
Changing the amyloid nucleation process using small molecules and substrates: a way to build two-dimensional materials†
Chao
Chen
a,
Chenyang
Wu
a,
Tiantian
Yang
a,
Wenhui
Zhao
 a,
Jiangtao
Lei
*b and
Dongdong
Lin
*a
a,
Jiangtao
Lei
*b and
Dongdong
Lin
*a
aSchool of Physical Science and Technology, Ningbo University, 818 Fenghua Road, Ningbo 315211, P. R. China. E-mail: lindongdong@nbu.edu.cn
bSchool of Physics and Materials Science, Nanchang University, Xuefu Avenue 999, Nanchang City 330031, P. R. China
First published on 24th January 2025
Abstract
The assembly of two-dimensional (2D) materials on substrates presents a wide range of potential applications in nanomaterials. However, there is limited information available in the literature regarding the tunable nucleation process in molecular assembly. In this paper, a neurodegenerative disease-related short peptide and a small molecule named Fast Green (FG) were selected for their binding affinity with mica/highly oriented pyrolytic graphite (HOPG) substrates. Based on atomic force microscopy (AFM) and molecular dynamics (MD) simulation, we investigated the control of 2D assemblies. By tuning FG small molecules and substrates, the assemblies grew epitaxially from nanosheets to nanofilms on mica and highly ordered nanofilaments on HOPG substrates. Notably, the nuclei formed an orderly array without a critical size or lag phase in the presence of FG molecules on the HOPG substrate, facilitating a quicker co-assembly of ordered filaments compared to bulk conditions. Our MD simulations further demonstrated that the interaction between Aβ16–22 molecules and the HOPG substrate was primarily due to π–π interactions between aromatic rings, which led to the formation of single-layer filaments by lying on the surface of HOPG. Additionally, parallel π–π stacking acted as the primary force to inhibit the aggregation of peptides into fibrils. Overall, our results provide a strategy for modulating the interaction of amyloid peptides with small molecules and substrates in the assembly of 2D nanomaterials.
 Dongdong Lin | Dr Dongdong Lin is an Associate Professor at the School of Physical Science and Technology, Ningbo University. He received his PhD in Condensed Matter Physics from Fudan University and completed a visiting fellowship at the Department of Health Sciences & Technology, ETH Zurich. His research focuses on biomaterials, liquid–liquid phase separation, surface wettability and condensed droplets. |
Introduction
The fibrillation behaviours of amyloid proteins have been extensively investigated to better understand the mechanism of various diseases, so-called amyloid diseases,1–3 in which it has been found that the amyloid peptides could misfold.4 They are associated with dozens of grievous diseases, including Alzheimer's and Parkinson's diseases.5 On the other hand, they are also integral to many bio-functional processes, such as mechanical support, signal transduction, and surface adhesion.6,7 The exploration of the structural information regarding amyloid fibrils and peptides with fibril-like characteristics, as determined by nuclear magnetic resonance (NMR) and X-ray diffraction methods, has resulted in growing sophisticated physical and pathological descriptions of these special aggregates.8 For example, the atomic model of an Aβ1–42 amyloid fibril presented from solid-state NMR (ssNMR) data shows common axial twofold symmetry and a similar protofilament structure with the same number of Aβ molecules per cross-β repeat.9,10 The central, hydrophobic core of the full-length peptide, Aβ16–22, one of the shortest reported amyloidogenic sequences, exhibits a well-ordered conformation in the fibrils.11,12 The ssNMR results show that the peptide adopts a β-sheet structure inside the fibrils, and more specifically, the fibrils are constructed by an antiparallel, in-register conformation.13,14The self-assembly process follows as a result of hydrogen bonding and side-chain interactions (e.g., hydrophobic interactions, π-stacking), which facilitate the orderly formation of β-sheet layers. In particular, short peptide fibrils (such as Aβ16–22) exhibit favourable features, including thermal stability, high bending rigidity, tunable self-assembly, homogeneous fabrication and integration of functionality, and high yield rates during bio-synthesis.12,15,16 These excellent properties promote the extensive exploitation of amyloid fibrils as a kind of potential bio-nanomaterial.17 Notably, the self-assembly process of amyloids is a cooperative process that contains two phases: the lag phase and the elongation phase.18 Soluble monomers of amyloid peptides self-aggregate into prefibrillar aggregates during the lag phase (nucleation), and the protofibrils rapidly extend to mature fibrils in the elongation phase. As a result, self-assembly is described using the concepts of the nucleated conformational conversion (NCC) mechanism and classical nucleation theory (CNT).19,20 The oligomers serve as precursors of the amyloid nucleation process and convert to amyloid nuclei under certain concentrations and interpeptide interactions. The state of nucleation has a fatal influence on the self-assembly process. For example, the small molecule 2,8-bis (2,4-dihydroxyphenyl)-7-hydroxyphenoxazin-3-one (termed O4) can decrease the concentration of toxic Aβ oligomers by accelerating nucleation-dependent polymerization and reducing the toxicity of Aβ1–42.21 Cyclohexanehexol stereoisomers inhibit aggregation of high-molecular-weight oligomers and protofibrils.22 Thus, small molecules can serve as powerful tools for modulating amyloid formation, stabilizing aggregates and influencing structural changes in aggregation pathways.
