
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn intramolecular FRET biosensor for the detection of SARS-CoV-2 in biological fluids†
Daniele
Montepietra‡
ab,
Lorenzo
Germelli‡
c,
Laura
Marchetti
 *c,
Valentina
Tozzini
de,
Elisa
Angeloni
c,
Chiara
Giacomelli
c,
Barbara
Storti
d,
Ranieri
Bizzarri
df,
Elisabetta
Barresi
c,
Sabrina
Taliani
c,
Giorgia
Brancolini
*a and
Eleonora
Da Pozzo
c
*c,
Valentina
Tozzini
de,
Elisa
Angeloni
c,
Chiara
Giacomelli
c,
Barbara
Storti
d,
Ranieri
Bizzarri
df,
Elisabetta
Barresi
c,
Sabrina
Taliani
c,
Giorgia
Brancolini
*a and
Eleonora
Da Pozzo
c
aInstitute Nanoscience – CNR-NANO, Center S3, via G. Campi 213/A, 41125, Modena, Italy. E-mail: giorgia.brancolini@nano.cnr.it
bDepartment of Physics, Computer Science and Mathematics, University of Modena and Reggio Emilia, Via Campi 213/A 4100 Modena, Italy
cDepartment of Pharmacy – University of Pisa, via Bonanno 6, 56127 Pisa, Italy. E-mail: laura.marchetti@unipi.it
dIstituto Nanoscienze – CNR-NANO, Lab NEST CNR and Scuola Normale Superiore, Piazza San Silvestro 12, 56127 Pisa, Italy
eINFN Sezione Pisa, Largo Bruno Pontecorvo, 56127 Pisa, Italy
fDepartment of Surgical, Medical and Molecular Pathology and Critical Care Medicine – University of Pisa,
via Roma 55, 56126 PISA, Italy
First published on 11th March 2025
Abstract
The development of a FRET-based sensor for detecting the Spike surface antigen of SARS-CoV-2 in biological fluids is described here, exploiting the fluorescence properties of Green Fluorescent Protein (GFP). Our design strategy combines experimental and molecular modeling and simulations to build a smart modular architecture, allowing for future optimization and versatile applications. The prototype structure incorporates two reporter elements at the N-terminus and C-terminus, with two interaction elements mediating their separation. This design supports two fluorescence measurement methods: direct measurement and the molecular beacon approach. The former detects changes in GFP fluorescence intensity due to interactions with the Spike protein, while the latter involves an organic quencher that restores GFP fluorescence upon Spike protein binding. In silico design of linkers, using molecular dynamics (MD) simulations, ensured optimal flexibility and stability. The AAASSGGGASGAGG linker was selected for its balance between flexibility and stability, while the LEAPAPA linker was chosen for its minimal structural impact on the interaction elements. Fluorophores’ behavior was analyzed, showing stable FRET efficiency, essential for reliable detection. Quenching efficiency calculations, based on Förster energy transfer theory, validated the sensor's sensitivity. Further, MD simulations assessed GFP stability, confirming minimal unfolding tendencies, which explains the sensor functioning mechanism. The sensor was successfully produced in E. coli, and functional validation demonstrated its ability to detect the Spike protein, with fluorescence recovery proportional to protein concentration, while the modular computer aided design allowed for sensitivity optimization. The developed biosensor prototype offers a promising tool for rapid and precise viral detection in clinical settings.
1. Introduction
Viral infections, especially acute respiratory ones, pose a major global health threat, causing around 15 million deaths annually.1 The emergence of the novel coronavirus SARS-CoV-2 since 2020 has profoundly impacted public health, leading to widespread disruptions and a new crisis.2 This situation has highlighted the critical need for reliable, swift diagnostic methods. Accurately measuring viral load, or titre, is essential for diagnosing infections, tracking disease progression, and evaluating treatment effectiveness.Traditional diagnostic methods typically involve detecting viral RNA using polymerase chain reaction (qRT-PCR) or rely upon identifying antibodies against the virus in serum samples obtained from a patient by ELISA-like immunoassays. These methods offer several advantages in terms of sensitivity (qRT-PCR) and affordability (immunoassays). However, viral RNA presence does not always indicate infectivity, and immunoassays are characterized by a significant fraction of false negatives.3 Moreover, these techniques are slow, in that antibodies appear days after symptoms, and PCR results take several hours, needing specialized labs and personnel.4
In this context, Förster Resonance Energy Transfer (FRET)-based biosensors offer promise due to their high sensitivity. FRET sensors exploit the energy transfer between two fluorophores, a donor and an acceptor, when they are in close proximity, detecting changes within 1–10 nanometers.5,6 This method quantitatively correlates changes in fluorescence with the presence of the target molecule, being therefore ideal for assessing viral titer of clinical samples.7 Accordingly, it represents a promising technology for the development of a high-performance biosensor capable of assessing viral titer directly from clinical samples.
FRET-based molecular assays have been proposed for detecting SARS-CoV-2's spike protein receptor-binding domain (S-RBD) by means of a “molecular beacon” architecture, and making use of Total Internal Reflection Fluorescence (TIRF) microscopy as read-out.8 These molecular beacons, containing two oligopeptides forming a heterodimer, namely an S-RBD ligand and fluorophore–quencher pair, reveal S-RBD presence by an increase of fluorescence, which stems from the conformationally-driven separation of the two coiled-coil oligopeptides triggered by S-RBD binding. Yet, these sensors do not rely on a specific binding displacement, allegedly depressing the sensor's specificity. Additionally, the reliance on TIRF microscopy for fluorescence signal detection limits the proposed method large-scale deployment.8
Here, we propose a much more versatile technology that in principle may be not limited to the detection of the sole SARS-CoV-2 in biological fluids. The biosensor prototype is a recombinant protein designed through rational modification of the Green Fluorescent Protein (GFP) sequence. The native GFP protein, naturally produced by the jellyfish Aequorea Victoria, consists of 238 amino acids and has a molecular weight of 27 kDa. It generates green fluorescence by converting blue light from the UV-visible spectrum.9 Studies have shown that GFP retains its optical properties when produced in biological systems other than Aequorea Victoria. Moreover, it can be engineered and modified to alter its fluorescence emission in response to specific parameters of interest, effectively serving as a fluorescence sensor for those parameters.10 To date, several GFP-based fluorescent sensors have been developed for detecting ions (such as Ca2+ and Cl−), pH levels, and molecular interactions. These sensors have primarily been used in basic research, as well as in preclinical in vitro and in vivo studies.9 Recent research has also explored the potential of GFP-based sensors for detecting environmental11 and clinical parameters.10 In line with these approaches, the prototype biosensor here proposed is designed with the aid of molecular dynamics and molecular modeling and analysis techniques to detect the presence of the Spike surface antigen of the SARS-CoV-2 virus by modifying GFP's fluorescence emission.
