
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of carbon dots from spent coffee grounds: transforming waste into potential biomedical tools†
Yingru
Zhou
 ab,
Adalberto
Camisasca
a,
Sofia
Dominguez-Gil
a,
Michał
Bartkowski
a,
Keith D.
Rochfort
bc,
Martina
Piletti
d,
Anita
White
bc,
Dorottya
Krizsan
e,
Robert
O'Connor
f,
Susan J.
Quinn
e,
Daniela
Iacopino
d,
Alex J.
Eustace
*bc and
Silvia
Giordani
*ab
ab,
Adalberto
Camisasca
a,
Sofia
Dominguez-Gil
a,
Michał
Bartkowski
a,
Keith D.
Rochfort
bc,
Martina
Piletti
d,
Anita
White
bc,
Dorottya
Krizsan
e,
Robert
O'Connor
f,
Susan J.
Quinn
e,
Daniela
Iacopino
d,
Alex J.
Eustace
*bc and
Silvia
Giordani
*ab
aSchool of Chemical Science, Dublin City University, Glasnevin, Dublin, Ireland. E-mail: silvia.giordani@dcu.ie
bLife Sciences Institute, Dublin City University, Glasnevin, Dublin, Ireland. E-mail: alex.eustace@dcu.ie
cSchool of Biotechnology, Dublin City University, Glasnevin, Dublin, Ireland
dTyndall National Institute, University College Cork, Cork, Ireland
eSchool of Chemistry, University College Dublin, Belfield, Dublin, Ireland
fSchool of Physical Sciences, Dublin City University, Glasnevin, Dublin, Ireland
First published on 3rd March 2025
Abstract
Carbon dots (CDs) are small-sized, spherical nanoparticles presenting amorphous carbon cores with nanocrystalline regions of a graphitic structure. They show unique properties such as high aqueous solubility, robust chemical inertness, and non-toxicity and can be manufactured at a relatively low cost. They are also well known for outstanding fluorescence tunability and resistance to photobleaching. Together, these properties boost their potential to act as drug delivery systems (DDSs). This work presents a low-cost synthesis of CDs by upcycling spent coffee grounds (SCGs) and transforming them into value-added products. This synthetic route eliminates the use of highly toxic heavy metals, high energy-consuming reactions and long reaction times, which can improve biocompatibility while benefiting the environment. A series of physico-chemical characterisation techniques demonstrated that these SCG-derived CDs are small-sized nanoparticles with tunable fluorescence. In vitro studies with 120 h of incubation of SCG-derived CDs demonstrated their specific antiproliferative effect on the breast cancer CAL-51 cell line, accompanied by increased reactive oxygen species (ROS) production. Importantly, no impact was observed on healthy breast, kidney, and liver cells. Confocal laser scanning microscopy confirmed the intracellular accumulation of SCG-derived CDs. Furthermore, the drug efflux pumps P-glycoprotein (P-gp) and the breast cancer resistance protein (BCRP) did not impact CD accumulation in the cancer cells.
Introduction
Cancer is a societal problem that accounted for almost 20 million new cases and 9.7 million deaths in 2022, according to the International Agency for Research on Cancer (IARC).1 The IARC estimates that there will be an increase in cancer diagnosis, resulting in over 32.6 million cases, which will result in the deaths of up to 16.9 million people annually by 2045.1 To address these issues, modern medicine is focusing its efforts on developing novel treatments and improving traditional therapies to boost the survival rate of cancer patients.2 Chemotherapy remains one of the most common treatment methods, aiming to stop cancer cells by inhibiting their division and growth. Chemotherapy can have good anti-cancer efficiency but is limited by several drawbacks, including multidrug resistance (MDR), the high incidence of side effects like pain and nausea, and the development of secondary cancers.3 To overcome limitations associated with chemotherapy and to improve patient outcomes, nanomedicine is evolving rapidly to address these issues.4,5Nanomedicine involves the use of nanomaterials for the treatment, diagnosis, monitoring, control, and prevention of diseases.6 Nanomaterials with three external dimensions <100 nm are referred to as nanoparticles (NPs).7 Due to their unique physico-chemical properties and large surface-to-volume ratios, the development of NPs as drug delivery systems (DDSs) has evolved rapidly in the past two decades. Using NPs as DDSs offers significant benefits compared to chemotherapy alone by reducing treatment-associated toxicity and side effects, improving targetability, and overcoming drug resistance.8 Since doxorubicin-loaded liposomal particles (Doxil™) were approved by the FDA in 1995,9 many types of organic, inorganic, and hybrid NPs have been investigated. Carbon dots (CDs) have garnered significant interest due to their excellent optical properties, stability, dispersibility, versatile surface modification, and biocompatibility.10
Discovered in 2004,11 CDs are quasi-spherical NPs (<10 nm) featuring a carbon core structure consisting of a mix of sp2/sp3 amorphous carbon forms and a graphite lattice with sp2 carbon.10 They can be further categorised into graphene quantum dots, carbon quantum dots, carbon nanodots, and carbonised polymer dots based on their core carbon structure, surface functional groups, and properties. CDs can be synthesised by various top-down and bottom-up methods.10 The precursors of CDs vary; interestingly, CDs can also be synthesised with “green” precursors, such as food waste, which allows the synthesis to be highly sustainable and eco-friendly.10
Spent coffee grounds (SCGs) are a common daily food waste, with over 6 million tons produced annually.12 Landfills remain the common method to handle such food waste, which can lead to the generation of methane that harms the environment.13 Therefore, numerous attempts have been made to convert SCGs into reusable and value-added products. They have been used in the energy industry to produce biodiesel, fuel pellets, bioethanol, and composting materials.14,15 In addition, SCGs have also been adopted for biomedical applications such as the synthesis of bioplastic composites16 and the production of other NPs like graphite oxide.17 SCGs are typically composed of 50% cellulose and hemicellulose, 10–15% fat, and 8.5 to 13.6% nitrogenous compounds18—because of their high carbon composition, SCGs are a viable carbon source for generating CDs for sensing,19 anti-counterfeiting,20 bioimaging,21 and nanomedicine.22
CDs have high biomedical potential.23 Numerous in vitro and in vivo studies have displayed their high tolerance and low toxicity in human cell lines, bacteria, mouse, and zebrafish models.10 They have been investigated as imaging agents due to their photoluminescence properties and can serve as DDSs to target cancer cells.24 Successful surface modification, drug loading, penetration of biological barriers, and potential for improving the delivery of chemo-drugs have been reported.25
Reactive oxygen species (ROS) refer to various oxygen-containing molecules generated through cellular metabolism that are highly reactive.26 ROS play a critical role in cellular processes, influencing cell survival, cell death, differentiation, signalling, and more. A balanced level of ROS supports cellular redox activities, aiding in homeostasis; however, excessive ROS production can induce oxidative stress, damaging organelles and ultimately leading to cell death.27 CDs have been shown to modulate ROS levels in different ways, either promoting or scavenging ROS, thereby protecting or damaging cells depending on the context.28,29
Multidrug resistance (MDR) is one of the common drawbacks of chemotherapy, caused by various factors like epigenetics and drug efflux, resulting in cancer cells being more tolerant to chemo-drugs. The ATP-binding cassette (ABC) transporter family of cell membrane proteins acts on drug efflux significantly. One of the members of the ABC transporter family, multidrug resistance protein 1 (MDR1), which produces P-glycoprotein (P-gp), has been indicated to be over-expressed in cancerous tissues and aids in drug efflux during the treatment.30 Various methods have been developed to overcome such issues in chemotherapy, such as modification of anti-cancer drugs, MDR modulators, RNA interference therapy, and multifunctional NPs.31 A wide range of NPs have been reported to be promising in overcoming drug resistance caused by P-gp through mechanisms including bypassing drug efflux, controlling drug release, and disturbing metabolism.32
Herein, intrinsically fluorescent CDs were synthesised from SCGs and isolated with a novel acetone-driven precipitation method, an eco-friendly approach. These SCG-derived CDs were investigated in terms of structure, size, physico-chemical properties, and optical properties by an array of spectroscopic techniques and atomic force microscopy. The in vitro studies were carried out to assess their antiproliferative effects, impact on ROS generation, potential anti-drug resistance effect, and cellular internalisation and localisation. Our study confirms that SCG-derived CDs are novel potential agents that may have biomedical applications as DDSs.
Experimental
Materials
Coffee beans from two brands, Lavazza Qualita Rossa (LAV) and KENCO Colombian (KEN), were purchased locally in Dublin, Ireland. Sulphuric acid (H2SO4, 95–98%), nitric acid (HNO3, ≥65%), sodium hydroxide (NaOH) pellets, and acetone (≥99.5%) were purchased from Sigma-Aldrich. Qualitative filter paper was purchased from VWR. Polyethersulfone (PES) filters (0.20 μm pore size) were purchased from GVS Filter Technology (USA). Deionised water (diH2O) was purified using a MilliQ® ultra-purification system to 18.2 MΩ·cm at 25 °C.Dulbecco's modified Eagle's medium (DMEM) and RPMI-1640 medium were purchased from Sigma-Aldrich. Epithelial cell medium and hepatocyte cell medium were purchased from Innoprot. MEGM™ (Mammary Epithelial Cell Growth Medium) SingleQuots™ Kit was purchased from Lonza. 4-Nitrophenyl phosphate disodium salt hexahydrate (pNPP) was purchased from Thermo Scientific. Sulindac and elacridar were purchased from Selleckchem. DRAQ5 was purchased from Abcam. SN-38 was purchased from Selleckchem. IncuCyte® Caspase- 3/7 Green apoptosis reagent was purchased from Essen Bioscience. Dihydroethidium was purchased from Merck.
Preparation of SCG-derived CDs
5 g of spent coffee grounds (SCGs) from Lavazza and KENCO were weighed accurately and added to a round-bottomed flask. A 10 mL 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v mixture of H2SO4:HNO3 (7.5 and 2.5 mL, respectively) was prepared. The acid solution was gently transferred to the flask containing the SCGs. After the acid addition, the mixture was sonicated in a Fisher Scientific FB 15050 ultra-sonic bath at 37 kHz (sweep mode) for 2 h. Then, the reaction mixture was refluxed at 100 °C for 3 h under stirring conditions. After the reaction ended, the flask was cooled down to room temperature. The solution was diluted in 20 mL of diH2O and neutralised using NaOH 8 M. CDs were isolated from the solution by two filtration steps (5 μm paper filter and 0.20 μm pore size PES filter).
:1 v/v mixture of H2SO4:HNO3 (7.5 and 2.5 mL, respectively) was prepared. The acid solution was gently transferred to the flask containing the SCGs. After the acid addition, the mixture was sonicated in a Fisher Scientific FB 15050 ultra-sonic bath at 37 kHz (sweep mode) for 2 h. Then, the reaction mixture was refluxed at 100 °C for 3 h under stirring conditions. After the reaction ended, the flask was cooled down to room temperature. The solution was diluted in 20 mL of diH2O and neutralised using NaOH 8 M. CDs were isolated from the solution by two filtration steps (5 μm paper filter and 0.20 μm pore size PES filter).
The purification of the CDs was achieved through two distinct methods. The first method employed was the acetone-driven precipitation (AP) of the filter solution with acetone in a 1 to 1 ratio. The precipitated CDs were further washed three times with acetone and dried in a vacuum desiccator. The second method employed dialysis (D); briefly, another batch of SCG-derived CDs was dialysed using a 1 kDa Pur-A-Lyzer™ Mega dialysis kit (Sigma-Aldrich). A 1 L beaker was filled with diH2O and the dispersion was kept under constant stirring for 3 days. The solution medium used for the dialysis was changed three times every 3 h, with the final medium left overnight to remove as much excess salts as possible (Na2SO4 and NaNO3). The final dark brown solution was filtered with a 0.20 μm pore size nylon filter (GVS Filter Technology) to remove further impurities and dried using a rotary evaporator.
Characterisation
UV–vis absorption spectroscopy was carried out on a Shimadzu UV-2600 using 1 mL quartz cuvettes. The reported concentrations, 5 to 250 μg mL−1, were prepared in diH2O, and the dispersions were achieved through sonication for 15 min at 37 kHz (sweep mode). Background correction was carried out. For all spectra, the 0.1% extinction coefficient at 265 nm is reported, with a linear regression R2 value of ≥0.999.Fluorescence emission spectroscopy was carried out on a PerkinElmer LS-55 spectrofluorometer with FL WinLab software used for instrument control. A xenon flash lamp, operating at 50 Hz with a peak power output equivalent to 20 kW during continuous operation, served as the light source for both emission studies and repeated sample irradiation. All samples were prepared and dispersed in diH2O and analysed at concentrations between 5 and 5000 μg mL−1 after 15 min of sonication. The analysis parameters optimised for the SCG-derived CDs were as follows: the excitation wavelengths were 350 and 390 nm, the emission scan range was set to 360–700 nm, and the excitation and emission slit widths were 15 nm, unless otherwise specified.
Attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) was conducted using PerkinElmer Spectrum Two and Nicolet iS5 instruments under ambient conditions. The ATR crystal was a diamond with a single reflection, the detector was a DTGS KBr, and a large aperture (100) was used. Spectral resolution was optimised for a data spacing of 0.482 cm−1, and 128 sample accumulations were taken over a wavenumber range of 4000–500 cm−1 for all SCG-derived CDs. Norton Beer Strong apodization and atmospheric suppression were applied by default. Baseline correction was not performed.
1H and 13C nuclear magnetic resonance (NMR) analyses were carried out on a 600 MHz Bruker Avance. AP-LAV-CDs were dissolved in D2O (4% w/v), sonicated for 5 min, and analysed at a high scan count to achieve a good S/N ratio, specifically, 1024 scans for 1H NMR, and 4096 scans for 13C NMR.
X-ray photoelectron spectroscopy (XPS) analysis was carried out on a Scienta Omicron XPS (monochromatic Al Kα 1486.7 eV X-ray source, base pressure 5 × 10−9 mbar) with a 128 channel Argus CU detector (54.7° source analyser angle). AP-LAV-CDs were deposited on carbon pads affixed to a Cu carrier wafer. CasaXPS software was used for data processing, quantification and visualisation. The binding energy (BE) scale has been calibrated to 284.5 eV with respect to the C 1s peak.
Dynamic light scattering (DLS) and zeta potential (ZP) analyses were conducted on all SCG-derived CDs at concentrations of 10, 20, 50, 100, 250, and 500 μg mL−1. Prior to analysis, each sample was dispersed in diH2O and sonicated for 15 min, then equilibrated to 25 °C. Measurements were performed using a Malvern Zen 3600 ZetaSizer instrument in backscatter (173°) mode. DLS data were collected using a PCS1115 cell, and ZP measurements were taken with a DTS1070 cell. Each sample underwent 6–9 rounds of DLS and ZP measurements. The Malvern ZetaSizer software was used to control the instrument, acquire data, and handle initial processing, including the conversion of intensity data to volume and number data. A refractive index (RI) value of 2.05 was used for this conversion. Although RI values of 1.55 and 2.55 were also tested, the small size of the SCG-derived CDs meant that the RI parameter had virtually no impact on the data's standard deviation, as confirmed by an eta-squared (η2) test, with η2 ≤ 0.0003. Further data handling, statistical analysis, and plotting were performed using a custom Python script incorporating the matplotlib package. Outliers in the datasets for each concentration were removed using the generalised extreme studentised deviate (ESD) test with a significance level of 0.05.
AFM images were taken in amplitude modulation mode under air using Tap150Al-G tip (Budget Sensors). The instrument used was Asylum Research Olympus AFM, at a scan rate of 0.8 Hz. The target amplitude and setpoint were adjusted to 1.0 V and 500.0 mV, respectively. The amplitude inverse optical laser sensitivity (Amp InvOLS) and the spring constant of the cantilever were determined to be 92.17 nm V−1 and 29.43 nM nm−1, respectively, using thermal calibration. The drive frequency and drive amplitude of the tip were determined to be 272.110 kHz and 1.07 V, respectively, using Tune calibration. A solution of AP-LAV-CDs was prepared in diH2O with an optical absorbance of 0.1 at 330 nm. A glass microscope slide was soaked in MeOH for 1 h, rinsed in EtOH and dried under a stream of nitrogen. A 1 × 1 cm mica disc was adhered onto the middle of the glass slide using superglue and allowed to set. The top layer of the mica was cleaved using Scotch tape. 100 μL of diH2O was dropped on the mica, and onto this, 20 μL of the AP-LAV-CDs solution was dropped and mixed well with a pipette tip. The prepared glass slide was allowed to dry on a hotplate set to 50 °C for 5 min, which was then turned up to 70 °C for a further 5 min. The slide was then removed and allowed to cool to room temperature for 5 min. NP population size was assessed using Gwyddion software with the line scan method, as shown in Fig. S8B†, based on n = 610 NPs. Data visualisation was achieved using a custom script written in Python.
Transmission electron microscopy (TEM) analysis was performed on a high-resolution JEOL 2100 electron microscope equipped with a LaB6 electron source and a Gatan DualVision 600 charge-coupled device (CCD), operating at an accelerating voltage of 200 keV. NP population size was assessed using ImageJ software with the equivalent spherical diameter (ESD) method, based on n = 1494 NPs.
The quantum yield (QY) of AP-LAV-CDs was determined using a relative method.33 Rhodamine 6G (QY = 95% in ethanol, excitation wavelength 410 nm) was selected as the reference. The QY of the AP-LAV-CDs was calculated using eqn (1) given below:
 | (1) |
The AP-LAV-CDs and Rhodamine 6G solutions were prepared at concentrations adjusted to ensure optical absorbance values between 0 and 0.5 at 330 nm. The photoluminescence (PL) spectra were measured at an excitation wavelength of 341 nm, and the emission intensity was integrated using Origin software. A Gaussian function was applied to fit the peak, and the integration was performed based on this fit.
Raman spectra were obtained by analysing AP-LAV-CDs deposited on a glass slide. Ten different points on the powder were selected, and spectra were acquired at each point using a 532 nm laser with a 10× objective. The laser power was set to 10%, and the acquisition time for each spectrum was 15 seconds. The measurements were conducted using an XploRA™ PLUS Raman spectrometer with an XploRA™ PLUS confocal Raman microscope (HORIBA). Raman data handling and processing were carried out in Python. Baseline correction was carried out using cubic spline interpolation. Smoothing was applied using the Savitzky–Golay filter, with a window size of 7 and a polynomial order of 3. Spectral normalisation was employed to facilitate comparison between spectra obtained from different points. Finally, data from multiple points were averaged to obtain a representative spectrum of the sample, with n = 4. A p = 0.05 confidence band was fitted to the data. To avoid overfitting of the data, deconvolution of the spectra was only on the most prominent bands, namely the D, A and G.
Biological studies
Human breast carcinoma cell line CAL-51, human ovarian carcinoma cell line SKOV-3, and human breast epithelial cell line MCF-10A were purchased from ATCC. Human renal epithelial cells (HREpiC) and human hepatocyte cells (HH) were purchased from Innoprot. CAL-51 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), SKOV-3 cells were cultured in RPMI-1640 medium supplemented with 10% FBS, MCF-10A cells were cultured in DMEM-F12 supplemented with 10% FBS and the MEGM™ (Mammary Epithelial Cell Growth Medium) SingleQuots™ Kit. HREpiC were cultured in epithelial cell medium supplemented with 2% FBS, 1% epithelial cell growth supplement, and 1% penicillin/streptomycin solution. HH cells were cultured in hepatocyte cell medium supplemented with 5% FBS, 1% hepatocyte growth supplement, and 1% penicillin/streptomycin solution. All cell lines were maintained at 37 °C under a humidified atmosphere with 5% CO2.The cell viabilities were measured using the acid phosphatase assay. Cells were seeded onto 96-well plates and incubated for 24 h to allow attachment. The following conditions were tested: AP-LAV-CDs treatment, drug efflux pump inhibitor treatment, and combination treatments of AP-LAV-CDs and drug efflux pump inhibitors.
AP-LAV-CDs were firstly prepared in ultra-pure water at a concentration of 5 mg mL−1 and further diluted to desired concentrations in cell media. In the AP-LAV-CDs treatment, the final concentrations of AP-LAV-CDs ranged from 250 μg mL−1 down to 1 μg mL−1. In the drug efflux pump inhibitor treatment, the concentrations of sulindac tested were 100, 50, 25, and 12.5 μM, and the concentrations of elacridar tested were 1, 0.5, 0.25, and 0.125 μM. In the combined treatment, the concentrations of sulindac and elacridar tested with the AP-LAV-CDs were 12.5 μM and 0.25 μM, respectively. The final concentrations of AP-LAV-CDs in the combined treatment were 250, 125, 62.5, and 31.25 μg mL−1.
After incubating for 120 h, acid phosphatase assays were performed. The plates were washed twice with PBS, and then 100 μL of 1 mg mL−1 PNPP solution was added to each well. The plates were incubated for 80 to 110 min, followed by the addition of 50 μL of 1 M sodium hydroxide solution. Absorbance was measured using a Biotek-Synergy HT Multi-detection microplate reader at 620 nm, subtracting the reading from 405 nm. Each test was repeated three separate times, and averages were used for analysis.
ROS production was assessed using a dihydroethidium (DHE) protocol. DHE is a superoxide indicator that stains the cytoplasm of living cells blue and emits red fluorescence upon oxidation by ROS. CAL-51 and MCF-10A cells were seeded and incubated under the same conditions as in the cell viability test, except black-walled 96-well plates were used. After 120 h of incubation, DHE was added to each well to a final concentration of 3 μM. The plates were incubated for 10 minutes before being read using a Tecan Infinite F200 microplate reader. Fluorescence emission at 590 nm was measured under excitation at 560 nm. The data were normalised to the control, and standardisation was made to account for changes in cell viability.
The apoptosis assay was performed using the Incucyte® live-cell analysis system. CAL-51 cells were seeded at a density of 1500 cells per well. The cells were treated with the final concentration of 250 μg mL−1 AP-LAV-CDs or 150 nM SN38. All wells were applied with the Caspase-3/7 Green Apoptosis Reagent diluted at a 1:2000 ratio. The plate was monitored for 120 h in Incucyte, with two images per well captured every 4 h to measure cell confluency and caspase-3/7 activation.
To conduct the cellular internalisation and localisation study of AP-LAV-CDs, SKOV-3 cells were seeded onto 8-well glass-bottom μ-slides (ibidi®) at a seeding density of 3600 cells per well and incubated for 24 h to allow attachment. AP-LAV-CDs were added to the wells at final concentrations of 50 and 250 μg mL−1, while the control was treated with cell medium only. After incubating for 24, 72, and 96 h, the wells were washed twice with PBS and fixed with 4% formaldehyde for 10 min at room temperature. Following fixation, formaldehyde was removed, and the wells were washed twice with PBS and then stained with 250 μL of DRAQ5 diluted in PBS at a ratio of 1:1000. The images were captured using a Leica SP8 STED confocal microscope. A UV laser (405 nm) and a white light laser (647 nm) were used to excite the AP-LAV-CDs and DRAQ5, respectively. A magnification of 40× was used for the time course cellular accumulation study, and 63× was used for the localisation study. Images were viewed and adjusted with consistent brightness parameters applied and post-processed for the reduction of autofluorescence and artifacts. Specifically, identical parameters were applied to all images for hot pixel removal to minimise cellular autofluorescence and artefacts. Additionally, exposure equalisation across all images was achieved by normalising the red channel of each individual image via white point reduction, applying the same adjustment to the green channel. Finally, all images were uniformly adjusted by increasing the black point of the green channel by 1.96%, a value that was optimised to maximise the clipping of background autofluorescence, and minimise the clipping of intracellular fluorescence.
Statistical analysis was performed using two-tailed t-tests. One-sample t-tests for cell viability and ROS production on AP-LAV-CDs, and drug efflux pump inhibitor studies; two-sample t-tests for the combined treatments in anti-drug resistance study were used. The significance threshold was set at p < 0.05. In the results, * denotes p < 0.05, ** denotes p < 0.01, and *** denotes p < 0.001.
Results and discussion
Preparation of SCG-derived CDs
SCG-derived CDs were prepared using an oxidative method focused on a top-down approach (Fig. 1), in which strong acids oxidise a carbon-rich precursor material. Herein, SCGs from Lavazza (LAV) and KENCO (KEN) were used as a precursor and source of carbon. We performed two purification methods on the SCG-derived CDs: the conventional combination of filtration and dialysis (D) and a novel acetone-precipitation (AP)-driven purification method. Both methods yield CDs with comparable characteristics based on the results of our analyses. Given the novelty of the AP method over the conventional approach, we focused our studies on the AP-LAV-CDs. | ||
| Fig. 1 Schematic synthesis of SCG-derived CDs and summary of differences between SCG-derived CDs on the precursor SCGs and the purification method. | ||
Different brands of coffee beans showed no significant differences during the synthesis procedure. The SCGs, Lavazza and KENCO, differ in the origin of the coffee beans. KENCO is composed entirely of Arabica beans, while Lavazza primarily consists of Southeast Asian Robusta beans. Based on our observations and results, which will be detailed in the next section, these two brands generated highly similar SCG-derived CDs. Therefore, we believe the brand of coffee beans has a limited impact on the synthesis process and the properties of the resulting SCG-derived CDs.
For the biological in vitro studies, we have selected the SCG-derived CDs purified through the AP method, specifically AP-LAV-CDs. We decided to further explore CDs purified by the novel AP method as an alternative to the commonly utilised dialysis approach as it offers comparable CDs with several benefits. For instance, in addition to expensive filter membranes, dialysis requires systematic water changes over several days, leading to significant deionised water consumption. These frequent water changes underscore a very significant drawback of dialysis: the extensive time requirement. In fact, with the AP method, the purification step in making the CDs is a question of a few minutes vs. several days.
Characterisation of SCG-derived CDs
Attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR) was conducted to identify functional groups on the surface of AP-LAV-CDs (Fig. 2A). The broad band at 3322 cm−1, associated with ν(O–H) stretching, indicates the presence of –COOH and –OH groups on the surface. The breakdown and partial oxidation of the carbon precursor result in a surface rich in hydroxyl and carboxylic acid groups, explaining the SCG-derived CDs’ excellent water solubility. The band centred at 1402 cm−1 corresponds to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) stretching, while the sharp band at 1598 cm−1 relates to ν(CO) stretching. The 1099 cm−1 peak is attributed to ν(C–O–C) stretching.34 ATR-FTIR spectroscopy was also carried out on D-LAV-CDs (Fig. S1A†) and D-KEN-CDs (Fig. S1B†). The spectra of these two dialysis-purified materials are similar to one another, with minor differences observed in their fingerprint regions. Further distinctions are evident when comparing these spectra to those of the AP-LAV-CDs; notably, the acetone-purified CDs do not exhibit a band in the aliphatic ν(C–H) region, while the dialysis-purified CDs both show a low-intensity band at 2925 ± 1 cm−1. This observation suggests that acetone precipitation may be more effective than dialysis in removing certain impurities. For a comprehensive comparison, the ATR-FTIR spectral data for all SCG-derived CDs are provided in (Table S1†).
C) stretching, while the sharp band at 1598 cm−1 relates to ν(CO) stretching. The 1099 cm−1 peak is attributed to ν(C–O–C) stretching.34 ATR-FTIR spectroscopy was also carried out on D-LAV-CDs (Fig. S1A†) and D-KEN-CDs (Fig. S1B†). The spectra of these two dialysis-purified materials are similar to one another, with minor differences observed in their fingerprint regions. Further distinctions are evident when comparing these spectra to those of the AP-LAV-CDs; notably, the acetone-purified CDs do not exhibit a band in the aliphatic ν(C–H) region, while the dialysis-purified CDs both show a low-intensity band at 2925 ± 1 cm−1. This observation suggests that acetone precipitation may be more effective than dialysis in removing certain impurities. For a comprehensive comparison, the ATR-FTIR spectral data for all SCG-derived CDs are provided in (Table S1†).
 | ||
| Fig. 2 Spectroscopic characterisation of AP-LAV-CDs. (A) ATR-FTIR spectra of AP-LAV-CDs. Prominent bands have been highlighted and labelled with the corresponding wavenumber. (B) Raman spectra of AP-LAV-CDs. The spectra (grey) represent a smoothed average of n > 3 scans, with a 95% confidence band shown (light grey). The fitted envelope (black) represents the deconvolution of the bands D, A and G; the individual traces are shown for each band (red) and labelled with the corresponding wavenumber. | ||
Raman spectroscopy was employed to examine the internal structure of the SCG-derived AP-LAV-CDs. The Raman spectrum (Fig. 2B) reveals three characteristic bands. Namely, the D-band at 1323 cm−1, A-band at 1469 cm−1, and G-band at 1567 cm−1, which are typical of CDs.35,36 The ID/IG ratio, calculated based on the integrated ratio of the area under the deconvolution curves, is 1.98.
The D-band at 1323 cm−1 is attributed to the vibrational modes of carbon atoms hybridised sp3, while the G-band at 1567 cm−1 corresponds to carbon hybridised sp2. The D-band is associated with the vibrations of carbon atoms with dangling bonds, unsaturated valence electrons on immobilised atoms,37 in the termination plane of disordered graphite or glassy carbon. These bonds are highly reactive, and at the nanoscale, their reactivity and concentration increase due to the larger surface-to-volume ratio. This reactivity explains the intense D-peak observed in the Raman spectrum in Fig. 2B. The G-band corresponds to the E2g mode of graphite and is related to the vibration of sp2 hybridised carbon atoms in a two-dimensional hexagonal lattice.38
The intensity ratio (ID/IG) is commonly used to measure the graphitisation or degree of disorder in CDs, as expressed by the sp3/sp2 carbon ratio. A high ID/IG ratio indicates a greater degree of disorder in the sample.39
The 980 cm−1 peak in the Raman spectrum arises from the glass substrate on which the sample was deposited, and it is attributable to the symmetric stretching vibrations of Si–O bonds. Since glass spectra typically do not exhibit peaks above 1200 cm−1, any peaks observed beyond this range unequivocally originate from the AP-LAV-CDs sample.40 A similar signal due to the Si/SiO2 substrate was reported by Qing Zhao and H. Daniel Wagner during their analysis of carbon-nanotube-based composites.41
At approximately 1800 cm−1, the so-called C-band can be observed.42 This band corresponds to the stretching mode of carbonyl groups present on the surface of the AP-LAV-CDs. This observation is consistent with the ATR-FTIR spectrum.43,44
1H and 13C nuclear magnetic resonance (NMR) spectroscopy of AP-LAV-CDs was performed. The 1H NMR spectrum (Fig. S2A†) confirms the presence of both sp3 and sp2 hybridised carbon atoms. Based on their chemical shifts (δ), distinct carbon types can be identified: sp2 hybridised carbons, associated with aromatic and vinylic groups, appear in the 9–6.5 ppm range, while sp3 hybridised carbon atoms are present in two key regions: one corresponding to aliphatic hydrocarbons attached to hydroxyl, ether, and carbonyl groups (4.5–3.5 ppm), and the other to sp3 C–H protons (2.5–1 ppm). The latter two regions, indicative of sp3 hybridised carbon, are more abundant than the regions of sp2 hybridised carbon, as shown in the spectrum.45
In addition, the 13C NMR spectrum (Fig. S2B†) provides further insights. It shows peaks in two main regions, the sp3 hybridised carbon and carbon attached to hydroxyl groups around 70 ppm, as well as sp2 hybridised carbon, particularly those in carbonyl groups (CO), observed near 180 ppm.46
X-ray photoelectron spectroscopy (XPS) was carried out to investigate the surface chemical states and composition of AP-LAV-CDs (Fig. 3). The survey spectrum (Fig. 3A) shows the presence of two predominant peaks, a C 1s peak at a binding energy (BE) of ∼284.5 eV, and an O 1s peak at 532.5 eV. The calculated atomic percentages for O, C and N are ∼43%, ∼54% and 2%, respectively. According to a recent meta-analysis of 115 different reported CDs,47 the ∼43% oxygen content of AP-LAV-CDs is high, but not unusual, and the ∼2% nitrogen content is normal. The XPS scans showed significant charging, even when using a charge neutralisation source. To account for this, the spectra were shifted such that the peak of the C 1s envelope appears at 284.5 eV.
 | ||
| Fig. 3 XPS survey and high-resolution spectra of AP-LAV-CDs. (A) Survey spectrum annotated with predominant peaks, including substrate-derived Si peaks. Binding energy (BE), atomic percentages, and ratios (relative to C) of shaded peaks are provided. High-resolution core-level spectra of (B) C 1s and (C) O 1s are deconvoluted, with distinct bonding environments labelled. | ||
The high-resolution XPS spectra of AP-LAV-CDs provide insights into the surface chemical states of carbon and oxygen (Fig. 3B and C). The deconvoluted C 1s spectrum (Fig. 3B) reveals distinct peaks corresponding to C sp2 and C sp3 bonding environments (∼284.0, ∼284.9 eV), as well as C–O (∼286.4 eV), CO (∼287.8 eV), and O–CO (∼289.2 eV), confirming the presence of diverse oxygen-containing functional groups. A notable feature of the C 1s peak is its broadness, suggesting a heterogeneous distribution of chemical states. Precise assignment of peak positions is difficult due to the charging effect mentioned above, and the C sp2 component appears at a slightly lower binding energy than is commonly reported. This lower binding energy is consistent with a defective structure, and this is further supported by the binding energy separation between C–O and sp2 components of the fit which shows a separation of 2.4 eV, which is larger than is typically reported for graphitic carbon.48
Similarly, the O 1s spectrum (Fig. 3C) is resolved into peaks at ∼531.6, ∼532.2 and ∼533.0 eV, attributed to CO, O–CO and C–O bonding environments, respectively. The predominance of carbon–oxygen functionalities in both spectra corroborates the high oxygen content revealed in the survey spectrum.
The photoluminescence excitation (PLE), photoluminescence (PL) and UV-Vis absorption spectra of the AP-LAV-CDs dispersed in diH2O at 250 μg mL−1 are reported in Fig. 4A. In the PLE spectrum, two excitation maxima are observed at 341 and 385 nm for an emission wavelength of 448 nm. The corresponding PL spectra confirm that the transition at 341 nm has the highest intensity. The UV-vis spectrum highlights a shoulder at 265 nm.
 | ||
| Fig. 4 Photophysical characterisation of AP-LAV-CDs. (A) Representative photoluminescence (PL), photoluminescence excitation (PLE) and UV-vis absorption spectra of AP-LAV-CDs in diH2O at 250 μg mL−1; (B) excitation emission matrix (EEM) fluorescence spectrum of AP-LAV-CDs. Relevant experimental conditions have been noted, and significant emission regions have been annotated as a, b and c, with the excitation wavelength (λex), emission wavelength (λem) and Stokes shift (Δλ) at the point of highest intensity emission (PL). First- and second-order Rayleigh scattering signals have been excluded (dark spaces). (C) Fluorescence emission concentration-dependence studies of AP-LAV-CDs (λex = 350 nm) dispersed in diH2O at the indicated concentrations. Due to spectral overlap, concentrations >250 μg mL−1 are shown as dashed lines. Arrow indicators (grey) are provided to track the changes in emission intensity with respect to concentration. (D) Fluorescence emission photostability study of AP-LAV-CDs dispersed in diH2O at 250 μg mL−1 (λex = 341 nm, from 0 to 120 min). Inset: scatter plot of relative fluorescence intensity (F/F0) at a fixed wavelength, showing a linear decrease over time. | ||
Further UV-vis absorption spectroscopy was performed on AP-LAV-CDs dispersed in diH2O at 5, 10, 25, 50, 100 and 250 μg mL−1 (Fig. S3C†). The UV-vis absorption spectra of AP-LAV-CDs present a tail in the visible range, with absorption increasing towards the UV region (<400 nm). A shoulder at 265 nm is observed at all concentrations, which is correlated to the π → π* transitions of CC bonds.49 The comparison of UV-vis spectra between acetone-precipitated and dialysed SCG-derived CDs, namely D-LAV-CDs (Fig. S3A†) and D-KEN-CDs (Fig. S3B†), suggests that acetone-precipitation is a more effective purification method in this context. Dialysis appears to leave behind small, absorbent impurities that obscure the characteristic shoulder at 265 nm.
The wavelength dependence of the AP-LAV-CDs is represented using the excitation–emission matrix (EEM) fluorescence spectrum, as shown in Fig. 4B. The AP-LAV-CDs exhibit multi-mode excitation-dependent emission, characterised by three distinct emission behaviours: the first region centred at λex/λem wavelengths of 230/434 nm, the second region at 341/448 nm, and the third peak at 390/478 nm.50,51 The first region (Fig. 4Ba) exhibits the smallest Stokes shift (Δλ) of 88 nm, followed by the second region (Fig. 4Bb) with a shift of 107 nm, while the third region (Fig. 4Bc) shows the largest Δλ at 204 nm. Similar behaviour is observed for D-KEN-CDs (Fig. S4A†) and D-LAV-CDs (Fig. S4B†). Due to the partial overlap of second-order Rayleigh scattering with the third emissive region at λex 210–250 nm for the three SCG-derived CDs, the EEM data have been visualised in an alternative manner that highlights the Rayleigh regions, allowing for clearer observation of the third emissive region (Fig. S5†). All EEM data have been summarised in Table S2†. These minimal variations in emission behaviour, including the wavelength shifts and changes in fluorescence intensity, can be attributed to factors such as differences in nanoparticle size, surface defects, and surface functionalisation, which influence the photophysical properties of the CDs.52,53
Φ FL is an important parameter that reflects the effectiveness of CDs in various applications. It is influenced by multiple factors, ranging from synthesis to surface functionalisation.53 The ΦFL (λex = 341 nm, diH2O) of AP-LAV-CDs is notably high at ∼16%. This aligns with the literature,19 and can be attributed to the substantial presence of oxygen-rich surface functional groups.53
Concentration-dependence studies (Fig. 4C) demonstrated that the maximum fluorescence intensity of AP-LAV-CDs dispersed in diH2O is obtained at 250 μg mL−1. Various processes can induce quenching in the fluorescence, such as energy transfer, ground state complex formation, and collisional quenching.54 We speculate that it could be due to an aggregation of the SCG-derived CDs at high concentrations, inducing a self-quenching,55 or the inner filter effect caused by the increased absorption of light.56
Finally, the fluorescence photostability studies (Fig. 4D) of AP-LAV-CDs dispersed in diH2O at the optimised concentration of 250 μg mL−1 were carried out using a xenon flash lamp (50 Hz, peak power output equivalent to 20 kW). After repeated sample irradiation over 120 min, the AP-LAV-CDs retained >94% of their initial fluorescence intensity at λex/λem of 341/448 nm (Fig. 4D inset), demonstrating good photobleaching resistance.
Dynamic light scattering (DLS) and zeta potential (ZP) were performed on the SCG-derived CDs. DLS provides insights into the particle size distribution, while ZP analysis reveals the surface electrostatic charge and colloidal stability. DLS results, represented by number distribution at the concentration of 500 μg mL−1, were 3.8 ± 1.5 nm, 43.8 ± 48.7 nm, and 6.2 ± 2.2 nm for AP-LAV-CDs, D-LAV-CDs, and D-KEN-CDs, respectively. Other concentrations were also tested, as shown in Fig. S6 and Table S3.† Among the three types of SCG-derived CDs, AP-LAV-CDs demonstrated the highest consistency across different concentrations and the smallest particle size. This characteristic is advantageous for biological applications, as smaller sizes facilitate cell membrane penetration and ensure proper clearance, which is critical to avoiding long-term toxicity.10 This observation further supports the selection of AP-LAV-CDs for in vitro investigations.
ZP was measured at a concentration of 100 μg mL−1, yielding results of −15 ± 6 mV, −23 ± 5 mV, and −14 ± 5 mV for AP-LAV-CDs, D-LAV-CDs, and D-KEN-CDs, respectively. All SCG-derived CDs exhibit a negative ZP, attributed to the oxidative synthetic procedure that results in deprotonated carboxyl and hydroxyl groups on the NPs’ surface.
TEM imaging was carried out to study the size and morphology of AP-LAV-CDs. The representative electron micrograph in Fig. 5A highlights two distinct populations of AP-LAV-CDs, the large AP-LAV-CD has a diameter of 8.4 nm, while the smaller AP-LAV-CDs range from 1.7 to 2.6 nm. To better understand the population distribution of NPs in the bulk sample, over 40 TEM images were taken at various locations and magnifications. Four representative images at different magnifications are displayed in Fig. S7.† Size distribution studies have been carried out on all acquired images to provide a comprehensive characterisation of the material across a significant portion of the sample. The results, summarised in Fig. 5C, further indicate the presence of two distinct populations of NPs: the smaller NPs measured at 2.1 ± 1.3 nm and the larger NPs measured at 16.3 ± 4.5 nm.
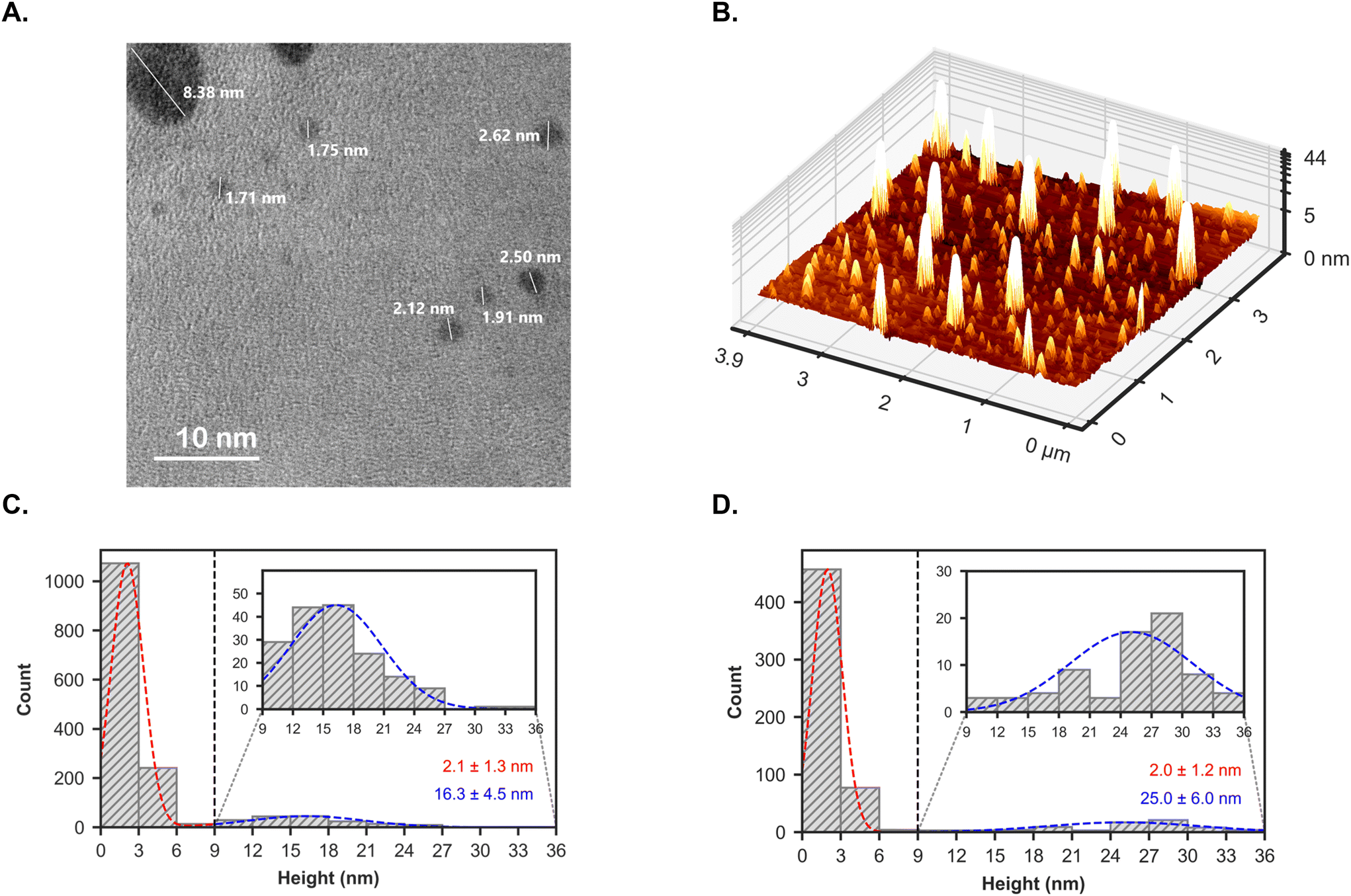 | ||
| Fig. 5 (A) Representative TEM image of AP-LAV-CDs. The diameter of smaller NPs ranges from 1.7 to 2.6 nm; the larger NP has a diameter of 8.38 nm (scale bar = 10 nm). (B) Representative AFM micrograph of AP-LAV-CDs presented as a 3D heightmap, with the y-axis presented in log form to better visualise the major population of small (<9 nm) NPs; (C, D) relative population distribution histogram representing n = 1494 NPs measured by TEM and n = 610 NPs measured by AFM, respectively. Normal distribution fitting of the data highlights two distinct NP populations; the major population (dashed red) and the minor population (dashed blue). Inset: a magnified representation of the NP distribution between 9 and 36 nm, highlighting the minor NP population. | ||
The morphology of AP-LAV-CDs was further characterised by atomic force microscopy (AFM), which revealed well-resolved individual NPs of varying sizes (Fig. 5B and Fig. S8A†). The overall size distribution, shown in Fig. 5D, was obtained by height profile analysis, as showcased in Fig. S8B.† This analysis identified two distinct NP populations, namely, a major population with a height of 2.0 ± 1.2 nm and a minor population with a height of 25.0 ± 6.0 nm.
Both TEM and AFM results show close agreement regarding the size of the smaller NPs, and both techniques identified a secondary population of larger NPs (>9 nm). In both cases, the smaller NPs represent the major population—with NPs over 9 nm constituting only 11% of the total measured by TEM and 12% by AFM.
Overall, the characterisation of SCG-derived CDs demonstrated that different brands of coffee beans have a minimal impact on their properties. Among the tested SCG-derived CDs, AP-LAV-CDs, isolated using the novel acetone precipitation method, displayed the most advantageous characteristics. AP-LAV-CDs are low-cost and require less energy and time for synthesis. They also exhibited superior physico-chemical properties, including smaller particle size as indicated by DLS and higher purity as shown by UV-vis absorption spectra and FTIR analyses. Based on these findings, we believe AP-LAV-CDs hold significant potential for biomedical applications, and therefore, we focused the in vitro studies on this material.
Cell viability studies
The study investigated the antiproliferative effects of AP-LAV-CDs across different cell lines to evaluate its potential as a therapeutic agent. Specifically, the cancer cell lines CAL-51 and SKOV-3, as well as the non-transformed MCF-10A, HH, and HREpiC cell lines, were tested. The results demonstrated a cell line-dependent response to the treatment, with a specific targetability towards CAL-51 at high concentrations (Fig. 6).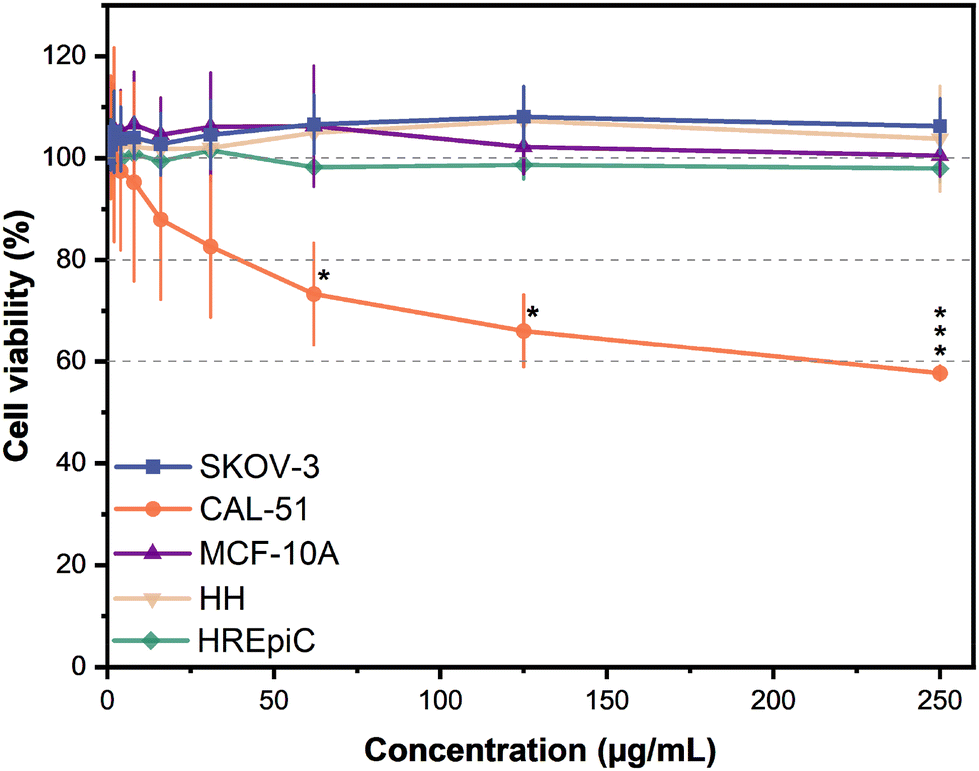 | ||
| Fig. 6 Cellular viability of SKOV-3, CAL-51, MCF-10A, HH, and HREpiC cells treated with various concentrations of AP-LAV-CDs for 120 h. Data were generated from the mean and standard deviations of three replicates. | ||
In the CAL-51 cell line, concentrations up to 31 μg mL−1 showed less than 20% inhibition of cell proliferation. However, at 62 and 125 μg mL−1 of AP-LAV-CDs, mild cytotoxicity was observed, reducing cell viability to 60–80%. At the highest concentration tested (250 μg mL−1), CAL-51 cell viability dropped to 60%, which falls at the threshold limit of the ‘moderate cytotoxicity’ range of 40–60%.57
In contrast, AP-LAV-CDs did not lead to significant cell viability change in the rest of the cell lines tested, even at the highest concentration of 250 μg mL−1. This result indicates AP-LAV-CDs alone had no impact on the cancerous SKOV-3 ovarian cell line, as well as three healthy cell lines: the MCF-10A breast epithelial cells, HH liver cells, and the HREpiC kidney cells.
AP-LAV-CDs demonstrated specific targetability toward CAL-51 cells while avoiding any impact on healthy cell lines in the study. Since in vivo studies have shown that CDs commonly localise in the kidneys and liver, it is crucial to ensure they do not cause toxicity to these organs before applying them in biomedical applications.10 Our results confirmed that AP-LAV-CDs had no adverse effects on kidney and liver cells, paving the way for their safe use in vivo without harming these critical organs.
In addition to sparing healthy cells, AP-LAV-CDs specifically reduced the viability of CAL-51 cells under high concentrations, highlighting their potential as DDSs, which are nanoparticles that carry and deliver drugs efficiently to improve treatment outcomes.10 This intrinsic ability to selectively eliminate breast cancer cells, even without surface functionalisation or drug loading, underscores their versatility and promise in therapeutic applications.
The differing behaviour of AP-LAV-CDs across cell lines could be attributed to various factors, such as the cell line's origin and the composition of the cell medium, which can influence CD uptake, intracellular trafficking, and exocytosis, ultimately leading to cell line-dependent behaviour.58 In other studies, CDs have also been reported to maintain over 80% cell viability in SKOV-3 cells with up to 1000 μg mL−159 and exhibit low toxicity in MCF-10A cells as well as in normal mouse liver cell lines.60 Interestingly, silver NPs displayed a concentration-dependent antiproliferative effect on CAL-51 cells,61 which is similar to what we discovered here.
Additionally, the toxicity of CDs can be influenced by their properties, such as charge, nitrogen content, size, synthesis precursor, and aggregation in the cell medium.62 The environmentally friendly approach of using SCGs as a precursor also contributes to low toxicity, as CDs synthesised from biomass are generally safer and offer greater affordability, stability, and eco-friendliness.63
A notable factor of our cell viability study is that it employed an incubation period of 120 h, which extends beyond the conventional 72 h. The extended incubation period allowed additional time for active cell division and proliferation, which allowed us to derive more information on AP-LAV-CDs’ cellular proliferation effects.
Overall, numerous factors contribute to the low toxicity of AP-LAV-CDs on healthy cell lines and a selective targetability towards breast cancer cells. The unique properties of the SCG-derived CDs in this study led to a minor impact on cell proliferation, suggesting their potential for development into low-toxicity DDSs and bioimaging tools for biomedical applications with a potential advantage in targeting breast cancer.
ROS production study
AP-LAV-CDs specifically target the breast cancer CAL-51 cell line without affecting other cell lines studied. Various mechanisms have been reported for how CDs induce cell death, including apoptosis, mitochondrial damage, metabolic inactivity, and oxidative stress.64–66 We conducted ROS production and apoptosis assays on AP-LAV-CDs to understand how this specific targetability eventually leads to cell death.We involved two cell lines in the ROS production study: one breast cancer cell line CAL-51, which displayed an anti-proliferative effect under high concentrations of AP-LAV-CDs, and another non-tumorigenic epithelial breast cell line MCF-10A, which does not respond to AP-LAV-CDs under all concentrations tested. The results revealed increased ROS production in CAL-51 cells but not in MCF-10A cells.
Under the same incubation conditions as the cell proliferation study, after 120 h of exposure to AP-LAV-CDs, ROS production in CAL-51 cells significantly increased with rising concentrations. At the highest concentration (250 μg mL−1), fluorescence intensity, which indicates the presence of ROS, increased by over 170%; at 31 μg mL−1, ROS levels also rose by more than 120%. In contrast, ROS production remained unaffected in MCF-10A cells at all tested concentrations, further confirming no impact on this healthy cell line (Fig. 7).
 | ||
| Fig. 7 ROS production on MCF-10A and CAL-51 cells treated with various concentrations of AP-LAV-CDs for 120 h. Data represent the mean and standard deviations of three replicates. The significance threshold was set at p < 0.05. * denotes p < 0.05, ** denotes p < 0.01, and *** denotes p < 0.001. | ||
The capability of CDs to induce ROS is closely linked to their surface properties and functional groups. For instance, oxygen-containing groups on CDs are believed to promote ROS generation.67 Moreover, ROS production by CDs can occur through various mechanisms, with photochemical pathways being commonly reported—specifically, upon light excitation, electrons transferred to oxygen can generate highly active ROS species.68 This ROS-induction capability not only enhances AP-LAV-CDs’ targetability towards breast cancer cells but also highlights their potential as photosensitisers for treating other diseases, such as Alzheimer's disease and acute kidney injury.68
From the results obtained, it is significant that ROS was induced in the CAL-51 cell line. To understand if the as-produced ROS caused cell death by apoptosis, we investigated the apoptosis effect of AP-LAV-CDs on CAL-51 cells. Our results showed that AP-LAV-CDs reduced cell confluency but did not increase apoptosis signals (Fig. S9†), indicating that ROS did not induce cell death via apoptosis. Since CDs have been reported to affect cell metabolism, they could disrupt glycolysis, tricarboxylic acid cycle, and lipid metabolism, affecting cellular signal transduction pathways such as Akt/mTOR signalling pathways, which are strongly interacting with ROS.51 Therefore, we suspect that the increased ROS induced by AP-LAV-CDs may affect cell metabolic activities and, thus, lead to decreased cell viability.
Overall, AP-LAV-CDs specifically triggered ROS production in the CAL-51 cell line, which we inferred to induce cell death by disrupting cellular metabolism. This process resulted in both cytotoxic and cytostatic effects. These findings highlight the potential of AP-LAV-CDs for cancer treatment and broader biomedical applications.
Anti-drug resistance study
We studied the impact of drug efflux pumps on AP-LAV-CDs’ cellular accumulation using sulindac and elacridar, both of which block the drug efflux pump MDR-1 (P-gp) and breast cancer resistance protein (BCRP). Sulindac is a nonsteroidal anti-inflammatory drug and a pro-drug of sulindac sulphide, which can block MDR-1 or P-gp. Sulindac is effective at decreasing MDR and increasing the sensitivity of chemo-drugs in various cell lines when administered at clinically achievable concentrations.69 Elacridar can act not only on P-gp but also on BCRP. It is an acridone carboxamide derivative that has proved capable of increasing intracellular chemo-drug concentration when targeting a range of cancers, such as small-cell lung cancer and gastric cancer.70,71We wanted to measure the impact of drug efflux pumps on the cellular accumulation of our SCG-derived CDs. Drug efflux is caused by the ABC transporter family proteins located on the cell membrane. The removal of drugs by drug efflux pumps is a significant cause of drug resistance in cancer treatment, leading to inefficient treatment outcomes.30 Therefore, we aimed to determine whether the drug efflux pump MDR-1 (P-gp) and BCRP would affect the cellular level of AP-LAV-CDs.
At the concentration of 12.5 μM, sulindac did not impact cell viability in either cell line. Previous studies indicated that the optimal concentration at which sulindac69 and elacridar inhibit P-gp, while not affecting cell viability, is 12.5 μM and 0.25 μM, respectively.70 We used these effective concentrations in the combined treatment studies, shown in Fig. 8 and Fig. S10.†
 | ||
| Fig. 8 Cellular viability of (A) CAL-51 and (B) SKOV-3 cells treated with various concentrations of AP-LAV-CDs (0, 31.5, 62.5, 125, and 250 μg mL−1) in combination with and without 12.5 μM sulindac and 0.25 μM elacridar for 120 h. Data were generated from the mean and standard deviations of three replicates. No statistical significance was found between AP-LAV-CDs alone and combined with drug treatment. | ||
The combination of elacridar and sulindac with the AP-LAV-CDs did not enhance the antiproliferative effects of the CDs alone. This finding suggests that drug efflux pump inhibitors (P-gp and BCRP) do not aid in the cellular accumulation of AP-LAV-CDs. Given that drug efflux pumps contribute to MDR and reduce the efficacy of chemotherapy treatments, a DDS not affected by the drug efflux pumps is highly desirable.32 This finding further supports the potential of SCG-derived CDs to overcome MDR when transporting chemo-drugs readily affected by drug efflux pumps.
In conclusion, AP-LAV-CDs are unaffected by the drug efflux pump MDR-1 (P-gp) and BCRP, which further paves the way for SCG-derived CDs to escape MDR in biomedical applications.
Cellular localisation and internalisation
Under the excitation wavelength of 405 nm, the green light fluorescent AP-LAV-CDs were observed in the SKOV3 cells (Fig. 9). It remains unclear whether the AP-LAV-CDs localise within endo-lysosomal structures or the cytoplasm. However, the punctuated fluorescence pattern with perinuclear localisation suggests endo-lysosomal confinement and thus active uptake. | ||
| Fig. 9 Cellular localisation of AP-LAV-CDs after 72 h of incubation in SKOV-3 cells treated with 250 μg mL−1 and untreated (control). Nuclei are stained with DRAQ5 (red). (Scale bar = 25 μm). | ||
Generally, most CDs have been proved to penetrate the cell membrane and localise in the cytoplasm, which has a minimal impact on the proliferation of cells.72 Some literature studies have also reported CDs penetrating the nuclear envelope, which highlights their potential to target diseases like Alzheimer's.73 However, the penetration of the nucleus by bare CDs is rarely reported, as additional properties induced by surface charges and functional groups are needed to improve the interaction of CDs with the nuclear envelope and, therefore, aid in penetration.72 Indeed, further functionalisation of the SCG-derived CDs could potentially aid their penetration into the nucleus or other organelles.
In the time course analysis (Fig. 10), we observed that higher AP-LAV-CD concentrations resulted in higher intracellular accumulation. This accumulation increased over time and was most significant after incubating the cells for 72 h.
 | ||
| Fig. 10 Time-course analysis of cellular accumulation of AP-LAV-CDs in SKOV-3 cells at concentrations of 0 (control), 50 and 250 μg mL−1 and incubation times of 24, 72 and 96 h. Nuclei are stained with DRAQ5 (red). (Scale bar = 50 μm). | ||
Studies also reported the rapid internalisation of certain CDs within 5 min.74 However, for CDs to function effectively as DDSs, prolonged intracellular retention is preferred to enable sustained drug release, a key factor in enhancing cancer treatment outcomes. Factors such as cell type, physico-chemical properties, size, and shape of NPs can affect their cell retention time.75 NPs within the size range of 10–100 nm are reported to exhibit enhanced permeability and prolonged retention.76 Our SCG-derived CDs include larger species (>9 nm), which favour intracellular retention. This characteristic likely explains the observed accumulation of AP-LAV-CDs within cells, peaking at 72 h of incubation, followed by a slight decrease. Overall, the highly emitting AP-LAV-CDs were observed with intracellular accumulation, with peak accumulation found at 72 h, demonstrating their potential in bioimaging applications and as DDSs.
Conclusions
In this study, we produced carbon dots (CDs) by upcycling a natural resource, specifically spent coffee grounds (SCGs). This oxidative method allowed the synthesis of small-sized and spherical CDs, presenting intrinsic and tunable fluorescence. The synthetic route eliminates the use of highly toxic, heavy metals and high energy-consuming reactions and yields SCG-derived CDs that display high cellular accumulation and limited toxicity.Differences were observed between SCG-derived CDs purified by acetone precipitation and those purified by dialysis. Minor variations were noted in ATR-FTIR spectra, particularly in band positions and relative intensities. Similarly, EEM analysis revealed slight differences in the exact properties of their emissive regions, such as variations in the wavelength of maximum emission and Stokes shift. UV-vis absorption spectroscopy also highlighted distinctions, with the acetone-precipitated AP-LAV-CDs displaying the π → π* transitions of CC bonds, which were absent in the dialysis-purified D-LAV-CDs and D-KEN-CDs. Furthermore, TEM and AFM imaging confirmed the presence of populations of two sizes in AP-LAV-CDs, with the smaller population being predominant. NMR and XPS analyses also detailed the hybridised carbon structures and the abundance of surface chemical compositions of AP-LAV-CDs.
Our in vitro studies focused on AP-LAV-CDs. Among the two cancerous and three healthy cell lines tested for antiproliferative effects, AP-LAV-CDs demonstrated toxic effects against the breast cancer CAL-51 cell line, reducing cell viability to below 60% after 120 h of incubation. At the highest concentration tested (250 μg mL−1), AP-LAV-CDs also significantly increased ROS production in CAL-51 to more than 170%, which is speculated to disrupt cellular metabolic activity and ultimately lead to reduced cell viability. Importantly, AP-LAV-CDs showed negligible effects on healthy breast, kidney, and liver cell lines, highlighting their potential to damage breast cancer cells while sparing healthy tissues.
Additionally, our results indicated that AP-LAV-CDs are not substrates for the ABC transporters MDR-1 and BCRP, a crucial advantage for their potential application as nanocarriers in anticancer drug delivery. AP-LAV-CDs were also observed to accumulate intracellularly, with peak accumulation occurring after 72 h of incubation. This suggests their potential for active uptake and sustained drug release when further developed into DDSs.
Overall, the physico-chemical and biological properties of SCG-derived CDs underscore their potential as a sustainable and effective nanoplatform for biomedical applications. The selective anticancer activity, low toxicity to healthy cells, resistance to drug efflux pumps, and ability to accumulate intracellularly highlight their promise as nanocarriers for targeted drug delivery. These findings establish a strong foundation for further development, including the optimisation of SCG-derived CDs as DDSs and the evaluation of their dosimetry in future studies.
Author contributions
The manuscript was written with contributions from all authors. All authors have approved the final version of the manuscript. Y. Zhou: investigation, formal analysis, validation, writing – original draft, and visualization. A. Camisasca: methodology, investigation, and writing – review & editing. S. Dominguez-Gil: investigation and writing – original draft. M. Bartkowski: software, validation, formal analysis, writing – original draft, and visualization. K. D. Rochfort: investigation. M. Piletti: investigation, formal analysis, and writing – review & editing. A. White: investigation and formal analysis. D. Krizsan: investigation. R. O'Connor: investigation. S. J. Quinn: writing – review & editing and supervision. D. Iacopino: writing – review & editing and supervision. A. J. Eustace: methodology, resources, writing – review & editing, and supervision. S. Giordani: conceptualization, methodology, resources, writing – review & editing, supervision, project administration, and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Science Foundation Ireland (22/FFP-A/11067 and 12/RC/2275_P2). Support from the School of Chemical Sciences at DCU (postdoctoral fellowship to A.C.), from the Irish Research Council and Breakthrough Cancer Research (EPSPG/2024/1846 to Y.Z., EPSPD/2023/309 to S.D.), and from the Irish Research Council (GOIPG/2017/1510 to D. K.) is greatly acknowledged. The authors would like to thank Mustafa Nabil Amin Mustafa for preliminary biological studies and Zengchun Xie (UCD) for preliminary TEM.References
- A. M. Filho, M. Laversanne, J. Ferlay, M. Colombet, M. Piñeros, A. Znaor, D. M. Parkin, I. Soerjomataram and F. Bray, Int. J. Cancer, 2024, 156(7), 1336–1346 CrossRef PubMed.
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, CA Cancer J. Clin., 2024, 74, 12–49 CrossRef PubMed.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu and J. M. Pérez De La Lastra, Genes Dis., 2023, 10, 1367–1401 CrossRef CAS PubMed.
- S. Soares, J. Sousa, A. Pais and C. Vitorino, Front. Chem., 2018, 6, 360 CrossRef PubMed.
- Y. He, X. Liu, Z. Xu, J. Gao, Q. Luo, Y. He, X. Zhang, D. Gao and D. Wang, J. Colloid Interface Sci., 2024, 663, 1064–1073 CrossRef CAS PubMed.
- F. Rehan, M. Zhang, J. Fang and K. Greish, Molecules, 2024, 29, 2073 CrossRef CAS PubMed.
- Z. Cheng, M. Li, R. Dey and Y. Chen, J. Hematol. Oncol., 2021, 14, 85 CrossRef PubMed.
- G. Liu, L. Yang, G. Chen, F. Xu, F. Yang, H. Yu, L. Li, X. Dong, J. Han, C. Cao, J. Qi, J. Su, X. Xu, X. Li and B. Li, Front. Pharmacol., 2021, 12, 735446 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- M. Bartkowski, Y. Zhou, M. N. A. Mustafa, A. J. Eustace and S. Giordani, Chem. – Eur. J., 2024, 30, e202303982 CrossRef CAS PubMed.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 Search PubMed.
- H. Ahmed, R. S. Abolore, S. Jaiswal and A. K. Jaiswal, Biomass, 2024, 4, 286–312 CrossRef CAS.
- A. Forcina, A. Petrillo, M. Travaglioni, S. di Chiara and F. De Felice, J. Cleaner Prod., 2023, 385, 135727 CrossRef.
- C. G. Malar, S. Muthulingam, M. Murugesan, G. Srinivasan and R. Sankar, J. Therm. Anal. Calorim., 2023, 148, 505–516 CrossRef CAS.
- T. C. Wareing, P. Gentile and A. N. Phan, ACS Nano, 2021, 15, 15471–15501 CrossRef CAS PubMed.
- S. Y. Masssijaya, M. A. R. Lubis, R. C. Nissa, Y. Nurhamiyah, P. Nugroho, P. Antov, S.-H. Lee, A. N. Papadopoulos, S. S. Kusumah and L. Karlinasari, J. Compos. Sci., 2023, 7, 512 CrossRef CAS.
- A. A. Challa, N. Saha, P. K. Szewczyk, J. E. Karbowniczek, U. Stachewicz, F. A. Ngwabebhoh and P. Saha, Mater. Today Commun., 2023, 35, 105974 CrossRef CAS.
- N. Zhao, Z. Liu, T. Yu and F. Yan, Trends Food Sci. Technol., 2024, 143, 104312 CrossRef CAS.
- A. I. Costa, P. D. Barata, B. Moraes and J. V. Prata, Chemosensors, 2022, 10, 113 CrossRef CAS.
- W. T. Hong and H. K. Yang, Optik, 2021, 241, 166449 CrossRef CAS.
- W. Zhang, L. Jia, X. Guo, R. Yang, Y. Zhang and Z. Zhao, Analyst, 2019, 144, 7421–7431 RSC.
- D. J. Kim, J. M. Yoo, Y. Suh, D. Kim, I. Kang, J. Moon, M. Park, J. Kim, K.-S. Kang and B. H. Hong, Nanomaterials, 2021, 11, 1423 CrossRef CAS.
- J. Wang, Y. Fu, Z. Gu, H. Pan, P. Zhou, Q. Gan, Y. Yuan and C. Liu, Small, 2024, 20, 2303773 Search PubMed.
- T. Bhattacharya, G. H. Shin and J. T. Kim, Pharmaceutics, 2023, 15, 1019 CrossRef CAS PubMed.
- A. Barhoum, A. Meftahi, M. S. K. Sabery, M. E. M. Heravi and F. Alem, J. Mater. Sci., 2023, 58, 13531–13579 CrossRef CAS.
- A. A. Dayem, M. Hossain, S. Lee, K. Kim, S. Saha, G.-M. Yang, H. Choi and S.-G. Cho, Int. J. Mol. Sci., 2017, 18, 120 CrossRef PubMed.
- H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef CAS PubMed.
- Y. Tu, G. Fan, N. Wu, B. Liu, H. Sun, Q. Wang, W. Zou, H. Xiao and S. Tan, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef PubMed.
- N. Peng, J. Wang, H. Zhu, Z. Liu, J. Ren, W. Li and Y. Wang, Int. Immunopharmacol., 2024, 131, 111871 CrossRef CAS PubMed.
- S. Abdullah, S. Mukherjee, Shweta and B. Debnath, Pharmacol. Res. – Mod. Chin. Med., 2024, 10, 100386 CrossRef.
- C. Duan, M. Yu, J. Xu, B.-Y. Li, Y. Zhao and R. K. Kankala, Biomed. Pharmacother., 2023, 162, 114643 CrossRef CAS PubMed.
- C. Wang, F. Li, T. Zhang, M. Yu and Y. Sun, Drug Delivery, 2022, 29, 1684–1697 CrossRef CAS PubMed.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360–5363 CrossRef CAS PubMed.
- A. Ventrella, A. Camisasca, A. Fontana and S. Giordani, RSC Adv., 2020, 10, 36404–36412 RSC.
- E. A. Stepanidenko, I. D. Skurlov, P. D. Khavlyuk, D. A. Onishchuk, A. V. Koroleva, E. V. Zhizhin, I. A. Arefina, D. A. Kurdyukov, D. A. Eurov, V. G. Golubev, A. V. Baranov, A. V. Fedorov, E. V. Ushakova and A. L. Rogach, Nanomaterials, 2022, 12, 543 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B:Condens. Matter Mater. Phys., 2001, 64, 075414 CrossRef.
- J. Zhou, G. Zheng, X. Liu, G. Dong and J. Qiu, Coord. Chem. Rev., 2021, 448, 214178 CrossRef CAS.
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., Int. Ed., 2012, 51, 12215–12218 CrossRef CAS PubMed.
- B. D. Mansuriya and Z. Altintas, Nanomaterials, 2021, 11, 2525 CrossRef CAS PubMed.
- T. Serbource, M. Courtial, M.-N. de Noirfontaine, S. Tusseau-Nenez, C. Sandt and L. Izoret, Cem. Concr. Res., 2024, 179, 107468 CrossRef CAS.
- Q. Zhao and H. D. Wagner, Philos. Trans. R. Soc., A, 2004, 362, 2407–2424 CrossRef CAS PubMed.
- C. S. Casari, A. Li Bassi, A. Baserga, L. Ravagnan, P. Piseri, C. Lenardi, M. Tommasini, A. Milani, D. Fazzi, C. E. Bottani and P. Milani, Phys. Rev. B:Condens. Matter Mater. Phys., 2008, 77, 195444 CrossRef.
- M. L. Frezzotti, Nat. Commun., 2019, 10, 4952 CrossRef PubMed.
- H. D. Ibarra-Prieto, A. Garcia-Garcia, F. Aguilera-Granja, D. C. Navarro-Ibarra and I. Rivero-Espejel, Nanomaterials, 2023, 13, 2753 Search PubMed.
- N. Sharma, I. Sharma and M. K. Bera, J. Fluoresc., 2022, 32, 1039–1049 Search PubMed.
- X. Jia, J. Li and E. Wang, Nanoscale, 2012, 4, 5572 RSC.
- R. de Boëver, J. R. Town, X. Li and J. P. Claverie, Chem. – Eur. J., 2022, 28, e202200748 Search PubMed.
- M. C. Biesinger, Appl. Surf. Sci., 2022, 597, 153681 Search PubMed.
- Y. Zhou, A. Desserre, S. K. Sharma, S. Li, M. H. Marksberry, C. C. Chusuei, P. L. Blackwelder and R. M. Leblanc, ChemPhysChem, 2017, 18, 890–897 CrossRef CAS PubMed.
- L. Zhang, X. Na, B. Lai, Y. Song, H. Wang and M. Tan, Food Chem., 2021, 338, 127832 Search PubMed.
- Y. Ding, J. Yu, X. Chen, S. Wang, Z. Tu, G. Shen, H. Wang, R. Jia, S. Ge, J. Ruan, K. W. Leong and X. Fan, Adv. Sci., 2021, 8, 2002404 CrossRef CAS PubMed.
- L. Bao, C. Liu, Z.-L. Zhang and D.-W. Pang, Adv. Mater., 2015, 27, 1663–1667 CrossRef CAS PubMed.
- Y. Deng, X. Chen, F. Wang, X. Zhang, D. Zhao and D. Shen, Nanoscale, 2014, 6, 10388–10393 RSC.
- A. Belay, H. K. Kim and Y.-H. Hwang, Luminescence, 2016, 31, 565–572 Search PubMed.
- H. J. Yoo, B. E. Kwak and D. H. Kim, J. Phys. Chem. C, 2019, 123, 27124–27131 CrossRef CAS.
- T. Weitner, T. Friganović and D. Šakić, Anal. Chem., 2022, 94, 7107–7114 CrossRef CAS PubMed.
- J. López-García, M. Lehocký, P. Humpolíček and P. Sáha, J. Funct. Biomater., 2014, 5, 43–57 CrossRef PubMed.
- J. Liu, Y.-Y. Liu, C.-S. Li, A. Cao and H. Wang, Nanomaterials, 2023, 13, 2215 CrossRef CAS PubMed.
- A. Bayat, S. Masoum and E. S. Hosseini, J. Mol. Liq., 2019, 281, 134–140 CrossRef CAS.
- C.-L. Li, C.-M. Ou, C.-C. Huang, W.-C. Wu, Y.-P. Chen, T.-E. Lin, L.-C. Ho, C.-W. Wang, C.-C. Shih, H.-C. Zhou, Y.-C. Lee, W.-F. Tzeng, T.-J. Chiou, S.-T. Chu, J. Cang and H.-T. Chang, J. Mater. Chem. B, 2014, 2, 4564 Search PubMed.
- R. H. Salih, W. O. Kachi and A. M. Al-Shammari, J. Nanomed. Res., 2022, 7, 37–48 Search PubMed.
- J. Fan, M. Claudel, C. Ronzani, Y. Arezki, L. Lebeau and F. Pons, Int. J. Pharm., 2019, 569, 118521 CrossRef CAS PubMed.
- H. Jing, F. Bardakci, S. Akgöl, K. Kusat, M. Adnan, M. Alam, R. Gupta, S. Sahreen, Y. Chen, S. Gopinath and S. Sasidharan, J. Funct. Biomater., 2023, 14, 27 CrossRef CAS PubMed.
- T.-H. Ku, W.-T. Shen, C.-T. Hsieh, G. S. Chen and W.-C. Shia, Int. J. Mol. Sci., 2023, 24, 4046 Search PubMed.
- T. Zhang, J. Qu, Y. Yao, Y. Zhang, Y. Ma, D. Wu, Y. Cao, M. Yang, Y. Zhang, M. Tang and Y. Pu, Chemosphere, 2020, 251, 126440 CrossRef CAS PubMed.
- Y. Wu, X. Song, N. Wang, S. Cong, X. Zhao, R. Rai and M. Tan, Food Funct., 2020, 11, 10105–10113 Search PubMed.
- E. Priyadarshini, R. Meena, H. B. Bohidar, S. K. Sharma, M. H. Abdellattif, M. Saravanan and P. Rajamani, Oxid. Med. Cell. Longevity, 2022, 1–11 Search PubMed.
- M. Kurian and A. Paul, Carbon Trends, 2021, 3, 100032 CrossRef CAS.
- D. Jason, B. Adam, D. Bernard, A. Larry, S. Kamlesh, C. Zhe-Sheng and A. Gary, J. Biomed. Res., 2016, 30, 120 Search PubMed.
- R. Alves, A. C. Gonçalves, J. Jorge, A. M. Almeida and A. B. Sarmento-Ribeiro, Biomedicines, 2022, 10, 1158 CrossRef CAS PubMed.
- D. Chen, X. Pan, F. Xie, Y. Lu, H. Zou, C. Yin, Y. Zhang and J. Gao, Int. J. Nanomed., 2018, 13, 6855–6870 CrossRef CAS PubMed.
- B. Unnikrishnan, R.-S. Wu, S.-C. Wei, C.-C. Huang and H.-T. Chang, ACS Omega, 2020, 5, 11248–11261 CrossRef CAS PubMed.
- M. Havrdová, I. Urbančič, K. B. Tománková, L. Malina, J. Štrancar and A. B. Bourlinos, Int. J. Mol. Sci., 2021, 22, 5608 Search PubMed.
- X.-W. Hua, Y.-W. Bao, Z. Chen and F.-G. Wu, Nanoscale, 2017, 9, 10948–10960 RSC.
- R. Sakhtianchi, R. F. Minchin, K.-B. Lee, A. M. Alkilany, V. Serpooshan and M. Mahmoudi, Adv. Colloid Interface Sci., 2013, 201–202, 18–29 CrossRef CAS PubMed.
- M. A. Subhan, S. S. K. Yalamarty, N. Filipczak, F. Parveen and V. P. Torchilin, J. Pers. Med., 2021, 11, 571 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05186f |
| This journal is © The Royal Society of Chemistry 2025 |