Self-assembly of short peptides on substrates has been widely applied as a biological model system to study protein folding and to design functional structures (such as fibrils, films, nanotubes, and liquid crystals) for use in biosensors, gene transfer, light harvesting, etc.16,23 For example, amyloid-like peptides (GAV-9) epitaxially self-assemble into 1D nanofilaments with an “upright” conformation on the mica surface.24 However, only under high-salt concentrations, the GAV-9 can self-assemble into highly ordered nanofilaments with upright conformations at the mica/water interface.25 Using peptides selected for their binding affinity to MoS2, amyloid aggregate arrays assembled in one row and the nuclei were ordered from the earliest stages, which could build two-dimensional materials.26 As a result, modulating the nucleation process by substrates could be a potential method to guide the amyloidogenic aggregates to transform into ordered arrays, which have potential applications in nanotechnology.
In this paper, the modulation of co-assembly of a short amyloid peptide (Aβ16–22) by Fast Green (FG) and substrates was investigated by atomic force microscopy (AFM) and molecular dynamics (MD) simulations. The hydrophilic (mica) and hydrophobic (HOPG) substrates were selected to study the construction of two-dimensional materials. We found that FG small molecules could modulate the assembly from nanosheets to nanofilms on the mica surface and highly ordered nanofilaments on the HOPG surface in aqueous solution. Comparatively, the nuclei formed an orderly array without a critical size or lag phase with the existence of FG molecules on the HOPG substrate. Additionally, we discovered that the ordered filaments could assemble on the peptide film induced by the HOPG substrate. Our MD simulation gave further evidence that the peptides tend to lie on the surface of HOPG, providing molecular mechanisms of Aβ16–22 co-assembly with FG molecules on substrates.
Results and discussion
Self-assembly of Aβ16–22 and FG on the mica surface
Our previous studies demonstrated the typical assembly of Aβ16–22 peptides into fibrils in solution.12,15 The process usually takes several days to form mature fibrils. In this study, we examined the aggregation of Aβ16–22 peptides on mica within a small water droplet. As shown in Fig. 1a, the peptide solution (5 μL) was applied on the mica surface and monitored by atomic force microscopy (AFM). The aggregation process of Aβ16–22 was characterized by an increasing number of flakes formed at a minute-level. Height measurements of these flakes revealed that as the residence time increased, single-, double-, and triple-layered structures were formed, measuring approximately 3 nm, 6 nm, and 9 nm in height, respectively. These phenomena indicated that the peptides arranged themselves in layers on the mica substrate. This could be explained by peptides growing layer by layer with the peptide backbone perpendicular to the substrate, which was similar to the reported GAV-9 peptides.27 Instead of forming chiral fibrils with a morphology of ∼4 nm in diameter,12 in this case, the electrostatic interaction between the negatively charged mica substrate and the positively charged N-terminal was the driving force leading to the ordered assembly of Aβ16–22 flakes and films. | ||
| Fig. 1 Self-assembly of Aβ16–22 and FG on the mica surface. (a) Aβ16–22 peptides grow on the mica surface. Representative AFM images and corresponding cross-sectional analyses. 5 μL Aβ16–22 (40 μM) solution was deposited on mica surfaces for 0, 10, and 20 minutes, respectively. (b) Aβ16–22 peptides and FG molecules co-assembled on the mica surface. AFM images of peptides and FG conjugates were captured at 0, 10, and 20 minutes, respectively. Corresponding cross-sectional analyses are shown in the right panel with marked lines. | ||
To alter the nucleation process of Aβ16–22, we investigated the co-assembly of the small molecule Fast Green (FG) and Aβ16–22 peptides on mica. Interestingly, large film-like structures, rather than flakes, were observed on the mica surface when the peptide and FG were in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. 1b). The heights of these new structures were about 1 nm, as determined from line profiles of the film when the incubation time reached 20 min. In the presence of FG molecules, Aβ16–22 peptides were constrained to a flat state. It is speculated that the peptides would lie on the mica surface due to the electrostatic shielding of FG molecules. To exclude the possibility that the structures were assembled solely by FG molecules, we examined the deposition of pure FG molecules on the mica surface. However, it was found that the FG molecules were homogeneously distributed on the substrate (Fig. S1†). These results demonstrated film-like two-dimensional structures on the hydrophilic mica under the regulation of FG molecules, providing a strategy to grow single-layer nanomaterials using peptides and small molecules, with its height tuning from ∼3 nm to ∼1 nm.
:1 ratio (Fig. 1b). The heights of these new structures were about 1 nm, as determined from line profiles of the film when the incubation time reached 20 min. In the presence of FG molecules, Aβ16–22 peptides were constrained to a flat state. It is speculated that the peptides would lie on the mica surface due to the electrostatic shielding of FG molecules. To exclude the possibility that the structures were assembled solely by FG molecules, we examined the deposition of pure FG molecules on the mica surface. However, it was found that the FG molecules were homogeneously distributed on the substrate (Fig. S1†). These results demonstrated film-like two-dimensional structures on the hydrophilic mica under the regulation of FG molecules, providing a strategy to grow single-layer nanomaterials using peptides and small molecules, with its height tuning from ∼3 nm to ∼1 nm.