2. Experimental
2.1. Construction of the molecular model: the FRET probe building blocks
In this section, we present the design and construction of a FRET-based sensor for detecting SARS-CoV-2 in biological fluids. The sensor integrates various molecular components engineered to interact with the virus receptor-binding domain (RBD). Key elements of this design, illustrated in Fig. 1, include:1. Reporter 1: a peptide sequence Gly(His)6, which consists of six histidine residues preceded by glycine. This sequence is essential for the biochemical purification of the sensor and can also be covalently conjugated to an organic quencher, exploiting a reaction recently reported in the literature14 (see Fig. S7†).
2. Interaction element 1: the receptor-binding motif (RBM) of SARS-CoV-2, a portion of the Spike protein that binds to the human ACE2 protein, facilitating viral entry into cells15 (see Fig. S1†).
3. Interaction element 2: a sequence called LCB1, designed to mimic the two alpha helices of the N-terminal end of ACE2, which are responsible for interacting with the RBM of SARS-CoV-2. This sequence was recently reported in the literature.12
4. Reporter 2: Enhanced Green Fluorescent Protein (EGFP), a bright variant of GFP, which provides the fluorescent signal.13
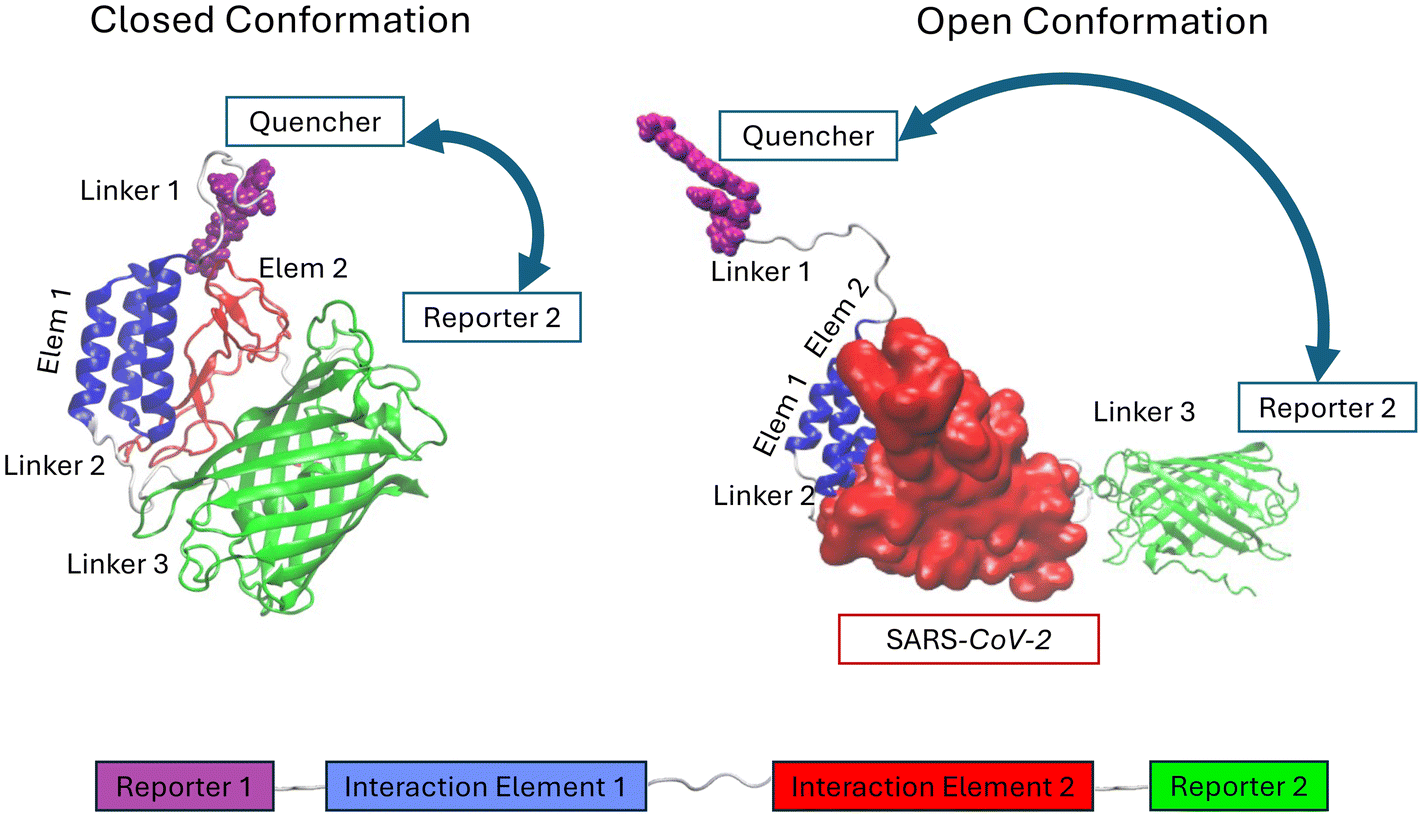 | ||
| Fig. 1 Schematic picture of the sensor prototype. Reporter 1: peptide Gly(His)6, a sequence of 7 amino acids consisting of a traditional His tag preceded by the amino acid Glycine. Interaction element 1: RBD or RBM domains of SARS-CoV-2. These are portions of the SARS-CoV-2 Spike protein that correspond to the amino acid sequence involved in binding with the human ACE2 protein, which the virus uses as an entry channel to infect cells. Interaction element 2: RBM interaction domain. It is a sequence called LCB1, recently reported in the literature.12 Reporter 2: EGFP, a particularly bright variant of the green fluorescent protein GFP.13 On the top-left: closed conformation (GFP-OFF) of the sensor, induced by the close contact of interaction elements 1 and 2 bringing the two reporter elements in proximity; on the top-right: open conformation (GFP-ON) of the sensor, induced by the binding of the SARS-CoV-2 spike protein and separating the two reporter elements. | ||
The sensor components, namely LCB1, RBM, EGFP, and a quencher, are connected via specific linkers to ensure proper function and stability. Through extensive molecular dynamics (MD) simulations, we optimized these components for high sensitivity and specificity in detecting viral presence. Below, we detail the construction and optimization process for each component of this FRET sensor.
The different protein linkers between LCB1–RBD and GFP-RBD were created with the MAESTRO and PyMOL22 software. The final optimized system, reported in Fig. 1 and experimentally tested, is composed of the quencher (Dabcyl molecule including linker), LCB1, AAASSGGGGGASGASGGAGG linker, RBM, LEAPAPA linker, and GFP. The complete FRET sensor system was simulated with a 500 ns MD to verify that RBM did not unfold due to the residue removal and that its interaction with LCB1 remained sufficiently stable.
