
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of Au13-based building block clusters for programmed dimer formation and Au13 cluster dimer photoexcitation properties†
Taiga
Kosaka
a,
Yoshiki
Niihori
 *b,
Tokuhisa
Kawawaki
c and
Yuichi
Negishi
*c
*b,
Tokuhisa
Kawawaki
c and
Yuichi
Negishi
*c
aDepartment of Chemistry, Graduate School of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
bResearch Institute for Science & Technology, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: niihori@rs.tus.ac.jp
cInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan. E-mail: yuichi.negishi.a8@tohoku.ac.jp
First published on 17th April 2025
Abstract
Recently, there has been increasing attention on the fabrication of ligand-protected metal clusters composed of a finite number of noble metal atoms and on their precise assembly to elicit novel properties that are not observed in individual metal clusters. In the present study, we investigated (1) the behavior of ligand exchange reactions and (2) the selective and efficient formation of dimers composed of Au13 clusters. Specifically, we focused on a gold cluster consisting of 13 atoms coordinated to diphosphine ligands (dppe) and either chloride (Cl) or acetylide, i.e., [Au13(dppe)5X2]3+ (X = Cl or acetylide). The findings showed that Au13 clusters containing the counter anion Cl− undergo a transformation under specific conditions, where Cl− acts as a ligand (rather than an anion) directly coordinated to the Au13 surface. The introduction of two types of ligands—chelating ligands that coordinate to the Au13 cluster surface and end-capping ligands that suppress polymerization—enabled the synthesis of building block molecules that are programmed to selectively and spontaneously form Au13-based dimers upon the addition of metal ions. The designed building block clusters indeed selectively and efficiently formed stable dimers composed of two Au13 clusters in the presence of iron ions. Furthermore, in the Au13-based dimer, the phosphorescent Au13 moiety is directly connected to a coordination site that exhibits quenching effects, enabling rapid intramolecular photoinduced charge transfer even with a small driving force.
Introduction
Ligand-protected metal clusters, which consist of a finite number of noble metal atoms (such as gold and silver), exhibit size- or composition-dependent luminescence properties, magnetic behavior, and catalytic activity. Owing to these characteristics, they are expected to be applied in luminescent materials, magnetic memory devices, and oxidation catalysts.1–6 The shape of the electronic orbitals in metal clusters resembles that of atomic orbitals (s, p, d, etc.). Therefore, metal clusters can be regarded as “superatoms”.7–17In recent years, three main approaches (Scheme S1†) have been explored for developing materials using superatoms as fundamental building units, i.e.: (1) fusing superatomic cores to form superatomic molecules;18–35 (2) arranging and crystallizing metal clusters into superlattice structures, driven by interactions between surface ligands of adjacent clusters;36–42 and (3) linking metal clusters through ligand exchange reactions with bridging ligands to form covalently bonded assemblies.43–50 The materials obtained through these approaches exhibit absorption and emission properties,19 as well as catalytic activity,51 that differ from those of individual metal clusters. Consequently, the fabrication of materials using metal clusters as building units has garnered attention as an effective strategy for unlocking novel properties based on metal clusters. However, core fusion between metal clusters in approach (1) is complex, thereby hindering its universal application to different systems. In approach (2), where crystal growth is driven by isotropic interactions between metal cluster surfaces, only a limited range of packing structures can be achieved.52,53 In approach (3), where metal clusters are linked via ligand exchange reactions, reaction control is challenging, often leading to the formation of randomly connected assemblies or amorphous polymers with multiple linkages.54 Therefore, designing and precisely synthesizing metal cluster assemblies with the desired structures using approaches (1)–(3) is a significant challenge.
In this context, to explore alternative methods of linking, research has recently focused on introducing acetylide ligands and their analogs into gold (Au) clusters. The Au clusters are coordinated by diverse types of ligands, such as phosphines and halogens, through ligand exchange reactions.55–67 For example, Tsukuda et al. synthesized a building block superatom by exchanging the two terminal chlorine ligands of [Au13(dppe)5Cl2]3+ (hereafter referred to as 1; dppe = 1,2-bis(diphenylphosphino)ethane) (Table 1 and Fig. S1A†) with terpyridine derivative acetylide ligands (chelating ligands), thereby introducing terpyridine (EPTpy = 4′-(4-ethynylphenyl)-2,2′:6′,2′′-terpyridine) at both ends of the Au13 cluster. This exchange subsequently enabled the formation of a one-dimensional polymer upon the addition of transition metal ions into the solution containing the building block superatom, wherein the metal ions coordinated with terpyridine, which led to the formation of Au13 cluster-linked polymer chains (Scheme 1A).68 Additionally, (IrAu12)2L dimers and (IrAu12)3L2 trimers were synthesized using the analogous compound [IrAu12(dppe)5(PA)2]+ (PA = phenylacetylide; Fig. S2A†) to control the cleavage of the Au–C bond (dissociation of PA) within the cluster and by introducing the diisocyanide linker L, which has isocyano groups at both ends (Scheme 1B).56 The approach shown in Scheme 1A affords the facile synthesis of one-dimensional assemblies by simply introducing transition metal ions. However, controlling the number of linkages is difficult. In contrast, the approach in Scheme 1B allows for control of the number of linkages in the product by combining clusters where PA is dissociated from the clusters, with one PA remaining as an end cap to suppress polymerization. However, the number of strong acid reagents suitable for PA dissociation is limited, which restricts the types of clusters that can be used for linking.
