
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Verifying the efficient functional N species of metal-free N-doped carbons for CO2-to-CO electrochemical conversion using zeolite-templated carbons with N species tuned by a recarbonization treatment†
Kotaro
Narimatsu
a,
Ryuji
Takada
*a,
Koji
Miyake
 *ab,
Yoshiaki
Uchida
a and
Norikazu
Nishiyama
ab
*ab,
Yoshiaki
Uchida
a and
Norikazu
Nishiyama
ab
aDivision of Chemical Engineering, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: kojimiyake@cheng.es.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita 565-0871, Japan
First published on 11th June 2025
Abstract
The electrochemical reduction reaction of carbon dioxide is expected as a potential solution to reducing CO2 emission and providing value-added chemicals. Carbon monoxide (CO) is a fundamental chemical precursor for industrially important reactions. Therefore, metal-free heteroatom-doped carbon materials have attracted increasing attention as electrocatalysts for CO2-to-CO conversion. Nitrogen (N)-doped carbon is a promising alternative catalyst. The introduction of N atoms modifies the electronic and chemical structures and leads to enhanced electrocatalytic performance. While the bonding states of N species influence the CO2RR activity has been discussed, no reports have demonstrated the relationship between the bonding state and activity by dividing graphitic N into two configurations (valley N and center N). In this study, we have controlled the local chemical state of N species of N-doped carbon materials through the combination of a zeolite templating method and a recarbonization treatment to investigate the effective functional N species for CO2-to-CO conversion. When the recarbonization temperature increased, the content of valley N increased, and the CO2-to-CO conversion was enhanced. The highest faradaic efficiency of CO was 76%. DFT calculations revealed that valley N and the edge site C atom adjacent to valley N stabilized *COOH intermediate, contributing to the enhanced CO2-to-CO conversion.
Introduction
The electrochemical carbon dioxide reduction reaction (CO2RR) operated using renewable energy has been regarded as an attractive solution to reducing the CO2 accumulation in the atmosphere and providing value-added chemicals.1–4 Among the CO2RR products, carbon monoxide (CO) is a fundamental chemical precursor for industrially important reactions, such as methanol synthesis and Fischer Tropsch synthesis.5–8 To date, great efforts have been made to develop metal-based electrocatalysts, such as Au,9 Ag,10 and Zn,11 for CO2 electroreduction into CO. However, the high cost or low durability of these metal-based catalysts prevent their widespread industrial implementation.12,13 The development of alternative catalysts for CO2-to-CO conversion is a highly desirable goal.In recent years, nitrogen (N)-doped carbon materials for CO2RR, such as single-atom catalysts14,15 and metal-free catalysts (e.g., nanodiamonds16 and carbon quantum dots17,18) have attracted attention as promising alternative catalysts. In particular, metal-free nitrogen (N)-doped carbon materials for CO2-to-CO conversion (e.g., N-doped graphene19,20 and carbon nanotubes21,22) have emerged as promising candidates for replacing metal-based catalysts. The introduction of N atoms modifies the electronic and chemical structure, leading to an enhancement of the electrocatalytic performance for the CO2RR.23,24 These materials have advantages: high stability, high conductivity, large specific surface areas and low cost.25–27 The N species incorporated in the carbon matrix are divided into four main configurations, depending on their structural and electronic properties: graphitic N, pyridinic N, pyrrolic N and oxidized N.28,29 Graphitic N is substituted for the C atom in the carbon matrix by bonding it to three C atoms, which should be differentiated into two configurations. One located in the edge site is called valley N and the other located on the graphene basal plane is called center N.30,31 However, no reports have suggested how the two configurations of graphitic N affect the catalytic activity for CO2RR. In addition, it is difficult to control the doping content because of the necessity of high-temperature heat treatment during the synthesis of carbon materials. Therefore, we focused on the zeolite templating method as an effective synthesis method to form abundant pores and defects for the control of the N-doping content.32 In prior research, we applied a zeolite templating method using pyridine (C and N precursors) for the synthesis of pyridinic N-rich carbon materials.33,34 Furthermore, to increase the valley N content, we also focused on recarbonization treatment. Generally, it is known that pyridinic N changes into valley N or center N, resulting in more stable N configurations owing to high-temperature heat treatment. Therefore, it should be possible to synthesize N-doped carbon materials with different content of pyridinic N, valley N and graphitic N by using both a zeolite templating method and a recarbonization treatment at the optimum temperature.