2.2 Molecular dynamics (MD) simulations and calculation of the quenching efficiencies
Classical MD simulations in explicit water solvent were carried out using GROMACS 2020.1 software,25 with CHARMM36 + TIP3P as force field and water model.26 All MD simulations were carried out using periodic boundary conditions in cubic boxes. After solvation, ions were added to obtain neutral systems, considering a neutral protonation state for HIS residues. The entire system was minimized using the steepest descent algorithm to remove van der Waals contacts of high potential energy, with the maximum force threshold value was set at 1000 kJ mol−1 nm−1. Minimization was followed by a 100 ps relaxation of the solvent around the position-restrained protein and a 100 ps NPT equilibration with isotropic Berendsen pressure coupling at 1 bar.27 The temperature was kept at 300 K using a velocity rescaling thermostat.28 The production run MD of the systems was performed using the leap-frog algorithm with a 2 fs time step, the Verlet cutoff scheme for van der Waals interactions, and the Particle Mesh Ewald (PME) method for the treatment of electrostatic interactions with 1.0 nm cutoff. The temperature coupling method used was velocity rescale, with 0.1 ps of time coupling constant. We used an isotropic Berendsen barostat with 1 bar of reference pressure and 1 ps of time constant. Covalent bonds involving hydrogen atoms were constrained using the LINCS algorithm. Positions and coordinates were saved every 20 ps. Standard structural analysis on the MD trajectories as RMSD and RMSF was performed using GROMACS 2020.1. Trajectories and structures were visualized using VMD29 and PyMOL.22 In Fig. 2 we computed the distance between the Dabcyl N2 atom and the Tyrosine Cα in the TYG sequence involved in the GFP fluorescence for each trajectory frame using the “compute_distance” function of the Python package MDTraj version 1.9.4.30 The analysis plots in Fig. 2 have been created using in-house Python codes. | ||
| Fig. 2 Analysis of the FRET sensor performance. (A) Distance between the Dabcyl N2 atom and the Tyrosine Cα in the TYG sequence involved in the GFP fluorescence during the 500 ns MD trajectory of the complete FRET sensor system. The red line shows the running average with a 100-frame window. Snapshots of the systems at 0 ns, 200 ns, and 500 ns are reported on the plot. (B) Positions of the Dabcyl N2 atom (purple spheres) and the Tyrosine TYG Cα atom (green spheres) during the 500 ns MD simulation. | ||
The collected trajectories from MD simulations were used to calculate the quenching efficiency E according to the Förster energy transfer theory
 | (1) |
 | (2) |
Besides the obvious symbols (Avogadro number, NAv and refractive index n), this formula includes the quantum yield of the donor (in this case the GFP) QD, which is measured, and two quantities, the overlap integral J between the GFP emission and quencher adsorption, and the geometric correction k which can depend on the conformation of the system and can be evaluated from the simulation. In particular κ is a geometric factor depending on the orientation of the transition dipoles of donor and acceptor of the photon where all involved vectors are unitary. This is a purely geometrical factor directly evaluable from the simulations once the directions and positions of transition dipoles are established (red and pink bars).
 | (3) |
If the system is rotationally free, this factor is (2/3)1/2. The transition dipole directions of the GFP chromophore and of the diazobenzene optically active moiety of the quencher are known from previous experimental31 and theoretical studies,32 and roughly oriented along the main axes of the molecules (see Fig. S5†). The non geometrical part of the R0 depends on the superposition integral of emission spectrum of donor (GFP) and adsorption spectrum of acceptor (quencher), that we estimated to 4.8 nm (see Fig. S6† for details).
2.3. Molecular cloning of the FRET sensor
The nucleotide sequences coding for the modular structure (Reporter Element 1) – (linker1) – (Interacting Element1) – (linker2) – (Interacting Element 2) – (linker 3) – (Reporter Element 2) were designed in silico and then purchased already inserted into a cloning plasmid (ThermoFisher, GeneArt-Gene Synthesis). These sequences were obtained in two forms, containing either the RBM or the RBD as Interacting Element 2 sequence. The RBD portion consists of approximately 220 amino acids (aa); the RBM portion, which is the part of the RBD most involved in recognition, contains 72 aa. To avoid folding problems, we included 3 aa upstream and downstream of the two ends of the RBM, resulting in a total of 78 aa. Importantly, we used the codon optimization tool of the GeneArt software to ensure optimal expression in the E. coli host. Subsequently, the two sequences were expanded and subcloned into the pET11a vector (Novagen) using the KpnI/XhoI restriction sites. Positive clones were screened by restriction analysis, and the full-length construct sequence was always verified by DNA sequencing before proceeding with the experiment.2.4. FRET sensor expression and purification
BL21(DE3) E. coli (Thermo Fisher) were transformed with the two constructs in pET11a plasmid, and protein expression was induced with 1 mM of isopropyl-β-thio-galactoside (IPTG). After overnight incubation at 30 °C, the bacteria were collected by centrifugation (6500g, 30 min at 4 °C), and the pellet was resuspended with 5 ml of Lysis Buffer per pellet gram (20 mM Tris pH8, 300 mM NaCl and 10 mM Imidazole), 0.5% TritonX-100, and a complete EDTA free protease inhibitors tablet (Roche). The suspension was incubated at room temperature for 1 h, and then sonicated on ice (30′′ pulse followed by 60′′ stop, repeated 6 times). The lysate was subjected to clarification (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g, 30 min at 4 °C) to collect the supernatant, which contained the induced protein. The supernatant was then mixed with a Ni2+ ion functionalized agarose resin (NiNTA, cat. no. 30120, Qiagen). This resin was previously equilibrated in Buffer A (20 mM Tris pH8, 300 mM NaCl and 10 mM Imidazole) and then packed into a gravity-flow column (cat. no. 34964, Qiagen). Under these conditions the resin was capable of complexing the histidine residues present in the His-Tag fragment at the N terminus of the protein. The resin was extensively washed (three washes with 10 column volumes of Buffer A) to remove undesired proteins. The protein was then eluted from the resin by adding increasing concentrations of Buffer B (20 mM Tris pH8, 300 mM NaCl and 250 mM Imidazole). Specifically, we performed a 4-step elution in 10 column volumes per step, with Buffer B concentrations progressively increased to 10%, 25%, 50% and 100%, respectively. A total of 12 fractions was collected. The fractions were subsequently checked by SDS-PAGE followed by Coomassie staining. Fractions containing a satisfactory amount of purified protein at the correct molecular weight were pooled and subjected to dialysis to replace the buffer with Storage Buffer (20 mM Tris pH7.5, 200 mM NaCl). The dialyzed protein was concentrated using an Amicon ultrafiltration membrane with 10 kDa cut-off (Merck-Millipore) and stored at –80 °C. Typically, a purification from one liter culture yielded 500–700 μl of a 10–40 μM concentration of protein, with an estimated purity degree of about 40%.
