
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmart polymer prodrugs via responsive prodrug-initiated ring-opening polymerization of lactide for improved drug delivery†
Shiwei
Fu‡
a,
Miao
Zhang‡
de,
Nicholas
Calzadilla‡
a,
Bowen
Zhao
a,
Xiao
Zhang
 a,
Bowei
Yang
de,
Victoria A. A.
McKenzie
a,
Ajay
Zheng
a,
Qianqian
Ni
*de and
Fuwu
Zhang
*abc
a,
Bowei
Yang
de,
Victoria A. A.
McKenzie
a,
Ajay
Zheng
a,
Qianqian
Ni
*de and
Fuwu
Zhang
*abc
aDepartment of Chemistry, University of Miami, 1301 Memorial Drive, Coral Gables, Florida 33146, USA. E-mail: fxz174@miami.edu
bThe Dr John T. Macdonald Foundation Biomedical Nanotechnology Institute, University of Miami, Miami, FL 33136, USA
cSylvester Comprehensive Cancer Centre, University of Miami Miller School of Medicine, Miami, Florida 33136, USA
dNanomedicine Translational Research Program, NUS Centre for Nanomedicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, Singapore. E-mail: qqian.ni@nus.edu.sg
eClinical Imaging Research Centre, Centre for Translational Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117599, Singapore
First published on 4th April 2025
Abstract
Smart polymer prodrugs, created via responsive prodrug-initiated controlled polymerization of lactide, demonstrated extremely high drug loading, tuneable stimuli-triggered drug release, and significant tumor growth inhibition and improved survival with minimal toxicity. This adaptable strategy can precisely tailor drugs’ physicochemical properties for optimal therapeutic efficacy, demonstrating great promise for cancer treatment.
Small molecular drugs are a key component in treating various diseases, including cancer, infection, cardiovascular diseases, and more.1,2 However, many of these small molecular drugs, such as camptothecin (CPT), monomethyl auristatin E (MMAE), and mertansine (DM1), suffer from limited solubility, severe side effects, and unsatisfactory efficacy due to unfavorable pharmacokinetics and inefficient drug delivery to diseased sites.3–5 Drug delivery systems, including polymeric micelles, liposomes, polymer–drug conjugates, antibody–drug conjugates, and nanoparticles (NPs), are widely used to enhance drugs’ solubility, stability, pharmacokinetics and biodistribution, ultimately increasing therapeutic efficacy and reducing adverse effects.6–9
Polymeric micellar structures consist of a hydrophobic core for drug loading and a hydrophilic shell, typically polyethylene glycol, for improved colloidal stability and stealth effects.10–13 Micellar formulations, like Genexol-PM, NK012, and NK105, have advanced to clinical trials, yet none has been approved in the United States.14 Challenges include low drug loading capacity, poor colloidal stability, premature drug release, and limited drug accumulation in the diseased tissues.15,16 Hydrophilic drugs’ poorly miscible core often results in low drug-loading efficiency. Moreover, unfavorable surface/shell drug loading in NPs can lead to premature ‘burst release’, causing undesirable side effects and reduced therapeutic efficacy. One approach to address these limitations is to optimize the drug carrier. Shell crosslinking of micelles has demonstrated controlled drug release and improved structural stability.16–19 However, the structural complexities of these advanced nanocarriers are stumbling blocks for clinical translation. Alternatively, the payloads can be engineered to improve drug delivery.20 We previously reported a glutathione-sensitive heterodimeric multifunctional prodrug that significantly increased the drug's hydrophobicity, allowing high drug loading and slow responsive drug release. This resulted in preferable tumor accumulation and significantly improved tumor growth inhibition.21 However, coupling of two drugs offers limited tunability of the prodrug's physicochemical properties, and the fixed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 drug ratio might not be ideal for clinical applications. Herein, we developed a responsive prodrug-initiated ring-opening polymerization (ROP) of polylactide (PLA) to engineer a therapeutic agent with a hydrophobic polymer chain, which allows for the systematic tuning of a drug's physicochemical properties to align with those of the carrier, optimizing drug loading and release and ultimately enhancing therapeutic efficacy while minimizing side effects.
