
Using a newly designed porphyrin photocatalyst based on triptycene to emulate natural photosynthesis for regioselective fixation of NAD(P)+ to NAD(P)H and synthesis of value-added chemicals†
Rehana
Shahin
a,
Rajesh K.
Yadav
 *a,
Shaifali
Mishra
a,
Kanchan
Sharma
a,
Wonil
Seo
b,
Joonghan
Kim
b,
Navneet K.
Gupta
c and
Jin Ook
Baeg
*d
*a,
Shaifali
Mishra
a,
Kanchan
Sharma
a,
Wonil
Seo
b,
Joonghan
Kim
b,
Navneet K.
Gupta
c and
Jin Ook
Baeg
*d
aDepartment of Chemistry and Environmental Science, Madan Mohan Malaviya University of Technology, Gorakhpur, U.P., India. E-mail: rajeshkr_yadav2003@yahoo.co.in
bDepartment of Chemistry, The Catholic University of Korea, Bucheon, Gyunggi-do 14662, South Korea
cCentre for Sustainable Technologies, Indian Institute of Science, Gulmohar Marg, Mathikere, Bengaluru 560012, India
dKorea Research Institute of Chemical Technology, N3, 141 Gajeong-ro, Yuseong-gu, Daejeon 34114, South Korea. E-mail: jobaeg@krict.re.kr
First published on 31st May 2025
Abstract
This work explores a novel porphyrin photocatalyst based on triptycene, designed to replicate natural photosynthesis and facilitate regioselective fixation of NAD(P)+ to NAD(P)H and convert organic molecules into value-added chemicals. By leveraging the unique structural characteristics of triptycene, the photocatalyst amplifies light absorption and expedites electron transfer processes. The system exhibits a high selectivity for NAD(P)+ reduction under visible light irradiation, enabling the efficient production of NAD(P)H with low by-product formation. Additionally, the photocatalyst effectively catalyses the conversion of a variety of organic substrates into useful, underexplored motifs in medicine, demonstrating notable increases in yield and reaction efficiency in comparison with those of conventional techniques. This study contributes to the development of green technologies that emulate the effectiveness of natural photosynthesis, highlighting the potential of manufactured photocatalytic systems to promote sustainable chemical transformations. These results highlight the potential of porphyrin photocatalysts based on triptycene for advancing the field of photochemical catalysis and organic synthesis.
Introduction
The goal of mimicking natural photosynthesis is to reproduce the mechanisms that turn sunlight into chemical energy, thereby offering long-term solutions for the production of chemicals and renewable energy. This goal is achieved by the photocatalytic process. Nicotinamide adenine dinucleotide (NAD(P)H) regeneration from NAD(P)+ is one of its many applications that is of great interest. As an electron donor in a number of biological processes, such as the reduction of organic molecules, NAD(P)H is a crucial cofactor in redox reactions.1,2 The creation of sustainable processes for the synthesis of useful compounds from renewable resources would be made possible by the capacity to replenish NAD(P)H effectively in the presence of sunlight.3,4 Conventional techniques for NAD(P)H regeneration generally depend on enzymatic processes, which can be efficiently constrained by variables such substrate supply, reaction conditions, and enzyme stability.5 As a result, there is increased interest in non-enzymatic methods of NAD(P)H regeneration, particularly those that make use of photocatalytic systems that are able to use solar energy to power redox processes.6 One of the primary challenges hindering the industrial use of enzymatic methods for new energy production is the high cost of commercial NAD(P)H, which is priced at approximately US$3000 per mole. This is because NAD(P)H is converted into NAD(P)+ during enzymatic reactions. To advance industrial applications, there is an urgent need for simple and efficient methods to regenerate NAD(P)H from the consumed NAD(P)+.7,8 Using triptycene-based porphyrin photocatalysts, which efficiently absorb light and aid in the regioselective reduction processes that convert NAD(P)+ to NAD(P)H, is one intriguing approach.9–12 Because of their well-established optical and electrical characteristics, porphyrins are a great choice for photocatalytic uses.13 Numerous photocatalytic reactions have extensively investigated their capacity to engage in electron transfer processes and absorb light across the visible spectrum.14 However, when exposed to sustained light irradiation, typical porphyrins frequently experience constraints with regard to their stability and reactivity.15 Triptycene integration into porphyrin structures provides a special remedy for these problems. The resulting hybrid photocatalysts are more stable and have better electrical characteristics when they contain triptycene, a polycyclic aromatic hydrocarbon with a stiff three-dimensional structure.16,17 This change enhances the plant's capacity to absorb light while simultaneously promoting effective charge separation and transfer, which is essential for triggering photoredox processes.18 In synthetic chemistry, the regioselective photoreduction of NAD(P)+ to NAD(P)H via a porphyrin photocatalyst based on triptycene offers a tactical advantage. The selective production of important compounds in organic transformations can be facilitated by controlling the regioselectivity of reductions, which can have a considerable impact on product distribution.19 Porphyrin photocatalysts based on triptycene have recently been synthesized, and their potential for effective electron transfer has been shown. This electron transfer can promote the reduction of NAD(P)+ and enhance the transformation of different organic substrates. According to studies, these photocatalysts can produce NAD(P)H with excellent selectivity and yield, which can help with later reactions that turn organic molecules into value added chemicals11,20,21Porphyrin photocatalysts based on triptycene have demonstrated potential not only in NAD(P)H regeneration but also in a variety of photochemical processes, such as carbonyl compound reduction and the conversion of basic substrates into more complex, high-value products. This feature highlights the potential of these photocatalysts as flexible catalysts for chemical synthesis and instruments for cofactor renewal. Porphyrin photocatalysts based on triptycene have the potential to create a sustainable pathway for the production of valuable compounds from renewable feedstocks, thereby aiding in the establishment of a circular economy, by combining effective NAD(P)H regeneration and cyclization reaction for the synthesis of 3,4-dihydropyrimidin-2-(1H)-one (DHP) derivatives. DHPs are a significant class of value-added chemicals that have garnered a lot of interest because of their biological and pharmacological properties, which include calcium channel modulators, and anti-inflammatory, antiviral, anti-hypersensitive, anti-tumour, antimicrobial, and anti-HIV medicines.22–29 An aldehyde, keto-ester, and urea undergo a one pot three component reaction under highly acidic conditions as part of the conventional process for creating DPH derivatives, which was first designated by Pietro Biginelli (Italian chemist) in 1893.30 Unfortunately, this original process had lengthy reaction times, poor product yields, and harsh reaction conditions. Due to the potential significance of DPH derivatives, a number of enhanced techniques for their synthesis in the presence of different catalysts have been discovered.31–45 Although the developments were made, a number of these techniques had drawbacks, including the use of costly catalysts and hazardous organic solvents, poor yields, prolonged reaction times, and laborious workup procedures. DHP derivative synthesis is therefore still one of the most significant problems in the field of value added DHP derivative chemical synthesis. To achieve this, we proposed a metal free, greener approach with a cost effective method by using solar light as a source of renewable energy. In this work, we investigated the synthesis and characterisation of porphyrin photocatalysts based on triptycene with the goal of converting organic molecules into value-added chemicals and regioselectivity reducing NAD(P)+ to NAD(P)H in the presence of visible light. To comprehend the photocatalysts’ performance and mechanisms in the regeneration of NAD(P)H and organic transformations for DHP derivatives, a range of characterization approaches, such as UV-visible spectroscopy, cyclic voltammetry, and photochemical analysis, will be employed in a thorough evaluation. The results of this study will contribute to our knowledge of artificial photosynthesis and offer guidance for creating sustainable, effective catalytic systems for organic synthesis (Scheme 1).
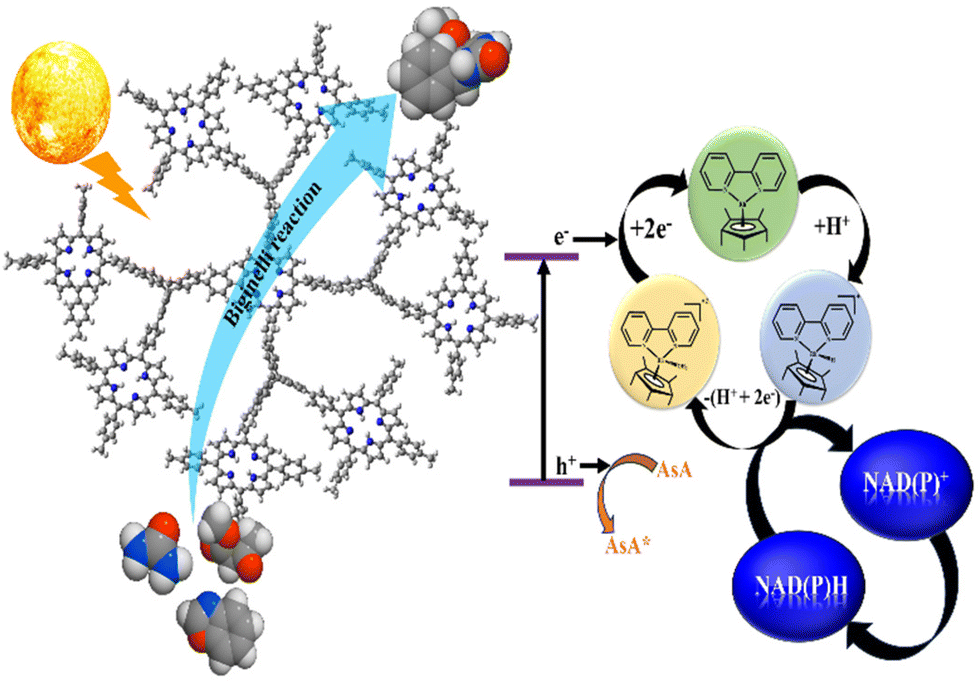 | ||
| Scheme 1 Diagrammatic representation of photocatalytic NAD(P)H regeneration via artificial photosynthesis and addition cyclization reaction. | ||
Experimental section
Photocatalytic experiment
A photocatalytic NAD(P)H regeneration experiment was conducted using a reaction medium containing NAD(P)+, ascorbic acid (AsA), sodium phosphate buffer (SPB) and a PBT photocatalyst. The reaction mixture was placed in a quartz cuvette and illuminated with visible light (λ ≥ 420 nm) from a xenon lamp. The distance between the light source and the reactor was 5.5 cm. The PBT photocatalyst's efficiency was evaluated by examining the absorbance at a 340 nm wavelength using an ultraviolet-visible spectrophotometer.8,46 The same experimentation was performed to check the photocatalytic activity of P in NAD(P)H regeneration. The photocatalytic activity of porphyrin (P) and PBT photocatalysts in NAD(P)H regeneration was assessed by monitoring the absorbance at 340 nm in the dark and under light at 30 minutes intervals using a UV-Visible spectrophotometer. The characteristic absorption peak of 340 nm, as described by the Lambert–Beer law, was used for the quantitative analysis of the NAD(P)H cofactor (molar extinction coefficient, ε = 6.22 mM−1 cm−1).47,48 This measurement allowed for the quantification of NAD(P)H production over time, providing insights into the photocatalytic efficiency of both P and PBT photocatalysts.49,50Green chemistry approach: Biginelli reaction
Aromatic aldehyde (1.0 mmol, 1), urea (1.5 mmol, 2), methyl acetoacetate (1.5 mmol, 3), and PBT (5 mg) in EtOH (3 mL) were stirred at room temperature under aerobic conditions. The progress of the reaction was recorded by TLC. After a reaction period of 5 minutes, the 3,4-dihydropyrimidin-2-(1H)-one product was recovered by a normal filtration technique and characterized by 1H-NMR and 13C-NMR spectroscopy. The ESI† contains comprehensive information51 (Fig. S5–S12†). The percentage yields were calculated by comparing the theoretical yield with the experimental yield, based on the mass of the purified product obtained after isolation (Fig. 1).52,53 | ||
| Fig. 1 Reaction conditions for green synthesis of DHPs with catalysis by the PBT photocatalyst under solar light. | ||
Results and discussion
In this work, we report the synthesis and characterization of porphyrin photocatalysts based on triptycene, with the aim of reducing NAD(P)+ to NAD(P)H regioselectively in the presence of visible light and transforming organic molecules into value-added chemicals. In order to fully understand the mechanisms and performance of the photocatalysts in the regeneration of NAD(P)H and organic transformations for DHP, a variety of characterization techniques including cyclic voltammetry, photochemical analysis, and UV-visible DRS spectroscopy were used in an extensive evaluation. The findings of this research enhance our understanding of synthetic photosynthesis and provide direction for the development of efficient and long-lasting catalytic systems for organic synthesis.Because of its extended conjugated structure, tetrabromophenylporphyrin (P), a brominated derivative of porphyrin, displays unique UV-visible absorption characteristics (Fig. 2a). Porphyrin derivatives are characterized by strong Soret bands (B-bands) in the range of 400–450 nm and low-energy Q bands in the region of 500–700 nm in the UV–visible spectrum.10 The electronic distribution of the porphyrin core is altered by the presence of bromine substituents, and this can cause red shifts in the absorption maxima.54 Studies have been shown that porphyrins influence the reactivity in photocatalytic processes and improve photochemical stability.13 The polycyclic aromatic hydrocarbon triptycene (T) is notable by its inflexible three-dimensional structure. Its UV-visible spectrum (Fig. S3†) typically shows very weak UV absorption bands, peaking at approximately 300–350 nm, corresponding to π–π* transitions.55 As a stand-alone photocatalyst, triptycene is less effective due to its limited absorption in the visible region. But when it is incorporated into porphyrin systems, its structural qualities play a major role in improving the overall photophysical properties. Porphyrin photocatalysts based on triptycene combine the benefits of porphyrins and triptycene (Fig. S3†). Due to the combined actions of the porphyrin core and the triptycene moiety, these hybrid systems usually exhibit higher absorption with a redshift in the visible region in their UV-visible spectra.56 The incorporation of triptycene frequently results in higher light absorption and enhanced photocatalytic performance, making these materials appropriate for a range of uses in synthetic photosynthesis and organic transformations. By altering the porphyrin and triptycene structures, these catalysts’ redox characteristics can be adjusted to enable certain photochemical processes.57
 | ||
| Fig. 2 (a) UV-visible DRS spectrum of the PBT photocatalyst with the inset showing the Tauc plot and(b) Fourier transform infrared spectra of T (red), P (green), and PBT photocatalysts (blue). | ||
Molecular vibrations and functional groups of P, T and PBT photocatalysts are revealed using FTIR spectroscopy. The FTIR spectrum of P corresponds to strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretches between 1500 and 1670 cm−1, CN stretching vibrations at approximately 1600–1680 cm−1, the pyrrolic ring's out-of-plane C–H bending vibrations located approximately at 750–900 cm−1, and C–Br stretching frequency approximately at 625 cm−1. These are typically observed in the region between 500 and 700 cm−1 and are known to exhibit characteristic absorption bands.58 Other characteristics brought about by the bromination include changes in vibrational modes as a result of alterations in the C–H stretching frequencies brought about by the bromine atoms’ ability to remove electrons.59 Three unique absorption bands are visible in the triptycene FTIR spectrum (Fig. 2b). These bands correspond to C–H stretching vibrations, which are roughly 2950 cm−1, and CC stretching vibrational modes around 1453 cm−1 due to the aromatic benzene ring.60 The distinct spectroscopic signature of triptycene is influenced by the overall vibrational pattern, which is a result of its rigidity.16 Porphyrin photocatalysts based on triptycene exhibit a combination of absorption characteristics from the triptycene and porphyrin components in their FTIR spectra. It is possible to see characteristic bands that correspond to the triptycene moiety and porphyrin core (CC, CN), showing that the two structures were successfully integrated. The electrical interactions between the two components are highlighted by the changes in absorption bands in comparison with those of pure porphyrins and triptycene, which may improve the hybrid materials’ photocatalytic qualities. To further enhance their stability and reactivity in photocatalytic applications, the emergence of additional bands may also signal the creation of hydrogen bonds around 2950–3300 cm−1.61–63
C stretches between 1500 and 1670 cm−1, CN stretching vibrations at approximately 1600–1680 cm−1, the pyrrolic ring's out-of-plane C–H bending vibrations located approximately at 750–900 cm−1, and C–Br stretching frequency approximately at 625 cm−1. These are typically observed in the region between 500 and 700 cm−1 and are known to exhibit characteristic absorption bands.58 Other characteristics brought about by the bromination include changes in vibrational modes as a result of alterations in the C–H stretching frequencies brought about by the bromine atoms’ ability to remove electrons.59 Three unique absorption bands are visible in the triptycene FTIR spectrum (Fig. 2b). These bands correspond to C–H stretching vibrations, which are roughly 2950 cm−1, and CC stretching vibrational modes around 1453 cm−1 due to the aromatic benzene ring.60 The distinct spectroscopic signature of triptycene is influenced by the overall vibrational pattern, which is a result of its rigidity.16 Porphyrin photocatalysts based on triptycene exhibit a combination of absorption characteristics from the triptycene and porphyrin components in their FTIR spectra. It is possible to see characteristic bands that correspond to the triptycene moiety and porphyrin core (CC, CN), showing that the two structures were successfully integrated. The electrical interactions between the two components are highlighted by the changes in absorption bands in comparison with those of pure porphyrins and triptycene, which may improve the hybrid materials’ photocatalytic qualities. To further enhance their stability and reactivity in photocatalytic applications, the emergence of additional bands may also signal the creation of hydrogen bonds around 2950–3300 cm−1.61–63
The molecular structure of the covalent organic framework (COF) was optimized by using the B3LYP/6-31G(d) method. All calculations were performed using the Gaussian16 program. We selected B3LYP due to its demonstrated effectiveness in yielding accurate results for organic molecules.64 Given the substantial size of the molecular systems under investigation, we employed a 6-31G(d) basis set to facilitate efficient quantum chemical calculations. The molecular structure tends to become severely distorted when performing general structure optimization. To prevent this, the carbon atoms of the methyl groups attached to the phenyl/aryl rings of the porphyrins were fixed. With this method, the optimized structure maintained the COF framework. The highest occupied molecular orbital (HOMO), HOMO−n (where n = 1–9), the lowest unoccupied molecular orbital (LUMO), and LUMO+n (where n = 1–9) were calculated and are presented in Fig. 3. The HOMO is localized on the central porphyrin ring, and the subsequent eight molecular orbitals (HOMO−n to LUMO+n) are each localized on one of the other eight porphyrin rings. The energies of these orbitals from the HOMO to HOMO−8 are nearly degenerate. From HOMO−9 onwards, the lower molecular orbitals of the porphyrin rings are observed. The molecular orbital energy levels show a drop in energy starting from HOMO−9. The LUMO is localized on the central porphyrin ring like the HOMO, but it is also conjugated with the adjacent phenyl ring. From the LUMO+1 to LUMO+9, the orbitals are localized on the nine porphyrin rings, with similar shapes and nearly degenerate energy levels.65–67
 | ||
| Fig. 3 Energy of molecular orbitals. The lowest transition from the HOMO to LUMO, with an energy of 2.25 eV, demonstrates the spatial change in electron concentrations associated with the charge transfer feature. | ||
XRD patterns were obtained for the structural characterization of the P, T and PBT photocatalysts (Fig. 4a). The XRD pattern of P (Fig. S4a†) showed two broad peaks with low intensity 2θ at 14.78 and 22.32°, representing the π–π stacking distance between nearby porphyrin cores perpendicular to the tetrapyrrole rings.68 The peak 2θ at 22.32° corresponds to a d-spacing of 0.397 nm. The diffraction peak 2θ at triptycene corresponds a rigid 3-D polycyclic aromatic hydrocarbon which exhibits unique structural properties due to its three-bladed propeller-like shape. The XRD pattern of T (Fig. S4b†) reveals a highly crystalline nature as it shows multiple high diffraction peaks mainly with 2θ at 11.56, 13.7, 17.46, 19.92, 23.04, 25.72, 28.02, 30.12, 33.78, and 37.35°. The diffractograms of PBT show broad bands around 15–25° which indicates π–π stacking interactions between porphyrin planes and many sharp peaks around 18.38, 23.88, 28.72, 34.72 and 39.22° represent unique reflections for well-ordered tritycene (T) crystals. The combination of the broad peak character from P and some sharp peaks from triptycene (T) in the PBT photocatalyst clearly indicates the coupling between P and T.63,69
 | ||
| Fig. 4 (a) XRD and (b) Raman spectra of the PBT photocatalyst. | ||
Raman spectroscopy is a versatile method for investigating the covalent modifications in the PBT photocatalyst (Fig. 4b). As per the reported literature, tetrabromophenylporphyrin exhibits distinct Raman bands that correspond to various vibrational modes of the porphyrin ring and bromine substituents. Key peaks are typically observed in the D band (1350 cm−1), the G band (1600 cm−1), and specific bands linked to C–N stretching (1100 cm−1) and CC stretching (1500–1600 cm−1).13,70 As per the reported literature,71 the Raman spectra of T show prominent bands associated with its inflexible polycyclic structure with aromatic CC stretching modes between 1400 and 1600 cm−1 and C–H bending modes at 750 cm−1. The coupling between P and T can enable effective charge transfer, as seen by the material's strong Raman signals at 1360 and 1800 cm−1. Combined Raman signals were obtained by π–π stacking between triptycene and the tertabromophenylporphyrin core. Improved charge separation and transfer efficiency, which are essential for photocatalytic activity, are frequently indicated by enhanced peaks in the Raman spectra of the PBT photocatalyst.
XPS is an invaluable technique for probing the element composition and electronic state of materials. In the case of P (Fig. 5c–e), deconvoluted XPS analysis reveals the presence of carbon, nitrogen, and bromine elements, with distinct binding energy values corresponding to the C 1s, N 1s, and Br 3d peaks.72 The C 1s peak is typically observed around 284.6 eV, while the N 1s peak appears around 399.6 eV and the Br 3d peak around 70.8 eV. The XPS spectra also provide insights into the oxidation-states of the elements, reflecting the electronic modifications induced by bromination.73 For triptycene, XPS studies (Fig. 5f) confirm the presence of carbon and hydrogen, with the C 1s peak observed at similar binding energy values to those in tetrabromophenylporphyrin (P). However, the absence of bromine signals confirms the purity of the triptycene structure.74 XPS can also be employed to assess the electronic interactions when triptycene is integrated into porphyrin systems, revealing shifts in binding energies that indicate changes in electronic states.18 The XPS analysis of triptycene-based porphyrin (PBT) photocatalysts shown in Fig. 5a and b provides insights into the electronic environment and interactions within the hybrid structure. The presence of both T and P components can be confirmed by the simultaneous detection of characteristic peaks with potential shifts in binding energies at 284.5, 286.6, and 285.5 eV for CC, CN, and C–C (sp3), respectively, indicating charge transfer or hybridization effects between the two moieties. This information is essential for understanding the electronic properties that govern the photocatalytic activities of these materials.61,75
 | ||
| Fig. 5 XPS deconvoluted spectra of the PBT photocatalyst: (a) C 1s, (b) N 1s and P (c) C 1s, (d) N 1s, (e) Br 3d, and T (f) C 1s. | ||
Gaining an understanding of the electrical and structural characteristics of PBT photocatalysts is essential for maximizing their performance in a variety of applications, namely catalysis and artificial photosynthesis. Because of their distinct photophysical characteristics, tetrabromophenylporphyrin, triptycene, and porphyrin-based triptycene photocatalysts have attracted a lot of attention.
EIS is a powerful technique for assessing the charge transport properties of photocatalysts. The Nyquist plots for tetrabromophenylporphyrin typically exhibit semicircular arcs, indicating the resistance and capacitance behaviour of the material. The charge transfer resistance can be extracted from the semicircle, providing insights into the kinetics of charge transfer processes during photocatalytic reactions.76 Studies have shown that tetrabromophenylporphyrin exhibits relatively low charge transfer resistance, suggesting its potential effectiveness as a photocatalyst. The EIS data (Fig. 6a) of T reveal higher charge transfer resistance compared to those of P due to its non-conductive nature. However, when T is incorporated into porphyrin systems, the charge transfer resistance can decrease significantly, indicating enhanced charge mobility and separation efficiency within the hybrid material. EIS studies of PBT photocatalysts typically demonstrate lower charge transfer resistance and increased conductivity compared to their individual components. This improvement is attributed to the favourable electronic interactions between triptycene and the porphyrin moiety, which facilitate efficient charge transport and separation. The enhanced conductivity is crucial for photocatalytic applications, allowing for improved efficiency in redox reactions under light irradiation. The structural characteristics, electronic states, and charge transport characteristics of these compounds are clarified by reviewing research conducted using Raman spectroscopy, XPS, and EIS.
 | ||
| Fig. 6 Electrochemical analysis: (a) Nyquist plot and (b) Tafel plot of P (green) and the PBT photocatalyst (blue). | ||
Fig. 6b shows the Tafel plot of P and PBT photocatalysts. For electrochemical study, the Tafel slope provides information about the kinetics and processes of electrochemical reactions. The Tafel slope is calculated using the Tafel plot, which depicts the relationship between the overpotential and the current. It shows how sensitive the current is to changes in potentials. A lower Tafel slope of the PBT photocatalyst implies that the reaction is more efficient (i.e., smaller increases in overpotentials result in bigger increases in current), whereas a higher Tafel slope of P shows that the reaction is slower and requires more potential to obtain a higher current. The current potential is directly related to the charge transfer capability of P and PBT photocatalysts; the lower the slope, the greater the photocatalytic activity like NAD(P)H regeneration.77–79
The ability to carry current was tested using chronoamperometry, which helps explain electron–hole pair separation and movement, which result in high photocatalytic activity.80 The high current density of the materials contributes to their ability to respond to solar light for photocatalytic activity.81Fig. 7 shows the current response vs. time graph of P, T and PBT. On solar light irradiation, the current of the PBT photocatalyst was found to be the highest (1.56 × 10−4 mA cm−2), as compared to those of P and T. Furthermore, the current response of PBT was reasonably reversible, as the current increased consistently with each irradiation and recovered immediately in the dark. The graph shows that PBT has a better ability to carry current compared to P and T, which indicates its improved conductivity due to the coupling between porphyrin and triptycene motifs. The enhanced charge transfer mechanism could also contribute to the increased NAD(P)H regeneration and the synthesis of value-added chemicals by the photocatalyst.
 | ||
| Fig. 7 Chronoamperometry graph of P (green), T (red) and PBT photocatalysts (blue). | ||
To provide detailed explanation of the electron transfer mechanism in the photocatalytic process, we performed cyclic voltammetry (CV) tests on the P, T, and PBT photocatalysts (Fig. 8a and b). The reduction peak potential of P, T and PBT photocatalysts was obtained approximately at −1.08, −1.10, and −1.06 V vs. Hg/Hg2Cl2, respectively. However, when EM (with a reduction potential of −0.86 V)82,83 was added to the solution containing the PBT photocatalyst, the potential shifted to around −1.18 V vs. Hg/Hg2Cl2. This shift happens because electrons are transferred from PBT to EM. According to the literature,84 EM speeds up the reduction rate significantly in the presence of NAD+. The potential increase in the reduction peak of the PTB photocatalyst at −1.24 V on NAD+ addition implied that the PTB-EM system was capable of catalyzing the photoreduction of NAD+. The nonappearance of any further oxidation peak further showed that the PBT photocatalyst followed the electrochemistry of EM.83,85 From these observations, it can be concluded that after the photo-excitation of the PBT photocatalyst, the electron is excited from the valence band/HOMO (E = −5.70 eV vs. vacuum level) to the conduction band/LUMO (E = −3.42 eV vs. vacuum level). The calculated energy band gap Eg = 2.28 eV, which discloses the semiconductor nature of the PBT photocatalyst. As shown in Fig. 3, the calculated Eg (HOMO–LUMO gap) value using B3LYP/6-31G(d) (2.25 eV) perfectly agrees with the experimental value (2.28 eV). However, when comparing the absolute values of the HOMO and LUMO energies to the experimental values, significant discrepancies are observed, suggesting that the agreement in Eg is likely coincidental. Therefore, the DFT calculation presented here provides qualitatively reliable results rather than quantitatively accurate ones. The proximity and potential gradient between visible light-responsive PBT and the electron mediator EM centre facilitate electron transfer further to NAD+. The resulting electrochemically reduced EM undergoes protonation in the buffer solution (SPB), followed by catalytic regeneration of enzymatically active 1,4-NADH with a high yield (73.59%). The same mechanistic explanation applies for 1,4-NADPH regeneration as described for 1,4-NADH.
 | ||
| Fig. 8 Cyclic voltammogram of (a) P (green), T (red) and PBT photocatalysts (blue) and (b) a mixture of two components PBT and EM with NAD+ (red) and without NAD+ (blue). | ||
High-resolution scanning electron microscopy (SEM) investigations, along with EDX elemental mapping, were performed for P, T, and PBT photocatalysts for morphological analysis. The SEM image of P (Fig. 9a) shows a rod-like, non-uniform, and irregular bulk morphology, which is attributed to the n to π* and π to π* charge transitions in the conjugated backbone of porphyrin rings.86 In contrast, the SEM image of T (Fig. 9e) exhibits a highly structured, crystalline surface with well-defined shapes due to its rigid, three-dimensional molecular framework. On the other hand, SEM images of PBT reveal a porous and smoother morphology compared to its starting materials, P and T. This change in the morphology of PBT (Fig. 10a) clearly indicates the coupling (Csp2–Csp2) between P and T, which is further confirmed by EDX elemental mapping analysis. The EDX elemental mapping visualizes the various elements present and their distribution throughout the constituents. The elemental mapping of the P (Fig. 9b–d), T (Fig. 9f) and PBT photocatalysts (Fig. 10b and c) shows that C and N are uniformly distributed and the absence of the Br element in PBT indicated the successful coupling between P and T. The EDS spectrum in Fig. 10d further confirms the elemental composition in the PBT photocatalyst.
 | ||
| Fig. 9 (a) The SEM image of P and the corresponding elemental mapping image (b–d) consisting of C, N, and Br and (e) the SEM image of T with the corresponding elemental mapping image of (f) C. | ||
 | ||
| Fig. 10 (a) SEM of PBT with the corresponding elemental mapping of elements (b) C and (c) N, and (d) EDX of the PBT photocatalyst. | ||
The primary objective of this research work was to compare the photocatalytic activity of the light responsive PBT photocatalyst with those of the monomers P and T. As a result, visible light-driven 1,4-NADH and 1,4-NADPH regeneration was examined. Over the course of 90 minutes, monomer P generated 6.27% NADH and 9.44% NADPH. In contrast, when the PBT photocatalyst was examined for photocatalytic activity, it caused 73.59% NADH and 76.08% NADPH regeneration (Fig. 11). In other words, the PBT photocatalyst regenerated roughly 10 times as much NAD(P)H as the monomer P in the same duration. In contrast, the monomer T had very low photocatalytic activity, which can be clearly attributed to its limited visible light absorption and poor charge transfer capabilities, as reported in the literature.75 The cycling stability and reusability of the PBT photocatalyst were analysed by five consecutive photocatalytic experiments for NAD(P)H regeneration (Fig. S14†). Furthermore, the chemical stability of the PBT photocatalyst was examined by XRD analysis before and after photocatalytic experiments (Fig. S15†). Scheme 2 shows the plausible photocatalytic pathway for NADH regeneration derived with the help of cyclic voltammetry studies.87,88
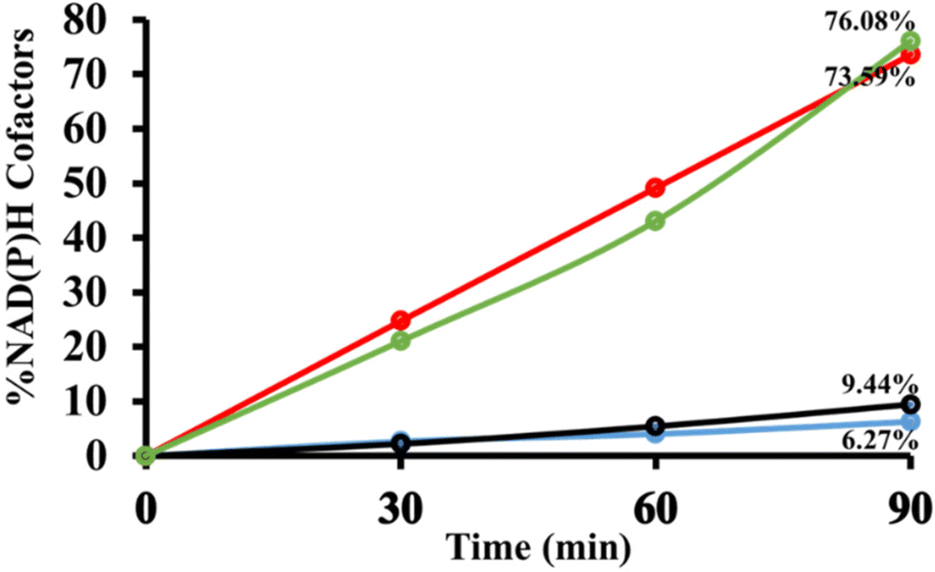 | ||
| Fig. 11 In 3.1 mL of SPB (100 mM, pH 7.0), the photocatalytic activities of P and PBT photocatalysts for NAD(P)H regeneration cofactors [β-NAD+/NADP+ (1.24 mmol), AsA (1.24 mmol), EM (0.62 mmol) and photocatalyst (0.5 mg)]. Note: blue/red line and black/green line for NADH and NADPH regeneration via newly designed porphyrin and PBT photocatalysts. | ||
 | ||
| Scheme 2 Potential energy diagram of the photocatalytic pathway. | ||
Mechanistic explanation of Biginelli reaction for the synthesis of value-added chemicals
Scheme 3 shows a detailed possible mechanistic pathway based on a literature survey.89–91 By employing the PCET strategy, the PBT photocatalyst utilizes visible light as a renewable energy source. On irradiation, the PBT photocatalyst undergoes a rapid transition from the ground state (PBT) into the excited state (PBT*), initiating electron transfer to generate a PBT radical anion and activating aromatic aldehydes to form radical anion species (1). The nucleophilic addition reaction of urea (2) and radical anions (1) leads to the formation of a reactive iminium intermediate (a). Meanwhile, the cation-radical (d) is generated from PBT* using the PCET method. The cation-radical (d) attacks the iminium intermediate (a) to form intermediate (c). A rapid cyclization reaction of (c) followed by dehydration gives the Biginelli product 3,4-dihydropyrimidin-2-(1H)-one (DHP) derivative (4). Under visible light irradiation, the bond with the PBT photocatalyst undergoes extremely rapid oscillation, facilitating efficient photocatalysis. The synergistic actions of the PBT photocatalyst and visible light radiation may be the cause of the increased chemical reaction rates and yield. | ||
| Scheme 3 Proposed mechanism for the Biginelli product under visible light irradiation. | ||
Control experiments
With optimum circumstances established, we decided to study the scope and limitations of the approach. Preliminary control experiments were conducted in the presence of different photocatalysts. Table S1† and Table 1 show the control experiments performed in different solvents and with different photocatalysts, respectively, for the synthesis of DHP. These substances comprised P, T, and PBT photocatalysts. In Table 1, the DHP synthesis without any photocatalyst in 30 minutes resulted in a trace amount of 4a at room temperature (entry 1). Similarly, a control test performed without a light source also led to the detection of 4a in trace amounts (entry 5). Using this approach, 4a could be synthesized with yields from 20 to 97%. These outcomes demonstrated that PBT (5 mg) was significantly more effective for the synthesis of DHP. Table 1, entry 3, indicates that utilizing 5 mg PBT resulted in a yield of 97%. Table S1† demonstrates that product yields were much lower when using solvents like DMF, DCM, THF, and DMSO. In contrast, solvents like H2O, CH3CN, CH3OH, and CH3CO2CH3 caused fast reaction time and better yield. Notably, in the presence of ethanol solvent, the product yield was 97% (entry 3) with no byproducts. The photocatalytic performance of the PBT photocatalyst was further examined using various substituted aromatic aldehydes (Table S2†). The results illustrate that both EWG and EDG on the aromatic aldehydes were synthesized effectively to obtain their corresponding products with excellent yield (up to 97%) and the observed melting points of the products were in good accordance with the literature.43,51,92,93| S. no. | Photocatalyst | Light | Solvent | Time (min) | Yield (%) |
|---|---|---|---|---|---|
| 1 | — | Blue light | EtOH | 30 | Trace |
| 2 | PBT (3 mg) | Blue light | EtOH | 5 | 90 |
| 3 | PBT (5 mg) | Blue light | EtOH | 5 | 97 |
| 4 | PBT (7 mg) | Blue light | EtOH | 5 | 97 |
| 5 | PBT (5 mg) | — | EtOH | 5 | Trace |
| 6 | P (5 mg) | Blue light | EtOH | 5 | 45 |
| 7 | T (5 mg) | Blue light | EtOH | 5 | 15 |
A plausible mechanistic pathway for 1,4-NAD(P)H photogeneration
The plausible mechanism for the photogeneration of the enzyme cofactor 1,4-NAD(P)H through an electron mediator organometallic rhodium complex (EM-complex) is delineated in Scheme S1,† which was performed as reported in the literature. On absorption of solar light by the donor–acceptor PBT framework photocatalyst, an electronic excitation takes place, leading to the transport of two photoexcited electrons through pi-channels of the PBT photocatalyst to an electron mediator cationic rhodium(III) complex, which subsequently reduced into a rhodium(I) complex. The cationic rhodium complex [Cp*Rh(bpy)Cl]+ hydrolyses in the presence of buffer solution into the water-coordinated oxidized rhodium complex, [Cp*Rh(bpy)H2O]2+(a). This rhodium complex (a) readily reacts with a hydride source methanoate ion (HCOO−) to form the rhodium complex (b), which further undergoes decarboxylation to give the [Cp*Rh(bpy)H]+ complex (c). This complex (c) undergoes reductive elimination to produce the rhodium complex (d). The natural enzyme cofactor NAD(P)+ directly coordinated with the reactive complex (d) to form the rhodium complex (e). This resulted in the coordination of the oxygen from the amide group of NAD(P)+, followed by selective hydride transfer forming complex (f). Finally, in situ formation of the regioselective cofactor 1,4-NAD(P)H takes place with the loss of the rhodium complex.Conclusion
In summary, we report the enrichment of the library of materials that exhibit strong interactions between porphyrin (P) and triptycene (T) and possess an architecture with a large porous surface, which may have useful applications in NAD(P)H regeneration, and the synthesis of value-added chemicals like 3,4-dihydropyrimidin-2-(1H)-ones (DHP). The photocatalysts have been characterized by using various spectroscopic techniques, including UV-visible DRS, FTIR, XPS, Raman, SEM, EDX and 1H-NMR. Furthermore, photoelectrochemical studies combined with DFT calculations have made it possible to analyse the photocatalyst's properties and understand the source of its photocatalytic activity. The presence of a bulky aryl substituent coupled with the triptycene unit reduces the axial π–π stacking and enhances the inter-layer spacing, resulting in extremely effective highly efficient photocatalytic activity. Thus, the present work successfully demonstrates a new and promising porphyrin photocatalyst based triptycene for the ultimate goal of regioselective NAD(P)H regeneration and one-pot Biginelli reaction for the synthesis of value-added chemicals.Author contributions
Rehana Shahin: original draft writing; Rajesh K. Yadav: supervision and conceptualization; Shaifali Mishra: editing and software; Kanchan Sharma: formal analysis and data curation; Wonil Seo: formal analysis; Joonghan Kim: investigation and data validation; Navneet K. Gupta: data curation and resources; and Jin-Ook Bag: supervision and visualization.Data availability
Supplementary data are available.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to MMMUT Gorakhpur, India, and Korea Research Institute of Chemical Technology, South Korea, for supporting this research work.References
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- S. Patel, C. J. Penny and T. Rahman, Trends Biochem. Sci., 2016, 41, 475–477 CrossRef CAS PubMed.
- M. Yuan, M. J. Kummer, R. D. Milton, T. Quah and S. D. Minteer, ACS Catal., 2019, 9, 5486–5495 CrossRef CAS.
- N. J. Turner and L. Humphreys, Biocatalysis in organic synthesis: The retrosynthesis approach, Royal Society of Chemistry, 2018 Search PubMed.
- S. Chan, S. Kanchanatawee, S. S. Jantama, K. Jantama, C. Joannis-Cassan and P. Taillandier, J. Chem. Technol. Biotechnol., 2018, 93, 600–608 CrossRef CAS.
- J. Preat, J. Phys. Chem. C, 2010, 114, 16716–16725 CrossRef CAS.
- Y. Wu, J. Ward-Bond, D. Li, S. Zhang, J. Shi and Z. Jiang, ACS Catal., 2018, 8, 5664–5674 CrossRef CAS.
- D. Yang, H. Zou, Y. Wu, J. Shi, S. Zhang, X. Wang, P. Han, Z. Tong and Z. Jiang, Ind. Eng. Chem. Res., 2017, 56, 6247–6255 CrossRef CAS.
- A. R. Sekhar, Y. Chitose, J. Janoš, S. I. Dangoor, A. Ramundo, R. Satchi-Fainaro, P. Slavíček, P. Klán and R. Weinstain, Nat. Commun., 2022, 13, 3614 CrossRef CAS PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- S. Chaubey, R. K. Yadav, S. K. Tripathi, B. C. Yadav, S. N. Singh and T. W. Kim, Photochem. Photobiol., 2022, 98, 150–159 CrossRef CAS PubMed.
- R. Singh, R. K. Yadav, S. Singh, R. Shahin, A. Umar, A. A. Ibrahim, O. Singh, N. K. Gupta, C. Singh and J. O. Baeg, et al. , Chemosphere, 2024, 353, 141491 CrossRef CAS PubMed.
- R. Guilard, K. M. Kadish, K. M. Smith and R. Guilard, The porphyrin handbook, Academic Press New York, 2003, vol. 18 Search PubMed.
- G. Zhao, C. Yang, W. Meng and X. Huang, J. Mater. Chem. A, 2024, 12, 3209–3229 RSC.
- B. Zhang, S.-X. Zhang, R. Yao, Y.-H. Wu and J.-S. Qiu, J. Electron. Sci. Technol., 2021, 19, 100080 CrossRef.
- M. Woźny, A. Mames and T. Ratajczyk, Molecules, 2021, 27, 250 CrossRef PubMed.
- J.-R. Mistry, S. Montanaro and I. A. Wright, Mater. Adv., 2023, 4, 787–803 RSC.
- G. M. Locke, K. J. Flanagan and M. O. Senge, Beilstein J. Org. Chem., 2020, 16, 763–777 CrossRef CAS PubMed.
- Y. Mikata, S. Aida, Y. Inaba and S. Yano, Org. Biomol. Chem., 2007, 5, 3834–3841 RSC.
- Y. Chai, Z. Pang, H. Jiang, C. C. Tsoi, L. Wan, Y. Du, H. Jia, Y. Zhu, D. Liu and F. Xie, et al. , Green Chem., 2025, 27, 623–632 RSC.
- Z.-W. Wang, X. Tan, Z.-H. Wu, J.-H. Chen, Y.-Z. Zhu and H.-F. Wang, J. Mater. Chem. A, 2025, 13, 8726–8733 RSC.
- K. Sujatha, P. Shanmugam, P. T. Perumal, D. Muralidharan and M. Rajendran, Bioorg. Med. Chem. Lett., 2006, 16, 4893–4897 CrossRef CAS PubMed.
- S. Wisén, J. Androsavich, C. G. Evans, L. Chang and J. E. Gestwicki, Bioorg. Med. Chem. Lett., 2008, 18, 60–65 CrossRef PubMed.
- L. Heys, C. G. Moore and P. J. Murphy, Chem. Soc. Rev., 2000, 29, 57–67 RSC.
- M. Ashok, B. S. Holla and N. S. Kumari, Eur. J. Med. Chem., 2007, 42, 380–385 CrossRef CAS PubMed.
- M. J. Genin, C. Biles, B. J. Keiser, S. M. Poppe, S. M. Swaney, W. G. Tarpley, Y. Yagi and D. L. Romero, J. Med. Chem., 2000, 43, 1034–1040 CrossRef CAS PubMed.
- E. W. Hurst and R. Hull, J. Med. Chem., 1960, 3, 215–229 CrossRef PubMed.
- Magerramov and R. A. Kurbanova, Russ. J. Appl. Chem., 2016, 42, 351–354 CAS.
- S. S. Bahekar and D. B. Shinde, Bioorg. Med. Chem. Lett., 2004, 14, 1733–1736 CrossRef CAS PubMed.
- P. Biginelli and P. Gazz, Gazz. Chim. Ital., 1893, 23, 360–416 Search PubMed.
- M. Nasr-Esfahani and M. Taei, RSC Adv., 2015, 5, 44978–44989 RSC.
- K. Gong, H. Wang, S. Wang and X. Ren, Tetrahedron, 2015, 71, 4830–4834 CrossRef CAS.
- M. Saikia, D. Bhuyan and L. Saikia, Appl. Catal., A, 2015, 505, 501–506 CrossRef CAS.
- J. Safari and S. Gandomi-Ravandi, J. Mol. Struct., 2014, 1065, 241–247 CrossRef.
- Y. Titova, O. Fedorova, G. Rusinov, A. Vigorov, V. Krasnov, A. Murashkevich and V. Charushin, Catal. Today, 2015, 241, 270–274 CrossRef CAS.
- C. G. S. Lima, S. Silva, R. H. Goncalves, E. R. Leite, R. S. Schwab, A. G. Correa and M. W. Paixao, ChemCatChem, 2014, 6, 3455–3463 CrossRef CAS.
- E. Kolvari, N. Koukabi, M. M. Hosseini, M. Vahidian and E. Ghobadi, RSC Adv., 2016, 6, 7419–7425 RSC.
- M. M. Hosseini, E. Kolvari, N. Koukabi, M. Ziyaei and M. A. Zolfigol, Catal. Lett., 2016, 146, 1040–1049 CrossRef CAS.
- C. V. Reddy, M. Mahesh, P. V. K. Raju, T. R. Babu and V. V. N. Reddy, Tetrahedron Lett., 2002, 43, 2657–2659 CrossRef CAS.
- K. Ramalinga, P. Vijayalakshmi and T. N. B. Kaimal, Synlett, 2001, 863–865 CrossRef CAS.
- D. Bhuyan, M. Saikia and L. Saikia, Microporous Mesoporous Mater., 2018, 256, 39–48 CrossRef CAS.
- D. Elhamifar, F. Hosseinpoor, B. Karimi and S. Hajati, Microporous Mesoporous Mater., 2015, 204, 269–275 CrossRef CAS.
- J. Pandey, N. Anand and R. P. Tripathi, Tetrahedron, 2009, 65, 9350–9356 CrossRef CAS.
- P. Li, S. Regati, R. J. Butcher, H. D. Arman, Z. Chen, S. Xiang, B. Chen and C.-G. Zhao, Tetrahedron Lett., 2011, 52, 6220–6222 CrossRef CAS PubMed.
- A. Wang, X. Liu, Z. Su and H. Jing, Catal. Sci. Technol., 2014, 4, 71–80 RSC.
- S. Singh, S. Y. Choi, R. K. Yadav, C. Y. Na, J. Kim, M. Y. Choi and T. W. Kim, Energy Fuels, 2025, 39, 1746–1758 CrossRef CAS.
- C. Singh, S. Chaubey, P. Singh, K. Sharma, A. Kumar, R. K. Yadav, D. K. Dwivedi, J.-O. Baeg, U. Kumar and B. C. Yadav, et al. , Diamond Relat. Mater., 2020, 101, 107648 CrossRef CAS.
- R. K. Yadav, J.-O. Lee, A. Kumar, N.-J. Park, D. Yadav, J. Y. Kim and J.-O. Baeg, Sci. Rep., 2018, 8, 16741 CrossRef PubMed.
- S. Choudhury, J.-O. Baeg, N.-J. Park and R. K. Yadav, Green Chem., 2014, 16, 4389–4400 RSC.
- R. Shahin, R. K. Yadav, R. K. Verma, C. Singh, S. Singh, R. Singhal, N. K. Gupta, J. O. Baeg, G. A. El-Hiti and K. K. Yadav, New J. Chem., 2024, 48, 12102–12111 RSC.
- F. Mohamadpour, ACS Omega, 2022, 7, 8429–8436 CrossRef CAS PubMed.
- S. Mishra, B. Prabhakar, P. S. Kharkar and A. M. Pethe, ACS Omega, 2022, 8, 1140–1145 CrossRef PubMed.
- M. I. Awad, Y. Makkawi and N. M. Hassan, ACS Omega, 2024, 9, 18654–18667 CrossRef CAS PubMed.
- R. Bonnett, A. Harriman and A. N. Kozyrev, J. Chem. Soc., Faraday Trans., 1992, 88, 763–769 RSC.
- B. S. Ghanem, M. Pi\cskin, M. Durmu\cs, M. E. El-Khouly and S. Y. Al-Raqa, Polyhedron, 2015, 90, 85–90 CrossRef CAS.
- M.-J. Gu, Y.-F. Wang, Y. Han and C.-F. Chen, Org. Biomol. Chem., 2021, 19, 10047–10067 RSC.
- S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2017, 117, 2910–3043 CrossRef CAS PubMed.
- H. Dogari, R. Peymanfar and H. Ghafuri, RSC Adv., 2023, 13, 22205–22215 RSC.
- H. He, Y. Zhong, L. Si and A. Sykes, Inorg. Chim. Acta, 2011, 378, 30–35 CrossRef CAS.
- A. Furlan, T. Fischer, P. Fluekiger, H. U. Guedel, S. Leutwyler, H. P. Luethi, M. J. Riley and J. Weber, J. Phys. Chem., 1992, 96, 10713–10719 CrossRef CAS.
- Y. Sun, J. Bai, P. Zheng, Q. Wu, Z. Cai, T. Han, L. Shan, S. Luo and S. Zhang, Chem. Eng. J., 2023, 471, 144414 CrossRef CAS.
- Q. Liang, G. Jiang, Z. Zhao, Z. Li and M. J. MacLachlan, Catal. Sci. Technol., 2015, 5, 3368–3374 RSC.
- S. Dey, A. Bhunia, D. Esquivel and C. Janiak, J. Mater. Chem. A, 2016, 4, 6259–6263 RSC.
- S. F. Sousa, P. A. Fernandes and M. J. Ramos, J. Phys. Chem. A, 2007, 111, 10439–10452 CrossRef CAS PubMed.
- P. Singh, R. K. Yadav, K. Kumar, Y. Lee, A. K. Gupta, K. Kumar, B. C. Yadav, S. N. Singh, D. K. Dwivedi and S.-H. Nam, et al. , Catal. Sci. Technol., 2021, 11, 6401–6410 RSC.
- C. Douvris, C. Lampropoulos, D. Matatov, D. J. Wink, A. E. Kuznetsov and D. Bussan, Polyhedron, 2022, 221, 115875 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- P. Zhu, F. Song, P. Ma, Y. Wang, C. Chen and J. Feng, J. Mater. Chem. C, 2016, 4, 10471–10478 RSC.
- A. Alam, A. Hassan, R. Bera and N. Das, Mater. Adv., 2020, 1, 3406–3416 RSC.
- A. Gorski, A. Starukhin, S. Stavrov, S. Gawinkowski and J. Waluk, Spectrochim. Acta, Part A, 2017, 173, 350–355 CrossRef CAS PubMed.
- S. Zhang, N. Fang, X. Ji, Y. Gu, Z. Xu, S. Jin and Y. Zhao, JACS Au, 2022, 2, 1638–1650 CrossRef CAS PubMed.
- Z. Wang, X. Wang, K. Chen, H. Yin, H. Su, Y. Wu, C. Ni and W. Liu, New J. Chem., 2024, 48, 8868–8876 RSC.
- A. Ghosh, J. Fitzgerald, P. G. Gassman and J. Almlof, Inorg. Chem., 1994, 33, 6057–6060 CrossRef CAS.
- S. Das, F. Ishiwari, Y. Shoji, T. Fukushima and M. Zharnikov, J. Phys. Chem. C, 2023, 127, 5178–5185 CrossRef CAS.
- R. K. Yadav, A. Kumar, D. Yadav, N. J. Park, J. Y. Kim and J. O. Baeg, ChemCatChem, 2018, 10, 2024–2029 Search PubMed.
- P. Xia, M. Liu, B. Cheng, J. Yu and L. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 8945–8953 CrossRef CAS.
- K. Park, B.-Y. Chang and S. Hwang, ACS Omega, 2019, 4, 19307–19313 CrossRef CAS PubMed.
- I. N. Reddy, A. Sreedhar, C. V. Reddy, J. Shim, M. Cho, D. Kim, J. S. Gwag and K. Yoo, J. Solid State Electrochem., 2018, 22, 3535–3546 CrossRef CAS.
- J. González, E. Laborda and Á. Molina, J. Chem. Educ., 2022, 100, 697–706 CrossRef PubMed.
- K. Zhou, H. Wang, S. Zhang, J. Jiu, J. Liu, Y. Zhang and H. Yan, J. Mater. Sci., 2017, 52, 12783–12794 CrossRef CAS.
- Y. Han, G. Wu, H. Li, M. Wang and H. Chen, Nanotechnology, 2010, 21, 185708 CrossRef PubMed.
- C.-C. Yang, J. Vernimmen, V. Meynen, P. Cool and G. Mul, J. Catal., 2011, 284, 1–8 CrossRef CAS.
- J. S. Lee, D. H. Nam, S. K. Kuk and C. B. Park, Chem. – Eur. J., 2014, 20, 3584–3588 Search PubMed.
- R. K. Yadav, G. H. Oh, N. Park, A. Kumar, K. Kong and J. Baeg.
- S. H. Lee, G. M. Ryu, D. H. Nam, J. H. Kim and C. B. Park, ChemSusChem, 2014, 7, 3007–3011 Search PubMed.
- Y.-C. Zhao, Q.-Y. Cheng, D. Zhou, T. Wang and B.-H. Han, J. Mater. Chem., 2012, 22, 11509–11514 RSC.
- S. Singh, R. K. Yadav, T. W. Kim, C. Singh, P. Singh, S. Chaubey, A. P. Singh, J.-O. Baeg, S. K. Gupta and D. Tiwary, Energy Fuels, 2022, 36, 8402–8412 CrossRef CAS.
- R. K. Yadav, J. O. Lee, A. Kumar, N. J. Park, D. Yadav, J. Y. Kim and J. O. Baeg, Sci. Rep., 2018, 8, 1–10 Search PubMed.
- F. Mohamadpour, RSC Adv., 2023, 13, 2514–2522 Search PubMed.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC.
- F. Mohamadpour, J. Photochem. Photobiol., A, 2021, 418, 113428 CrossRef CAS.
- H. Nagarajaiah, A. Mukhopadhyay and J. N. Moorthy, Tetrahedron Lett., 2016, 57, 5135–5149 CrossRef CAS.
- F. Mohamadpour, Sci. Rep., 2023, 13, 10262 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04461d |
| This journal is © The Royal Society of Chemistry 2025 |