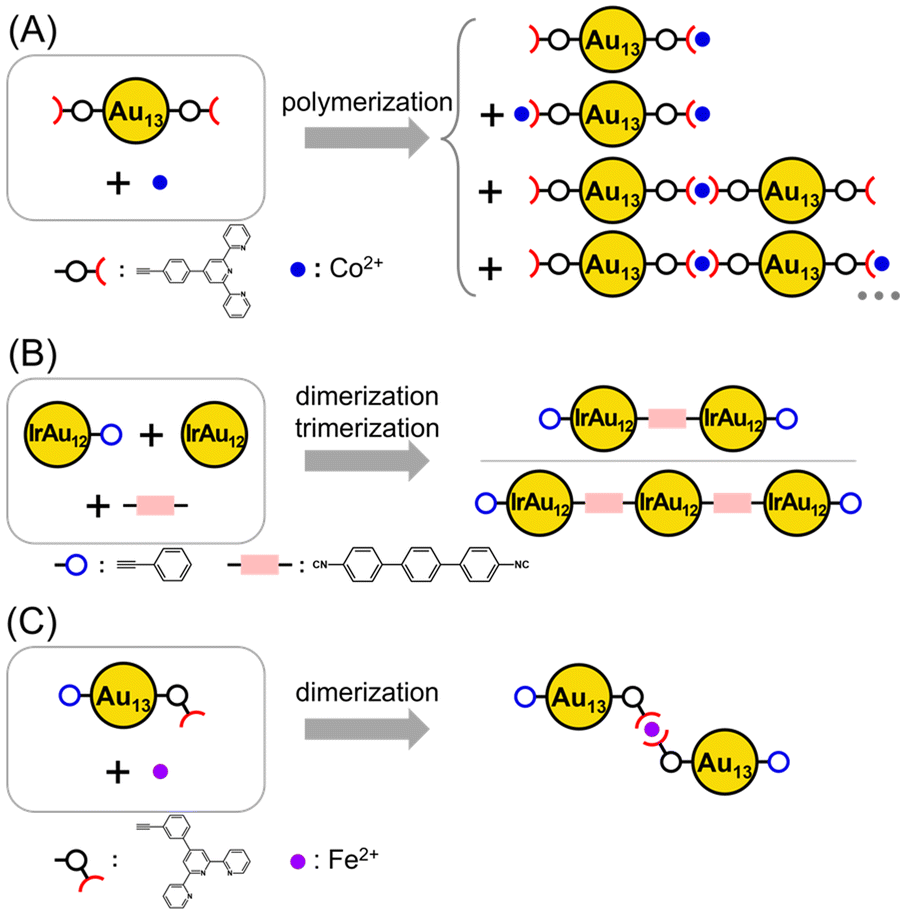 | ||
| Scheme 1 Synthesis strategies of the Au13-based cluster linkage. (A) Polymer formation through coordination bonds between transition metal ions and chelating ligands.68 (B) Au atom exposure by selective desorption of halogen and dimer and trimer formation by reaction with an isocyanide linker.56 (C) Selective dimer formation by complexation with building block molecules containing end-cap ligands and chelating ligands programmed to form dimers (this work). | ||
| Number | Chemical formula |
|---|---|
| 1 | [Au13(dppe)5Cl2]3+ |
| 20 | [Au13(dppe)5(PA)2]3+ |
| 21 | [Au13(dppe)5(PA)(C2PhTpy)]3+ |
| 22 | [Au13(dppe)5(C2PhTpy)2]3+ |
| 3 | [Fe(TpyPhC2H)2]2+ |
In the present study, we developed an alternative approach that focuses on driving the formation of oligomers through simple coordination between chelating ligands and transition metal ions. By introducing chelating ligands to the Au cluster surface and end-capping ligands to suppress polymerization, we developed building block molecules programmed to self-assemble into dimers upon the addition of metal ions (Scheme 1C). Indeed, the designed building block clusters efficiently formed stable dimers consisting of two Au clusters in the presence of iron ions. Furthermore, the dimers have been shown to exhibit a photo-induced charge transfer property.
Experimental
Synthesis of 1·(BF4−)3
First, the precursor complex Au2Cl2(dppe) was synthesized using a method reported by Corma et al.69 Specifically, 350 μmol of tetrachloroauric acid was dissolved in 2.5 mL of methanol. To this solution, 109 μL (1.05 mmol) of thiodiglycol, dissolved in 500 μL of methanol, was added, and the reaction mixture was stirred at room temperature for 1 h. After the solution changed from yellow to colorless and transparent, 69.7 mg (175 μmol) of dppe, dissolved in 4 mL of a chloroform![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene (1:1 v/v) mixed solvent, was added, and the mixture was stirred at room temperature for 2 h. The resulting white precipitate was washed with methanol to yield Au2Cl2(dppe).
:toluene (1:1 v/v) mixed solvent, was added, and the mixture was stirred at room temperature for 2 h. The resulting white precipitate was washed with methanol to yield Au2Cl2(dppe).
Next, 1·(Cl−)3 (Fig. S1A†) was synthesized using a method from the literature13 but without the addition of sodium sulfide. Specifically, 126 mg (100 μmol) of Au2Cl2(dppe) was added to 20 mL of dichloromethane. To this solution, a sodium borohydride aqueous solution (3.8 mg (100 μmol) per 1 mL) at 0 °C was added. The solution was then stirred at room temperature for 24 h. After evaporating the solvent, the resulting solid was washed with water, a hexane/dichloromethane mixture, and acetone to yield 1·(Cl−)3 (Fig. S3 (curve a)†). The obtained 1·(Cl−)3 was dissolved in ethanol, and an excess of sodium tetrafluoroborate was added to exchange the counter anion from Cl− to BF4−, yielding 1·(BF4−)3 as a powder (Fig. S3 (curve b)†).12,13
Formation of dimers via complex formation
The acetylene derivative 4′-(3-ethynylphenyl)-2,2′:6′,2′′-terpyridine (H-C2PhTpy) (Fig. S2B†), which contains a terpyridine unit that can coordinate with transition metal ions, was synthesized in the following three steps.70–72 The details of the organic synthesis are provided in the ESI.† First, the terpyridine framework (–PhTpy) was formed via Kröhnke pyridine synthesis using 3-bromobenzaldehyde, 2-acetylpyridine, and ammonia (Fig. S4A†). Next, the Sonogashira coupling reaction was used to introduce a trimethylsilylethynyl group at the 3-position of the phenylene ring (Fig. S4B†). Finally, the trimethylsilyl protecting group was removed using fluoride ions, yielding the white target compound H-C2PhTpy (Fig. S4C†) with a final yield of 13%.To prevent the formation of polymers larger than dimers, owing to coordination, we synthesized a Au13 cluster (building block cluster; BB cluster) containing H-C2PhTpy and the end-cap ligand phenylacetylene (H-PA). Specifically, 6.8 mg (1.39 μmol) of 1·(BF4−)3 was dissolved in 33.4 mL of acetonitrile. To this solution, 19.4 μL (139 μmol) of triethylamine, 13.8 μL (126 μmol) of H-PA, and 4.6 mg (13.8 μmol) of H-C2PhTpy were added, and the solution was stirred (concentration ratio 1:H-PA:H-C2PhTpy = 1:90:10 or 1:74:26). Using this method, the BB cluster [Au13(dppe)5(PA)2−x(C2PhTpy)x]3+ (where x = 0–2; hereafter referred to as 20 (x = 0; Fig. S1B†), 21 (x = 1; Fig. S1C†), and 22 (x = 2); Table 1) was obtained, in which two types of acetylide ligands were introduced to the surface of the Au13 cluster.