:1 drug ratio might not be ideal for clinical applications. Herein, we developed a responsive prodrug-initiated ring-opening polymerization (ROP) of polylactide (PLA) to engineer a therapeutic agent with a hydrophobic polymer chain, which allows for the systematic tuning of a drug's physicochemical properties to align with those of the carrier, optimizing drug loading and release and ultimately enhancing therapeutic efficacy while minimizing side effects.
As proof of concept, we used CPT as a model therapeutic agent. CPT is a potent topoisomerase I inhibitor with remarkable anticancer activities, but its clinical use is limited due to severe toxicity, lactone ring instability, and water insolubility.22–27 Effective drug carriers to overcome these limitations are needed. However, most CPT nanoformulations face challenges such as low drug loading, uncontrolled release, and poor NP stability, partly due to CPT's planar five-ring aromatic structure.28–31 In this study, we present a straightforward method for developing CPT-based responsive polymer prodrugs by using a disulfide-containing CPT prodrug to initiate the ROP of lactide. The obtained polymer prodrug had narrow dispersity, high drug loading efficiency, controlled drug release, and improved tumor growth inhibition compared to CPT. The prodrug's physicochemical and biological properties could be finely tuned by varying the polymer chain length. Our prodrug engineering strategy provides a simple and effective approach to optimize drugs’ physicochemical properties for high drug loading, controlled drug release, and improved in vivo efficacy.
An advantage of our strategy is the facile construction of a responsive polymer prodrug from commercially available CPT and lactide (Fig. 1). CPT was treated with triphosgene to activate its hydroxyl group which then reacted with a disulfide-containing linker to yield CPT-ss-OH which then initiated the ROP with D/L-lactide in the presence of the organocatalyst 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU). After 5 min, the reaction was quenched with acetic acid and purified by flash chromatography on a silica column. The prodrugs were characterized by 1H NMR spectroscopy (Fig. 2a). Characteristic peaks of PLA appeared at ∼1.56 ppm (methyl) and ∼5.17 ppm (methine) (Fig. 2a), which gave an average degree of polymerization (DP) of 16 after integration. Polymer prodrugs with DPs of 5 and 36 (CPT-ss-PLA5, CPT-ss-PLA36) were also synthesized. CPT-cc-PLA18, a control lacking a disulfide bond, was similarly synthesized. Gel permeation chromatography (GPC) was also used to analyze prodrug's size distribution and dispersity (Đ) and average molecular mass. All polymer prodrugs exhibited monomodal molar mass distributions, with Đ values of less than 1.1 (Table S1†). As anticipated, polymer prodrugs with longer chain lengths showed shorter retention times, indicating higher molar masses (Fig. 2b). Furthermore, we found out that longer PLA chains increased retention factors in thin-layer chromatography (Fig. 2c) and retention times in high-performance liquid chromatography (HPLC) (Fig. 2d), indicating that the hydrophobic polylactide systematically enhanced the overall hydrophobicity of CPT.