In this study, we controlled the local chemical state of N species in N-doped carbon materials through a combination of a zeolite templating method with pyridine and a recarbonization treatment. This is the first study to demonstrate that metal-free N-doped carbon materials prepared by zeolite templating can electrochemically catalyze CO2 into CO by controlling the proportion of N species. In addition, experimental analysis and density functional theory (DFT) calculation provided new insights into the effect of valley N on CO2RR activity.
Experimental
Materials
NaOH (4 M), HCl (5 M), pyridine and Nafion (5 wt%) were obtained from Wako Pure Chemical Industries. Protonated FAU-type zeolite (HY) with a Si/Al = 3.6 was obtained from Tosoh.Preparation of N-doped carbons (NDCs)
The overall synthetic strategy is based on our previous studies.33,34 HY (0.3 g) was annealed under N2 flow at 800 °C for 1 h. The chemical vapor deposition treatment of a pyridine vapor at the saturated vapor pressure at 0 °C was performed for 5 h. The obtained zeolite-carbon composite was named “HY/NDC”. HY/NDC was treated at 180 °C in NaOH (4 M) aqueous solution overnight. Then, the suspension was filtered and the collected solid was washed by deionized water. Then, the resultant powder was rinsed in HCl (5 M) aqueous solution. Moreover, until the solution became neutral, the solid powder was repeatedly rinsed by deionized water. Finally, the solid powder was dried at 90 °C overnight and was carbonized under N2 flow at T °C (T = 900, 1000, and 1100) for 3 h. The carbonization treatment is called recarbonization in this study. After the recarbonization, the samples are denoted as “NDC-T”.Characterizations
Scanning electron microscope-energy dispersive X-ray spectrometry (SEM-EDX) was performed using a JCM-7000 (JEOL, Japan) and Transmission electron microscope (TEM) was carried out using an H800 (Hitachi, Japan). X-ray diffraction (XRD) was conducted on a PANalytical X'Pert-MDR diffractometer using Cu Kα radiation. N2 adsorption isotherms at 77 K were recorded using a BELSORP MINI X (Microtrac MRB, Japan). Consequently, pore volumes, pore size distributions, and the specific surface areas were determined based on the BJH (Barrett–Joyner–Halenda), MP-Plot and Brunauer Emmett Teller (BET) methods. Analysis for C, H and N contents was conducted using FlashEA (Thermo Fisher Scientific, USA). X-ray photoelectron spectroscopic (XPS) was conducted using a Kratos Ultra 2 (Shimadzu, Japan).Electrochemical measurements
Isopropanol, Nafion (5 wt% in alcohol and water) and water were mixed in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:8. A catalyst (5 mg) was suspended in the solution (500 μL), and the suspension was ultrasonicated to obtain the catalyst ink. The ink (100 μL) was pipetted on a carbon paper. The mass loading of a catalyst on the carbon paper was fixed at 1 mg cm−2. A Biologic SP-50e potentiostat (TOYO, Japan) was used as an electrochemical analyzer. An as-prepared working electrode, Ag/AgCl (reference electrode), and a platinum wire (counter electrode) were equipped at a H-type cell. The potentials in this study are relative to the reversible hydrogen electrode (RHE), based on the equations below:
:1:8. A catalyst (5 mg) was suspended in the solution (500 μL), and the suspension was ultrasonicated to obtain the catalyst ink. The ink (100 μL) was pipetted on a carbon paper. The mass loading of a catalyst on the carbon paper was fixed at 1 mg cm−2. A Biologic SP-50e potentiostat (TOYO, Japan) was used as an electrochemical analyzer. An as-prepared working electrode, Ag/AgCl (reference electrode), and a platinum wire (counter electrode) were equipped at a H-type cell. The potentials in this study are relative to the reversible hydrogen electrode (RHE), based on the equations below:| ERHE = EAg/Agcl + 0.208 + 0.0591 × pH |
Linear sweep voltametric (LSV) measurements with a scan rate of 5 mV s−1 were performed in CO2-saturated and N2-saturated 0.1 M KHCO3 solution, and the potential range was from 0.15 V to −1.2 V vs. RHE. During the LSV measurements, CO2 and N2 keep flowing. CO2 splitting experiments were carried out in a CO2-saturated 0.1 M KHCO3 solution with stirring at 350 rpm. Before CO2 splitting experiments, the 0.1 M KHCO3 solution was pre-saturated with CO2 for at least 30 minutes. The gas-phase products were quantified by offline gas chromatography (GC) (GC-8A, Shimadzu, Japan) using a Shincarbon-ST column (SHINWA, Japan). The products in a liquid phase were detected using an 1H NMR (ECS, JEOL, Japan). The below equations were applied to calculate the faradaic efficiencies of the gas products:
 485.33 C mol−1
485.33 C mol−1
Density functional theory (DFT) calculations
B3YLYP hybrid DFT of Gaussian 16W35 was conducted with a basis set of 6-31G (d, p) to obtain the stabilized model, the atomic population, and the various thermodynamic energy. Several possible heteroatoms doped carbon configurations, containing pyridinic N, center N, and valley N, were used as calculated models in this study. The ground state structures of *COOH and *CO adsorbed on heteroatoms doped carbon model were identified by finding the lowest energy one among several possible configurations on N atoms and C atoms adjacent to N atoms as possible active sites.The below equation was applied for calculating adsorption energy:
| Eads = Ecomplex − Esubstrate − Eadsorbate |
According to computational hydrogen electrode (CHE) model, the Gibbs free energy was calculated by the following equation:
| ΔG = ΔEads + ΔEZPE − TΔS |
In this work, the reaction pathway of CO2 electroreduction into CO was based on following elementary step:
| *+ CO2 (g) + H+ + e− → *COOH |
| *COOH + H+ + e− → *CO + H2O (l) |
| *CO → *+ CO (g) |
As the first intermediate of CO2RR, not only *COOH but also *CO2- and *OCHO are considered, however, as regards CO production, *COOH is considered as the main first intermediate of CO2-to-CO conversion.3,36,37 So, *COOH was chosen as the first intermediate of CO2RR in this work.
Results & discussion
NDCs were prepared by the zeolite templating method using Y zeolite as a template and pyridine as C and N precursors. Y zeolite was heated under N2 atmosphere at 800 °C, and the CVD treatment of the pyridine vapor was conducted for 5 h. The obtained zeolite-carbon composite was denoted as HY/NDC. First, XRD measurement was performed to investigate the crystal structures of HY/NDC. HY/NDC exhibited a peak similar to those of a pristine protonated FAU-type zeolite (HY) as shown in Fig. S1,† suggesting that the crystal structure of zeolite template was not decomposed during the CVD process. The EDX analysis indicated that there is hardly any detection of Si and Al in NDC-T, suggesting that base and acid treatments removed zeolite contained in HY/NDC (Table S1†). In addition, the morphologies and pores of synthesized NDC-T were investigated by TEM and SEM-EDX. As shown in Fig. 1a and Fig. S2,† the morphology of each sample was similar to that of the zeolite template. Moreover, the pore structures of NDC-T were observed by TEM (Fig. 1b and Fig. S3†), it was revealed that the structure of NDC-1100 was sparser than that of HY/NDC, as shown in Fig. S3.†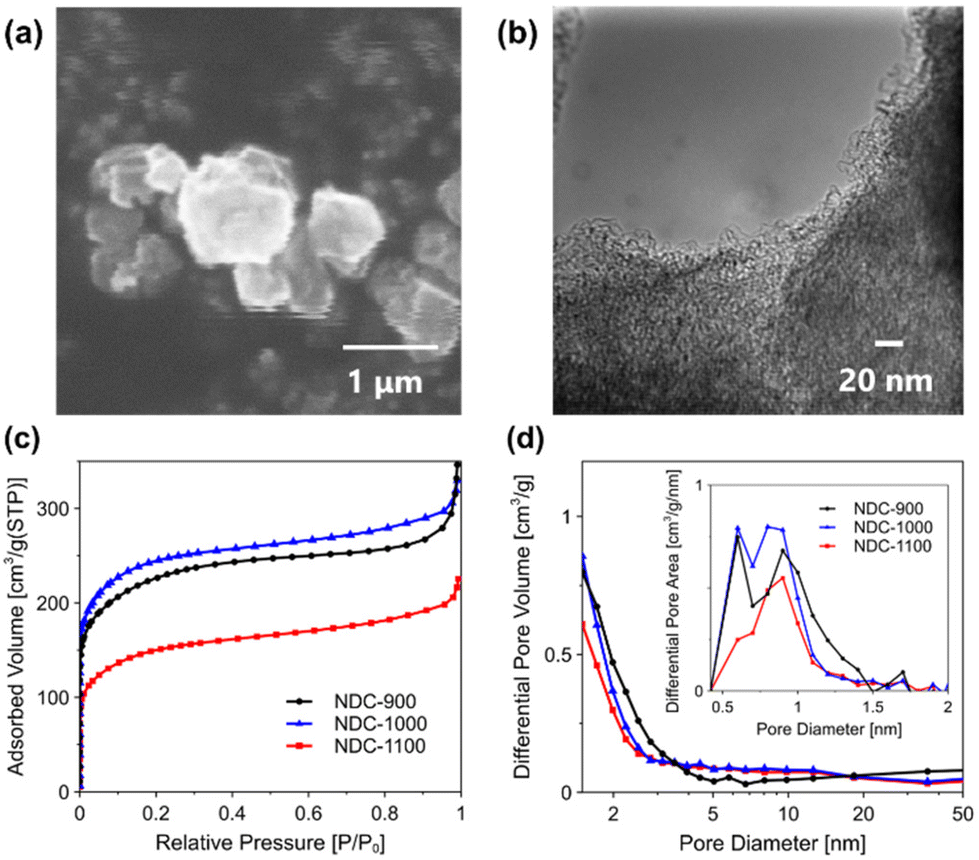 | ||
| Fig. 1 (a) SEM and (b) TEM images of NDC-1100. (c) N2 adsorption isotherms and (d) pore size distributions of NDC-T. | ||
As shown in Fig. 1c, the result of N2 adsorption isotherms indicated that the adsorption volume in the low relative pressure region (lower than 0.01), which is assigned to the adsorption of micropores, declined on NDC-1100 compared to NDC-1000. The high-temperature recarbonization treatment may cause a decline in the microporosity. The graphene microscale defects on a graphene lattice are aligned during high-temperature recarbonization treatment, leading to shrinking of the micropores. Moreover, Fig. 1d shows the pore size distributions based on MP-plot and BJH methods. Micropores (∼2 nm) were identified in NDC-T. The most abundant micropore family was centered at 0.9 nm. It is known that the space originally occupied by the zeolite framework provides a microporous structure with a uniform micropore size (theoretically approximately 1.2 nm).38 The mode pore diameters were close to the theoretical pore sizes. These results confirmed that pores derived from the zeolite template were formed in NDC-T. Moreover, Table S2† shows the BET areas, and each pore volume based on MP-plot and BJH methods. There was no correlation between the pore volume and recarbonization temperature. Fig. S4† shows the XRD patterns of NDC-T. All samples exhibited two broad peaks at 23.8° and 43.4° assigned to the (002) and (100) planes of carbon, which originated from the crystalline nature of carbon.39,40
The amount of nitrogen in NDC-T determined by The CHN elemental analysis (Table 1) was approximately 2–3 wt%. The results showed that sufficient nitrogen was present in the carbon materials after recarbonization at T °C. However, similar nitrogen contents were obtained for any of the samples.
| Sample | C [wt%] | H [wt%] | N [wt%] |
|---|---|---|---|
| NDC-900 | 67.10 | 1.97 | 2.75 |
| NDC-1000 | 72.63 | 1.18 | 2.64 |
| NDC-1100 | 76.84 | 1.55 | 2.77 |
The bonding states of the NDC-T surfaces were investigated using XPS measurements. As displayed in Fig. 2a, it was carried out regarding the XPS on N 1s spectra that a deconvolution into five peaks in the half-width at half-maximum range from 0.8 eV to 1.2 eV, so that each peak position is aligned for our samples (pyridinic N: ∼398.1 eV, pyrrolic N: 399.7 eV, center N: 401.0 eV, valley N: 402.2 eV, oxidized N: 404.0∼ eV).41,42 In addition, as shown in Fig. S5,† it was identified that each proportion of pyrrolic N and pyridinic N declined by carbonization. These results revealed that recarbonization caused a change in the carbon structure, and the proportion of N species was changed by recarbonization at various temperatures. Generally, pyrrolic N is known to be more unstable than other N species, such as pyridinic N and graphite N (center N and valley N) at relatively high temperature (>873 K), is converted to pyridinic N and graphitic N.31 Furthermore, Fig. 2b shows the proportion of N species in NDC-T. The results indicate that the bonding states of NDC-T differed with respect to the recarbonization temperature. With increasing recarbonization temperature, the proportion of center N, valley N, and oxidized N increased, while those of pyridinic N and pyrrolic N decreased.
 | ||
| Fig. 2 (a) XPS spectra (N 1s) of NDC-T and (b) the rate of N species in NDC-T. | ||
The electrocatalytic performance on the CO2RR was evaluated by electrochemical measurements in an H-type cell. LSV measurements were conducted in both N2-saturated and CO2-saturated 0.1 M KHCO3 solution using NDC-1100. NDC-1100 showed a higher current density in CO2-saturated 0.1 M KHCO3 solution than that in N2-saturated 0.1 M KHCO3 solution at a positive potential than −0.9 V vs. RHE (Fig. 3a), suggesting high catalytic activity for CO2RR. On the other hand, a higher current density was obtained in N2-saturated 0.1 M KHCO3 solution than that in CO2-saturated 0.1 M KHCO3 solution at a negative potential than −0.9 V vs. RHE, suggesting the hydrogen evolution reaction (HER) is dominant. Moreover, comparing the current density of NDC-T in CO2-saturated 0.1 M KHCO3 solution, it was found that the current density in CO2-saturated 0.1 M KHCO3 solution increased by increasing recarbonization temperature (Fig. 3b). It is expected that the differences in the proportion of N species in NDC-T cause changes in the charge density distribution, providing high electrical conductivity and catalytically active sites.
 | ||
| Fig. 3 (a) LSV curves in N2-saturated and CO2-saturated 0.1 M KHCO3 solution using NDC-1100 as an electrocatalyst. (b) LSV curves in CO2-saturated 0.1 M KHCO3 solution using each catalyst. (c) faradaic efficiencies of CO and H2 using NDC-T within the potential window from −0.5 V to −0.8 V vs. RHE in CO2-saturated 0.1 M KHCO3 solution. (d) Chronoamperometric responses and faradaic efficiency of CO and H2 on NDC-1100 at −0.7 V vs. RHE for 8 h in CO2-saturated 0.1 M KHCO3 solution. (e) Double-layer capacitance values of each catalyst. (f) The comparison of partial current density of CO (jCO; black bar) and the jCO normalized by ECSA (normalized jCO; red bar) of each catalyst at −0.7 V vs. RHE. | ||
Furthermore, the selectivity of electroreduction products for NDC-T at various potentials was investigated using a controlled potential electrolysis method. The measurement was conducted at potentials in 0.1 V increments from −0.5 to −0.8 V vs. RHE for 1.5 h. The products in liquid phase obtained for the experiment were investigated by 1H NMR, but it was below the detection limit. The electroreduction products were confirmed to be only gas-phase products. Products in the gas phase, mainly CO and H2, were yielded within the potential window from −0.5 V to −0.8 V vs. RHE. The FECO of NDC-T was the highest at −0.7 V vs. RHE (Fig. 3c), suggesting that NDC-T promoted the CO2RR and suppressed the HER. Whereas the FECO decreased gradually as the applied potential changed (less or more than −0.7 V vs. RHE), which means the dominance of the HER over the CO2RR. Moreover, comparing the FECO at −0.7 V vs. RHE, it was identified that the faradaic efficiency of CO (FECO) for NDC-T increased by carbonization (Fig. S6†). In addition, the FECO for NDC-T increased with increasing recarbonization temperature, suggesting that center N and valley N could contribute to the improvement of CO2RR performance. NDC-1100 exhibited the highest FECO of 76%, which is consistent with the results of the LSV measurements (Fig. 3a). Compared to our best catalyst (NDC-1100, the FECO of 76%), other metal-free N-doped carbon materials reported recently, such as GM2,43 PPc/CNT,36 and ZNMC-30,44 exhibited the highest FECO of 87.6%, 98.8%, and 88.9%. Therefore, our catalysts still have the potential to improve CO2-to-CO conversion performance. However, this catalyst synthesis method represents a breakthrough in that it enables the precise control of the local chemical state of N species in N-doped carbon while maintaining a nearly constant nitrogen content. To confirm the carbon source of products by CO2RR, the electrolysis experiment was conducted on NDC-1100 in N2-saturated 0.1 M KHCO3 solution at −0.7 V vs. RHE. Only H2 is detected in the gas-phase product (Fig. S7†), suggesting that the CO2 dissolved in the electrolyte was the only carbon source for producing CO. Furthermore, to investigate the influence of carbon paper, the electrolysis experiment was conducted in CO2-saturated 0.1 M KHCO3 solution at −0.7 V vs. RHE. In the gas-phase product, only H2 was detected (Fig. S8†). The results confirmed that carbon paper itself did not produce CO in the CO2RR electrolysis experiment.
Besides the selectivity of electroreduction products, the durability of the CO2RR catalysts is a crucial parameter for practical applications. The chronoamperometric response was performed on NDC-1100 at −0.7 V vs. RHE for 8 h (Fig. 3d). The current density slightly declined under continuous operation for up to 8 h because of catalyst stripping from the carbon paper and coating of the catalyst surface with gaseous products. Meanwhile, no decay of FECO was identified after continuous test for 8 h, suggesting that the NDC-1100 is a stable electrocatalyst for CO2RR.
Various comparisons were conducted to clarify the effects by the difference of the N species proportion based on experimental results. As shown in Table S2,† although NDC-1100 had a lower BET surface area than that of NDC-900 and NDC-1000, the FECO of NDC-1100 was higher than that of NDC-900 and NDC-1000 (Fig. 3c). In addition, Table 1 shows no significant differences in the nitrogen content of the NDC-T. Therefore, it is expected that the difference in the proportion of N species has a more significant effect on activity than the other properties. To verify this, the double-layer capacitance derived from cyclic voltammetry at different scan rates was measured for estimating the electrochemical surface area (ECSA) as shown in Fig. S9.† As shown in Fig. 3e, NDC-1100 exhibited the smallest ECSA of all catalysts. Furthermore, the current density generated by the production of CO (jCO) at −0.7 V vs. RHE normalized by ECSA was calculated (Fig. 3f). The normalized jCO of NDC-T increased with the recarbonization temperature. We also investigated the relationship between jCO and the content of each N species. As shown in Fig. S10a,†jCO decreased with increasing pyridinic N. Meanwhile, there is a positive correlation between jCO and the content of center N and valley N (Fig. S10b and c†). In particular, the best relationship was obtained between jCO and the content of valley N. For these reasons, it is considered that valley N should favorably contribute to the improvement of CO2RR activity.
DFT calculations using Gaussian were performed to gain further insight into the CO2RR mechanisms on N-doped carbon. Introducing N atom into carbon matrix could modify the electronic and chemical structure.23,24 It is suggested that center N and valley N had a good influence on the CO2RR activity based on experimental analysis (Fig. 2b, 3c and Fig. S10†). In addition, pyridinic N is regarded as an active site for several electrochemical reactions.20,21,45 For these reasons, three N-doped carbon configurations, containing pyridinic N, center N, and valley N each, were used as calculated models (Fig. 4a). The reaction pathways for the electroreduction of CO2 to CO are shown in Fig. S11.† The ground state structures of *COOH and *CO adsorbed on N-doped carbon models were identified by finding the lowest free energy one among several possible configurations on N atoms and C atoms adjacent to N atoms as possible active sites.
 | ||
| Fig. 4 (a) The computational structure models of three N-doped carbon configurations, containing pyridinic N, center N, and valley N each. (b) Gibbs free energy diagrams for CO2RR to CO on N atom on three N-doped carbon configurations. (c) Gibbs free energy diagrams for CO2RR to CO on three C atom configurations adjacent to valley N. | ||
The corresponding Gibbs free energy diagrams and reaction pathways for CO2RR to CO are shown in Fig. 4b and Fig. S11.† First, N atoms on N-doped carbon models were employed as the active sites. The free energy diagrams were constructed at 0 V vs. RHE according to the computational hydrogen electrode model.46 As shown in Fig. 4b, it was revealed that the rate-determining step on N-doped carbon models was *COOH adsorption of the first step. Moreover, the free energy barriers of 1.72 eV on model valley N and 1.79 eV on model pyridinic N were much lower than that of 3.79 eV on model center N, suggesting that the edge site N species contributed to stabilizing *COOH intermediate. Therefore, in addition to pyridinic N, valley N is also regarded as a candidate active site on N-doped carbon. Considering based on experimental results in addition to DFT calculations results, valley N is considered to be a more favorable active site rather than pyridinic N because the FECO for NDC-T increased as the increase of valley N and the decrease of pyridinic N (Fig. 2b and 3c).
Furthermore, to gain further insight into the effect of valley N on CO2RR, C atoms adjacent to valley N were employed as active sites. As shown in Fig. 4a, three C atom configurations, C1 located on edge planes and C2 and C3 located on the basal planes, were used as the calculated models. The corresponding Gibbs free energy diagrams and reaction pathways for CO2RR to CO are displayed in Fig. 4c and Fig. S12.† The free energy barriers of 1.55 eV on C1 located on edge planes were lower than that of 2.05 eV on C2 and 3.07 eV on C3 located on basal planes, suggesting that the edge site C atom adjacent to valley N stabilized *COOH intermediate. In contrast, the *CO adsorption energy of the second step was the same (0.86 eV) for all C configurations. Based on these DFT calculation results, a new insight into the effect of N species on CO2RR activity was provided that valley N and edge site C atoms adjacent to valley N can be more favorable active sites than other N atoms on N species.
Conclusions
N-doped carbons (NDC-T) were synthesized with various edge site N contents by combining a zeolite templating method and a recarbonization treatment. We succeeded in controlling the local chemical state of N species of N-doped carbons by changing recarbonization temperatures. The obtained NDC-T electrochemically catalyzed CO2 to CO, and the best catalyst, NDC-1100, exhibited the highest FECO of 76%. Furthermore, experimental analysis and DFT calculations revealed that valley N and the edge site C atom adjacent to valley N stabilized *COOH intermediate, contributed to improvement of CO2RR activity. Our work provides not only a rational design method for N-doped carbons to control the proportion of N species but also new insights into the effect of valley N on CO2RR activity.Author contributions
Kotaro Narimatsu: investigation, visualization, writing – original draft. Ryuji Takada: conceptualization, investigation, visualization, methodology, writing – review & editing. Koji Miyake: conceptualization, supervision, visualization, methodology, writing – review & editing. Yoshiaki Uchida: writing – review & editing. Norikazu Nishiyama: supervision, resources, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was partially conducted using a facility at the Research Center for Ultra-High Voltage Electron Microscopy, Osaka University, Suita, Japan.References
- Q. Lin, X. Zhang, T. Wang, C. Zheng and X. Gao, Engineering, 2022, 14, 27–32 CrossRef CAS.
- X. Tan, C. Yu, Y. Ren, S. Cui, W. Li and J. Qiu, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- M.-Y. Lee, K. T. Park, W. Lee, H. Lim, Y. Kwon and S. Kang, Crit. Rev. Environ. Sci. Technol., 2020, 50, 769–815 CrossRef CAS.
- S. Lu, Y. Shi, N. Meng, S. Lu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2020, 1, 100237 CrossRef.
- L. Delafontaine, T. Asset and P. Atanassov, ChemSusChem, 2020, 13, 1688–1698 CrossRef CAS PubMed.
- K. Gan, H. Li, R. Li, J. Niu, J. He, D. Jia and X. He, Inorg. Chem. Front., 2023, 10, 2414–2422 RSC.
- L. Luo, Z. Huang and Q. Tang, J. Mater. Chem. A, 2024, 12, 5145–5155 RSC.
- J. Fu, Y. Wang, J. Liu, K. Huang, Y. Chen, Y. Li and J.-J. Zhu, ACS Energy Lett., 2018, 3, 946–951 CrossRef CAS.
- J. Gao, S. Zhao, S. Guo, H. Wang, Y. Sun, B. Yao, Y. Liu, H. Huang and Z. Kang, Inorg. Chem. Front., 2019, 6, 1453–1460 RSC.
- X. Y. Zhang, W. J. Li, J. Chen, X. F. Wu, Y. W. Liu, F. Mao, H. Y. Yuan, M. Zhu, S. Dai, H. F. Wang, P. Hu, C. Sun, P. F. Liu and H. G. Yang, Angew. Chem., Int. Ed., 2022, 61, e202202298 CrossRef CAS PubMed.
- I. Masood ul Hasan, L. Peng, J. Mao, R. He, Y. Wang, J. Fu, N. Xu and J. Qiao, Carbon Energy, 2021, 3, 24–49 CrossRef CAS.
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1701784 CrossRef.
- Y. Zhang, J. Zhao and S. Lin, Chin. J. Struct. Chem., 2024, 43, 100415 CAS.
- M. Humayun, M. Bououdina, A. Khan, S. Ali and C. Wang, Chin. J. Struct. Chem., 2024, 43, 100193 CAS.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- T. Zhang, W. Li, K. Huang, H. Guo, Z. Li, Y. Fang, R. M. Yadav, V. Shanov, P. M. Ajayan, L. Wang, C. Lian and J. Wu, Nat. Commun., 2021, 12, 5265 CrossRef CAS PubMed.
- R. M. Yadav, Z. Li, T. Zhang, O. Sahin, S. Roy, G. Gao, H. Guo, R. Vajtai, L. Wang, P. M. Ajayan and J. Wu, Adv. Mater., 2022, 34, 2105690 CrossRef CAS PubMed.
- H. Wang, Y. Chen, X. Hou, C. Ma and T. Tan, Green Chem., 2016, 18, 3250–3256 RSC.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X.-D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2016, 16, 466–470 CrossRef CAS PubMed.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- K.-H. Liu, H.-X. Zhong, X.-Y. Yang, D. Bao, F.-L. Meng, J.-M. Yan and X.-B. Zhang, Green Chem., 2017, 19, 4284–4288 RSC.
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Angew. Chem., 2020, 132, 11216–11222 CrossRef.
- B. Zhang, J. Zhang, F. Zhang, L. Zheng, G. Mo, B. Han and G. Yang, Adv. Funct. Mater., 2020, 30, 1906194 CrossRef CAS.
- G. An, K. Wang, Z. Wang, M. Zhang, H. Guo and L. Wang, ACS Appl. Mater. Interfaces, 2024, 16, 29060–29068 CrossRef CAS PubMed.
- Y. Dong, Q. Zhang, Z. Tian, B. Li, W. Yan, S. Wang, K. Jiang, J. Su, C. W. Oloman, E. L. Gyenge, R. Ge, Z. Lu, X. Ji and L. Chen, Adv. Mater., 2020, 32, 2001300 CrossRef CAS PubMed.
- Q. Zhai, H. Huang, T. Lawson, Z. Xia, P. Giusto, M. Antonietti, M. Jaroniec, M. Chhowalla, J.-B. Baek, Y. Liu, S. Qiao and L. Dai, Adv. Mater., 2024, 36, 2405664 CrossRef CAS PubMed.
- M. Inagaki, M. Toyoda, Y. Soneda and T. Morishita, Carbon, 2018, 132, 104–140 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- H. Kiuchi, R. Shibuya, T. Kondo, J. Nakamura, H. Niwa, J. Miyawaki, M. Kawai, M. Oshima and Y. Harada, Nanoscale Res. Lett., 2016, 11, 127 CrossRef PubMed.
- T. Sharifi, G. Hu, X. Jia and T. Wågberg, ACS Nano, 2012, 6, 8904–8912 CrossRef CAS PubMed.
- H. Nishihara and T. Kyotani, in Novel Carbon Adsorbents, ed. J. M. D. Tascón, Elsevier, Oxford, 2012, pp. 295–322 Search PubMed.
- Y. Taniguchi, S. Kokuryo, R. Takada, X. Yang, K. Miyake, Y. Uchida and N. Nishiyama, Electrochem. Commun., 2024, 160, 107665 CrossRef CAS.
- Y. Taniguchi, S. Kokuryo, R. Takada, X. Yang, K. Miyake, Y. Uchida and N. Nishiyama, Carbon Rep., 2024, 3, 11–17 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, F. Williams, F. Ding, F. Lipparini, J. Egidi, B. Goings, A. Peng, T. Petrone, D. Henderson, V. G. Ranasinghe, J. Zakrzewski, N. Gao, G. Z. Rega, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Rev. C. 01, Wallingford, CT, 2016 CrossRef CAS PubMed; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed https://gaussian.com/news/ (accessed February 7, 2025).
- W. Li, C. Yu, X. Song, X. Tan, H. Xu, Y. Ren, S. Cui, Y. Zhang, J. Chang, Y. Ding, Y. Xie and J. Qiu, Chem. Mater., 2024, 36, 1602–1611 CrossRef CAS.
- W. Zheng, D. Wang, Y. Zhang, S. Zheng, B. Yang, Z. Li, R. D. Rodriguez, T. Zhang, L. Lei, S. Yao and Y. Hou, Nano Energy, 2023, 105, 107980 CrossRef CAS.
- H. Nishihara and T. Kyotani, Chem. Commun., 2018, 54, 5648–5673 RSC.
- L.-L. Ling, L. Jiao, X. Liu, Y. Dong, W. Yang, H. Zhang, B. Ye, J. Chen and H.-L. Jiang, Adv. Mater., 2022, 34, 2205933 CrossRef CAS PubMed.
- W. Wang, L. Shang, G. Chang, C. Yan, R. Shi, Y. Zhao, G. I. N. Waterhouse, D. Yang and T. Zhang, Adv. Mater., 2019, 31, 1808276 CrossRef PubMed.
- Q. Lai, J. Zheng, Z. Tang, D. Bi, J. Zhao and Y. Liang, Angew. Chem., 2020, 132, 12097–12104 CrossRef.
- J. Quílez-Bermejo, E. Morallón and D. Cazorla-Amorós, Chem. Commun., 2018, 54, 4441–4444 RSC.
- S. Liu, H. Yang, X. Huang, L. Liu, W. Cai, J. Gao, X. Li, T. Zhang, Y. Huang and B. Liu, Adv. Funct. Mater., 2018, 28, 1800499 CrossRef.
- J. Zhang, K. Wang, X. Wang and X. Li, ACS Appl. Mater. Interfaces, 2024, 16, 54092–54104 CrossRef PubMed.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X.-D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00846h |
| This journal is © The Royal Society of Chemistry 2025 |