The obtained BB cluster was dissolved in acetonitrile, and a solution of iron(II) tetrafluoroborate (Fe(BF4−)2) in acetonitrile (12.3 μM) was added. Complex formation was monitored using ultraviolet–visible (UV–vis) absorption spectroscopy. The chemical composition of the products after complexation was evaluated by mass spectrometry. For products obtained by adding Fe(BF4−)2 in excess of the equivalence point, mass spectrometry was conducted after removing excess Fe2+.
Evaluation of luminescence lifetime and quenching experiments
The luminescence decay curves of the dimers obtained from complexation were obtained using a pico-TAS system from UNISOKU (Hirakata City, Osaka, Japan). Excitation light with a wavelength of 520 nm was used. Prior to the measurements, the solutions were deoxygenated by argon bubbling. The primary objective of the measurement was to observe the photoinduced charge transfer behavior of the BB cluster. The Fe2+ terpyridine complex [Fe(TpyPhC2H)2](BF4−)2, hereafter referred to as 3·(BF4−)2 (Table 1 and Fig. S5†), was used as the quenching agent to quench the luminescence of the Au13 component.Formation of dimers via the introduction of a diyne
An attempt was also made to synthesize a linkage of Au13 clusters by reacting compound 1 with a diyne having ethynyl groups at both ends (Scheme S2B†). Specifically, 49 mg (10 μmol) of 1·(Cl−)3 was dissolved in 33 mL of acetonitrile. To this solution, 1.3 mg (10 μmol) of 1,4-diethynylbenzene (deb-H2; Fig. S2C†) and 14 μL (100 μmol) of triethylamine were added, followed by stirring the solution ([1]:[deb-H2] = 1:1). The solution was then dried, and excess ligands, bases, and impurities were washed with water, hexane, and toluene. The chemical composition of the obtained product was evaluated by mass spectrometry.
Results and discussion
Influence of counter ions on ligand exchange reactions
We first discuss the effect of the counteranion type on the ligand exchange reaction.Fig. 1 shows the electrospray ionization (ESI) mass spectra of 20·(Cl−)3 (Fig. S1B and S3 (curve c)†) and the product obtained by mixing 20·(Cl−)3 with Fe(BF4−)2. The mass spectrum of the product featured peaks corresponding to [Au13(dppe)5(PA)2−xClx](BF4−)3 (x = 0–2). This finding indicates that during the reaction, ligand exchange occurred between the counteranion Cl− of the Au13 cluster and PA (eqn (1)).
 | (1) |
 | ||
| Fig. 1 Positive-ion ESI mass spectra of (A) 20·(Cl−)3 and (B) the product obtained by mixing 20·(Cl−)3 and [BF4]− in acetonitrile solution. | ||
Thus, (1) the Cl− changes its role from a counteranion to a ligand depending on the reaction conditions and (2) under conditions where Cl− ions are present, ligand exchange occurs from the acetylide ligand to Cl−.
To achieve dimerization via complexation, it is essential to introduce only Fe(BF4−)2 into the system, while maintaining the structure of the BB cluster. To suppress the occurrence of the aforementioned ligand exchange during mixing, [BF4]− was consistently used as a counteranion of the precursor Au13 cluster (e.g., 1·(BF4−)3 and 20–1·(BF4−)3). In the experiment, the addition of NaBF4 to an ethanol solution of 1·(Cl−)3 induced counteranion exchange and changes in the solubility of the cluster, resulting in the precipitation of 1·(BF4−)3 (Fig. S3 (curve b)†).
Formation of dimers via complex formation
Fig. 2A (curve a) shows the ESI mass spectrum of the product obtained by adding H-PA and H-C2PhTpy to an acetonitrile solution of 1·(BF4−)3 in the presence of a base (triethylamine). The mass spectrum mainly exhibited peaks attributed to 20 and 21. This result confirms that two Cl− ligands on the Au13 cluster surface were substituted by PA or C2PhTpy. When C2PhTpy was introduced more excessively, 22 formed and the subsequent addition of Fe2+ could result in the formation of dimers or larger assemblies (Scheme S2A†). Therefore, during the synthesis of the BB cluster, the molar ratio of acetylide ligands to H-PA:H-C2PhTpy was set to 0.9:0.1 to minimize the formation of 22, while achieving the desired formation of 21 (Fig. S6 and Table S1†).
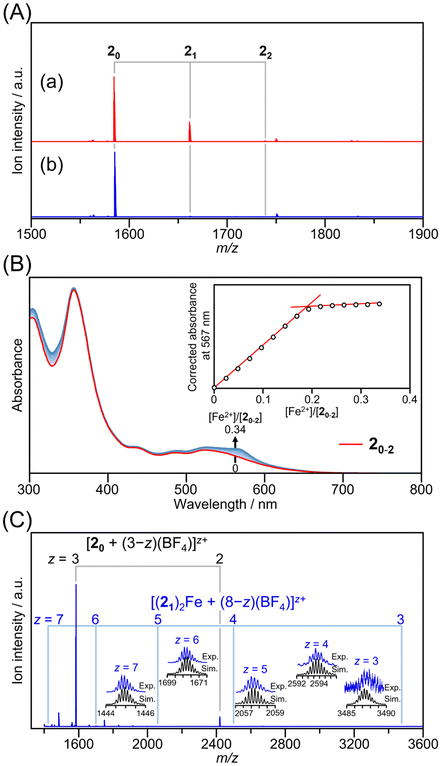 | ||
| Fig. 2 (A) ESI mass spectra of the BB cluster containing 20–2 prepared at a H-PA:H-C2PhTpy molar ratio of 0.9:0.1 (a) and the product obtained from the reaction between the BB cluster containing 20–2 and Fe(BF4−)2 (b). (B) UV–vis absorption spectra of the BB cluster containing 20–2 as a function of concentration of Fe(BF4−)2. The inset shows the titration curve monitored at the characteristic absorption peak of 567 nm, which can be attributed to the MLCT transition of the [Fe(Tpy)2]2+ moiety. (C) Wide-range ESI mass spectrum of the product obtained from the reaction between the BB cluster containing 20–2 and Fe(BF4−)2. | ||
The composition distribution of the ligands in the obtained products differed from the binomial distribution predicted from the molar ratio of ligands added during synthesis (Fig. S6 and Table S2†). This result indicates that there is a difference in the ligand exchange ability between the two types of acetylide ligands used in this study. The observed deviation suggests that C2PhTpy is more likely to replace the Cl− ligand of the Au13 cluster than PA. C2PhTpy has a bulkier geometric structure than PA (Fig. S2A and B†). Nevertheless, C2PhTpy is more easily introduced into the Au13 cluster than PA, which is attributed to the difference in the ease of proton dissociation of the acetylene groups (pKa) of both ligands. The 1H NMR measurements revealed that the NMR shifts of the acetylene (H–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) protons in H-C2PhTpy and H-PA were 3.15 ppm (Fig. S4C†) and 3.07 ppm (TCI Product Number: 3RJHI), respectively, indicating that the electron density of the hydrogen in H-C2PhTpy is lower than in H-PA. This suggests that H-C2PhTpy more readily deprotonates than H-PA. This difference makes H-C2PhTpy more nucleophilic than H-PA and more likely to attack the Au13 core; therefore, H-C2PhTpy is more readily incorporated into the Au13 cluster than H-PA.
C) protons in H-C2PhTpy and H-PA were 3.15 ppm (Fig. S4C†) and 3.07 ppm (TCI Product Number: 3RJHI), respectively, indicating that the electron density of the hydrogen in H-C2PhTpy is lower than in H-PA. This suggests that H-C2PhTpy more readily deprotonates than H-PA. This difference makes H-C2PhTpy more nucleophilic than H-PA and more likely to attack the Au13 core; therefore, H-C2PhTpy is more readily incorporated into the Au13 cluster than H-PA.
Fig. 2B shows the optical absorption spectrum of the BB cluster (red line). The absorption profile is comparable to those previously reported for 20·(PF6−)3 and [Au13(dppe)5(EPTpy)2]·(PF6)3.55,68 This similarity indicates that the structure of the terminal functional groups of the acetylide ligands has little effect on the optical absorption properties of the cluster.
The addition of an Fe(BF4−)2 acetonitrile solution to the BB cluster containing 20–2 resulted in an increase in absorbance at ∼567 nm (Fig. 2B). The peak displayed a comparable shape and absorption wavelength to the metal-to-ligand charge transfer (MLCT) absorption peak of [Fe(Tpy)2]2+, which is formed by Fe2+ and two Tpy molecules. This result suggests that the addition of Fe2+ led to the formation of a complex between Fe2+ and the Tpy sites of 21.
The inset in Fig. 2B illustrates the correlation between the concentration ratio [Fe2+]/[20–2] and the absorbance at ∼567 nm. As the [Fe2+]/[20–2] ratio increased, the absorbance increased linearly up to [Fe2+]/[20–2] = 0.18, after which there was no increase or decrease in absorbance. This result suggests that [Fe2+]/[20–2] = 0.18 represents the equivalence point of complex formation ([Fe2+]/[20–2]eq.). Typically, for the formation of [Fe(Tpy)2] from Fe2+ and two Tpy molecules, the equivalence point is at [Fe2+]/[Tpy]eq. = 0.5.73 However, because the products obtained in this study mainly consist of 20 and 21, the equivalence point is lower. By introducing the composition distribution obtained from the ESI mass spectrometry data into the stoichiometric equation (eqn (S10)†), a value of [Fe2+]/[20–2]eq. = 0.13 is obtained. This value is in relatively good agreement with the value of [Fe2+]/[20–2]eq. = 0.18 determined from the absorbance measurements, suggesting that 21, formed through ligand exchange, efficiently complexes with Fe2+.
Fig. 2A (curve b) shows the mass spectrum of the product obtained after adding an excess amount of Fe(BF4−)2 beyond the equivalence point and removing excess Fe2+. The peak corresponding to 21 was absent and only the peak corresponding to unreacted 20 remained. When examining the mass spectrum over a broader range, peaks attributed to [(21)2Fe + (8 − z)(BF4)]z+ (z = 3–7) were observed in the high mass-to-charge (m/z) ratio region (Fig. 2C). These peaks correspond to Au13 dimers formed by the complexation of Fe2+ with two 21 units. For example, the charge state of +7 of the dimer molecule can be explained by a combination of two Au13 units with a charge of +3, one Fe ion with a charge of +2, and one counteranion [BF4]−. Since the product used in this study (Fig. 2A, curve a) contains little of compound 22, which is capable of forming higher-order assemblies, oligomers such as trimers (n = 3) or tetramers (n = 4), represented as (21)2(22)n−2Fen−1 (n ≥ 3), were scarcely observed in the mass spectrum. These results indicate that BB clusters were successfully designed to selectively form dimers as the connected structure by simply adding Fe2+ (Fig. S7†).
The Au13 cluster carries a charge of +3, and during dimer formation, there should be electrostatic repulsion between the Au13 units as well as between the Fe2+ ions. However, the stability constant (β) for the complexation between Fe2+ and Tpy, Fe2+ + 2Tpy ⇄ [Fe(Tpy)2], is large (β = 1020.9 M−2),74 indicating that complex formation is thermodynamically favorable. This large stability constant likely contributes to the efficient formation of dimers through complexation despite electrostatic repulsion. This thermodynamic driving force enables the successful formation of dimers despite competing repulsive interactions.
The formation of dimers via complexation was achieved but the ion intensity of the dimer in the mass spectrum was weak. These dimers have a significantly larger mass than the monomer, which reduces their detection efficiency in mass spectrometry. Additionally, because [BF4]− attached to the dimer with various combinations, the peaks originating from the dimer were observed across a range of m/z ratios. Furthermore, the addition of [BF4]− causes a widening of the isotope distribution, further decreasing the apparent intensity of the peaks. For example, in Fig. 2C, peaks corresponding to the addition of 1–5 [BF4]− ions to the dimer were observed, resulting in charge states ranging from +3 to +7. These factors contribute to the apparent low yield of the dimer in the mass spectrum. However, from the data in Fig. 2A (curve b) and Fig. 2B, we estimate that all 21 units included in the mixture with a 24.4% molar ratio were likely forming dimers at the equivalence point of [Fe2+]/[20–2] = 0.18.
Photoexcitation characteristics of dimers
To investigate the photophysical properties of the synthesized (21)2Fe dimer, its luminescence decay curve was evaluated. Fig. 3A shows the emission decay curve of the sample containing 20 and (21)2Fe. The experimentally obtained decay curve could be fitted to a biexponential decay model with short and long lifetime components of 7.8 ns and 3.5 μs, respectively. To identify these two emission species, the luminescence lifetime of the BB cluster was evaluated. The emission decay of the BB cluster could be fitted to a nearly single exponential decay, with a lifetime of 3.5 μs (Fig. 3B). This value is consistent with the long-lived component observed in the sample containing 20 and (21)2Fe. Therefore, the luminescence lifetime of (21)2Fe can be assigned as 7.8 ns. This value is comparable to that reported by Tsukuda et al. for a one-dimensional polymer composed of Au13 and Co2+, which supports the validity of this assignment.68 From these results, the following conclusions can be drawn: (1) the luminescence lifetime of the BB cluster remains roughly constant and is independent of the type of acetylide ligand;55 (2) quenching of the Au13-derived luminescence occurs upon complex formation between 21 and Fe2+; and (3) in the presence of (21)2Fe containing Fe(Tpy)2 moieties, the 20 luminescence is not quenched. | ||
| Fig. 3 (A) Photoluminescence decay curve of the sample containing the 20 monomer and the (21)2Fe dimer. The inset shows an enlarged decay curve around 0–80 ns. (B) Photoluminescence decay curve of the building block cluster 20–2. The inset shows the Stern–Volmer plot based on the luminescence lifetimes of 20–2 in the presence of 3 as a quencher. (C) Redox potentials of 20–2·(BF4−)3 and 3·(BF4−)2. | ||
Previous reports on Au13 clusters, such as Au13(dppe)5(EPTpy)2 and transition metal ions (Ni2+, Co2+, Cu2+), have shown that the luminescence intensity and lifetime of Au13 decrease upon complexation.68 This phenomenon is believed to be due to the energy transfer from the excited state of Au13 to the transition metal ion. In our study of the complexation between 21 and Fe2+, the MLCT transition of [Fe(Tpy)2]2+ was observed in the long-wavelength region, but there was no significant overlap with the Au13 emission spectrum in that absorption wavelength region (Fig. S8†). Therefore, the quenching observed upon Fe2+ complexation of 21 is likely due to photoinduced charge transfer, rather than Förster-type resonance energy transfer.75 Our present study reveals that [Fe(Tpy)2]2+ can act as a charge transfer quencher for Au13 luminescence. However, as mentioned earlier, 20 is not quenched by the coexisting complex component (21)2Fe dimer.
To confirm this, a quenching experiment was conducted using 3·(BF4−)2, which has a similar structure to that of the constituent unit of the dimer, as the quencher against the 20–2-containing BB cluster (Fig. 3B inset and Fig. S9†). Although attempts were made to observe quenching using steady-state luminescence, the absorption band of 3 overlapped with the entire absorption spectrum of 20–2, resulting in a decrease in emission intensity owing to the excitation inner filter effect. Therefore, steady-state emission quenching experiments were deemed inappropriate. In contrast, time-correlated single-photon counting measurements of the emission decay confirmed that the emission lifetime of 20–2 remained constant regardless of the concentration of 3 (Fig. S10†). This finding indicates that 3 does not act as a quencher for 20–2, and considering the redox potentials of 20–2·(BF4−)3 and 3·(BF4−)2, it is evident that the driving force for photoinduced electron transfer from 20–2 to 3 is very small (Fig. 3C and Fig. S11†). Therefore, the drastic decrease in emission lifetime due to dimerization can be attributed to the shortening of the excited-state of Au13, which results from the chemical direct binding of a metal complex to the emission site of Au13. Although the driving force is small, the metal complex has the potential to act as a quenching site. This finding demonstrates that even combinations of clusters and transition metal ions that would not typically cause quenching can cause photoinduced charge transfer in molecules created by the linkage of these moieties, thereby enabling luminescence quenching.
Formation of dimers via the introduction of a diyne
We also examined a different method to synthesize the linkage. An attempt was made to synthesize a linkage of Au13 clusters by reacting compound 1 with a diyne having ethynyl groups at both ends (Scheme S2B†). Fig. 4 shows the ESI mass spectrum of the product obtained by adding deb-H2 (Fig. S2C†) to an acetonitrile solution of 1·(BF4−)3 in the presence of a base (triethylamine). The peak with the strongest ion intensity was attributed to [Au13(dppe)5(deb-H)2]3+. This result indicates that the Cl ligands on the Au13 surface were substituted by the [deb-H]− anion generated through deprotonation of one of the hydrogens of deb-H2 by the base. In contrast, no distinct peaks were observed in the expected m/z range (1550, 1565, 1580) associated with dimer formation. If both terminal hydrogens of [deb-H2] are deprotonated by the base, [deb]2− is expected to replace one Cl on each Au13 cluster, forming the desired dimer. However, if both Cl ligands on each Au13 cluster are replaced by [deb]2−, this would lead to the formation of a polymer connecting multiple Au13 clusters (Scheme S2B†). As the reaction proceeded to form structures linking many Au13 clusters, we conclude that selective formation of the desired dimer cannot be achieved using this method. | ||
| Fig. 4 Positive-ion ESI mass spectrum of the product obtained by mixing 1 and deb-H2 in acetonitrile solution in the presence of a base. | ||
Accordingly, we speculated that selective dimer formation might be possible by replacing one of the Cl ligands of 1 with PA, forming Au13(dppe)5Cl(PA). However, the findings revealed that ligand exchange occurred between the halogen and acetylide ligands, as well as between the different acetylides on the Au13 cluster (Fig. S12†). To examine the ligand exchange ability of the acetylide ligands, a model experiment was conducted by adding deb-H2 and triethylamine to 20·(Cl−)3 (Fig. S1B and S3 (curve c)†). As observed in Fig. S12,† the mass spectrum of the product featured peaks corresponding to [Au13(dppe)5(PA)2−x(deb-H)x]3+. This indicates that ligand exchange reactions occur even between acetylide ligands, and that ligand exchange reactions involving halogens or acetylides on the Au13 cluster are reversible, similarly to thiol or thiolate exchange reactions in thiolate-protected Au clusters such as Au25(SR)18.57,76–80 Therefore, control over the reaction and selective formation of the desired linkage structure is difficult using bridging ligands such as deb-H2. In contrast, the approach using the aforementioned complexation and end-cap ligands is a more straightforward and precise synthetic approach.
Conclusions
In this study, we focused on Au13 clusters with phosphine ligands and chlorine or acetylide as the coordinating ligands, [Au13(dppe)5X2]3+ (X = Cl or acetylide), to investigate (1) the behavior of ligand exchange reactions and (2) the development of selective and efficient methods for the formation of dimers using Au13 clusters. The following key findings were obtained. (1) Under certain conditions, Au13 clusters with Cl− as a counteranion can undergo a transformation where Cl− no longer acts as the counteranion but instead directly binds to the Au13 surface as a coordinating ligand. (2) The introduction of two types of ligands—chelating ligands that can complex and end-cap ligands that can suppress polymerization—resulted in the development of building block clusters that were programmed to form dimers consisting of two Au13 clusters. By applying Fe2+ ions to these designed building block clusters, we succeeded in efficiently forming a dimer consisting of two Au13 molecules. Furthermore, these dimers, having a charge donor and acceptor chemically bound together, displayed a higher rate constant for light-induced charge transfer than typical endothermic complexes. Future studies are expected to further investigate the new properties resulting from dimerization.Author contributions
Y. Niihori and Y. Negishi conceived the idea for this study and supervised the project. T. Kosaka and Y. Niihori conducted the synthesis and characterization. T. Kosaka, T. Kawawaki and Y. Niihori wrote the manuscript and Y. Negishi revised the manuscript. All authors discussed the results and approved the final version of the manuscript.Data availability
Relevant data are available from the corresponding authors (Y. Niihori and Y. Negishi) upon reasonable request.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We gratefully acknowledge Mr Ryusei Kaneko and Prof. Shinichi Saito (Tokyo University of Science) for supporting the synthesis of terpyridine derivatives. We thank Dr Tatsuo Nakagawa and Toshiaki Suzuki in UNISOKU Co., Ltd, for conducting the measurement of the photoluminescence decay curves. Computation was performed at the Research Center for Computational Science, Okazaki, Japan (project no. 24-IMS-C282). This research was financially supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant no. 22K04858, 22K19012, 23KK0098, and 23H00289), the Yazaki Memorial Foundation for Science and Technology, and The Sumitomo Foundation.References
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS PubMed.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS PubMed.
- Y. Negishi, T. Nakazaki, S. Malola, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206–1212 CrossRef CAS PubMed.
- K. Kwak, Q. Tang, M. Kim, D.-E. Jiang and D. Lee, J. Am. Chem. Soc., 2015, 137, 10833–10840 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- J.-I. Nishigaki, K. Koyasu and T. Tsukuda, Chem. Rec., 2014, 14, 897–909 CrossRef CAS PubMed.
- D. M. P. Mingos, Dalton Trans., 2015, 44, 6680–6695 RSC.
- L. Cheng and J. Yang, J. Chem. Phys., 2013, 138, 141101 CrossRef PubMed.
- B. K. Teo and H. Zhang, Coord. Chem. Rev., 1995, 143, 611–636 CrossRef CAS.
- C. E. Briant, B. R. C. Theobald, J. W. White, L. K. Bell, D. M. P. Mingos and A. J. Welch, J. Chem. Soc., Chem. Commun., 1981, 201–202 RSC.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- J. Zhang, Y. Zhou, K. Zheng, H. Abroshan, D. R. Kauffman, J. Sun and G. Li, Nano Res., 2018, 11, 5787–5798 CrossRef CAS.
- R. C. B. Copley and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1996, 491–500 RSC.
- R. C. B. Copley and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1992, 1755–1756 RSC.
- M. Laupp and J. Strähle, Angew. Chem., Int. Ed. Engl., 1994, 33, 207–209 CrossRef.
- X. Kang, L. Xiong, S. Wang, Y. Pei and M. Zhu, Chem. Commun., 2017, 53, 12564–12567 RSC.
- Y. Niihori, S. Miyajima, A. Ikeda, T. Kosaka and Y. Negishi, Small Sci., 2023, 3, 2300024 CrossRef CAS PubMed.
- S. Miyajima, S. Hossain, A. Ikeda, T. Kosaka, T. Kawawaki, Y. Niihori, T. Iwasa, T. Taketsugu and Y. Negishi, Commun. Chem., 2023, 6, 57 CrossRef CAS PubMed.
- K. Nobusada and T. Iwasa, J. Phys. Chem. C, 2007, 111, 14279–14282 CrossRef CAS.
- B. K. Teo, X. Shi and H. Zhang, J. Chem. Soc., Chem. Commun., 1992, 1195–1196 RSC.
- B. K. Teo, X. Shi and H. Zhang, J. Am. Chem. Soc., 1991, 113, 4329–4331 CrossRef CAS.
- B. K. Teo, H. Zhang and X. Shi, Inorg. Chem., 1990, 29, 2083–2091 CrossRef CAS.
- B. K. Teo, X. Shi and H. Zhang, J. Cluster Sci., 1993, 4, 471–476 CrossRef CAS.
- Z. Qin, J. Zhang, C. Wan, S. Liu, H. Abroshan, R. Jin and G. Li, Nat. Commun., 2020, 11, 6019 CrossRef CAS PubMed.
- B. K. Teo and H. Zhang, Angew. Chem., Int. Ed. Engl., 1992, 31, 445–447 CrossRef.
- C. Zhou, P. Pan, X. Wei, Z. Lin, C. Chen, X. Kang and M. Zhu, Nanoscale Horiz., 2022, 7, 1397–1403 RSC.
- B. K. Teo, H. Dang, C. F. Campana and H. Zhang, Polyhedron, 1998, 17, 617–621 CrossRef CAS.
- X. Lin, J. Tang, J. Zhang, Y. Yang, X. Ren, C. Liu and J. Huang, J. Chem. Phys., 2021, 155, 074301 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- L. Xu, Q. Li, T. Li, J. Chai, S. Yang and M. Zhu, Inorg. Chem. Front., 2021, 8, 4820–4827 RSC.
- Y. Negishi, K. Igarashi, K. Munakata, W. Ohgake and K. Nobusada, Chem. Commun., 2012, 48, 660–662 RSC.
- E. Ito, S. Ito, S. Takano, T. Nakamura and T. Tsukuda, JACS Au, 2022, 2, 2627–2634 CrossRef CAS PubMed.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- R. Saito, K. Isozaki, Y. Mizuhata and M. Nakamura, J. Am. Chem. Soc., 2024, 146, 20930–20936 CrossRef CAS PubMed.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Science, 1995, 270, 1335–1338 CrossRef CAS.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- B. Yoon, W. D. Luedtke, R. N. Barnett, J. Gao, A. Desireddy, B. E. Conn, T. Bigioni and U. Landman, Nat. Mater., 2014, 13, 807–811 CrossRef CAS PubMed.
- H. Li, X. Kang and M. Zhu, Acc. Chem. Res., 2024, 57, 3194–3205 CrossRef CAS PubMed.
- C. Wang, X. Bu, N. Zheng and P. Feng, J. Am. Chem. Soc., 2002, 124, 10268–10269 CrossRef CAS PubMed.
- D. Wang, P. Wang, Z. Liang, Z. Li, N. Liu and Q. Ma, Chem. Eng. J., 2023, 478, 147512 CrossRef CAS.
- N. K. Chaki and K. P. Vijayamohanan, J. Phys. Chem. B, 2005, 109, 2552–2558 CrossRef CAS PubMed.
- Y. Saito, Y. Shichibu and K. Konishi, Nanoscale, 2021, 13, 9971–9977 RSC.
- T. Sekine, J. Sakai, Y. Horita, H. Mabuchi, T. Irie, S. Hossain, T. Kawawaki, S. Das, S. Takahashi, S. Das and Y. Negishi, Chem. – Eur. J., 2023, 29, e202300706 CrossRef CAS PubMed.
- S. Das, T. Sekine, H. Mabuchi, S. Hossain, S. Das, S. Aoki, S. Takahashi and Y. Negishi, Chem. Commun., 2023, 59, 4000–4003 RSC.
- J. Sakai, S. Biswas, T. Irie, H. Mabuchi, T. Sekine, Y. Niihori, S. Das and Y. Negishi, Nanoscale, 2023, 15, 12227–12234 RSC.
- R. Nakatani, S. Biswas, T. Irie, J. Sakai, D. Hirayama, T. Kawawaki, Y. Niihori, S. Das and Y. Negishi, Nanoscale, 2023, 15, 16299–16306 RSC.
- Z. Wang, Y.-J. Zhu, Y.-Z. Li, G.-L. Zhuang, K.-P. Song, Z.-Y. Gao, J.-M. Dou, M. Kurmoo, C.-H. Tung and D. Sun, Nat. Commun., 2022, 13, 1802 CrossRef CAS PubMed.
- Y.-P. Xie, J.-L. Jin, G.-X. Duan, X. Lu and T. C. W. Mak, Coord. Chem. Rev., 2017, 331, 54–72 CrossRef CAS.
- L.-M. Zhang and T. C. W. Mak, J. Am. Chem. Soc., 2016, 138, 2909–2912 CrossRef CAS PubMed.
- T. Higaki, Y. Li, S. Zhao, Q. Li, S. Li, X.-S. Du, S. Yang, J. Chai and R. Jin, Angew. Chem., Int. Ed., 2019, 58, 8291–8302 CrossRef CAS PubMed.
- T. Chen, S. Yang, Q. Li, Y. Song, G. Li, J. Chai and M. Zhu, Nanoscale Horiz., 2021, 6, 913–917 RSC.
- T. Higaki, C. Liu, C. Zeng, R. Jin, Y. Chen, N. L. Rosi and R. Jin, Angew. Chem., Int. Ed., 2016, 55, 6694–6697 CrossRef CAS PubMed.
- Z.-R. Wen, Z.-J. Guan, Y. Zhang, Y.-M. Lin and Q.-M. Wang, Chem. Commun., 2019, 55, 12992–12995 RSC.
- M. Sugiuchi, Y. Shichibu, T. Nakanishi, Y. Hasegawa and K. Konishi, Chem. Commun., 2015, 51, 13519–13522 RSC.
- Y. Fukumoto, T. Omoda, H. Hirai, S. Takano, K. Harano and T. Tsukuda, Angew. Chem., Int. Ed., 2024, 63, e202402025 CrossRef CAS PubMed.
- C. A. Hosier, I. D. Anderson and C. J. Ackerson, Nanoscale, 2020, 12, 6239–6242 RSC.
- X.-K. Wan, J.-Q. Wang, Z.-A. Nan and Q.-M. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef PubMed.
- Q. Tang and D.-E. Jiang, J. Phys. Chem. C, 2015, 119, 10804–10810 CrossRef CAS.
- Q. Tang and D.-E. Jiang, Chem. Mater., 2017, 29, 6908–6915 CrossRef CAS.
- F. Bejarano, I. J. Olavarria-Contreras, A. Droghetti, I. Rungger, A. Rudnev, D. Gutiérrez, M. Mas-Torrent, J. Veciana, H. S. J. van der Zant, C. Rovira, E. Burzurí and N. Crivillers, J. Am. Chem. Soc., 2018, 140, 1691–1696 CrossRef CAS PubMed.
- X.-K. Wan, Z.-J. Guan and Q.-M. Wang, Angew. Chem., Int. Ed., 2017, 56, 11494–11497 CrossRef CAS PubMed.
- T. Zaba, A. Noworolska, C. M. Bowers, B. Breiten, G. M. Whitesides and P. Cyganik, J. Am. Chem. Soc., 2014, 136, 11918–11921 CrossRef CAS PubMed.
- K. P. Hall and D. M. P. Mingos, Prog. Inorg. Chem., 1984, 32, 237–325 CAS.
- P. Maity, T. Wakabayashi, N. Ichikuni, H. Tsunoyama, S. Xie, M. Yamauchi and T. Tsukuda, Chem. Commun., 2012, 48, 6085–6087 RSC.
- P. Maity, S. Takano, S. Yamazoe, T. Wakabayashi and T. Tsukuda, J. Am. Chem. Soc., 2013, 135, 9450–9457 CrossRef CAS PubMed.
- N. Kobayashi, Y. Kamei, Y. Shichibu and K. Konishi, J. Am. Chem. Soc., 2013, 135, 16078–16081 CrossRef CAS PubMed.
- N. Kito, S. Takano, S. Masuda, K. Harano and T. Tsukuda, Bull. Chem. Soc. Jpn., 2023, 96, 1045–1051 CrossRef CAS.
- A. Leyva-Pérez, A. Doménech-Carbó and A. Corma, Nat. Commun., 2015, 6, 6703 CrossRef PubMed.
- L. Wang, R. Liu, J. Gu, B. Song, H. Wang, X. Jiang, K. Zhang, X. Han, X.-Q. Hao, S. Bai, M. Wang, X. Li, B. Xu and X. Li, J. Am. Chem. Soc., 2018, 140, 14087–14096 CrossRef CAS PubMed.
- S.-Y. Zhang, H.-Y. Sun, R.-G. Wang, Y.-S. Meng, T. Liu and Y.-Y. Zhu, Dalton Trans., 2022, 51, 9888–9893 RSC.
- E. C. Constable, J. Lewis, M. C. Liptrot and P. R. Raithby, Inorg. Chim. Acta, 1990, 178, 47–54 CrossRef CAS.
- J. Kožíšek, J. Svoboda, J. Zedník, B. Vlčková and I. Šloufová, J. Phys. Chem. B, 2021, 125, 12847–12858 CrossRef PubMed.
- R. H. Holyer, C. D. Hubbard, S. F. A. Kettle and R. G. Wilkins, Inorg. Chem., 1965, 4, 929–935 CrossRef CAS.
- B. Valeur and M. N. Berberan-Santos, in Molecular Fluorescence, Wiley-VCH, 2012, ch. 8, pp. 213–261 Search PubMed.
- Y. Niihori, W. Kurashige, M. Matsuzaki and Y. Negishi, Nanoscale, 2013, 5, 508–512 RSC.
- Y. Niihori, Y. Kikuchi, A. Kato, M. Matsuzaki and Y. Negishi, ACS Nano, 2015, 9, 9347–9356 CrossRef CAS PubMed.
- Y. Niihori, M. Eguro, A. Kato, S. Sharma, B. Kumar, W. Kurashige, K. Nobusada and Y. Negishi, J. Phys. Chem. C, 2016, 120, 14301–14309 CrossRef CAS.
- Y. Wang and T. Bürgi, Nanoscale Adv., 2021, 3, 2710–2727 RSC.
- W. Suzuki, R. Takahata, Y. Chiga, S. Kikkawa, S. Yamazoe, Y. Mizuhata, N. Tokitoh and T. Teranishi, J. Am. Chem. Soc., 2022, 144, 12310–12320 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00724k |
| This journal is © The Royal Society of Chemistry 2025 |