 | ||
| Fig. 1 Synthesis of glutathione-sensitive polymer prodrugs toward improved drug delivery to tumours. | ||
 | ||
| Fig. 2 (a) 1H NMR spectrum of the polymer prodrug CPT-ss-PLA16. (b) GPC traces of CPT-ss-PLA5 (green line), CPT-ss-PLA16 (blue line) and CPT-ss-PLA36 (black line). (c) Thin-layer chromatography of CPT and CPT polymer prodrugs. Lanes 1–5 are CPT, CPT-ss-PLA5, CPT-ss-PLA16, CPT-ss-PLA36, and CPT-cc-PLA18 under a UV 254 nm lamp, respectively. (d) The HPLC traces of CPT polymer prodrugs. Characterization by DLS (e) and TEM (f) of CPT-ss-PLA16 loaded NPs. (g) A photograph of CPT-ss-PLA16 NPs in water in the dark with green laser light passing through. | ||
Following the synthesis of prodrugs, CPT-ss-PLA16 was selected for preliminary drug loading tests using biodegradable PEG-b-PLA as an amphiphilic drug carrier (Table 1). CPT-ss-PLA16 and PEG-b-PLA were mixed in THF and added dropwise to water, initiating NP self-assembly. All trials with CPT-ss-PLA16 produced NPs with high colloidal stability, without visible precipitation during preparation or storage. Dynamic light scattering (DLS) demonstrated a monomodal distribution with a hydrodynamic diameter of 41 ± 13 nm (Fig. 2e). When green laser light passed through the formulation, a uniform light path was observed, indicating homogeneous NPs (Fig. 2g). Transmission electron microscopy (TEM) images of CPT-ss-PLA16 loaded NPs suggested the formation of spherical particles with dry-state diameters of 40 ± 6 nm (Fig. 2f). At a feed ratio of 4 (mass of polymer prodrug CPT-ss-PLA16 to mass of PEG-b-PLA), the loading capacity of the prodrug reached 80 wt%, corresponding to a CPT loading of 16.6 wt% (Table 1). Similar results were observed with other CPT polymer prodrugs, including CPT-ss-PLA5, CPT-ss-PLA36, and CPT-cc-PLA18. In contrast, a mixture of CPT and PEG-b-PLA in DMSO (as CPT is poorly soluble in THF) with a low ratio of 1:20 formed visible precipitates within 2 hours, with no purification possible. The high drug loading efficiency and capacity are likely due to the increased hydrophobicity and improved miscibility of the prodrug PLA chains with the PLA chains of the nanocarrier, which highlights the advantages of the CPT polymer prodrug toward developing an effective drug delivery system compared to free CPT.
| Entry | Ratioa | DL-Pb | DL-Cc | Sized | Stabilitye |
|---|---|---|---|---|---|
| a Mass of the polymer prodrug to the mass of PEG-b-PLA. b Drug loading prodrug (DL-P) was calculated as DL-P = mass of the polymer prodrug/mass of (polymer drug + PEG-b-PLA) × 100%. c Drug loading CPT (DL-C) was calculated as DL-C = mass of CPT/mass of (CPT-ss-PLA + mass of PEG-b-PLA) × 100%. d The size of the drug-loaded NPs was measured by DLS in terms of number distribution (nm). e Stable means no visual precipitation over 1 month. | |||||
| CPT-ss-PLA16 | 1:4 |
20 | 4.2 | 58 ± 21 | Stable |
| CPT-ss-PLA16 | 1:2 |
33 | 6.9 | 41 ± 13 | Stable |
| CPT-ss-PLA16 | 1:1 |
50 | 10.4 | 52 ± 15 | Stable |
| CPT-ss-PLA16 | 2:1 |
67 | 13.8 | 53 ± 16 | Stable |
| CPT-ss-PLA16 | 4:1 |
80 | 16.6 | 61 ± 19 | Stable |
| CPT-ss-PLA5 | 1:2 |
33 | 13.1 | 91 ± 32 | Stable |
| CPT-ss-PLA36 | 1:2 |
33 | 3.7 | 44 ± 14 | Stable |
| CPT-cc-PLA18 | 1:2 |
33 | 6.5 | 68 ± 27 | Stable |
| CPT | 1:20 |
— | 4.8 | 101 ± 22 | Not stable |
Another critical feature of CPT-ss-PLA NPs is their responsive release of intact drugs due to disulfide cleavage. HPLC traces clearly demonstrate that long polymer chains resulted in longer retention times due to increased hydrophobicity (Fig. 2d). In the presence of 40 mM GSH, the concentration of CPT-ss-PLA16 displayed a gradual decrease accompanied by an increase of CPT concentration due to the cleavage of disulfide bonds (Fig. 3a). It was observed that longer PLA chains significantly slowed the CPT release (Fig. 3b). Specifically, within 8 hours, CPT-ss-PLA5 NPs released ∼95% of CPT, while CPT-ss-PLA16 and CPT-ss-PLA36 released ∼26% and ∼7%, respectively. Over a prolonged incubation for 48 hours, CPT-ss-PLA16 NPs released 78% of CPT, while CPT-ss-PLA36 NPs released only 30%. In contrast, the CPT-cc-PLA18 NPs, without a disulfide bond, showed negligible CPT release (Fig. 3b), which clearly demonstrates the need for a labile disulfide bond to release intact CPT. Furthermore, minimal CPT release was observed for CPT-ss-PLA36 while up to 40% for CPT-ss-PLA5 in 144 h in PBS without GSH (Fig. S4†). Together, these results indicate that the CPT polymer prodrug, designed to systematically tune the physicochemical properties of CPT, achieves high drug loading and offers responsive tunable release capabilities. The ability to fine-tune the drug's release rate by altering the polymer's chain length lays the foundation for further studies into the optimization of effectiveness of cancer therapies.