000 g, 30 min at 4 °C) to collect the supernatant, which contained the induced protein. The supernatant was then mixed with a Ni2+ ion functionalized agarose resin (NiNTA, cat. no. 30120, Qiagen). This resin was previously equilibrated in Buffer A (20 mM Tris pH8, 300 mM NaCl and 10 mM Imidazole) and then packed into a gravity-flow column (cat. no. 34964, Qiagen). Under these conditions the resin was capable of complexing the histidine residues present in the His-Tag fragment at the N terminus of the protein. The resin was extensively washed (three washes with 10 column volumes of Buffer A) to remove undesired proteins. The protein was then eluted from the resin by adding increasing concentrations of Buffer B (20 mM Tris pH8, 300 mM NaCl and 250 mM Imidazole). Specifically, we performed a 4-step elution in 10 column volumes per step, with Buffer B concentrations progressively increased to 10%, 25%, 50% and 100%, respectively. A total of 12 fractions was collected. The fractions were subsequently checked by SDS-PAGE followed by Coomassie staining. Fractions containing a satisfactory amount of purified protein at the correct molecular weight were pooled and subjected to dialysis to replace the buffer with Storage Buffer (20 mM Tris pH7.5, 200 mM NaCl). The dialyzed protein was concentrated using an Amicon ultrafiltration membrane with 10 kDa cut-off (Merck-Millipore) and stored at –80 °C. Typically, a purification from one liter culture yielded 500–700 μl of a 10–40 μM concentration of protein, with an estimated purity degree of about 40%.
2.5. Click-chemistry conjugation of Dabcyl quencher to the FRET sensor
We used a labeling approach via click chemistry reaction to attach a Dabcyl quencher molecule (Thermo Fisher, cat. no. D2245) to the first residue of the Gly-His tag present in the RBM version of the FRET probe (named S probe). We adopted a recently described procedure,14 with some modifications. Briefly, the methoxyphenyl ester containing an N3 azide group was first synthesized, following protocol steps previously described.14 The protein labeling reaction was carried out following 3 steps:1. Synthesis of Dabcyl-DBCO: 25 mg of Dabcyl-NHS ester (Thermo Fisher) was reacted with an excess of the NH2-DBCO crosslinker (Sigma Aldrich). The reaction was monitored by TLC, and the product was precipitated by the addition of ice.
2. Preparation of the clickable quencher: 100 μL of 10 mM Dabcyl-DBCO was reacted with an equimolar amount of the methoxyphenyl ester containing an N3 azide group14 for 1 hour at room temperature, to obtain the final clickable quencher (Q);
3. Conjugation of the quencher to the S probe: 20 to 40 equivalents of Q were reacted with the S probe in acetonitrile solvent, for one to two overnights at 4 °C, respectively, obtaining the quenched biosensor, called SQ.
SQ preparations were subsequently dialyzed in Storage Buffer for 24 hours at 4 °C and concentrated. The final concentration was then estimated as previously described. The efficacy and stoichiometry of the conjugation was verified by performing a mass spectrometry proteomic analysis of the unconjugated versus conjugated versions of the S sensor.
2.6 Fluorescence intensity measurements and fitting
The S probe was first tested to determine its minimum detectable concentration in a biological fluid sample. For this purpose, a plate assay was set up in which the fluorescence intensity was evaluated following the addition of different S probe concentrations to a saliva sample, and measured using an EnSight multiplate reader (PerkinElmer). 5 mL of saliva was collected in a 50 mL test tube approximately 12 hours before the experiment and stored at 4 °C. Particular precautions for the collection were adopted:(I) Avoiding the consumption of alcohol, nicotine and caffeine by donors at least in the two hours before the collection and at least one hour after consuming the meal.
(II) Ensuring the sample was taken before daily dental hygiene.
On the morning of the experiment, the biological fluid was centrifuged at 1200 rpm for 5 minutes and the supernatant was collected and distributed into the wells.
 | (4) |
In eqn (4), F0 and F∞ represent the initial fluorescence (i.e. zero Spike concentration) and the maximum fluorescence when the sensor is fully saturated by the Spike, respectively. Data fitting to eqn (4) affords the Kd value. For the estimation of the dynamic range of the sensor, i.e. (F∞ − F0), in order to exclude F0 fluctuations due to different S and SQ preparations, eqn (4) was applied to normalized data as reported in Fig. 6.
3. Results and discussion
3.1 Rational design of the sensor
The sensor has been developed aiming at detecting the Spike surface antigen of the SARS-CoV-2 virus by using the fluorescence properties of Green Fluorescent Protein (GFP). The sensor was designed with modular architecture to facilitate future optimization and versatile applications. The prototype structure incorporates two “Reporter” elements, strategically positioned at the N-terminus and C-terminus of the protein, with two “interaction elements” mediating the reporter proximity separation (Fig. 1). This design allows for compatibility with two distinct fluorescence measurement methods, both potentially exploitable for detecting SARS-CoV-2 in biological fluids:3.2 Calculation of quenching efficiency
We explored thoroughly the sensor functioning by calculating the quenching efficiency according to the Förster energy transfer theory, incorporating parameters such as the distance between GFP and the quencher, quantum yield of GFP, and geometric factors derived from MD simulations. Fig. 3A–D shows the quenching efficiency calculated along the simulated trajectories on four possible variants of the sensor including RBM/RBD and LEGASA/LEAPAPA (structures in expanded form reported in each panel). The quenching efficiency calculated according to eqn. (1) with (black curves) or without (magenta) the correct geometric factor evaluated as in (3) are reported in the upper plots. It is apparent that the combinations with RBM show a larger dynamical range of efficiency, while those with RBD display low efficiencies. The reason is shown by the images under the plots, representing in purple the volumes spanned by the quencher bearing extremity around the GFP (in green) during the simulation. It is clear that in the RBD combinations (C and D) the quencher extremity spans a spherical shell around the GFP, at quite a large distance that maintains the efficiency low, without being able to get nearer to the GFP. Conversely in the RBM combinations (A and B) the construct is flexible enough to allow more compact configurations, allowing the quencher to efficiently act on GFP and exploring a large dynamical range of efficiencies. Without the geometric correction (magenta lines) LEGASA (B) linker seems to work slightly better, allowing a closer conformation with respect to LEAPAPA, but as the geometric correction is added (black) the efficiency turns out on average lower or comparable to LEGASA (A). These observations point to the choice of RBM as preferred for the design with the quencher.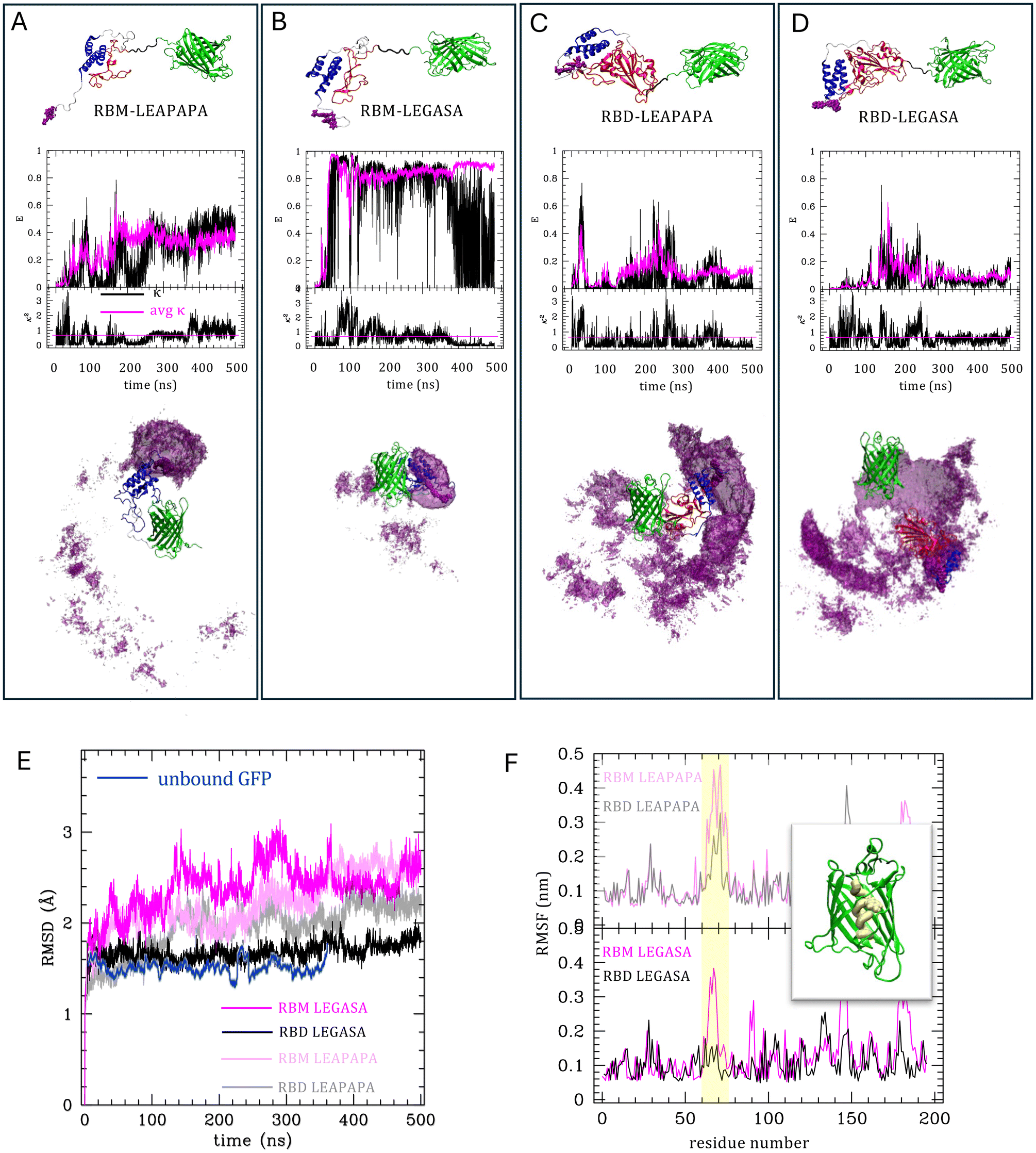 | ||
| Fig. 3 MD simulations of the molecular constructs. Panels (A–D) reports the four molecular constructs used in simulations, as per legends. The structures are sketched on the top, with the GFP in green, the quencher in purple, the LCB in blue, RBM/RBD in red, LEAPAPA/LEGASA in black and other linkers in white. The four plots are divided in two parts, the lower one reporting the geometrical factor κ along the trajectory (in black) and its average value (in magenta), the upper one the quenching efficiency E (eqn (1) and (3), with R0 = 4.74 nm), either using the average value (magenta) or the actual value of κ (black line). Under the plots the space distribution of the quencher around the GFP is reported as an iso-surface of the volume density of the quencher mediated over the trajectories. Panel (E) reports the root mean squared deviation of the GFP structure in time, in the four simulations (in black and magenta shades, as per legend) compared to the unbound GFP (blue). Panel (F) reports the root mean squared fluctuations of GFP residues along the chain, in each simulation as per legends. The chromophore region is highlighted in yellow in the plot and in the structure reported therein. | ||
To explore other intrinsic quenching mechanisms, we also analyzed the internal fluctuations of the GFP in the four simulations. These, specifically the root mean squared deviation with respect to the starting configuration (RMSD) are reported along the trajectories in panel E. The combinations with RBD (black and grey) produce less internal fluctuations than those with RBM (shades of magenta). This is consistent with the fact that with RBD the open structures are favored: the internal fluctuations of the GFP are due to the disturbance produced by the other elements of the construct as they close and bind to its surface. This effect is stronger with RBM, which allows closed and compact structures. We also observe that the difference RBM/RBD is larger with the LEGASA linker. In fact, the combination RBD-LEGASA displays RMSD as low as the one of the GFP alone evaluated on a reference simulation (blue). The connection of this behavior with the sensor functioning is understood by looking at the fluctuations averaged in time and evaluated along the chain of the GFP, namely the Root Mean Square Fluctuations (RMSF) (panel F). These are in agreement with previous similar analysis in the literature,34 with peaks on the loops and lower fluctuations in the strands, but with a noticeable exception: the area round the chromophore (residues 60–70 according to the standard numbering in GFP, highlighted in yellow) are normally little fluctuating, because the GFP structure is optimized to maintain rigid its chromophore to favor fluorescence.35 This is the case in the RBD-LEGASA combination, in which the GFP is almost always “unbound”. In the other cases, conversely, the dynamical formation of compact states greatly enhances the fluctuations in the chromophore regions. This indicates that quenching may occur even in absence of a quencher at the extremity, due to the formation of compact closed states of the GFP with the other elements. Again, the combinations with RBM enhance this intrinsic quenching effect.
These calculations provided insights into the sensor's performance under varying conditions, validating its potential for sensitive and specific detection of SARS-CoV-2 antigens.
3.3 Production of the designed sensor in E. coli
The sensor design was successfully translated into practical applications through cloning and expression in Escherichia coli. The two versions of the designed sensor (S), differing for the presence of the RBM (RBM-S) and RBD (RBD-S) domains as Interacting Element 1, were cloned in a prokaryotic expression system and produced in BL21DE3 E. coli strain. As shown in Fig. 4A, both proteins can be produced following IPTG induction. After overnight (ON) growth, the bacteria were pelleted, lysed, and the cell lysate was clarified. Notably, only the RBM extract retained EGFP spectral properties, whereas the RBD extract did not show these characteristics (Fig. 4B and C). Based on these results, we decided to proceed with the purification of the RBM-S sensor only. This decision is also supported by the simulations, which indicate an enhanced propensity for the RBD to unfold compared to the more stable RBM when embedded within the sensor architecture, as far as a larger dynamical range for quenching efficiency and an enhanced intrinsic quenching mechanism in the RBM simulated constructs compared to those with RBD. | ||
| Fig. 4 Expression in E. coli and purification of the RBM-S/RBD-S protein. (A) SDS-PAGE showing the time-course of RBM-S and RBD-S production. 1 ml of E. coli growth were pelleted at time 0, 1 h, 2 h and overnight after IPTG induction, denatured in Laemmli buffer and loaded in different wells of the gel. The gel was stained using Coomassie blue reagent. (B) Representative picture of the clarified lysates of RBM-S and RBD-S transformed E. coli obtained from a bacteria growth after an overnight of IPTG protein expression induction. (C) Fluorescence intensity quantification of the RBM-S and RBD-S lysates. Results are represented as mean ± SEM of three independent experiments and statistical analysis was performed by Student's t-test: ** p < 0.01. (D) SDS-PAGE showing the purification of RBM-S. The gel was stained using Coomassie blue reagent. Load: fraction of the extract loaded on the column; FT: flowthrough of the loaded column; W1–3: washes of the loaded column; El1–11: fractions eluted from the column at increasing concentrations of Imidazole. The red square represents the band of interest and the respective eluted fractions were pooled and dialyzed. (E) SDS-PAGE showing quantification of the purified RBM protein by comparison with known amounts of purified BSA loaded in the same gel as reference. | ||
3.4 Conjugation of the designed sensor to the Dabcyl quencher probe
To enhance the detection sensitivity, the sensor was stoichiometrically labelled with a Dabcyl quencher probe (Fig. S7A†), by using a reported protocol of click-chemistry conjugation.14 We decided to prove the efficacy and stoichiometry of conjugation by performing a mass spectrometry proteomic analysis of the unconjugated (RBM-S) versus conjugated (RBM-SQ) versions of the RBM-S protein (Fig. 5A). Theoretical evaluation of the MW of the unconjugated and conjugated proteins revealed that the conjugation should add ∼610.7 Dalton to the MW of the protein, estimated to be 46652 Dalton. The MALDI spectra of proteins preliminarily run on RP-HPLC revealed the presence of proteins with MW compatible with the aforementioned ones. However, the main peak of RBM-S was compatible with a protein displaying ∼15 amino acids less than the theoretical sequence, probably due to the loss of the last residues in the GFP protein (Fig. S8†). Nevertheless, as reported in Fig. 4B and C, this phenomenon did not lead to significant loss in the intrinsic emission capacity of the EGFP protein. Furthermore, RBM-SQ displayed a MW compatible with the introduction of two rather than one quencher groups. Aware of the presence of other possible residues accessible in the protein for quencher conjugation, we thereafter adopted a milder protocol of conjugation using a lower amount of reacting quencher. Thereafter, we evaluated the fluorescence intensity of EGFP before and after conjugation with the quencher (Fig. 5B). Notably, a significant reduction in fluorescence in the SQ protein was observed for both mild and strong protocols, providing evidence that the click-chemistry approach provides effective quenching of EGFP. Overall, these data helped us in the optimization of the subsequent labelling reactions.
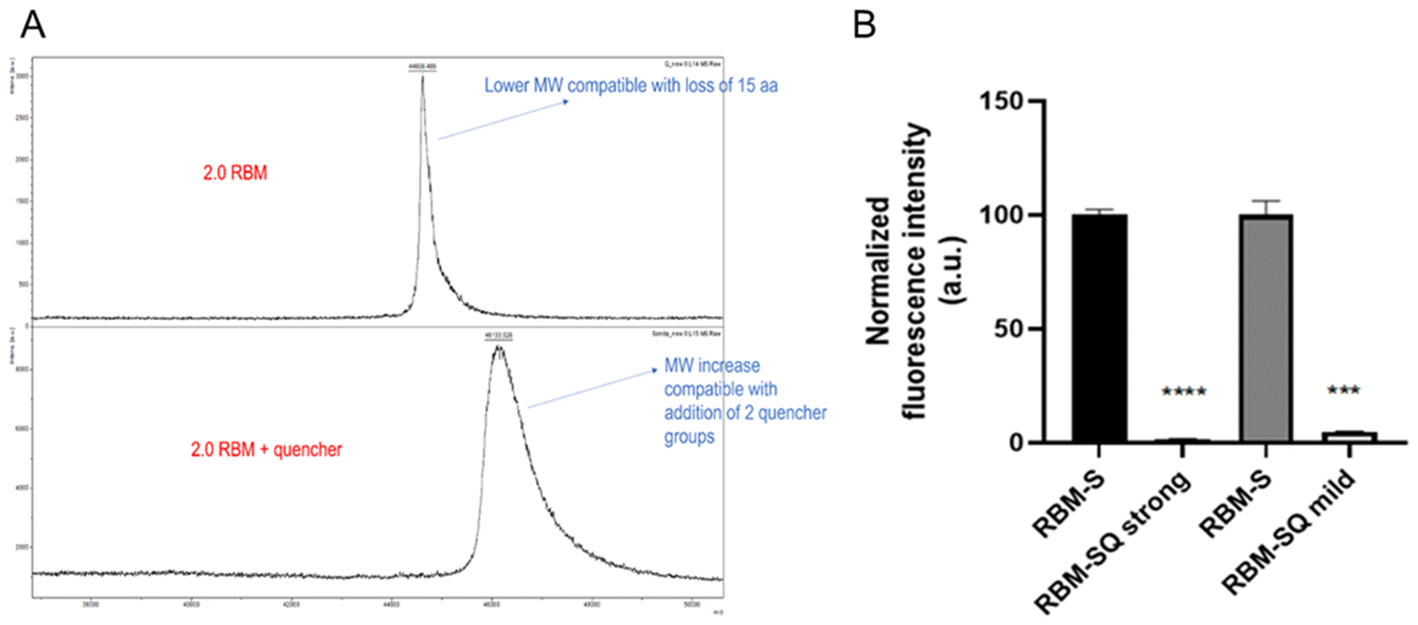 | ||
| Fig. 5 Validation of the conjugation of RBM-S with a quencher molecule. (A) MALDI spectra of RBM-S before (top) and after (bottom) conjugation to the quencher group. (B) Graphs reporting the results of two independent measures of SQ versus S fluorescence intensity, where SQ was produced using strong (left) and mild (right) conjugation conditions. Data are reported as mean ± SD of two or three different replicas. | ||
3.5 Test RBM-S and RBM-SQ fluorescence recovery in the presence of Spike protein
Functional validation assay was performed to demonstrate the sensor ability to recover fluorescence in the presence of Spike protein, indicative of its utility in detecting SARS-CoV-2 antigens (Fig. 6A and B). S or SQ were incubated with increasing amounts of Spike purified protein, or with BSA as a control. The study was conducted at a 10 nM concentration, because at this concentration the probe was detectable also in saliva biological fluid (Fig. S9†). The proteins were mixed directly in the well, in PBS buffer, and were incubated for 30 minutes, then fluorescence intensity was measured with a multi plate fluorescence reader. Fluorescence intensity was normalized to the value of 10 nM S or SQ devoid of any protein addition. Notably, results showed that a recovery of fluorescence could be appreciated for both S and SQ proteins, incubated with a 5 and 10 molar excess of Spike, but not of BSA protein used as a control. These data suggest that S and SQ probes may be efficient in recognizing Spike protein, at selected concentrations, and this may in turn result in a de-quenching of EGFP fluorescence, which corresponds to the opening of the molecular beacon. | ||
| Fig. 6 Functionality assays of RBM-S against SARS-CoV-2 Spike protein. Graphs reporting the results of S (A) versus SQ (B) fluorescence intensity in a multi-well format, obtained by incubating 10 nM S and SQ with 10, 50 and 100 nM of Spike (Sp) and BSA proteins. Data are reported as mean ± SEM of the values pooled from 3 independent experiments performed on two different batches of S and SQ preparations. The plotted fluorescence intensity has been subtracted from the contribution of buffer and Spike or BSA proteins and normalized to the S or SQ fluorescence. **p < 0.01, *p < 0.05, according to a One-Way ANOVA with Dunnett's multiple comparison of means. | ||