FG controls the epitaxial assembly of Aβ16–22 on the HOPG surface
To investigate the two-dimensional materials constructed by amyloids on a hydrophobic substrate, highly oriented pyrolytic graphite (HOPG) was selected. As shown in Fig. S1c,† the morphology of the HOPG surface exhibited a smooth surface but with steps between single layers. AFM images (Fig. 2a) revealed that pure Aβ16–22 peptides self-assembled into two-dimensional film structures of various sizes on HOPG. However, the morphologies were different compared to the peptide conjugated with FG on the mica surface. As the incubation period increased on the HOPG surface, the film structures gradually developed edges, transitioning from a disordered state to rectangular shapes. Additionally, phase images provided further information that the assembly after 10 min exhibited a large phase shift, which would be an intermediate state in growth. In contrast, the well-constructed films after 20 min exhibited a small phase shift. Interestingly, the film structures on the surface could cross the steps of HOPG in the process of expansion, a behaviour not observed previously in amyloid systems. These findings suggested that the HOPG surface with its strong hydrophobic properties altered the nucleation of peptides/FG, inducing the formation of nanofilms.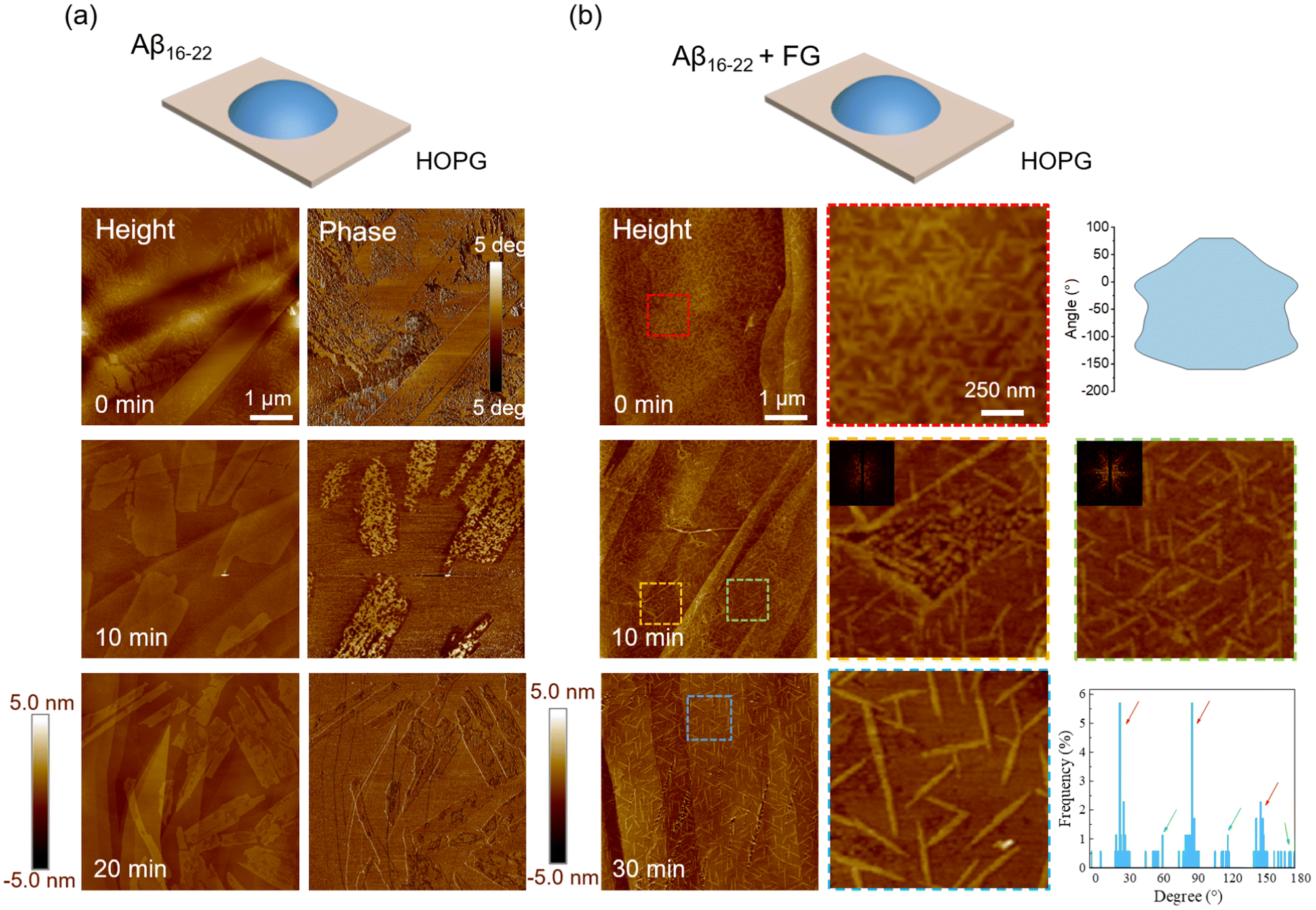 | ||
| Fig. 2 Self-assembly of Aβ16–22 and FG on the HOPG surface. (a) Aβ16–22 peptides grow on the HOPG surface. Representative AFM images and corresponding phase images. 5 μL Aβ16–22 (40 μM) solution was deposited on HOPG surfaces for 0, 10, and 20 minutes, respectively. (b) Aβ16–22 peptides and FG molecules co-assembled on the HOPG surface. AFM images of peptides and FG conjugates were captured at 0, 10, and 20 minutes, respectively. Zoomed images are shown from the dashed line regions. The distribution of orientation was calculated at 0 min and 20 min, respectively. At 10 min, the two-dimensional Fourier transform was applied to each zoomed image. | ||
We further examined the modulation properties of the co-assembly of FG molecules on the HOPG substrate. The nucleation process changed further when the system was added with FG molecules (peptide and FG = 1:1). This time, the small nanofilaments were found after the deposition (Fig. 2b), with a random distribution as shown in the high-resolution image. As freshly prepared (termed 0 min of incubation), the average length of the fibrils was only ∼200 ± 47 nm. As the incubation time increased, the length of the short filaments gradually increased and finally stabilized at about 400 ± 26 nm (20 min). It is found that the filaments after 10 min were primarily oriented in three fixed directions on the planar HOPG surface, but random in some defect regions (yellow zoomed-in region). These orientations became more apparent as the residence time increased to 20 min. The orientations of aggregates mainly along the three directions were reflected in the characteristic 3-fold symmetry of the two-dimensional Fourier transform image (inset), consistent with the underlying atomic lattice of the substrates (Fig. S2a†).24
Most of the filaments aligned with the orientations of 30°, 90° and 150°. However, we found that a small fraction of filaments were also oriented at 60°, 120° and 180°, suggesting that the modulation of HOPG would also have another rotational (30°) packing mode as shown in Fig. S2b.† Notably, there were no oligomers, protofibrils and other intermediates in the quick (minute-level) self-assembly process of nanofilaments, indicating that the presence of FG molecules changed the nucleation process of the peptide without a critical size or lag phase, and the nuclei were arranged in an orderly manner by the atomic lattice. These findings suggested that FG small molecules could modulate the assembly transition from a special film structure to highly ordered nanofilaments on HOPG. This epitaxial nanofilament structure aligned with the lattice structure with a height of ∼1 nm. They were roughly equivalent to the inter-sheet distance of Aβ16–22, indicating that the Aβ16–22 molecules tended to lie on HOPG with their hydrophobic side chains in contact with the HOPG surface. In addition, the samples incubated for a longer time (48 min) were studied. On the HOPG surface, the filaments exhibited a crowded arrangement with an average height of ∼1 nm (Fig. S3†) and were connected to each other. The filaments exhibited a predominant length of approximately 300 nm, suggesting that an increase in incubation time does not alter the lengths of the filaments but promotes their aggregation into nest-like structures. These findings demonstrate that the FG molecule can regulate Aβ16–22 to form completely different epitaxial nanofilaments on the hydrophobic substrate compared to those on the hydrophilic mica. This structure may become an ideal template for functional biological nanomaterials.
Based on the modulation behaviours of FG molecules and the substrate, we further designed a protocol to grow two-layer 2D materials. As shown in Fig. 3, we first grew a single-layer film on the HOPG substrate, as discussed previously. It can be seen that the single-layer peptide film had a thickness of ∼1 nm (line profile in Fig. 3b). Based on this film-covered substrate, we further grew the peptides and FG conjugates. As depicted in the AFM image and schematic in Fig. 3c, the filaments formed both on the substrate and the existing peptide films with highly ordered orientation (Fig. 3c). As a result, we obtained complex double-layer aggregates where the ordered filaments formed on the surface of the peptide film. In addition, this phenomenon indicated that the orientation of the filaments on the surface could still be driven by the atomic lattice interaction adjacent to the peptide film, enriching the construction of 2D biomaterials.
 | ||
| Fig. 3 Two-step growth of peptide film and peptide/FG filaments. (a) AFM image of a cleaner HOPG surface, with its line profile shown in the lower panel. (b) Aβ16–22 peptides were deposited and grew on HOPG surface. The cross-sectional analysis showed a film height of ∼1 nm. (c) Aβ16–22 peptides and FG was deposited and grew on peptide film from b. The cartoon illustrates the double-layer growth of conjugates. The corresponding cross-sectional analysis is shown below with marked lines. | ||
In situ recording of the formation of filaments on the HOPG surface
To investigate the dynamic details of the ordered assembly process, in situ AFM studies were also carried out to capture the assembly at the nanoscale (Fig. 4). We found that the assembly is a dynamic process where the peptides or FG molecules move to different locations, even after being deposited on the substrate. As shown in Fig. 4b, it can be clearly seen that the process of fibril growth occurred in the marked circular region, with their growth directions restricted to three epitaxial orientations, indicating that the self-assembly of Aβ16–22 peptides and FG proceeded along the lattice orientation of the substrate surface. With the help of in situ AFM captures, it gave direct evidence that Aβ16–22 peptides and FG molecules self-assembled into filaments on the surface of the HOPG substrate in the liquid phase without the formation of rich intermediates, indicating that the nuclei formed in an orderly array were different compared with bulk solution conditions. | ||
| Fig. 4 In situ capture of the growth of peptide/FG filaments on the HOPG surface. (a) Schematic diagram of the AFM tip deposited in solution. The temperature was 24 °C and the humidity was 60%. (b) Captured AFM images at the start, 5 min and 10 min, respectively. The dashed line circle and arrays show the dynamic formation of filaments. | ||
Additionally, we observed that, over time, more and more filaments accumulated on the substrate (red arrays). These filaments can extend to lengths of ∼150 nm in 5 min. The results presented a quick in situ nucleation and growth of peptides and FG molecule in our system. The speed would be much faster than the previous measurements.28 Here we did not obtain a large number of highly ordered filaments. The reason would be the disturbance of the AFM tip in solution, leading to a disturbance in assembly. Nevertheless, the in situ measurements provided directional information that the filaments formed along the fixed orientation through nucleation and growth at a high speed.
Interaction between peptides, FG and substrate
To comprehensively understand the molecular mechanisms underlying amyloid aggregation, molecular dynamics simulations were conducted to examine the nucleation process of Aβ16–22 both in solution and on the HOPG surface. Initially, we constructed a system comprising six individual Aβ16–22 peptide chains to investigate the self-assembly characteristics of Aβ16–22 in free solution. As depicted in Fig. 5a, the six randomly distributed peptide chains gradually converged during the assembly process. This interaction ultimately led to the formation of a stable structure with β-sheets. Consistent with the previous AFM results, Aβ16–22 peptides commonly aggregated into fibrils in solution (Fig. S4†). To investigate their binding properties on different amino acid residues, we calculated the contact numbers between different types of residues (Fig. 5b). The phenylalanine residues on two separate peptide chains exhibit the most intimate contact with each other. The result indicated that the aggregation of Aβ peptides primarily depends on the π–π interactions between FF residue pairs, which would be one of the key factors. | ||
| Fig. 5 MD simulation of nucleation with free Aβ16–22 peptides. (a) The contact number calculated between single chains versus simulation time. The inset cartoons show the initial state and final state. The simulation time was 1000 ns. (b) The contact map between Aβ16–22 residues. (c) The β-sheet probability in the system as a function of the number of π–π stacking interactions between aromatic rings. (d) Potential mean force (PMF, in kcal mol−1) as a function of the centroid distance of two aromatic rings and the angle of the two rings; the snapshot shows the arrangement order of the aromatic rings. | ||
We found that β-sheet structures were formed in the system, as shown in Fig. 5c, and the probability of β-sheet formation was proportional to the number of π–π stacking interactions. The result further confirmed that π–π interactions are the primary force driving the close contact and fibril formation between peptide chains. To elucidate the π–π stacking patterns of the aromatic rings of each peptide chain, we analysed the potential mean force (PMF) as a function of the distance between the center of mass of the two aromatic rings and the angle between the two rings (Fig. 5d). The results clearly demonstrated a limited number of aromatic rings forming angles ranging from 30° to 90° within the peptide chains. In contrast, the majority of aromatic rings between peptide chains exhibited a concentrated distribution of angles within 0 to 10°, with distances between these rings ranging from 0.4 to 0.8 nm. A representative snapshot in the inset of Fig. 5d provided a clear visualization of the parallel stacking pattern exhibited by the aromatic rings.
In the next step, we examined Aβ16–22 self-assembly on hydrophobic substrates by placing thirty Aβ16–22 peptide chains on a graphene substrate for simulation (500 ns, Fig. 6). The snapshots clearly showed the dynamic evolution of peptides on the HOPG surface, from randomly distributed to film-like aggregates. However, due to the strong interaction with the HOPG substrate, the β-sheet structures were hard to be formed (see secondary structural map in Fig. S5†), dominated by coil, bend and turn structures. First, we analyzed the changes in the contact number between the peptide and the substrate surface. The results exhibited that the molecules of the peptide chains interacted with the atoms on the HOPG surface, from an upright state to lie down on the surface. The height of the main chain reached about 1 nm. The simulation results were in accordance with our experimental observation. The analysis of the contact atoms further showed that the side chains of F and L residues exhibited the largest contact probability. The result indicated that Aβ16–22 bound to atoms on the surface of graphene mainly through π–π stacking and hydrophobic interactions. In order to analyse the interactions of π–π stacking between Aβ16–22 and the substrate, we conducted the PMF map between the peptide and HOPG during a simulation duration of 500 ns. The angles between the aromatic rings in the residues and the HOPG substrate were concentrated within 15°, and the distance was concentrated at 0.35 nm, indicating that a parallel π–π stacking mode had been mainly formed between the diphenylalanine residues and the substrate. Our results confirmed that the Aβ16–22 molecules tend to lie on the surface of the HOPG substrate, driving the formation of various 2D nanofilms.
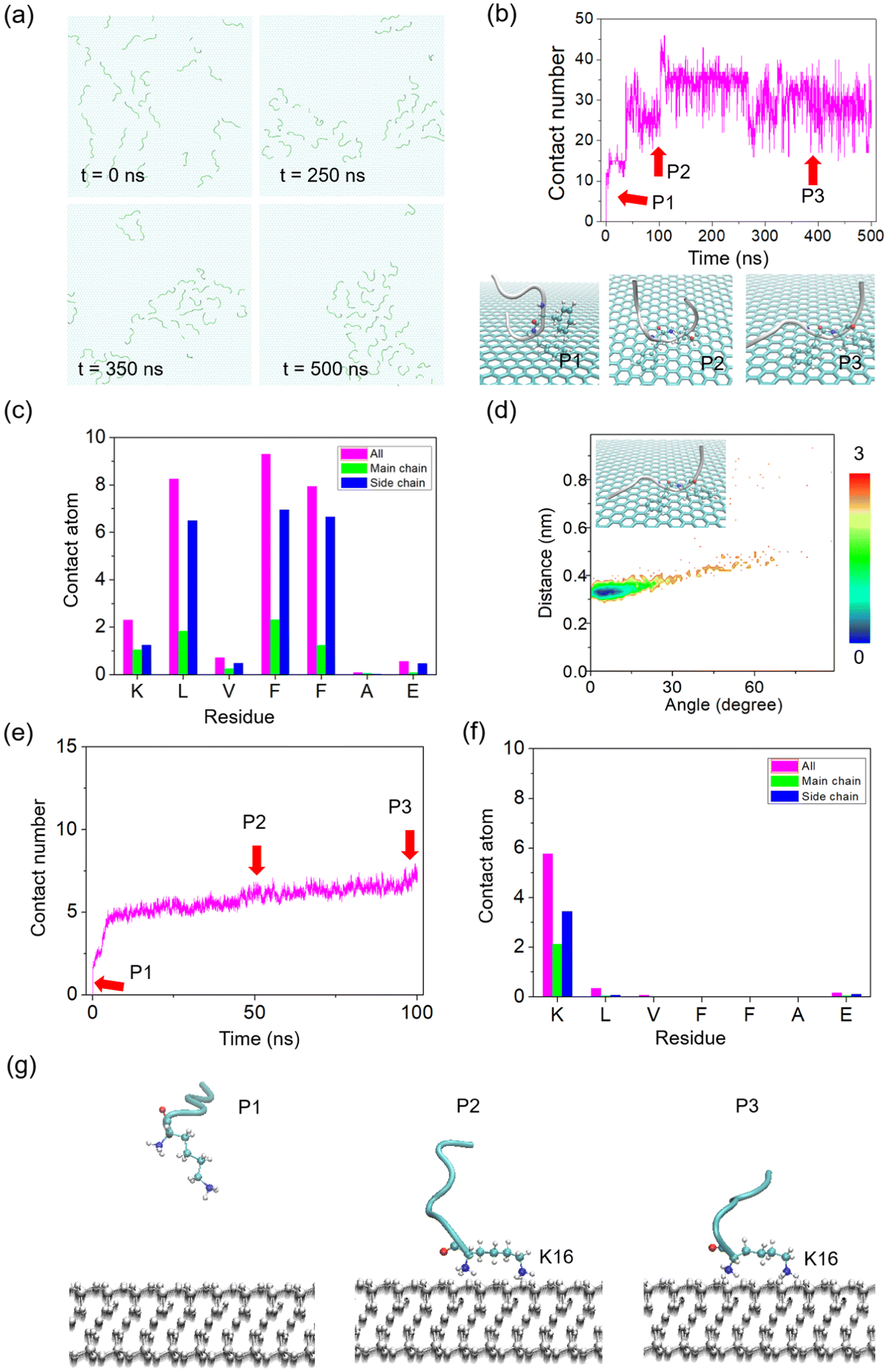 | ||
| Fig. 6 MD simulation of Aβ16–22 peptide nucleation on HOPG and mica surfaces. (a) The snapshots of the growth state (0 ns, 250 ns, 350 ns and 500 ns, respectively) on the HOPG surface. 30 chains were used in the system. (b) Time evolution of the contact number between Aβ16–22 and the HOPG surface. Three typical snapshots were exhibited with different postures. (c) Analysis of the contact number between the substrate surface and each residue of Aβ16–22. (d) Potential mean force (PMF, in kcal mol−1) as a function of the centroid distance of two aromatic rings and the angle of the two rings, with one ring from the amyloid and the other from graphene. (e) Time evolution of the contact number between Aβ16–22 and the mica surface. (f) Time evolution of the contact number between Aβ16–22 and the mica surface. (g) Three typical snapshots were exhibited with different postures from (e) with red arrows with key points. | ||
In the mica system, the interactions were also investigated. Similarly, thirty Aβ16–22 peptide chains were placed on a mica substrate and randomly distributed initially. Due to the strong electrostatic interaction between the N-terminal of Aβ16–22 peptides and mica substrate with negative charges, the peptides show upright postures with the N-terminal connected to the mica surface through the simulation (Fig. S6†). In order to check the binding information, the contact number was calculated, exhibiting that the peptides quickly adsorbed onto the mica surface from 5.4 ns (Fig. 6e). The analysis of the contact atoms further exhibited that the side chains of K and L residues have the only contact probability with the substrate (Fig. 6f), proving the upright state. To show the states visually, three representative points were captured in Fig. 6g. The results indicated that Aβ16–22 peptides interacted with the surface of mica in an upright way by the pinning of K residues. In the presence of FG molecules, the nucleation process was further explored. As shown in Fig. S7,† thirty peptides and thirty FG molecules were designed in the MD system. Instead of interacting with the mica surface, the FG molecules prefer to bind to the peptides, as shown by the result of contact analysis that the FG molecules had a lower contact number value than the peptides (Fig. S7b†). This time, the upright state was disturbed with the existence of FG molecules, showing full contact with the substrate (Fig. S7c†). We speculate that the electrostatic tunability from the FG molecules provides the possibility of formation of 2D nanofilms.
Conclusion
In summary, we investigated the nucleation of 2D assemblies on hydrophilic (mica) and hydrophobic (HOPG) substrates through AFM and MD simulations. FG small molecules and substrates can modulate the assembly transitions from nanosheets to nanofilms on the mica surface and highly ordered nanofilaments on the HOPG surface. The peptides and FG conjugates grew epitaxially on the HOPG surface without a lag phase. In situ AFM images directly exhibited the dynamic process of epitaxial growth of Aβ16–22 on the HOPG surface. Using MD simulation, we observed that the close contact between Aβ16–22 and the HOPG surface mainly adopted the parallel π–π stacking. The Aβ16–22 peptide exhibited a “lying” configuration on the surface of the HOPG substrate, suggesting a favourable two-dimensional alignment and adhesion. Our findings enrich the understanding of the amyloid aggregation mechanisms and pave the way for nanoscale manipulation of peptide self-assembly by introducing small molecules and alterations in the substrate, enabling the production of highly ordered and controllable 2D nanomaterials.Materials and methods
Materials and sample preparation
Synthetic Aβ16–22 peptide composed of seven amino acids (KLVFFAE) was purchased from GL Biochem Ltd (Shanghai, China). Fast Green (FG) was purchased from Hefei Qiansheng Biotechnology Co., Ltd (Anhui, China). The final purity of FG was greater than 98%. Hydrophilic muscovite was sourced from Sichuan Ya'an Mica Co., Ltd, and hydrophobic highly oriented pyrolytic graphite (HOPG) was procured from Shanghai Onway Technology Co., Ltd. All deionized water used in this work was made using a Millipore purification system with a minimum resistivity of 18.2 MΩ cm.To prepare the Aβ16–22 solution, 1 mg of the peptide dry powder, stored at −20 °C, was meticulously dissolved in 14.7 mL of deionized water for separation. Immediately, the solution was diluted to achieve a final concentration of 80 μM. The remaining solution was stored at −20 °C. 8 mg FG dry powder was precisely weighed and dissolved to prepare a 10 mM FG solution. The final diluted concentration was 40 μM.
Atomic force microscopy
The clean and smooth mica and HOPG substrates were stripped to expose fresh surfaces. A 5 μL aliquot of the uncultured mixed solution was then carefully dispensed onto the surface of either the mica or HOPG substrate. After the solution has been allowed to dwell on the surface for a designated time, the liquid droplet was absorbed through a pipette and then dried with a gentle nitrogen flow. The growth temperature on the substrate was 24 °C. A high-humidity environment was provided to avoid the quick evaporation of the droplet. The evaporation in 15 min could be neglected as the whole evaporation of the droplet takes about 120 min. The samples were observed using a Multimode VIII AFM (Bruker, USA). Tapping mode was applied in air, with a scan rate of 1.0 Hz. We used the NanoScope Analysis 1.5 and FiberAPP software29 to analyse the AFM images.For in situ AFM measurement, a homemade device was used. It can maintain the temperature and solution concentration automatically during the entire imaging period. Real-time AFM images were acquired in fluid with a soft tip at a scan rate of 2.5 Hz.
MD simulation
We constructed the Aβ16–22 fibril based on X-ray diffraction data (PDB ID: 3OW9). Atomistic MD simulations were performed in the isothermal-isobaric (NPT) ensemble using GROMACS 2018 software.30 The GROMOS53A6 force field31 is applied to model the Aβ structure as it can obtain Aβ conformational propensities in agreement with nuclear magnetic resonance results.32 The graphite sheet is 16 nm × 15.8 nm and the mica sheet is 14 nm × 13.5 nm in size, providing sufficient area for the peptides to adsorb. During the initial configuration, the minimum distance between the peptides/FG molecules and the graphite/mica sheet is 1 nm. The graphite system and the mica system were solvated in a 16 nm × 15.8 nm × 6 nm and a 14 nm × 13.5 nm × 7 nm SPC33 (simple point charge model) water box, respectively. The total number of water molecules in the systems is 37434 and 34844, respectively. All MD simulations were performed under periodic boundary conditions. The position of the graphite/mica sheet was fixed during all of the MD simulations. The time step is 2 fs, and peptide bonds are constrained by the LINCS algorithm.34 The temperature is maintained at 310 K using the velocity rescaling method,35 and the pressure is kept at 1 bar using the isotropic Parrinello–Rahman method.36,37 Long-range electrostatic interactions are calculated using the PME method38 with a real-space cutoff of 1.0 nm, and van der Waals interactions are calculated using a cutoff of 1.4 nm. Secondary structure analyses were performed using the DSSP method.39
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
D. L. would like to acknowledge the financial support from the National Natural Science Foundation of Ningbo (Grant No. 2024J198). W. Z. would like to acknowldge the financial support from the National Natural Science Foundation of Ningbo (Grant No. 2022J127). J. L. would like to acknowledge the financial support from the National Natural Science Foundation of China (12264027) and the Natural Science Foundation of Jiangxi Province (20242BAB20030).References
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188–1203 CrossRef CAS PubMed.
- D. J. Selkoe, Cell, 1989, 58, 611–612 CrossRef CAS PubMed.
- J. C. Polanco, C. Li, L.-G. Bodea, R. Martinez-Marmol, F. A. Meunier and J. Götz, Nat. Rev. Neurol., 2018, 14, 22–39 CrossRef CAS PubMed.
- J. E. Maggio, E. R. Stimson, J. R. Ghilardi, C. J. Allen, C. E. Dahl, D. C. Whitcomb, S. R. Vigna, H. V. Vinters, M. E. Labenski and P. W. Mantyh, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5462–5466 CrossRef CAS PubMed.
- D. J. Irwin, V. M. Y. Lee and J. Q. Trojanowski, Nat. Rev. Neurosci., 2013, 14, 626–636 CrossRef CAS PubMed.
- J. Li, T. McQuade, A. B. Siemer, J. Napetschnig, K. Moriwaki, Y.-S. Hsiao, E. Damko, D. Moquin, T. Walz, A. McDermott, F. K.-M. Chan and H. Wu, Cell, 2012, 150, 339–350 CrossRef CAS PubMed.
- S. K. Maji, M. H. Perrin, M. R. Sawaya, S. Jessberger, K. Vadodaria, R. A. Rissman, P. S. Singru, K. P. R. Nilsson, R. Simon, D. Schubert, D. Eisenberg, J. Rivier, P. Sawchenko, W. Vale and R. Riek, Science, 2009, 325, 328–332 CrossRef CAS PubMed.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef PubMed.
- M. Schmidt, C. Sachse, W. Richter, C. Xu, M. Fändrich and N. Grigorieff, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19813–19818 CrossRef CAS PubMed.
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schröder, Science, 2017, 358, 116–119 CrossRef CAS PubMed.
- D. Lin, Y. Luo, S. Wu, Q. Ma, G. Wei and X. Yang, Langmuir, 2014, 30, 3170–3175 CrossRef CAS PubMed.
- D. Lin, Z. Qian, M. Bagnani, M. A. Hernández-Rodríguez, J. Corredoira-Vázquez, G. Wei, L. D. Carlos and R. Mezzenga, ACS Nano, 2023, 17, 9429–9441 CrossRef CAS PubMed.
- J. J. Balbach, Y. Ishii, O. N. Antzutkin, R. D. Leapman, N. W. Rizzo, F. Dyda, J. Reed and R. Tycko, Biochemistry, 2000, 39, 13748–13759 CrossRef CAS PubMed.
- S. A. Petty and S. M. Decatur, J. Am. Chem. Soc., 2005, 127, 13488–13489 CrossRef CAS PubMed.
- D. Lin, J. Lei, S. Li, X. Zhou, G. Wei and X. Yang, J. Phys. Chem. B, 2020, 124, 3459–3468 CrossRef CAS PubMed.
- D. Lin, M. Bagnani, H. Almohammadi, Y. Yuan, Y. Zhao and R. Mezzenga, Adv. Mater., 2024, 36, 2312564 CrossRef CAS PubMed.
- C. A. E. Hauser, S. Maurer-Stroh and I. C. Martins, Chem. Soc. Rev., 2014, 43, 5326–5345 RSC.
- A. S. DeToma, S. Salamekh, A. Ramamoorthy and M. H. Lim, Chem. Soc. Rev., 2012, 41, 608–621 RSC.
- R. D. Hills and C. L. Brooks, J. Mol. Biol., 2007, 368, 894–901 CrossRef CAS PubMed.
- E. Chatani and N. Yamamoto, Biophys. Rev., 2018, 10, 527–534 CrossRef CAS PubMed.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez del Amo, B. A. Grüning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoegl, M. Fändrich, R. F. Frank, B. Reif, S. Günther, D. M. Walsh and E. E. Wanker, Nat. Chem. Biol., 2012, 8, 93–101 CrossRef CAS PubMed.
- J. McLaurin, M. E. Kierstead, M. E. Brown, C. A. Hawkes, M. H. L. Lambermon, A. L. Phinney, A. A. Darabie, J. E. Cousins, J. E. French, M. F. Lan, F. Chen, S. S. N. Wong, H. T. J. Mount, P. E. Fraser, D. Westaway and P. S. George-Hyslop, Nat. Med., 2006, 12, 801–808 CrossRef CAS PubMed.
- T. Yang, T. Yu, W. Zhao and D. Lin, Chin. Phys. B, 2021, 30(8), 088701 CrossRef CAS.
- F. Zhang, H.-N. Du, Z.-X. Zhang, L.-N. Ji, H.-T. Li, L. Tang, H.-B. Wang, C.-H. Fan, H.-J. Xu, Y. Zhang, J. Hu, H.-Y. Hu and J.-H. He, Angew. Chem., Int. Ed., 2006, 45, 3611–3613 CrossRef CAS PubMed.
- B. Dai, S.-G. Kang, T. Huynh, H. Lei, M. Castelli, J. Hu, Y. Zhang and R. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8543–8548 CrossRef CAS PubMed.
- J. Chen, E. Zhu, J. Liu, S. Zhang, Z. Lin, X. Duan, H. Heinz, Y. Huang and J. J. De Yoreo, Science, 2018, 362, 1135–1139 CrossRef CAS PubMed.
- B. Dai, S.-G. Kang, T. Huynh, H. Lei, M. Castelli, J. Hu, Y. Zhang and R. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8543–8548 CrossRef CAS PubMed.
- P. N. Nirmalraj, J. List, S. Battacharya, G. Howe, L. Xu, D. Thompson and M. Mayer, Sci. Adv., 2020, 6, eaaz6014 CrossRef CAS PubMed.
- I. Usov and R. Mezzenga, Macromolecules, 2015, 48, 1269–1280 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- O. O. Olubiyi and B. Strodel, J. Phys. Chem. B, 2012, 116, 3280–3291 CrossRef CAS PubMed.
- H. Berendsen, J. P. M. Postma, W. van Gunsteren and J. Hermans, Intermol. Forces, 1981, 11, 331–342 Search PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. Nosé and M. L. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04624b |
| This journal is © The Royal Society of Chemistry 2025 |