 | ||
| Fig. 3 (a) HPLC traces of intact CPT released from the prodrug CPT-ss-PLA16 in PBS containing 40 mM GSH at different time points. (b) Intact CPT released in the presence of 40 mM GSH. (c) In vitro cytotoxicity of CPT and CPT prodrugs against BxPC-3 and (d) 4T1 cancer cells. | ||
In vitro cytotoxicity of CPT-ss-PLA NPs was assessed against 4T1, PC-3, and BxPC-3 cancer cells (Fig. 3c, d and S6†). CPT-ss-PLA NPs showed concentration-dependent inhibition of cell proliferation. For BxPC-3 cells, CPT-ss-PLA5 showed an IC50 of 1.78 μM, slightly higher than free CPT's IC50 of 0.41 μM. CPT-ss-PLA16 and CPT-ss-PLA36 showed IC50 values of 4.62 μM and 14.1 μM, indicating reduced potency for longer PLA chains. CPT-cc-PLA18, lacking the disulfide bond, exhibited much lower cytotoxicity. Similar trends appeared in 4T1 and PC-3 cells (Fig. 3c, d, S6 and 7†) (Table S2†). These data revealed that longer PLA chains resulted in lower cytotoxicity which was probably due to decreased drug release. The disulfide bond was essential for maintaining prodrugs’ in vitro cytotoxicity.
Building on promising in vitro findings, we progressed to in vivo mouse studies to evaluate therapeutic efficacy and safety in a more complex biological setting to verify our polymer prodrug strategy. Tumor-bearing mice (BxPC-3 model) were treated with formulations every 3 days, 5 times. Body weight and tumor size were monitored every 2 days (Fig. 4a). As expected, mice treated solely with PBS experienced a substantial increase in tumor size, highlighting the aggressive nature of pancreatic cancer. In contrast, the groups treated with drug loaded NPs exhibited much slower tumor growth rates (Fig. 4b and e). CPT-ss-PLA5 and CPT-ss-PLA16 NPs reduced tumor size by half compared to CPT-loaded NPs on day 26. CPT-ss-PLA36, with slower drug release, resulted in similar tumor size to CPT loaded NPs. After 32 days, all mice treated with PBS had succumbed, while the CPT-ss-PLA36 group exhibited a 60% survival rate, and the CPT-cc-PLA18 group showed a 40% survival rate. Impressively, the CPT-ss-PLA5 and CPT-ss-PLA16 groups achieved an 80% survival rate (Fig. 4d). No significant differences in body weight (Fig. 4c) or histopathological abnormalities (e.g., hemorrhage, necrosis, inflammation) were observed (Fig. S8†). These findings emphasize the exceptional therapeutic efficacy and safety profile of CPT-ss-PLA NPs, underscoring their potential as a treatment for pancreatic cancer. More importantly, this study demonstrates the capability of these polymer prodrugs to tune their PLA chain length, enabling tunable responsive drug release kinetics to optimize treatment outcomes against different cancers.