According to experimental data, the analytical parameters were determined. To calculate LOD and determine the sensitivity of biosensor, a linear correlation was established with Spike protein concentrations, up to 75 nM for S probe and to 50 nM for SQ probe. Thus, for the S probe, LOD and LOQ resulted in 4.5 and 29.7 nM, respectively. For the SQ probe, LOD and LOQ were 3.2 and 4.2 nM, respectively. Furthermore, by means of eqn (4), we found Kd = 25.0 ± 6.2 nM and Kd = 9.7 ± 3.5 nM (mean ± SD) for S and SQ, respectively. While this mechanism can be expected for SQ protein, the reason why this occurs also for S protein is less obvious, but coherent with the reported simulations. As explained in section 3.3, the closed conformations of the construct display larger fluctuations especially in the region of the GFP chromophore, which is likely to lead to fluorescence loss opening the way to non-radiative paths for the ground state recovery alternative to photon emission. The normal fluorescence can be recovered once the Spike binding leads to the open conformation. In any case, the Spike-induced recovery in fluorescence turned out to be more statistically robust for S than for SQ protein (F∞ − F0 = 1.4 ± 0.4 and F∞ − F0 = 3.6 ± 2.6 (mean ± SD) for S and SQ respectively and Fig. 6A and B), although the former was less in absolute fold-change with respect to the control than the latter. This behavior is also coherent with the simulations: although the dynamic range spanned by the quenching efficiency is potentially very large (panels A and B in Fig. 3), simulations also show that maximum effect requires that the closed conformation occurs with a specific orientation, which may not be always achieved. Conversely, the intrinsic quenching only due to the disturbance of the chromophore region in closed conformation does not require specific closed structures and is therefore statistically more robust.
Another possibility is that this might be due to the lower stability displayed in solution by the SQ probe when compared to the S counterpart. Indeed, once prepared and stored at 4 °C, SQ could be used for no more than 1–2 weeks, to avoid protein precipitation occurring and interfere with the assay. An optimization of SQ preparations, in terms of both higher purity degree and monovalent labelling of the quencher at N-term position, may help to fully exploit the potential of the proposed diagnostic tool.
4. Conclusions
This study presents the design, construction, and validation of a novel FRET-based sensor for detecting the Spike surface antigen of SARS-CoV-2 in biological fluids. The novelty of our biosensor lies in the proposed workflow, which was carefully developed following criteria defined by molecular dynamics (MD) simulations. This approach ensured that the sensor could be effectively produced in E. coli, demonstrating its practical applicability. By harnessing the fluorescence properties of GFP, entirely genetically encoded, we have created a sensor that offers versatile, modular architecture. This structure allows for future optimization and customization for a variety of diagnostic applications.The modularity of the sensor is one of its key advantages. It incorporates two reporter elements and two interaction elements, offering flexibility and scalability for a range of diagnostic needs. Additionally, our design supports two distinct fluorescence measurement approaches: direct GFP fluorescence intensity measurements and a molecular beacon method utilizing an organic quencher. This dual approach enhances the sensor's versatility and sensitivity.
Furthermore, both measurement methods can be easily adapted to widely available multi-plate fluorescence readers, as well as sensor chip formats, or cell-based assays which are commonly found in both research and healthcare settings. This versatility makes the sensor practical for widespread deployment in diverse environments, further enhancing its value for current and future diagnostic and therapeutic applications.
Molecular dynamics simulations were crucial in selecting optimal linkers that balance flexibility and stability, ensuring the functional integrity of the sensor. The AAASSGGGASGAGG and LEAPAPA linkers were identified as the best choices for maintaining stable configurations conducive to reliable FRET signal generation. In particular, calculations based on Förster energy transfer theory validated the sensor's quenching efficiency, providing insights into its performance under various conditions and indicating as the RBM the optimal portion of Spike domain for the construct to function. An alternative mechanism for intrinsic quenching due to the disturbance of the GFP chromophore region in close conformation is clarified through the analysis of the MD simulations.
Following the criteria defined by MD simulations, the sensor was effectively produced in E. coli, demonstrating its practical applicability. Functional validation assays confirmed the sensor ability to recover fluorescence in the presence of the Spike protein, indicative of its potential for sensitive and specific detection of SARS-CoV-2 antigens. To increase sensitivity, the sensor was also stoichiometrically labeled with a Dabcyl quencher probe, as confirmed by mass spectrometry. Although variations in conjugation stoichiometry were observed depending on the click-chemistry protocol, a procedure for the putative 1:1 Dabcyl:GFP conjugation was identified. The performance of the SQ probe turned out to be promising, but also highlighted areas for refinement. Addressing SQ stability challenges and optimizing linker designs will be essential for improving the sensor robustness and longevity. Future work should focus on enhancing the purity and stability of the quenched probe to fully exploit the sensor diagnostic potential.
In summary, this study demonstrates the feasibility and potential of a GFP-based FRET sensor for SARS-CoV-2 detection. The sensor modular design, combined with in silico optimization prodromal to practical validation, lays a strong foundation for developing reliable, sensitive, and specific diagnostic tools for current and future pandemic preparedness.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Spark Global, Proof-of-Concept SPARK PISA 2020-2022, Fret sensor for the Assessment of Coronavirus Titre (FACT). GB acknowledges the funding by DFM.AD004.324. PRIN2020 – 2020LW7XWH “Early Phase Preclinical Development of PACECOR, a Mutation-Independent Anti-SARS-CoV-2 Therapeutic Strategy” and support by EU within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project no. PE00000007, INF-ACT). VT and EDP knowledge the support of Next Generation-EU (Piano Nazionale di Ripresa e Resilienza (PNRR), Missione 4, Componente 2, Ecosistemi dell'Innovazione) through the project Tuscany Health Ecosystem (THE-Spoke 1 and 6, grant ECS_00000017).References
- C. Dye, After 2015: infectious diseases in a new era of health and development, Philos. Trans. R. Soc., B, 2014, 369(1645), 20130426, DOI:10.1098/rstb.2013.0426.
- Q. Rafique, et al., Reviewing methods of deep learning for diagnosing COVID-19, its variants and synergistic medicine combinations, Comput. Biol. Med., 2023, 163, 107191, DOI:10.1016/j.compbiomed.2023.107191.
- M. Pretorius and M. Venter, Diagnosis of Viral Infections, in Viral Infections in Children, ed. R. J. Green, Springer, Cham, 2017, vol. I, DOI:10.1007/978-3-319-54033-7_6.
- N. Younes, et al., Challenges in Laboratory Diagnosis of the Novel Coronavirus SARS-CoV-2, Viruses, 2020, 12(6), 582, DOI:10.3390/v12060582.
- V. Naresh and N. Lee, A Review on Biosensors and Recent Development of Nanostructured Materials-Enabled Biosensors, Sensors, 2021, 21(4), 1109, DOI:10.3390/s21041109.
- L. Liu, F. He, Y. Yu and Y. Wang, Application of FRET Biosensors in Mechanobiology and Mechanopharmacological Screening, Front. Bioeng. Biotechnol., 2020, 8, 595497, DOI:10.3389/fbioe.2020.595497.
- A. K. Verma, A. Noumani, A. K. Yadav and P. R. Solanki, FRET Based Biosensor: Principle Applications Recent Advances and Challenges, Diagnostics, 2023, 13(8), 1375, DOI:10.3390/diagnostics13081375.
- S. P. Tripathy, M. Ponnapati, S. Bhat, J. Jacobson and P. Chatterjee, Femtomolar detection of SARS-CoV-2 via peptide beacons integrated on a miniaturized TIRF microscope, Sci. Adv., 2022, 8(34), eabn2378, DOI:10.1126/sciadv.abn2378.
- H. Kim, J. Ju, H. N. Lee, H. Chun and J. Seong, Genetically Encoded Biosensors Based on Fluorescent Proteins, Sensors, 2021, 21(3), 795, DOI:10.3390/s21030795.
- K. Y. Wen, et al., A Cell-Free Biosensor for Detecting Quorum Sensing Molecules in P. aeruginosa -Infected Respiratory Samples, ACS Synth. Biol., 2017, 6(12), 2293–2301, DOI:10.1021/acssynbio.7b00219.
- D. Zappi, et al., A microbial sensor platform based on bacterial bioluminescence (luxAB) and green fluorescent protein (gfp) reporters for in situ monitoring of toxicity of wastewater nitrification process dynamics, Talanta, 2021, 221, 121438, DOI:10.1016/j.talanta.2020.121438.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, Open Babel: An open chemical toolbox, J. Cheminf., 2011, 3(1), 33, DOI:10.1186/1758-2946-3-33.
- B. P. Cormack, R. H. Valdivia and S. Falkow, FACS-optimized mutants of the green fluorescent protein (GFP), Gene, 1996, 173(1), 33–38, DOI:10.1016/0378-1119(95)00685-0.
- M. C. Martos-Maldonado, et al., Selective N-terminal acylation of peptides and proteins with a Gly-His tag sequence, Nat. Commun., 2018, 9(1), 3307, DOI:10.1038/s41467-018-05695-3.
- J. Lan, et al., Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor, Nature, 2020, 581(7807), 215–220, DOI:10.1038/s41586-020-2180-5.
- E. Vanquelef, et al., R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments, Nucleic Acids Res., 2011, 39(suppl_2), W511–W517, DOI:10.1093/nar/gkr288.
- G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussiañ 16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- W. D. Cornell, et al., A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- A. D. MacKerell, et al., All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins, J. Phys. Chem. B, 1998, 102(18), 3586–3616, DOI:10.1021/jp973084f.
- K. Vanommeslaeghe, et al., CHARMM general force field: A force field for drug–like molecules compatible with the CHARMM all–atom additive biological force fields, J. Comput. Chem., 2010, 31(4), 671–690, DOI:10.1002/jcc.21367.
- L. Cao, et al., De novo design of picomolar SARS-CoV-2 miniprotein inhibitors, Science, 2020, 370(6515), 426–431, DOI:10.1126/science.abd9909.
- L. Schrödinger and W. DeLano, PyMOL. Version 2.4.0, 2020 Search PubMed.
- A. Waterhouse, et al., SWISS-MODEL: homology modelling of protein structures and complexes, Nucleic Acids Res., 2018, 46(W1), W296–W303, DOI:10.1093/nar/gky427.
- D. P. Barondeau, C. D. Putnam, C. J. Kassmann, J. A. Tainer and E. D. Getzoff, Mechanism and energetics of green fluorescent protein chromophore synthesis revealed by trapped intermediate structures, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(21), 12111–12116, DOI:10.1073/pnas.2133463100.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, GROMACS: A message-passing parallel molecular dynamics implementation, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- J. Huang and A. D. MacKerell, CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data, J. Comput. Chem., 2013, 34(25), 2135–2145, DOI:10.1002/jcc.23354.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126(1), 014101, DOI:10.1063/1.2408420.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics, 1996, 14(1), 33–38, DOI:10.1016/0263-7855(96)00018-5.
- R. T. McGibbon, et al., MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories, Biophys. J., 2015, 109(no. 8), 1528–1532, DOI:10.1016/j.bpj.2015.08.015.
- P. Crisalli and E. T. Kool, Multi-Path Quenchers: Efficient Quenching of Common Fluorophores, Bioconjugate Chem., 2011, 22(11), 2345–2354, DOI:10.1021/bc200424r.
- R. Nifosí, P. Amat and V. Tozzini, Variation of spectral, structural, and vibrational properties within the intrinsically fluorescent proteins family: A density functional study, J. Comput. Chem., 2007, 28(14), 2366–2377, DOI:10.1002/jcc.20764.
- Eurachem Guide: The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, ed. H. Cantwell, 3rd edn, 2025, Available from: https://www.eurachem.org Search PubMed.
- F. Trovato, R. Nifosì, A. D. Fenza and V. Tozzini, A minimalist model of protein diffusion and interactions: the green fluorescent protein within the cytoplasm, Macromolecules, 2013, 46, 8311–8322, DOI:10.1021/ma401843h.
- R. Nifosì and V. Tozzini, Cis–trans photoisomerization of the chromophore in the green fluorescent protein variant E2GFP: A molecular dynamics study, Chem. Phys., 2006, 323, 358–368, DOI:10.1016/j.chemphys.2005.09.032.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05040a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |