 mL, and 1.7 mL for x = 0.38. The pH of the suspensions was ∼8.5 and, under these slightly alkaline conditions, H2O2 underwent disproportionation to produce oxygen gas, as evidenced by the formation of bubbles in the suspensions, according to eqn (3):| 2H2O2 → 2H2O + O2 | (3) |
The nanoparticle suspensions changed color from black to brown, indicating oxidation of Fe(II) to Fe(III) by both H2O2 and O2. After equilibration in the presence of H2O2 overnight, the nanoparticles were washed to remove any excess H2O2 and resuspended in DIW. Qualitative examination of the particles using TEM revealed that oxidation did not significantly change the particle shape or surface structure (Fig. 1B), as also observed in studies by Jungcharoen et al. (2021)18 and Demangeat et al. (2018).43Fig. 1A and B show representative particles from the x = 0.15 sample, before and after oxidation, respectively. The euhedral particles were similar in size (∼9 nm in equivalent diameter), and exhibited atomically flat, common cubic faces.
While the amounts of Fe(II) (mmol g−1) measured by acid digestion and ferrozine assay were similar to the Fe(II) (mmol g−1) calculated from XRD data for the as-synthesized samples, the amounts measured by digestion for the H2O2 oxidized samples were substantially lower than the amounts calculated from the XRD data, and the amounts measured by digestion for the Fe2+ reduced samples were substantially higher than the amount calculated from the XRD data, for most of the samples (Table 2). Possibly, the results were impacted by some residual H2O2 and Fe2+, despite replacing the solution with water, that is why the Fe(II)/Fe(III) ratio determined by chemical digestion was not included in Table 2.
| Sample | Fe(II) (mmol g−1) Chem. | Fe(II) (mmol g−1) XRD | Fe(II)/Fe(III) XRD ± 0.05 | Fe(II)/Fe(III) XMCD ± 0.02 | Particle Size (nm) | Ti/Fe Chem. | Ti/Fe XAS |
|---|---|---|---|---|---|---|---|
| x = 0.00 | |||||||
| Before oxidation | 4.42 | 4.64 | 0.556 | 0.604 | 10.2 | 0.0 | 0.0 |
| Oxidation with 0.063 mL of H2O2 | 2.42 | 3.11 | 0.320 | 0.447 | 9.6 | 0.0 | 0.0 |
| Recharge with 0.05 mmol g−1 Fe2+ | 7.52 | 4.37 | 0.509 | 0.630 | 10.6 | 0.0 | 0.0 |
| Oxidation with 0.162 mL of H2O2 | 2.24 | 3.25 | 0.339 | 0.493 | 9.3 | 0.0 | 0.0 |
| Oxidation with 1.62 mL of H2O2 | 0.92 | 2.76 | 0.274 | 0.382 | 9.2 | 0.0 | 0.0 |
| Recharge with 2.5 mmol g−1 Fe2+ | 5.35 | 4.25 | 0.489 | 0.523 | 10.6 | 0.0 | 0.0 |
| x = 0.15 | |||||||
| Before oxidation | 4.44 | 4.90 | 0.656 | 0.667 | 9.5 | 0.045 | 0.049 |
| Oxidation with 0.0725 mL of H2O2 | 1.71 | 3.25 | 0.364 | 0.552 | 9.0 | 0.047 | 0.055 |
| Recharge with 0.05 mmol g−1 Fe2+ | 5.20 | 4.34 | 0.526 | 0.638 | 9.9 | 0.033 | 0.043 |
| Oxidation with 0.145 mL of H2O2 | 1.11 | 2.35 | 0.241 | 0.425 | 8.9 | 0.047 | 0.035 |
| Recharge with 2.5 mmol g−1 Fe2+ | 4.34 | 4.18 | 0.516 | 0.625 | 10.4 | 0.032 | 0.031 |
| Oxidation with 1.45 mL of H2O2 | 1.31 | 2.54 | 0.266 | 0.411 | 8.6 | 0.046 | 0.050 |
| x = 0.38 | |||||||
| Before oxidation | 4.76 | 5.14 | 0.829 | 0.878 | 10.7 | 0.133 | 0.120 |
| Oxidation with 0.085 mL of H2O2 | 2.10 | 2.98 | 0.368 | 0.690 | 10.6 | 0.120 | 0.104 |
| Recharge with 0.05 mmol g−1 Fe2+ | 5.01 | 4.02 | 0.561 | 0.800 | 12.0 | 0.084 | 0.117 |
| Oxidation with 0.17 mL of H2O2 | 0.73 | 2.03 | 0.228 | 0.526 | 10.6 | 0.118 | 0.085 |
| Recharge with 2.5 mmol g−1 Fe2+ | 3.66 | 4.02 | 0.561 | 0.735 | 11.6 | 0.086 | 0.090 |
| Oxidation with 1.7 mL of H2O2 | 0.51 | 2.16 | 0.246 | 0.500 | 10.0 | 0.130 | 0.072 |
The nanoparticles were also measured under anaerobic conditions and in aqueous suspension using capillary μ-XRD before and after exposure to H2O2 to determine the effect of oxidation on the crystal structure of the nanoparticles. The background subtracted diffraction patterns for x = 0.15 nanoparticles before and after reaction with different concentrations of H2O2 (Fig. S1†) show that no new phases were produced, and the only peaks present corresponded to titanomagnetite. A shift of peak position towards higher two-theta values after exposure to increasing concentrations of H2O2 was observed with all values of x, demonstrating a decrease of unit cell parameter. These observations are consistent with the previously published studies on this material.8,10
Due to the systematic relationship between unit-cell parameter and Fe(II)/Fe(III) ratio in these phases, defined as the “Master Curve” in Pearce et al. (2012),8 the change in Fe(II)/Fe(III) ratio in the Fe3−xTixO4 nanoparticles because of oxidation with H2O2 could be determined. The unit cell parameter was obtained using whole pattern fitting and Rietveld refinement which also provided crystallite particle size. The unit cell parameters, derived Fe(II)/Fe(III) ratios and particle size for the Fe3−xTixO4 (x = 0.00, 0.15 and 0.38) nanoparticles, before (samples with 0 mL of H2O2) and after oxidation (variable volumes of H2O2 depending on the extent of Ti substitution) are given in Table 2. Oxidation at each value of x led to slight decrease in crystallite particle size due to: (i) contraction of the lattice due to the presence of larger amounts of the smaller Fe(III) cation; and (ii) removal of Fe from the structure through dissolution (Table 2). Fig. 2A shows the change in XRD-derived Fe(II)/Fe(III) ratio, as a function of x, with different concentrations of H2O2. The initial Fe(II)/Fe(III) ratio increased with x (expected due to charge balance for Ti(IV) in the lattice) and resulted in a greater potential for oxidation, as shown by the increasing impact of the different H2O2 concentrations on the extent of oxidation in the Fe3−xTixO4 nanoparticles. There was, however, a limit to the extent of oxidation under these conditions, as shown by the relatively small change in Fe(II)/Fe(III) ratio for the most oxidized samples, despite exposure to 10 times higher concentration of H2O2. Jungcharoen et al. (2022)44 investigated magnetite oxidation under different pH and redox conditions and also found that 100% oxidation was not achievable. This suggests that the extent of oxidation is controlled by the solid-state characteristics of the particles and not by the concentration of H2O2 in solution. The physical limit on the extent of oxidation of titanomagnetites (x > 0.00), representing the Fe(II) that must remain in the Fe3−xTixO4 structure to balance the Ti(IV), is shown as the dashed line in Fig. 2.9 Only the most oxidized x = 0.38 sample approaches this physical limit (Fig. 2A) suggesting higher availability of structural Fe(II). Worm and Banerjee (1984)9 experimentally demonstrated migration of Fe(II) out of the lattice with oxidation of titanomagnetite; here, higher substitution of Ti(IV) into magnetite, x, resulted in higher mobilization of the structural Fe(II).
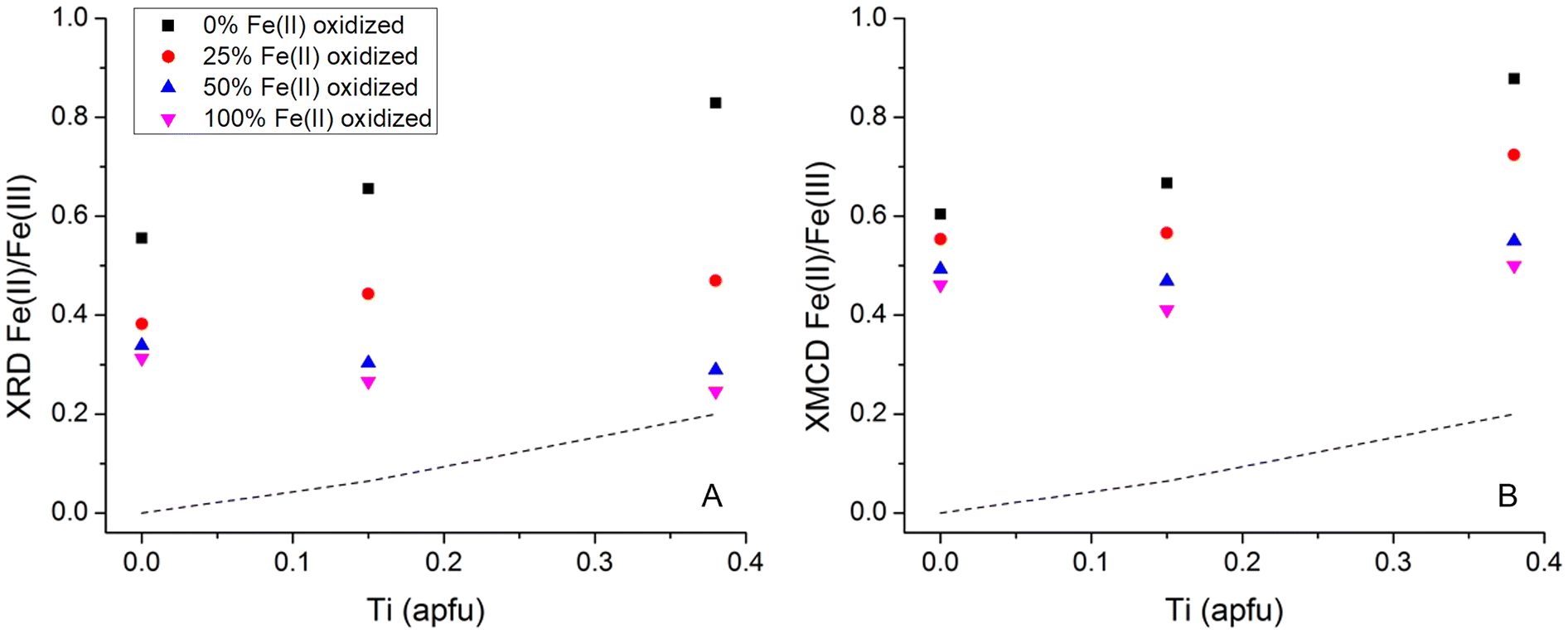 | ||
| Fig. 2 Change in (A) XRD-derived Fe(II)/Fe(III) ratio and (B) XMCD-derived Fe(II)/Fe(III) ratio, as a function of x, with a theoretical 0%, 25%, 50%, and 100% of the Fe(II) oxidized using different concentrations of H2O2 (exact volumes of H2O2 used are provided in Table 2). The dashed line represents the Fe2+ in the Fe3−xTixO4 structure to charge balance Ti(IV) (Worm and Banerjee, 1984).9 | ||
The Fe3−xTixO4 (x = 0.00, 0.15 and 0.38) nanoparticles before and after reaction with H2O2 were also analyzed using XA (total Fe and Ti) and XMCD (magnetic component of the sample) at the Fe L2,3-edge and the Ti L2,3-edge. The XA and XMCD spectra for x = 0.15 nanoparticles before and after reaction with different concentrations of H2O2 are shown in Fig. 3A and B respectively. The L2,3-edge for Fe shifts to higher energy if oxidation state increases.45 In Fig. 3A, the overall peak intensity has been normalized to one and the lower energy Fe L2,3-edge peak at 707.8 eV decreases with exposure to increasing H2O2 concentrations, due to oxidation. Measured XMCD spectra for the nanoparticles have three main peaks: octahedral Fe(II) (negative peak at ∼708 eV), tetrahedral Fe(III) (positive peak at ∼709.5 eV), and octahedral Fe(III) (negative peak at ∼710.5 eV). According to calculated spectra for these three main components in magnetite, the experimental XMCD spectrum can be fit to produce occupancy ratios for the proportions of Fe(II) and Fe(III) in the octahedral site, and of Fe(III) in the tetrahedral site. The Fe(II)/Fe(III) ratios for titanomagnetite nanoparticles before and after oxidation, calculated from the XMCD spectra, are given in Table 2. The Fe(II)/Fe(III) ratio at the surface decreased after reaction with different concentrations of H2O2 for all values of x, indicating that magnetically ordered surface Fe(II) was oxidized by H2O2. This can be seen in Fig. 3B by a decrease in the peak corresponding to Fe(II) in the octahedral sublattice. Fig. 2B shows the change in XMCD-derived Fe(II)/Fe(III) ratio at the surface of the nanoparticles, as a function of x, with different concentrations of H2O2. The trend in XMCD-derived Fe(II)/Fe(III) ratios is similar to that for the XRD-derived Fe(II)/Fe(III) ratios (Fig. 2A), with extent of oxidation increasing as a function of x. As with the XRD-derived Fe(II)/Fe(III) ratios, there is little difference between the two most oxidized samples, therefore the extent of oxidation at the nanoparticle surface is also limited by the characteristics of the particles and not by the concentration of H2O2 in solution.
 | ||
| Fig. 3 XA (A) and XMCD (B) spectra for x = 0.15 nanoparticles before and after reaction with different concentrations of H2O2. (C) XMCD spectra for the x = 0.15 sample before oxidation, after oxidation with 0.0725 H2O2 and after resupply with 1 mmol FeCl2. | ||
The major difference between oxidation within the nanoparticle structure (XRD, Fig. 2A) and oxidation at the nanoparticle surface (XMCD, Fig. 2B) is that the Fe(II)/Fe(III) ratios at the surface are higher for all values of x. This is additional evidence that oxidation involves the solid-state migration of Fe(II) from the interior to the particle surface, as described in Pearce et al. (2012).8 The loss of the larger Fe(II) cation from the structure decreases the size of the unit cell of Fe3−xTixO4 nanoparticles, as measured by XRD, and localization of the Fe(II) to the surface is reflected by the higher XMCD Fe(II)/Fe(III) ratio.
The fact that the surface remains enriched in Fe(II) relative to the interior after oxidation is interesting and counterintuitive. Surface enrichment of Fe(II), even after reaction of Fe3−xTixO4 nanoparticles with an electron acceptor, i.e., the redox active aqueous contaminant pertechnetate, TcO4−, was also observed by Liu et al. (2012).10 We hypothesize that Ti(IV) may be enriched at the surface of the nanoparticles because of the fast precipitation kinetics during synthesis. Charge balance requires that the surface will also be enriched in Fe(II), despite there being sufficient oxidant in solutions to completely oxidize the nanoparticles. This goes some way to explaining the consistently higher XMCD-derived surface Fe(II)/Fe(III) ratio (vs. XRD-derived bulk Fe(II)/Fe(III) ratio) under different conditions. However, as will be shown below, the surfaces of unsubstituted Fe3O4 nanoparticles are also enriched in Fe(II) relative to the interior upon synthesis.
To test the hypothesis that the titanomagnetite nanoparticle surfaces are enriched in both Ti(IV) and Fe(II), STEM, in combination with EELS mapping, was used to examine the spatial distribution of Ti(IV), Fe(II) and Fe(III) within individual nanoparticles (Fig. 4). STEM-EELS mapping of the x = 0.15 sample after synthesis showed that a core–shell compositional structure is present, in which the shell tends to be relatively enriched in Ti relative to the core. The shell can be described as ∼1 to 4 nm thick, consisting of higher Ti(IV) concentrations and corresponding enrichment in Fe(II). Fig. 4B shows that oxygen K-edge peaks in the EELS collected at the edge of the particle (red spectrum) are shifted to higher energy. The inset in Fig. 4B shows that the peak in the EELS corresponding to the Fe L-edge shifts to lower energy, indicating that the iron is more reduced in the shells of the nanoparticles.
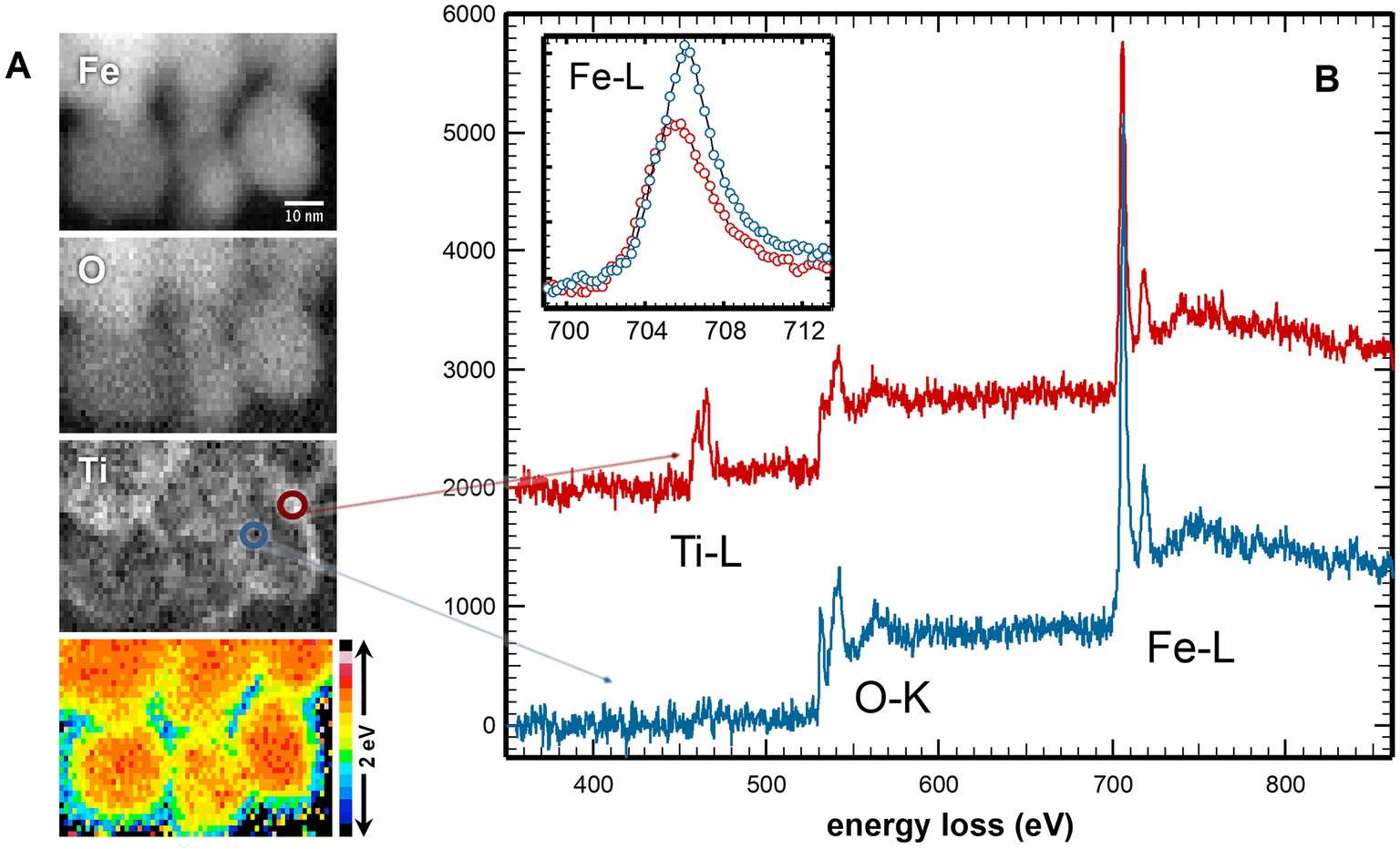 | ||
| Fig. 4 STEM-EELS of as-synthesized titanomagnetite nanoparticles (x = 0.15): (A) STEM images of nanoparticles (scale bar is 10 nm); (B) EELS spectra collected in the core (blue) and on the edge (red) of a nanoparticle with inset of the enlarged Fe L-region. | ||
Bowles et al. (2019)46 reported a clustered distribution of Ti in titanomagnetite nanoparticles, similarly synthesized, and studied by Mössbauer and XMCD. To investigate the local environment of Ti atoms, Ti and Fe K-edge XANES (Fig. S2†) and EXAFS data were collected for the as-synthesized x = 0.00 and x = 0.15 nanoparticles, and compared to calculated EXAFS (Fig. S3 and Table S1†). Two titanomagnetite models were considered: (i) a magnetite cell with one Ti atom substituting for octahedral Fe(III); and (ii) a magnetite cell with two Ti atoms at octahedral Fe(III) sites. The first model predicts only Ti–Fe paths in the second shell, and the second model has a Ti–Ti path in addition to Ti–Fe paths in the second shell. The first shell is composed of 6 O atoms in both models. The Ti K-edge EXAFS data (x = 0.15 sample) were noisy, and fitting did not yield reliable parameters to compare with the model that included the Ti–Ti path (fit parameters to the magnetite structure are summarized in Table S1†).
The Fe K-edge data were of better quality, but it was not possible to discern the presence of Fe–Ti paths from the fit to the EXAFS. The reason for this is revealed by a comparison between the calculated Fe K-edge EXAFS for magnetite, with the Fe–Fe path averaged for the tetrahedral and octahedral sites, and the calculated Ti K-edge EXAFS for titanomagnetite, with Ti in two octahedral Fe sites. The χ(k) functions are very similar, irrespective of whether the absorber is Fe of Ti (Fig. S4†), making it difficult to discern the presence of Fe–Ti paths in the magnetite structure.
Fe K-edge EXAFS data were then compared with FEFF simulated Fe K-edge EXAFS for a magnetite cell with one or two substituting octahedral Ti atoms. Each of the two simulated spectra was an average of 20 simulations from different Fe atoms (12 in tetrahedral sites and 8 in octahedral sites) within 3.5 Å of a Ti atom. The spectrum simulated for the magnetite structure with two Ti atoms in octahedral Fe3+ sites more closely resembles the data, as seen from the EXAFS plot and more evident from the Fourier transform imaginary part plot, suggesting clustered Ti distribution in the titanomagnetite nanoparticles (Fig. 5). This is a preliminary result, however, as the ensemble of neighboring Fe atoms was chosen randomly and does not fully represent the Fe local environment of the titanomagnetite cell.
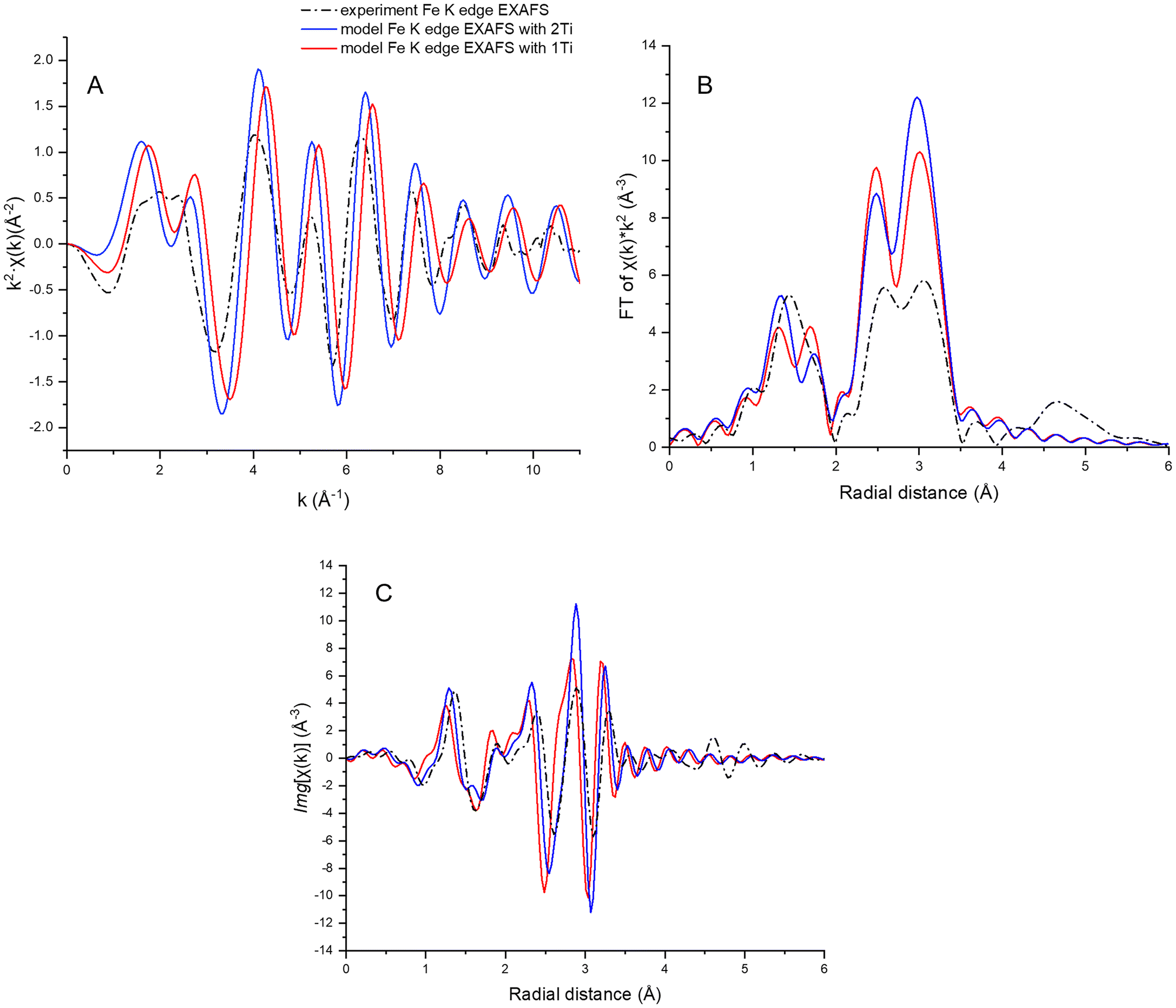 | ||
| Fig. 5 Fe K-edge (A) EXAFS data, (B) Fourier transform magnitude, and (C) imaginary part for as-synthesized titanomagnetite (x = 0.15) nanoparticles (dotted black line); calculated Fe K-edge EXAFS for magnetite cell with one substituting octahedral Ti atom (red) and with two substituting octahedral Ti atoms (blue). | ||
The combination of STEM-EELS results with Ti and Fe K-edge XANES and EXAFS data, shows that the Ti-containing nanoparticles have a core–shell-like structure consisting of a hyperstoichiometric magnetite core, with Ti(IV) and charge balancing Fe(II) enriched at the surface, and that isolation of the Ti to the surface results in a clustered distribution of Ti atoms in the shell of the Fe3−xTixO4 (x > 0.00) nanoparticles. The unit cell parameter of titanomagnetite is larger than that of stoichiometric magnetite,8 and this may induce strain gradients in the confined Ti(IV)/Fe(II)-enriched shell. Strain in Fe/Fe-oxide core–shell nanoparticles has been shown to enhance ionic transport and increase the rate of diffusion-limited oxidation reactions,47 therefore, strain could play a role in the redox reactivity of these Fe3−xTixO4 (x > 0.00) nanoparticles.
3.3 Recharge of oxidized titanomagnetite nanoparticles with Fe2+
To investigate the potential for recharge after oxidation, the oxidized Fe3−xTixO4 (x = 0.00, 0.15 and 0.38) nanoparticle suspensions were exposed to FeCl2 solution (4 mmol of Fe2+ per gram of nanoparticles). Fig. S5† shows the uptake of Fe2+ from the solution by x = 0.00, 0.15 and 0.38 nanoparticles with two different extents of oxidation. Fe2+ uptake from the solution was almost instantaneous, with 50–80% of uptake occurring within the first 15 min of exposure to the Fe2+ solution (Fig. S5†).For a particular value of x, the extent of Fe(II) uptake from solution was related to the XMCD-derived surface Fe(II)/Fe(III) ratio. For example, the x = 0.15 nanoparticles after oxidation with 0.145 mL of H2O2, with a starting surface Fe(II)/Fe(III) ratio of 0.425, could uptake 3.47 mmol g−1 Fe2+ from solution (as determined by ferrozine assay), whereas the x = 0.15 nanoparticles after oxidation with 0.0725 mL of H2O2, with a starting surface Fe(II)/Fe(III) ratio of 0.552 could only uptake 2.54 mmol g−1. However, the extent of Fe2+ uptake from solution by oxidized Fe3−xTixO4 nanoparticles with different values of x was not well correlated with the XMCD-derived surface structural Fe(II)/Fe(III), as suggested by the poor linear fit to the data in Fig. 6A (R2 = 0.44). Over the range of three x values, the extent of Fe2+ uptake from the solution was instead better correlated with the XRD-derived whole particle structural Fe(II)/Fe(III) ratio of the oxidized material (Fig. S5†), as supported by a linear fit with an R2 value of 0.95 (Fig. 6B).
 | ||
| Fig. 6 Linear regression plots for Fe2+ uptake (mmol g−1) from the solution (as determined by ferrozine assay) for Fe3−xTixO4 nanoparticles (x = 0.00, 0.15 and 0.38), plotted as one dataset in each figure, vs. (A) XMCD-derived surface Fe(II)/Fe(III) ratios after oxidation, (B) XRD-derived whole particle Fe(II)/Fe(III) ratios after oxidation, and (C) XRD-derived whole particle Fe(II) (mmol g−1) after oxidation; (D) Fe(II) recharge (mmol g−1) estimated from the XRD-derived Fe(II) in the recharged nanoparticles minus the Fe(II) in the oxidized nanoparticles. | ||
The linear fit to the overall trend for Fe2+ uptake vs. the amount of Fe(II) in the solid (derived from the XRD Fe(II)/Fe(III) ratio) was not as good (R2 = 0.71), as shown in Fig. 6C. Thus, it is the Fe(II)/Fe(III) ratio in the whole particle that controls the amount of Fe2+ uptake, and not the actual Fe(II) amount in the bulk solid. This is supported by Gorski and Scherer (2009)19 who showed that Fe2+ uptake depends on magnetite stoichiometry, not initial Fe(II) content, as Fe(II) is not stable and transfers electrons to Fe(III). A thermodynamic model for the magnetite-maghemite solid solution was developed by Jungcharoen et al. (2022)44 to predict the behavior and transformation of magnetite nanoparticles in aqueous solutions, and the results in Fig. 6C suggest that this model could be extended to tianomagnetite-titanomaghemite solid solution. However, modelling the behavior of the Fe3−xTixO4 nanoparticles studied here has some additional complications in that the crystallite size increases after Fe(II) recharge (Table 2), and the Ti is not evenly distributed throughout the nanoparticles (Fig. 4). Therefore, Fe2+ uptake does not correlate as well with the XMCD derived surficial Fe(II)/Fe(III), as was shown in Jungcharoen et al. (2022),44 but instead correlates with the XRD-derived whole particle Fe(II)/Fe(III). Given the trend in Fig. 6C, the stoichiometry of the Fe3−xTixO4 nanoparticles exposed to pH 8 conditions, partially oxidized by H2O2 and subsequently recharged by Fe2+, could potentially be predicted by an equilibrium model, if the XRD-derived Fe(II)/Fe(III) is used.
The amount of structural Fe(II) recharge in the Fe3−xTixO4 nanoparticles (x = 0.00, x = 0.15 and x = 0.38) was calculated by using the Fe(II) concentrations derived from the XRD Fe(II)/Fe(III) ratio and subtracting the amount of Fe(II) in the oxidized nanoparticles from the amount of Fe(II) in the recharged nanoparticles. Fig. 6D shows that the amount of Fe2+ uptake from solution can be related to the amount of Fe(II) recharge in the solid by a linear fit with an R2 value of 0.90. However, the amount of Fe2+ uptake from the solution is approximately twice the amount of Fe(II) recharge in the solid. The crystallite size, as determined by XRD, increased by 1–2 nm after Fe(II) recharge, suggesting that the extra Fe2+ uptake from the solution could be explained by growth of the nanoparticles (Table 2). The Ti/Fe ratio at the surface of the Fe3−xTixO4 nanoparticles (x > 1) was calculated from the integrated area of the XA spectra for the Fe and Ti L2,3-edges taken at the same point on the sample, after background subtraction as described in Droubay (1999).48 In general, there was a small decrease in Ti/Fe ratio after Fe(II) recharge, indicating that the surface was slightly enriched in Fe. However, the Ti/Fe ratio did not change significantly, suggesting that the composition of the surface was not drastically altered by Fe(II) recharge. This was also confirmed by TEM of a representative particle from the x = 0.15 sample after Fe(II) recharge (Fig. 1C). The particle in Fig. 1C has a similar morphology to those in Fig. 1A and B, with continuous lattice fringes to the edge of the particle, suggesting that it is structurally homogeneous with no evidence of a different surface phase.
To obtain further information on the Fe(II)/Fe(III) ratio and the Fe site occupancy at the surface of the recharged Fe3−xTixO4 nanoparticle suspensions, the samples were analyzed by XMCD. Fig. 3C shows the XMCD spectra for the x = 0.15 sample before oxidation, after oxidation with H2O2 and after resupply with FeCl2, normalized to the intensity of the octahedral Fe(III) peak. A significant amount of Fe(II) was lost from the B-site during oxidation and Fig. 3C shows that this site was refilled because of Fe2+ resupply. Despite the presence of a higher Fe2+ concentration in the solution than would be required to refill the B-site, it is not restored to its original site occupancy in any of the samples.
Fig. 7 shows the different steps in the treatment of the Fe3−xTixO4 nanoparticle suspensions, characterized by their Fe(II)/Fe(III) ratio in the whole particle from XRD and at the surface from XMCD, for the as-synthesized nanoparticles (step 1), nanoparticles oxidized with lower concentrations of H2O2 (step 2), nanoparticles oxidized with higher concentrations of H2O2 (step 3), oxidized nanoparticles recharged with lower concentrations of Fe2+ (step 4), and oxidized nanoparticles recharged with higher concentrations of Fe2+ (step 5). For all nanoparticle samples, oxidation decreased the XRD-derived whole particle Fe(II)/Fe(III) ratio to a greater extent than the XMCD-derived surface Fe(II)/Fe(III) ratio, producing a discrepancy between the two. For as-synthesized x = 0.00 nanoparticles, the Fe(II)/Fe(III) ratio should be close to that for stoichiometric Fe3O4 (0.5). However, Fig. 7 (x = 0.00) shows that as-synthesized x = 0.00 nanoparticles (step 1) had excess Fe(II) in the whole particle (XRD Fe(II)/Fe(III) = 0.556) and at the surface (XMCD Fe(II)/Fe(III) = 0.604), possibly due to rapid precipitation kinetics during synthesis. After oxidation then resupply with high concentrations of Fe2+ (step 5), both the XRD and XMCD derived Fe(II)/Fe(III) became close to 0.5 (step 5). For as-synthesized Fe3−xTixO4 nanoparticles (x = 0.15 and x = 0.38), both XRD Fe(II)/Fe(III) and XMCD Fe(II)/Fe(III) increased with x, as replacement of Fe(III) by Ti(IV) in the magnetite structure is accompanied by reduction of structural Fe(III) to Fe(II) for change balance, yielding a proportional increase in Fe(II)/Fe(III).
 | ||
| Fig. 7 XMCD (red) and XRD (black) Fe(II)/Fe(III) ratios for different steps of Fe3−xTixO4 nanoparticle suspensions treatment, starting from synthesized material (step 1) and proceeding through oxidation with lower concentrations of H2O2 (step 2), oxidation with higher concentrations of H2O2 (step 3), recharge with 0.05 mmol Fe2+ (step 4) to recharge with 2.5 mmol Fe2+ (step 5). | ||
For the x = 0.15 and x = 0.38 nanoparticles, the surface remained enriched with Fe(II) after the oxidation-recharge cycle (XMCD Fe(II)/Fe(III) of ∼0.6 and ∼0.7, respectively, in step 5). However, the whole particle Fe(II) was never refilled to the same extent as at the surface (XRD Fe(II)/Fe(III) of 0.5 for both x = 0.15 and x = 0.38 in step 5), and the discrepancy increases with x. Redistribution of cations in the lattice during magnetite oxidation is known,49 and similar observations were reported for synthesized 10 nm magnetite nanoparticles which were recrystallized into stoichiometric magnetite after an oxidation-recharge cycle.18 Results presented in Fig. 7 show that, while Fe(II) enrichment at the surface is preserved for the titanomagnetite nanoparticles after the oxidation and recharge cycle, the final whole particle XRD-derived Fe(II)/Fe(III) ratio approaches that for stoichiometric magnetite (0.5) for both magnetite and titanomagnetite nanoparticles.
3.4 Reaction of oxidized and recharged titanomagnetite nanoparticles with TcO4−
Reduction experiments were conducted using Fe3−xTixO4 nanoparticles (x = 0.15) to evaluate the effect of the oxidation-recharge cycle on reduction of Tc(VII)O4−. Dependence of TcO4− reduction on the amount of Ti (x = 0.00, 0.15, 0.25, 0.38, and 0.53) in as-synthesized Fe3−xTixO4 nanoparticles was investigated previously at initial TcO4− concentrations of 10 and 30 μM,10 which showed higher TcO4− reduction rates with larger x. After 4 hours, x = 0.00 nanoparticles only reduced 30% of the TcO4−,10 in solutions with the same starting concentration as used here (10 μM). Reduction of TcO4− by as-synthesized x = 0.15 nanoparticles increased to ∼70%, showing that doping magnetite nanoparticles with Ti(IV) increases their redox reactivity. However, a study of the reduction of U(VI), as uranyl acetate, by x = 0.5 nanoparticles with different extents of oxidation, revealed the controlling effect of Fe(II)/Fe(III) ratio over variable Ti(IV) content.12 A higher Fe(II)/Fe(III) ratio enhances the accessibility of reducing equivalents localized on octahedral Fe(II)–Fe(III) pairs.19,50 Here, TcO4− removal from the solution was the highest with the as-synthesized hyperstoichiometric x = 0.15 nanoparticles (Fig. 7B, step 1), but was negligible with oxidized nanoparticles, and was partially restored after recharge, depending on the amount of Fe2+ that was resupplied (0.5 mmol vs. 1 mmol of FeCl2) (Fig. 8). The highest redox reactivity can be attributed to the excess of Fe(II), in the whole particle and at the surface, that was present in the x = 0.15 nanoparticles because of rapid precipitation during synthesis. The partially restored redox reactivity of the x = 0.15 nanoparticles after recharge can be explained by the lower, near stoichiometric structural Fe(II)/Fe(III) ratio of 0.5 (Fig. 7, step 5). Hence, while the redox reactivity of x = 0.15 nanoparticles is affected by the additional Fe(II) required to charge balance Ti(IV) substitution, it is mainly controlled by the electron transfer through structural Fe(II)–Fe(III) pairs, enhanced by the excess Fe(II) in the rapidly precipitated hyperstoichiometric x = 0.15 starting material. Even though surface Fe(II)/Fe(III) ratio was restored during recharge, the whole particle Fe(II)/Fe(III) ratio was not restored (Fig. 7). The lower reduction capacity of the x = 0.15 nanoparticles after recharge, compared to that for starting material, demonstrates that reduction of TcO4− requires the resupply of Fe(II) from the nanoparticle interior, to replenish the oxidized Fe(II) at the surface. The whole particle Fe(II)/Fe(III) ratio is only recharged to 0.5, i.e., that for x = 0.00, resulting in a smaller pool of reactive Fe(II), and a reduction capacity that is closer to the x = 0.00 nanoparticles in Liu et al. (2012)10 than to the x = 0.15 starting material (Fig. 8). | ||
| Fig. 8 TcO4− reduction kinetics in x = 0.15 nanoparticles: as-synthesized material, oxidized material, and oxidized material recharged with 0.5 and 1 mmol FeCl2. | ||
In addition, the Fe3−xTixO4 (x > 0.00) nanoparticles have a core–shell-like structure consisting of a hyperstoichiometric magnetite core, with Ti(IV) and charge balancing Fe(II) enriched at the surface, and may exhibit strain-enhanced ionic conduction, resulting in increased reactivity.47 During oxidation and recharge, diffusion of Fe(II) cations and vacancies can give rise to structural reorganization and a lower degree of structural disorder, as shown by a smaller spin canting of partially oxidized magnetite compared to the pristine stoichiometric magnetite.18 This structural reorganization could relieve some of the strain in the Fe3−xTixO4 (x > 0.00) nanoparticles and explain why the reduction capacity of the x = 0.15 nanoparticles after recharge is lower compared to that for starting material.
4. Conclusions
The series of synthetic Fe3−xTixO4 nanoparticles with different amounts of substituted Ti(IV) (x = 0.00, 0.15, 0.38) were investigated to determine the potential for Fe(II) recharge and restoration of reduction capacity after oxidation with H2O2. We found that rapid precipitation during Fe3−xTixO4 nanoparticle synthesis led to enrichment of substituted Ti(IV) at the surface. The Fe(II)/Fe(III) ratio was determined to be: (i) dependent on x (amount of Ti(IV) requiring Fe(II) for charge balance); (ii) higher at the surface of nanoparticles, determined by surface sensitive XMCD analysis vs. whole particle XRD analysis; (iii) higher at the surface of titanomagnetite nanoparticles after oxidation, compared to magnetite nanoparticles after oxidation; (iv) representative of nearly stoichiometric magnetite for the whole titanomagnetite nanoparticle while remaining higher at the surface after nanoparticle recharge. Ti(IV) substitution increased the amount of Fe(II) available for TcO4− reduction, and the reduction capacity was controlled by electron transfer from Fe(II)–Fe(III) pairs in the interior to the surface of the Fe3−xTixO4 nanoparticles.This study shows that titanium substitution increases the solid-state Fe(II)/Fe(III) ratio in titanomagnetite, making them promising low-cost, high-efficiency reductants with ‘tunable’ reactivity for environmental remediation applications. In addition, natural titanomagnetites exhibit a range of Ti contents, and their structural Fe(II)/Fe(III) ratio can be modified by changing redox conditions resulting from fluctuating water levels. The results presented here show that these materials also display reversibility with respect to reincorporation of Fe(II) into the structure upon exposure to reducing microenvironments creating, for example, by microbial activity. This new understanding of the effects of changing titanomagnetite stoichiometry on redox reactivity is of relevance to remediation of redox active contaminants, e.g., technetium, uranium, and chromium, present in the subsurface at legacy nuclear sites associated with past nuclear weapons production, testing, and energy research.
Conflicts of interest
There are no conflicts of interest to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This material is based upon work supported by the U.S. Department of Energy (DOE) Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division through its Geosciences program at Pacific Northwest National Laboratory (PNNL) (FWP 56674). The authors gratefully acknowledge Bernd Kabius and Robert Colby for TEM/EELS mapping data. A portion of the work was carried out in the Environmental and Molecular Sciences Laboratory (EMSL), a national scientific user facility at PNNL sponsored by the DOE Office of Biological and Environmental Research. XA and XMCD measurements were performed at the Advance Light Source supported by the DOE Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-05CH11231. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. PNNL is a multiprogram national laboratory operated by Battelle Memorial Institute under contract no. DE-AC05-76RL01830 for the DOE.References
- A. Thompson, O. A. Chadwick, D. G. Rancourt and J. Chorover, Iron-oxide crystallinity increases during soil redox oscillations, Geochim. Cosmochim. Acta, 2006, 70(7), 1710–1727 CrossRef CAS.
- J. J. Papike, J. M. Karner and C. K. Shearer, Comparative planetary mineralogy: Valence state partitioning of Cr, Fe, Ti, and V among crystallographic sites in olivine, pyroxene, and spinel from planetary basalts, Am. Mineral., 2005, 90(2–3), 277–290 CrossRef CAS.
- Z. Wang, J. Ma, J. Li, T. Zeng, Z. Zhang, X. He, L. Zhang and G. Wei, Effect of Fe–Ti oxides on Mo isotopic variations in lateritic weathering profiles of basalt, Geochim. Cosmochim. Acta, 2020, 286, 380–403 CrossRef CAS.
- O. Orlický, F. Caňo, J. Lipka, A. Mihaliková and B. Toman, Fe-Ti magnetic minerals of basaltic rocks: a study of their nature and composition, Geol. Carpathica, 1992, 43(5), 287–293 Search PubMed.
- W. O'reilly and S. K. Banerjee, Cation distribution in titanomagnetites (1-x)Fe3O4 - xFe2TiO4, Physics Letters, 1965, 17(3), 237–238 CrossRef.
- S. K. Banerjee, W. O'reilly, T. C. Gibb and N. N. Greenwood, The behaviour of ferrous ions in iron-titanium spinels, J. Phys. Chem. Solids, 1967, 28(7), 1323–1335 CrossRef CAS.
- C. I. Pearce, C. M. Henderson, N. D. Telling, R. A. Pattrick, J. M. Charnock, V. S. Coker, E. Arenholz, F. Tuna and G. van der Laan, Fe site occupancy in magnetite-ulvospinel solid solutions: A new approach using X-ray magnetic circular dichroism, Am. Mineral., 2010, 95(4), 425–439 CrossRef CAS.
- C. I. Pearce, O. Qafoku, J. Liu, E. Arenholz, S. M. Heald, R. K. Kukkadapu, C. A. Gorski, C. M. Henderson and K. M. Rosso, Synthesis and properties of titanomagnetite (Fe3− xTixO4) nanoparticles: A tunable solid-state Fe(II/III) redox system, J. Colloid Interface Sci., 2012, 387(1), 24–38 CrossRef CAS PubMed.
- H. U. Worm and S. K. Banerjee, Aqueous low–temperature oxidation of titanomagnetite, Geophys. Res. Lett., 1984, 11(3), 169–172 CrossRef CAS.
- J. Liu, C. I. Pearce, O. Qafoku, E. Arenholz, S. M. Heald and K. M. Rosso, Tc(VII) reduction kinetics by titanomagnetite (Fe3− xTixO4) nanoparticles, Geochim. Cosmochim. Acta, 2012, 92, 67–81 CrossRef CAS.
- R. Marsac, M. Pasturel and K. Hanna, Reduction kinetics of nitroaromatic compounds by titanium-substituted magnetite, J. Phys. Chem. C, 2017, 121(21), 11399–11406 CrossRef CAS.
- D. E. Latta, C. I. Pearce, K. M. Rosso, K. M. Kemner and M. I. Boyanov, Reaction of UVI with titanium-substituted magnetite: Influence of Ti on UIV speciation, Environ. Sci. Technol., 2013, 47(9), 4121–4130 CrossRef CAS PubMed.
- E. M. Wylie, D. T. Olive and B. A. Powell, Effects of titanium doping in titanomagnetite on neptunium sorption and speciation, Environ. Sci. Technol., 2016, 50(4), 1853–1858 CrossRef CAS PubMed.
- R. Ding, C. Guida, C. I. Pearce, E. Arenholz, J. M. Grenèche, A. Gloter, A. C. Scheinost, K. O. Kvashnina, K. Wang, A. Fernandez-Martinez, Y. Mu, K. M. Rosso and L. Charlet, Single rhenium atoms on nanomagnetite: Probing the recharge process that controls the fate of rhenium in the environment, Sci. Adv., 2025, 11(20), eadq3650 CrossRef CAS PubMed.
- M. L. Peterson, A. F. White, G. E. Brown and G. A. Parks, Surface passivation of magnetite by reaction with aqueous Cr(VI): XAFS and TEM results, Environ. Sci. Technol., 1997, 31(5), 1573–1576 CrossRef CAS.
- J. Scaria, M. Pédrot, L. Fablet, T. Yomogida, T. T. Nguyen, Y. Sivry, C. Catrouillet, A. E. Pradas Del Real, F. Choueikani, D. Vantelon, A. Dia, A. Groleau and R. Marsac, Magnetite Stoichiometry (Fe(II)/Fe(III)) Controls on Trivalent Chromium Surface Speciation, Environ. Sci. Technol., 2025, 59(11), 5747–5755 CrossRef CAS PubMed.
- M. L. Peterson, G. E. Brown Jr and G. A. Parks, Direct XAFS evidence for heterogeneous redox reaction at the aqueous chromium/magnetite interface, Colloids Surf., A, 1996, 107, 77–88 CrossRef CAS.
- P. Jungcharoen, M. Pédrot, F. Choueikani, M. Pasturel, K. Hanna, F. Heberling, M. Tesfa and R. Marsac, Probing the effects of redox conditions and dissolved Fe 2 + on nanomagnetite stoichiometry by wet chemistry, XRD, XAS and XMCD, Environ. Sci.: Nano, 2021, 8(7), 2098–2107 RSC.
- C. A. Gorski and M. M. Scherer, Influence of magnetite stoichiometry on FeII uptake and nitrobenzene reduction, Environ. Sci. Technol., 2009, 43(10), 3675–3680 CrossRef CAS PubMed.
- J. M. Byrne, N. Klueglein, C. Pearce, K. M. Rosso, E. Appel and A. Kappler, Redox cycling of Fe(II) and Fe(III) in magnetite by Fe-metabolizing bacteria, Science, 2015, 347(6229), 1473–1476 CrossRef CAS PubMed.
- J. M. Byrne, G. Van Der Laan, A. I. Figueroa, O. Qafoku, C. Wang, C. I. Pearce, M. Jackson, J. Feinberg, K. M. Rosso and A. Kappler, Size dependent microbial oxidation and reduction of magnetite nano-and micro-particles, Sci. Rep., 2016, 6(1), 30969 CrossRef CAS PubMed.
- K. Schwochau, Technetium: chemistry and radiopharmaceutical applications, John Wiley & Sons, 2008 Nv 21 Search PubMed.
- L. L. Stookey, Ferrozine–a new spectrophotometric reagent for iron, Anal. Chem., 1970, 42(7), 779–781 CrossRef CAS.
- E. Arenholz and S. O. Prestemon, Design and performance of an eight-pole resistive magnet for soft X-ray magnetic dichroism measurements, Rev. Sci. Instrum., 2005, 76(8), 083908 CrossRef.
- S. Gota, M. Gautier-Soyer and M. Sacchi, Fe 2 p absorption in magnetic oxides: Quantifying angular-dependent saturation effects, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62(7), 4187 CrossRef CAS.
- R. A. Pattrick, G. Van Der Laan, C. M. Henderson, P. Kuiper, E. Dudzik and D. J. Vaughan, Cation site occupancy in spinel ferrites studied by X-ray magnetic circular dichroism: developing a method for mineralogists, Eur. J. Mineral., 2002, 14(6), 1095–1102 CrossRef CAS.
- G. Van der Laan and I. W. Kirkman, The 2p absorption spectra of 3d transition metal compounds in tetrahedral and octahedral symmetry, J. Phys.: Condens. Matter, 1992, 4(16), 4189 CrossRef CAS.
- G. Van der Laan and B. T. Thole, Strong magnetic X-ray dichroism in 2p absorption spectra of 3d transition-metal ions, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43(16), 13401 CrossRef CAS PubMed.
- V. S. Coker, N. D. Telling, G. Van Der Laan, R. A. Pattrick, C. I. Pearce, E. Arenholz, F. Tuna, R. E. Winpenny and J. R. Lloyd, Harnessing the extracellular bacterial production of nanoscale cobalt ferrite with exploitable magnetic properties, ACS Nano, 2009, 3(7), 1922–1928 CrossRef CAS PubMed.
- B. Ravel and M. A. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, Synchrotron Radiat., 2005, 12(4), 537–541 CrossRef CAS PubMed.
- C. Haavik, S. Stølen, H. Fjellvag, M. Hanfland and D. Hausermann, Equation of state of magnetite and its high-pressure modification: Thermodynamics of the Fe-O system at high pressure, Am. Mineral., 2000, 85(3–4), 514–523 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+ U study, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57(3), 1505 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953 CrossRef PubMed.
- J. J. Kas, F. D. Vila, C. D. Pemmaraju, T. S. Tan and J. J. Rehr, Advanced calculations of X-ray spectroscopies with FEFF10 and Corvus, Synchrotron Radiat., 2021, 28(6), 1801–1810 CrossRef CAS PubMed.
- B. A. Wechsler, D. H. Lindsley and C. T. Prewitt, Crystal structure and cation distribution in titanomagnetites (Fe3−xTixO4), Am. Mineral., 1984, 69(7–8), 754–770 CAS.
- F. Bosi, U. Hålenius and H. Skogby, Crystal chemistry of the magnetite-ulvospinel series, Am. Mineral., 2009, 94(1), 181–189 CrossRef CAS.
- G. Van der Laan, J. Zaanen, G. A. Sawatzky, R. Karnatak and J. M. Esteva, Comparison of X-ray absorption with X-ray photoemission of nickel dihalides and NiO, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33(6), 4253 CrossRef CAS PubMed.
- C. T. Chen, Y. U. Idzerda, H. J. Lin, N. V. Smith, G. Meigs, E. Chaban, G. H. Ho, E. Pellegrin and F. Sette, Experimental confirmation of the X-ray magnetic circular dichroism sum rules for iron and cobalt, Phys. Rev. Lett., 1995, 75(1), 152 CrossRef CAS PubMed.
- J. Stöhr, X-ray magnetic circular dichroism spectroscopy of transition metal thin films, J. Electron Spectrosc. Relat. Phenom., 1995, 75, 253–272 CrossRef.
- M. Sassi, C. I. Pearce, P. S. Bagus, E. Arenholz and K. M. Rosso, First-principles Fe L2, 3-edge and O K-edge XANES and XMCD spectra for iron oxides, J. Phys. Chem. A, 2017, 121(40), 7613–7618 CrossRef CAS PubMed.
- E. Demangeat, M. Pédrot, A. Dia, M. Bouhnik-le-Coz, F. Grasset, K. Hanna, M. Kamagate and F. Cabello-Hurtado, Colloidal and chemical stabilities of iron oxide nanoparticles in aqueous solutions: the interplay of structural, chemical and environmental drivers, Environ. Sci.:Nano, 2018, 5(4), 992–1001 RSC.
- P. Jungcharoen, M. Pédrot, F. Heberling, K. Hanna, F. Choueikani, C. Catrouillet, A. Dia and R. Marsac, Prediction of nanomagnetite stoichiometry (Fe(II)/Fe(III)) under contrasting pH and redox conditions, Environ. Sci.: Nano, 2022, 9(7), 2363–2371 RSC.
- P. A. Van Aken, B. Liebscher and V. J. Styrsa, Quantitative determination of iron oxidation states in minerals using Fe L 2, 3-edge electron energy-loss near-edge structure spectroscopy, Phys. Chem. Miner., 1998, 25, 323–327 CrossRef CAS.
- J. A. Bowles, S. C. Lappe, M. J. Jackson, E. Arenholz and G. van der Laan, Curie temperature enhancement and cation ordering in titanomagnetites: evidence from magnetic properties, XMCD, and Mössbauer spectroscopy, Geochem., Geophys., Geosyst., 2019, 20(5), 2272–2289 CrossRef.
- A. Pratt, L. Lari, O. Hovorka, A. Shah, C. Woffinden, S. P. Tear, C. Binns and R. Kröger, Enhanced oxidation of nanoparticles through strain-mediated ionic transport, Nat. Mater., 2014, 13(1), 26–30 CrossRef CAS PubMed.
- T. C. Droubay, Soft X-ray spectroscopy and microspectroscopy of magnetic and naturally occurring materials, The University of Wisconsin-Milwaukee, 1999 Search PubMed.
- R. Bliem, E. McDermott, P. Ferstl, M. Setvin, O. Gamba, J. Pavelec, M. A. Schneider, M. Schmid, U. Diebold, P. Blaha and L. Hammer, Subsurface cation vacancy stabilization of the magnetite (001) surface, Science, 2014, 346(6214), 1215–1218 CrossRef CAS PubMed.
- C. A. Gorski, J. T. Nurmi, P. G. Tratnyek, T. B. Hofstetter and M. M. Scherer, Redox behavior of magnetite: Implications for contaminant reduction, Environ. Sci. Technol., 2010, 44(1), 55–60 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00989h |
| This journal is © The Royal Society of Chemistry 2025 |