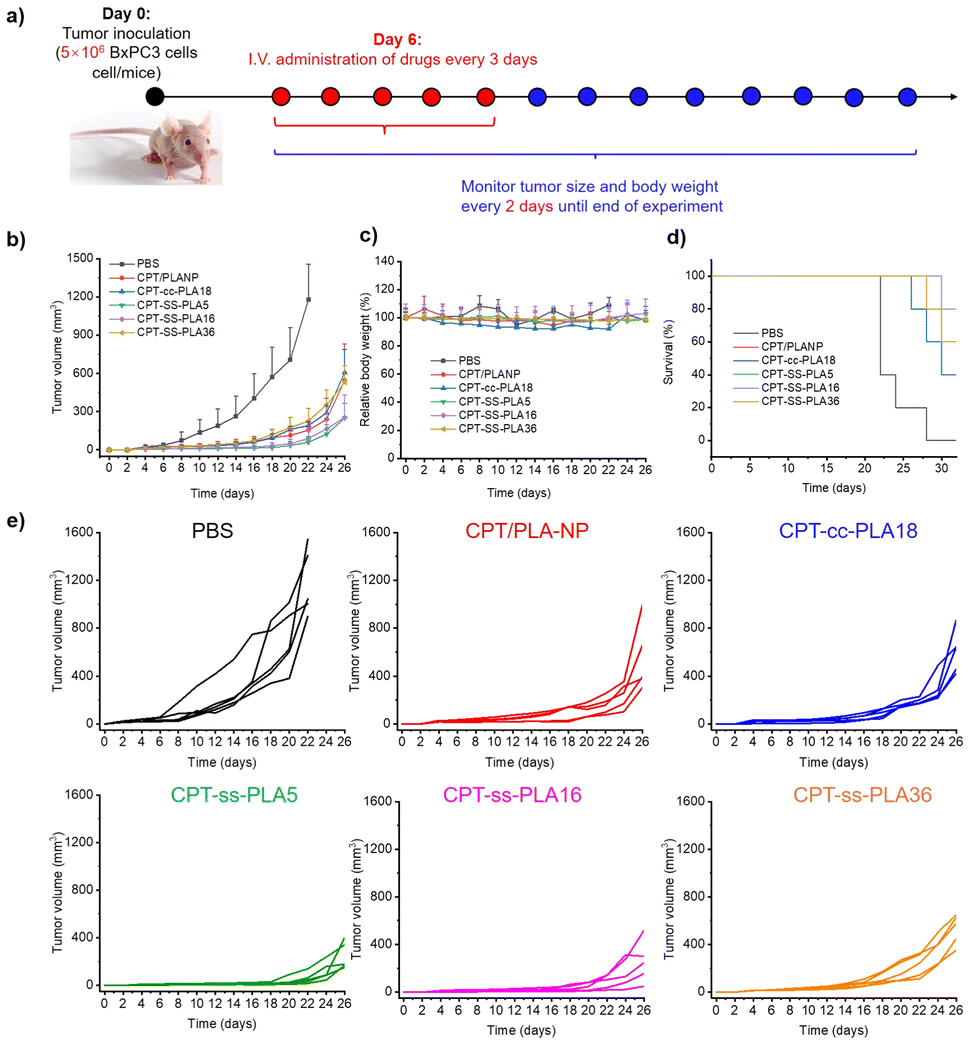 | ||
| Fig. 4 In vivo antitumor activities of various treatments on BxPC-3 tumour bearing BALB/c nude mice. (a) Scheme of the experiment. (b–d) Tumour inhibition curve, relative body weight, and survival rate of tumour-bearing mice (n = 5 biologically independent mice per group). (e) Individual tumour growth curve after each treatment. Results are presented as the mean ± standard deviation (SD). *P < 0.05 indicates a statistically significant difference between CPT-ss-PLA16 and CPT-cc-PLA18 groups. | ||
In summary, we developed a simple and efficient method to construct a polymer prodrug through direct, responsive prodrug-initiated polymerization of lactide. The resulting prodrugs achieve exceptionally high drug loading efficiency and capacity, stimuli-responsive drug release, and enhanced tumour inhibition with minimal systemic toxicity, showcasing significant potential for cancer therapy. Adjusting the polymer chain length allows effective tailoring of drug's physicochemical properties for optimized therapeutic outcomes. This versatile strategy offers new opportunities for delivering various drugs, both hydrophobic and hydrophilic, such as DM1, MMAE, and paclitaxel.
Author contributions
S. F., M. Z., and N. C. contributed equally to this work. S. F.: methodology and investigation, writing – original draft. M. Z.: methodology and investigation. N. C.: methodology and investigation. B. Z.: methodology and investigation. X. Z.: methodology and investigation. B. Y.: methodology and investigation. V. A. A. M.: methodology and investigation. A. Z.: methodology and investigation. Q. N.: conceptualization, supervision, writing – original draft. F. Z.: conceptualization, supervision, writing – original draft.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. Z. gratefully acknowledges 2023 Provost's Research Awards (UM PRA 2022-3284) from the University of Miami. M. Z. is thankful for a Singapore Open Fund Young Individual Research Grant (MOH-001581-01). We thank Vania Almeida and the University of Miami Transmission Electron Microscopy Core for EM sample preparation and assistance with the generation of EM images.References
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang and S. Yang, Signal Transduction Targeted Ther., 2021, 6, 201 CrossRef PubMed.
- H. Beck, M. Härter, B. Haß, C. Schmeck and L. Baerfacker, Drug Discovery Today, 2022, 27, 1560–1574 CrossRef CAS PubMed.
- L. Zhou, Y. Lu, W. Liu, S. Wang, L. Wang, P. Zheng, G. Zi, H. Liu, W. Liu and S. Wei, Exp. Hematol. Oncol., 2024, 13, 26 CrossRef PubMed.
- S. Fu, A. Zheng, L. Wang, J. Chen, B. Zhao, X. Zhang, V. A. A. McKenzie, Z. Yang, R. M. Leblanc, R. Prabhakar and F. Zhang, J. Mater. Chem. B, 2024, 12, 6563–6569 RSC.
- X. Zhang, X. Li and X. Zhang, Eur. J. Med. Chem., 2017, 139, 542–563 CrossRef CAS PubMed.
- T. A. Seidu, P. T. Kutoka, D. O. Asante, M. A. Farooq, R. N. Alolga and W. Bo, Pharmaceutics, 2022, 14, 1113 CrossRef CAS PubMed.
- L. Huang, X.-H. Huang, X. Yang, J.-Q. Hu, Y.-Z. Zhu, P.-Y. Yan and Y. Xie, Pharmacol. Res., 2024, 201, 107100 CrossRef CAS PubMed.
- O. Guliy, A. Fomin, E. Zhnichkova, S. V. Kozlov, S. Staroverov and L. Dykman, in Pharmaceutical Nanobiotechnology for Targeted Therapy, ed. H. Barabadi, E. Mostafavi and M. Saravanan, Springer, Cham, 2022, pp. 521–559 Search PubMed.
- R. Prasad, A. Ghosh, V. Patel, B. Peng, B. B. Mendes, E. H. A. Win, L. G. Delogu, J. Y. Wong, K. J. Pischel, J. R. Bellare, A. Bar-Shir, A. S. Thakor, W. J. Parak, Z. M. Bhujwalla, Y. S. Zhang, N. Kommineni, V. M. Rotello, W. Cai, T. Lammers, T. W. Odom, G. Padmanaban, D. Peer, J. F. Lovell, R. Srivastava, R. Langer and J. Conde, ACS Nano, 2025, 19, 2979–2991 CrossRef CAS PubMed.
- S. Schottler, K. Landfester and V. Mailander, Angew. Chem., Int. Ed., 2016, 55, 8806–8815 CrossRef PubMed.
- F. Jia, X. Lu, X. Tan, D. Wang, X. Cao and K. Zhang, Angew. Chem., Int. Ed., 2017, 56, 1239–1243 CrossRef CAS PubMed.
- H. Sun and Z. Zhong, Biomacromolecules, 2025, 26, 33–42 CrossRef CAS PubMed.
- J. Li, Y. Hao, H. Wang, M. Zhang, J. He and P. Ni, ACS Appl. Mater. Interfaces, 2024, 16, 51876–51898 CrossRef CAS PubMed.
- C. Oerlemans, W. Bult, M. Bos, G. Storm, J. F. W. Nijsen and W. E. Hennink, Pharm. Res., 2010, 27, 2569–2589 CrossRef CAS PubMed.
- A. G. Cheetham, P. C. Zhang, Y. A. Lin, L. L. Lock and H. G. Cui, J. Am. Chem. Soc., 2013, 135, 2907–2910 CrossRef CAS PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- F. W. Zhang, S. Y. Zhang, S. F. Pollack, R. C. Li, A. M. Gonzalez, J. W. Fan, J. Zou, S. E. Leininger, A. Pavia-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. P. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 2056–2066 CrossRef CAS PubMed.
- X. Y. Tan, B. B. Li, X. G. Lu, F. Jia, C. Santori, P. Menon, H. Li, B. H. Zhang, J. J. Zhao and K. Zhang, J. Am. Chem. Soc., 2015, 137, 6112–6115 CrossRef CAS PubMed.
- F. Zhang, S. Khan, R. Li, J. A. Smolen, S. Zhang, G. Zhu, L. Su, A. A. Jahnke, M. Elsabahy, X. Chen and K. L. Wooley, Nanoscale, 2017, 9, 15773–15777 RSC.
- F. W. Zhang, Q. Q. Ni, O. Jacobson, S. Y. Cheng, A. Liao, Z. T. Wang, Z. M. He, G. C. Yu, J. B. Song, Y. Ma, G. Niu, L. J. Zhang, G. Z. Zhu and X. Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 7066–7070 CrossRef CAS PubMed.
- F. Zhang, Q. Ni, O. Jacobson, S. Cheng, A. Liao, Z. Wang, Z. He, G. Yu, J. Song, Y. Ma, G. Niu, L. Zhang, G. Zhu and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 7066–7070 CrossRef CAS PubMed.
- C. Bailly, Pharmacol. Res., 2019, 148, 104398 CrossRef CAS PubMed.
- F. Li, T. Jiang, Q. Li and X. Ling, Am. J. Cancer Res., 2017, 7, 2350 CAS.
- R. H. Mathijssen, R. J. van Alphen, J. Verweij, W. J. Loos, K. Nooter, G. Stoter and A. Sparreboom, Clin. Cancer Res., 2001, 7, 2182–2194 CAS.
- E. Martino, S. Della Volpe, E. Terribile, E. Benetti, M. Sakaj, A. Centamore, A. Sala and S. Collina, Bioorg. Med. Chem. Lett., 2017, 27, 701–707 CrossRef CAS PubMed.
- W. J. Slichenmyer, E. K. Rowinsky, R. C. Donehower and S. H. Kaufmann, J. Natl. Cancer Inst., 1993, 85, 271–291 CrossRef CAS PubMed.
- A. Lorence and C. L. Nessler, Phytochemistry, 2004, 65, 2735–2749 CrossRef CAS PubMed.
- R. Tong, N. P. Gabrielson, T. M. Fan and J. Cheng, Curr. Opin. Solid State Mater. Sci., 2012, 16, 323–332 CrossRef CAS PubMed.
- R. Satchi-Fainaro, R. Duncan and C. M. Barnes, in Polymer Therapeutics II, ed. R. Satchi-Fainaro and R. Duncan, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 1–65. DOI:10.1007/12_024.
- R. B. Greenwald, J. Controlled Release, 2001, 74, 159–171 CrossRef CAS PubMed.
- K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00780a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |