
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dissolution–precipitation reactions of blast furnace slag and sodium carbonate with 2,3-dihydroxynaphthalene†
Rajeswari
Ramaswamy
a and
Juho
Yliniemi
 *b
*b
aFibre and Particle Engineering Research Unit, University of Oulu, P.O. Box 4300, 90014, Oulu, Finland. E-mail: rajeswari.ramaswamy@oulu.fi
bFibre and Particle Engineering Research Unit, University of Oulu, P.O. Box 4300, 90014, Oulu, Finland. E-mail: juho.yliniemi@oulu.fi
First published on 11th March 2025
Abstract
Chemical admixtures are needed to enhance the reactivity of the industrial waste by-products to expand their utilization in the cement and concrete industry to create low CO2 sustainable binders. One such chemical admixture which is a complexing ligand (2,3-dihydroxynaphthalene) has been shown to accelerate the hydration kinetics and enhance the mechanical strength (from 2 MPa to 40 MPa) of sodium carbonate-activated blast furnace slag binder. This study aims to understand the working mechanism of 2,3-dihydroxy naphthalene as an accelerator and the formation of the micro- and nano-surface precipitates for sodium carbonate-activated slag through batch dissolution experiments. Both solution (pH evolution, element concentration, organic and inorganic carbon content) and solid (identification and chemical composition of the phases) chemistry were investigated using various analytic and microscopic techniques. The results showed that the ligand significantly increased the extent of the slag dissolution, which affected the solution chemistry, consequently accelerating the precipitation kinetics. Further, the ligand affected the amount of precipitation (calcite and gaylussite) and the modification of Mg–Al–Si–Na–Ti-rich nano- and micro-sized precipitate morphology in the ligand system compared to the reference system. This work provides important information on ligand–carbonate–slag reactions, which pave the way for developing new chemical admixtures for a sustainable future.
1. Introduction
Cement and concrete sectors are increasingly using and searching for alternative binders to replace Portland cement, as it causes approximately 8% of global CO2 emissions.1,2 Alkali-activated materials (AAMs) and supplementary cementitious materials (SCMs) have shown potential as alternative binders, thereby decreasing the CO2 footprint of concretes.3–5 The scientific community has investigated various industrial by-products, such as metallurgical slags, ashes, and mine tailings, as emerging precursors for AAMs and SCMs.6–8 However, almost all the industrial by-product-based precursors have low hydraulic reactivity, which lowers their potential as alternative binders. In this context, “reactivity” indicates how much of the precursor dissolves during the early stages of the reaction and provides elements into a pore solution for the formation of cementitious phases.9–11 Even one of the most reactive and commonly used SCM, ground granulated blast furnace slag (GBFS), lowers the early-age strength development of concretes because its reactivity is lower than that of Portland cement. For AAMs, aggressive activators such as NaOH and sodium silicate are used to accelerate the reactivity; however, they diminish the potential CO2 reduction because of the CO2 emission from the production of NaOH and sodium silicate.3,12,13 Thus, an in-depth understanding of the reactivity of sustainable precursors is needed14 to utilize them efficiently, obtain desired engineering properties for concretes, and create a sustainable binder.Chemical admixtures have been necessary ingredients in any concrete formulation for over 70 years. Commonly used admixtures in Portland cement-based concretes are polycarboxylate ethers as superplasticizers and inorganic/organic salts as retarders and accelerators, respectively.15–17 As Portland cement is a Ca-rich material, the working mechanism of conventional admixtures is often related to the adsorption effect on the surface of cement grains. This controls particle segregation, lowers water demand, improves workability, and influences Ca (but also other elements such as Al) chemistry during cement hydration.8,16,18,19 However, for the various new precursors and alternative binder mixes with variable chemical compositions, mineralogical characteristics, and reactivities, their chemistry is more complicated than that in Portland cement, which decreases the effectiveness of conventional admixtures.19,20 Thus, new types of admixtures are needed, with a special focus on accelerating admixtures to improve the reactivity of industrial by-product-based precursors.
We have investigated the concept of using organic ligands as accelerating admixtures for alternative binders.21–23 Organic ligands are the conjugate bases of organic acids or salts with complexing abilities. While conventional concrete admixtures also have the complexing ability (for Ca), that aspect has been investigated systematically only.24–27 Ligands can strongly affect underlying chemical reactions in a pore solution and between a solid–liquid interface.22 These influences range from alternating the speciation and chemical activity of the ions to changing the solubility of hydrates (forward reaction) and the saturation levels and thermodynamic properties of the precipitation products.24,28,29 Complexing ligands and other admixtures are typically evaluated by studying their effects on the engineering properties of the fresh and hardened state of hydrated binders. However, little attention has been paid to understanding the change in the underlying chemical and physical mechanism of the system introduced by these complexing ligands.30 Understanding the chemistry behind the working principle of a ligand under specific conditions will facilitate the successful engineering of a sustainable binder system.
Sodium carbonate-activated GBFS is a more environmentally friendly binder than the GBFS activated by NaOH and sodium silicates, as the CO2 footprint of Na2CO3 is considerably low.31,32 However, this binder has a long setting time (≥1 d) and slow strength evolution at early stages (<7 d), thereby preventing its use on an industrial scale. Previously,21 we investigated the effect of various ligands with different functional groups and at different ligand dosages on the early-stage strength development of Na2CO3-activated GBFS by studying their workability, hydration kinetics, strength development, and phase identification. Among the ligands, 2,3-dihydroxynaphthalene (DHNP) was observed to accelerate the hydration kinetics by 28 h by producing a 2-day strength of 40 MPa, as compared with that of the reference system of 2 MPa. However, the reaction pathway with DHNP, which accounted for the acceleration of hydration, remained unclear, and only a hypothesis could be derived. This indicated the need for understanding the initial reaction chemistry at nano- and micro-levels and the succeeding evolution of precipitation products. In general, the investigation of the mechanism of ligands in the pH range of 11–13 and with carbonates is considered useful. This is because that is the typical pH range of various cementitious systems, and the competition of the complexation of metal ions by hydroxyls, carbonates, and organic ligands is crucial for the effectiveness of the ligand admixtures.
Thus, in this study, we attempt to address the key questions on the reactions behind the acceleration of the hydration kinetics and phase assemblage of Na2CO3-activated GBFS with DHNP through batch dissolution experiments. The liquid-to-solid ratio (L/S) for the dissolution experiments is designed to investigate and understand the effect of ligands on the initial dissolution–precipitation reactions, which involve carbonate precipitation. The solution chemistry evolution is studied by pH measurements, elemental release determinations through inductively coupled optical emission spectrometry (ICP-OES), and concentration investigation of carbonate ions and organic ligand (i.e., DHNP) by total inorganic carbon (TIC) and total organic carbon (TOC) analyses. The formation of precipitation products is investigated by intensive scanning and transmission electron microscopy equipped with energy-dispersive X-ray spectroscopy analysis (SEM/TEM-EDS), and qualitative and quantitative X-ray diffraction (XRD) was employed to identify phases and their composition.
2. Materials and methods
2.1 Sample characterization
GBFS with the product name KJ 400 (Finnsementti, Finland) was used as the solid precursor for the dissolution experiments. The reactivity of the GBFS varies according to the chemical composition and particle size.33,34 Thus, we conducted isothermal calorimetry experiments to check the reactivity of the GBFS batch used in this study and compared it with that of a previous study.21 The results showed that the GBFS batch used in this study was more reactive; however, DHNP had a similar accelerating effect (ESI, Fig. S1†).The chemical composition of the GBFS was determined by X-ray fluorescence from a melt-fused tablet, and the particle size distribution with median size d50 (Fig. 1a) was determined using the Beckman Coulter LS 132 320 multiwavelength Laser diffraction particle size analyzer. The specific surface area of the GBFS was determined by the multipoint Brunauer–Emmett–Teller method using N2-adsorbent gas. The results are presented in Table 1. The XRD analysis using external standard (as discussed in section 2.3) showed that the GBFS (Fig. 1b) was mostly amorphous with a typical hump at approximately 25°–45° 2θ. This indicated a glassy phase structure (approximately 99 wt%) with a small amount of calcite (0.5 wt%) and quartz (0.4 wt%).
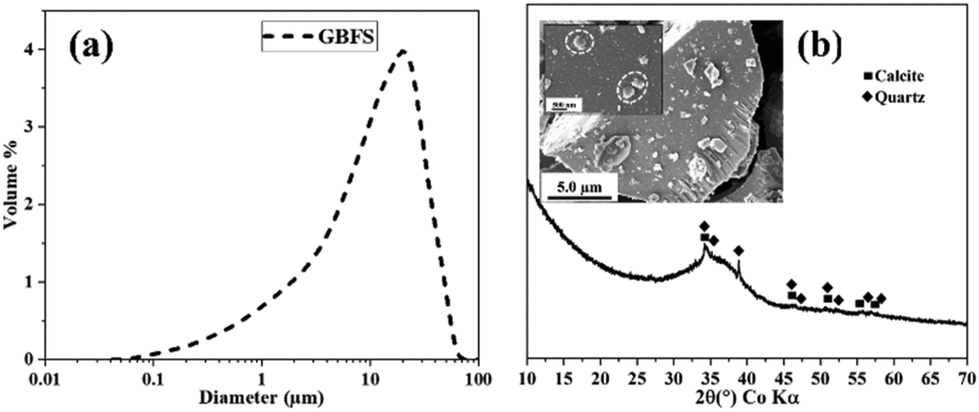 | ||
| Fig. 1 (a) PSD of raw GBFS and (b) XRD with SEM images of raw GBFS, where dashed white circles in the enclosed SEM image show the nucleated masses on GBFS particle surface identified as calcite phase which will be discussed in later sections. | ||
| Oxides (wt.%) | D50 (μm) | BET (m2 g−1) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CaO | SiO2 | Al2O3 | Fe2O3 | Na2O | K2O | MgO | TiO2 | MnO | SO3 | V2O5 | Cl | ||
| 39.4 | 34.4 | 9.2 | 0.7 | 0.4 | 0.8 | 9.5 | 1.2 | 0.3 | 1.9 | 0.1 | 0.1 | 11.2 | 0.807 |
2.2 Batch dissolution experiments
Dissolution experiments were carried out at room temperature (22 °C) with a maximum duration of 60 d. Polypropylene bottles used for the dissolution experiments were prewashed with 2 cycles of dilute HNO3/HCl and deionized water; thereafter, they were dried before use. Na2CO3 solutions were made at a concentration of 2.13 M using analytical-grade sodium carbonate powder from Sigma Aldrich and deionized water. The analytical grade of DHNP (Sigma Aldrich) was added to the Na2CO3 solution 5 min before mixing with GBFS to attain a concentration of 25 mM. However, the ligand did not dissolve fully in the Na2CO3 solution as observed in.21 The GBFS powder was added to Na2CO3-containing bottles with and without DHNP at L/S of 50, and the mixture was stirred using a horizontal shaker at 300 rpm.After specific dissolution times, the bottles were removed from the shaker and the suspension was quickly filtered first using vacuum suction with a 3–5 μm-pore-diameter polypropylene filter paper. The filtered solid residue was immediately rinsed twice using isopropanol at a 1g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ml ratio to stop further dissolution and avoid any formation of phases because of the drying process. Thereafter, the washed solids were air-dried for a few minutes for the rinsed solvent to evaporate, and they were stored in a desiccator. The filtrate solution was further filtered using a 0.45 μm-pore-diameter filter syringe and a polyether sulfone filter paper. The dried solids and the double-filtered filtrate solution were prepared for further analysis as explained in detail in the next section.
:50 ml ratio to stop further dissolution and avoid any formation of phases because of the drying process. Thereafter, the washed solids were air-dried for a few minutes for the rinsed solvent to evaporate, and they were stored in a desiccator. The filtrate solution was further filtered using a 0.45 μm-pore-diameter filter syringe and a polyether sulfone filter paper. The dried solids and the double-filtered filtrate solution were prepared for further analysis as explained in detail in the next section.
Samples prepared with only Na2CO3 are termed “Reference” or “R”, whereas samples prepared with Na2CO3 and DHNP are termed “Ligand” or “L”. Sample codes (sample term-mixing time; for example, R-1 d or L-1 d) are used throughout this paper. The normalized mass loss values (NL) for each element (i) (in μg cm−2) and the extent of dissolution (E) (in %) were calculated according to eqn (1) and (2).
 | (1) |
 | (2) |
2.3 Characterization of filtrate solution after dissolution
The pH of the filtrate solutions after the respective dissolution times was determined using Hach IntelliCAL PHC705 pH probes with an alkalinity error of <0.3 pH units. The replicability of the samples showed a variation of ≤0.03 pH units. The elemental concentration after the respective dissolution times was determined using ICP-OES. The filtrate solution was diluted and acidified with 2% HNO3 solution, and afterward, the metal ion concentration of each filtrate was determined following standard SFS-EN ISO 11885:2009. The reliability of the ICP results was tested by choosing a specific dissolution time and reproducing those experiments three times. TIC and TOC analyses were carried out to determine the carbon content corresponding to the inorganic carbon of the Na2CO3 solution and the carbon content corresponding to DHNP, respectively. Filtrate solutions were determined using a Shimadzu TOC analyzer. The TOC and TIC were determined by following standard ISO 20236:2018.2.4 Characterization of solids after dissolution
The high-resolution images of the precipitation products and their morphology were analyzed using a Zeiss Sigma field emission scanning electron microscope equipped with EDS for elemental analysis. Acceleration voltages of 5 and 15 kV were used for imaging and EDS analysis, respectively. For SEM-EDS analysis, dried solids of the sample were spread on a carbon tape and coated with platinum or carbon, depending on the analysis. Some SEM images exhibited charging-like effects, which were due to the refraction of the beam from the crystals present in the sample. A JEOL JEM-2200FS scanning transmission electron microscope equipped with EDS with an acceleration voltage of 200 kV was used for the elemental composition characterization of the surfaces of the GBFS and precipitation products by square and area analysis or elemental mapping. For scanning transmission electron microscopy analysis, sample grids were prepared by adding a droplet of gently ultrasonicated solid suspension (dried solid samples mixed with 99.5% purity ethanol) on a 200 mesh Cu grid with carbon film. Thereafter, it was air dried (where the alcohol immediately evaporated) and cleaned using JOEL EC-52000IC ion cleaner to prevent contamination during the TEM investigation. Powder XRD was performed using Rigaku SmartLab with a Co source lamp at 40 kV and 135 mA. An external standard (CaF2) was measured along the samples for the quantitative phase analysis. The phases were identified and quantified by performing Rietveld refinement using Highscore software version 5.1 (Malvern Panalytical) with the ICDD PDF4+ 2023 database.3. Results
3.1 Solution chemistry (pH, ICP-OES, TIC, and TOC)
The results of the pH of the solutions, NLi, TIC (carbonate ion concentration), and TOC (DHNP concentration) showed a distinction between the reference and ligand system.From 2.5 h onward, the pH was observed to be higher for the ligand system than that for reference until 30 d (Fig. 2). The pH decrease observed in the ligand system at the initial hours (<2.5 h) was attributed to the deprotonation of the ligand by the carbonate and hydroxyl ions. Notably, the real pH values of high alkaline solutions were underestimated by 0.3 pH units in contrast to the measured pH values using pH meters with a built-in calibration process.35
 | ||
| Fig. 2 pH of the solution after respective dissolution time for both reference and ligand system (magnified image inside the graph represents pH in initial hours (until 5 h) of reaction). Replicability of the pH results had a standard deviation of ≤0.032 pH units. | ||
As the GBFS used in this study is considered a Ca-rich Si glass, Si, which is a network former in glasses, can be used as an indicator for the further dissolution of the slag. However, under the given experimental conditions, precipitation reactions are possible. This indicates that the NLi and ESi may not be true at all reaction times if the ions participate in precipitation and adsorption reactions, consequently lowering the determined NLi.
The extent of the dissolution at 24 h for the reference system was calculated to be approximately 5.4%, whereas it was 7.6% for the ligand system. This indicated that with the ligand, the extent of dissolution of the GBFS increased. Particularly in the initial hours (until 5 h), the dissolution extent in the ligand system was roughly 12 times greater than that in the reference system. As shown in Fig. 3, the forward reaction slope (NLSi trend between 0.5 h and 1 d) was significantly steeper for the ligand system than for the reference system, indicating that the dissolution was accelerated with the ligand.
 | ||
| Fig. 3 Normalised mass loss (NLi) versus dissolution time (a) reference (b) ligand (magnified images inside respective graph represents the initial hours of reaction). Based on the replicated dissolution experiments, NLi results had a standard deviation of ≤0.4 μg cm−2. | ||
In the NLi trends of the reference sample, divalent ions like Ca (for the first 2.5 h) and Mg (for the first 5 h) showed higher leaching than Si as their respective NLCa,Mg was higher than NLSi (Fig. 3a). Although the high leaching of network modifiers from glasses is predominantly observed under low pH conditions, it reportedly occurs also under high pH conditions.11,36 However, this was not observed in the ligand system (Fig. 3b), which can be due to the instant precipitation of the solid phases associated with dissolved ionic species or because no leaching occurred with the ligands. For both systems, NLCa,Mg (between 5 h and 30 d) and NLAl (for the first 30 d) were observed to remain under 0.5 and 2 μg cm−2, respectively, and showed a similar mass loss trend. However, NLSi increased until 1 d and remained constant (i.e., so-called “residual mass loss regime”) for the first 30 d. This indicates the participation of Ca, Mg, and Al ionic species in the precipitation reactions along with dissolution reactions. The residual mass loss regime of NLSi observed in both systems may be due to the retardation of dissolution from 1 to 30 d or continuous Si-phase precipitation with ongoing dissolution reactions. In reference sample, NLTi was observed to be lower than NLSi during 5 h to 30 d, indicating precipitation, whereas, in the ligand system, high Ti leaching was observed during 2.5 h to 5 d. This indicated that with the ligand, soluble Ti concentration was increased, which can be attributed to the complexation reactions. Additionally, NLS (Fig. S2†) exhibited a high leaching behavior starting at 0.5 h and lasting until 30 d in both systems.
Fig. 4a shows a fluctuating carbonate ion concentration in both systems, indicating the occurrence of reversible precipitation reactions or structural changes of the precipitation products. The ligand system contained fewer free CO32− ions than the reference system, indicating that the carbonate ions participated more in the dissolution and precipitation reactions in the ligand system. Another interesting observation is that the TIC concentration trend was similar for both systems, except during the initial hours (for the first 2.5 h). This partly indicated that the reaction pathway involving the carbonate ions was not affected by the ligand; however, the concentration of the carbonate ions taking part in the reaction was affected. These observations are supported with the XRD results which will be discussed in the upcoming sections.
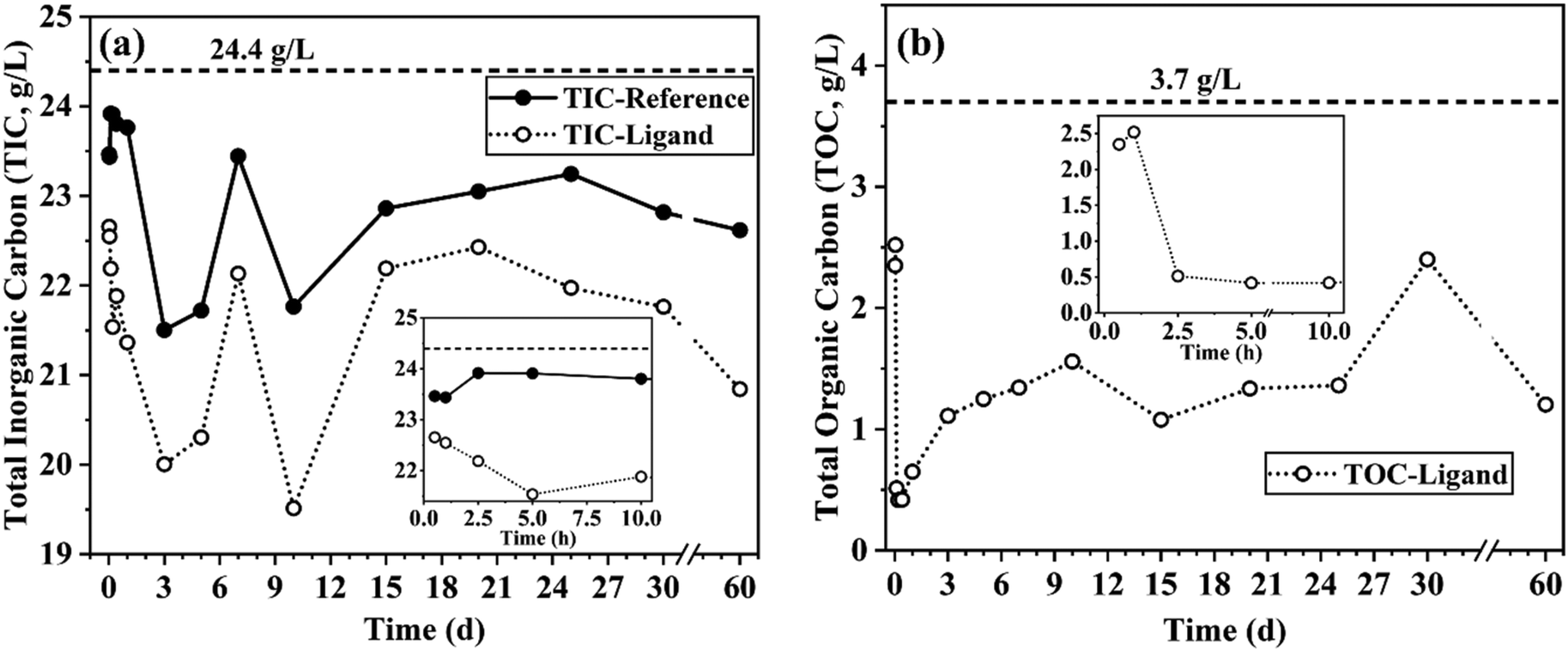 | ||
| Fig. 4 (a) TIC and (b) TOC values for the filtrate solution obtained from reference and ligand system in respect to dissolution time (magnified images inside respective graph represents the initial hours of reaction). TIC and TOC values had a standard deviation of ≤0.3 g L−1 and ≤0.03 g L−1, respectively. Values above the dashed lines represents the TIC of the Na2CO3 solution (left side) and TOC values of the completely dissolved ligand in solution (right side). | ||
Similar to the carbonate consumption trend by time, the results shown in Fig. 4b may indicate that the ligand is consumed in the dissolution process with time (particularly during the first 2.5 h) and then released back into the solution. The ligand consumption was possibly caused by the ligand adsorption onto the GBFS surface or incorporation into the precipitation products, or the formation of metastable metal complex precipitates.
3.2 Solids after dissolution experiments
As shown in Fig. 5 and 6, the SEM images show significant differences in the precipitate morphologies between the systems. Reference sample had three types of nano- and microstructures: (a) nano-sized nucleated mass on the surface of the GBFS (Fig. 5a), which originates from either the reaction of the slag with Na2CO3 or the GBFS (i.e., calcite, as shown in Fig. 1b). Further, it had (b) smectite or layered double hydroxide (LDH)-like structures on the GBFS surface, which started to form after 5 h (Fig. 5b and c); further, they completely covered the GBFS particle surface after 1 d and were present for the entire 60 days (Fig. 5d). Finally, it had (c) micro sized crystal-like structures with defined edges (Fig. 5e) with varying lengths of pyramidal shapes were observed to form after 5 h and were present until the 60 d. | ||
| Fig. 5 SEM images of surface morphology of the GBFS particles and precipitation products (a–f) of the reference system (R), where the number indicates reaction time in the dissolution experiments in hours (h) or days (d). | ||
 | ||
| Fig. 6 SEM images of surface morphology of the GBFS particle and precipitation products (a–f) of the ligand system (L), where the number indicates reaction time in the dissolution experiments in hours (h) or days (d). | ||
Additionally, the ligand system showed three different types of nano- and microstructures: (a) nanosized clustered rhombohedral-like structures formed on an altered gel on top of the GBFS surface, which started to form after 1 h and were observable for the first 10 h (Fig. 6a and b). Here, the term “altered” gel could be the etched layer of the GBFS surface due to a leaching phenomenon or precipitate formation. This rhombohedral morphology was similar to a well-crystallized CaCO3 morphology, which could be formed in the reaction with dissolved Ca from the GBFS and sodium carbonate.37 (b) Micro sized crystal-like pyramidal-shaped particles with varying sizes (Fig. 6c), as in the reference system, were observed from 2.5 h to 60 d. Further, (c) rod-like structures loosely adhered to or separated from the GBFS surface (Fig. 6d and e) were formed after 5 h and were present until the 60 d duration. However, on the nano-level, the GBFS particle in both systems exhibited a similar thin nano-layered precipitate covering the surface (Area II in Fig. 5f, 6f and 6e) of the slag particle (Area I in Fig. 5f and 6f) before any micro sized precipitates were observed.
The qualitative XRD phase analysis of the systems revealed that both systems contained (a) calcite CaCO3 (pdf#086-4273), (b) gaylussite Na2Ca(CO3)2(H2O)5 (pdf#012-6195), and (c) natrite Na2CO3 (pdf#009-8706) (not a reaction product but originated from sample drying pre-treatment). The main difference observed in the XRD patterns (Fig. S3a and b†) between the reference and ligand systems is the peak at approximately 12.6°–13° 2θ in the reference sample, which was present from 1 d onward, belonging to hydrotalcite Mg0.75Al0.25(CO3)0.125(OH)2(H2O)0.5 (pdf#026-3610). However, no such prominent peak was observed to form in the ligand system.
Quantitative XRD analysis revealed a significant difference in the quantity of the precipitated phases between the systems with respect to the dissolution reaction times (Fig. 7). The quantities of calcite and gaylussite were greater in the ligand system, particularly during the initial stages of the reaction. After 1 d, the precipitation of gaylussite slowed in the ligand system, whereas that in the reference system continuously increased throughout 60 d of the reaction. The calcite quantity in the reference system was similar to that in unreacted GBFS (approximately 0.5 wt%), whereas that in the ligand system was approximately 4 wt% after 5 h of reaction. This indicated that calcite precipitation was favored in the ligand system.
 | ||
| Fig. 7 Crystalline phase contents based on Q-XRD analysis after respective dissolution reaction times in reference and ligand systems. Natrite was present ≤2 wt% in both system throughout the reaction time where ≤0.2 wt% of hydrotalcite was present from 1 d in the reference. | ||
The elemental composition of the nano- and micro sized precipitation products on the surface or those separated from the slag particles were characterized by TEM-EDS (Fig. 8 and Table 2). Notably, the elemental compositions collected by TEM-EDS are only reliable for flat thin samples.38 Thus, for slag particles with micrometer-scale thickness and a random three-dimensional morphology, the atomic percentages obtained, particularly for light elements like C and O, may not be completely accurate and should be viewed from a qualitative aspect. However, the elemental analysis for precipitates carried out here should be reliable because of their relatively small size and low thickness.
 | ||
| Fig. 8 TEM-images and elemental mapping of the (a) unreacted GBFS and (b–g) are solids after dissolution experiments for both reference and ligand system. Red squares and dashed red circle in the TEM images are the representative areas analysed with mapping tool (values are presented in Table 2). | ||
| Fig. 8 | Sample | Elements (at.%) | |||||
|---|---|---|---|---|---|---|---|
| Si | Ca | Al | Mg | Ti | Na | ||
| A | GBFS (raw) | 21.7 ± 0.8 | 34.1 ± 1.9 | 7.0 ± 0.3 | 8.0 ± 0.2 | 0.8 ± 0.1 | 0.4 ± 0.1 |
| B | R-5 h | 22.6 ± 0.5 | 30.9 ± 1.8 | 7.0 ± 0.1 | 8.3 ± 0.2 | 0.9 ± 0.1 | 0.5 ± 0.1 |
| C | L-5 h | 13.9 ± 3.8 | 0.5 ± 0.3 | 2.0 ± 0.5 | 7.0 ± 1.3 | 0.2 ± 0.1 | 12.7 ± 1.2 |
| D | R-1 d | 13.1 ± 0.7 | 0.4 ± 0.1 | 3.3 ± 0.1 | 9.4 ± 1.0 | 0.4 ± 0.03 | 1.1 ± 0.03 |
| E | R-30 d | 18.5 ± 3.7 | 0.3 ± 0.3 | 4.1 ± 1.0 | 11.1 ± 1.4 | 0.6 ± 0.1 | 6.5 ± 0.6 |
| F | R-60 d | 16.1 ± 1.4 | 0.2 ± 0.02 | 3.5 ± 0.6 | 9.2 ± 1.3 | 0.5 ± 0.02 | 9.6 ± 2.4 |
| G | L-5 h-60 d | 11.4 ± 2.6 | 0.1 ± 0.01 | 1.7 ± 0.3 | 7.4 ± 1.5 | 0.2 ± 0.1 | 11.1 ± 2.6 |
The elemental mapping results revealed that unreacted GBFS particles had homogenous chemical composition. However, the presence of small high-Ca- and C-containing particulates could be observed on the surface of the GBFS, as shown in the Ca and C maps presented in Fig. 8a. They are likely CaCO3 formed on the surface because of Ca leaching and carbonation with the atmospheric air, as detected by XRD analysis. After 5 h of reaction, there was a clear difference in the elemental composition between the reference (Fig. 8b) and ligand systems (Fig. 8c). The elemental mapping and composition showed that the R-5 h sample was identical to the raw GBFS sample, whereas L-5 h showed nano-sized clustered rhombohedral-like structures formed on an altered gel-like surface on the GBFS particle (Fig. 6a and b). The rhombohedral-like structures were rich in Ca, C, and O i.e., they were calcites, as confirmed by XRD results; further, the altered gel was rich in Si, Mg, Al, and Na and had depleted Ca and Ti around the core of the unreacted GBFS. This indicated that the altered gel could be the leached layer from the GBFS surface rather than a precipitation product. The drastic elemental composition difference of the GBFS surface in the systems demonstrated the acceleration of the reaction with the ligand.
After 1 d of reaction, the reference sample, as presented in Fig. 8d, showed that the smectite or LDH-like structures on the GBFS surface were rich in Mg, Si, Al, and Na, i.e., they were hydrotalcite, as identified by the XRD analysis. As hydrotalcite is composed of only Mg, Al, and C, the presence of Na and Si indicates that the smectite or LDH-like precipitate can be the mixture of two phases: one rich in Mg and Al and the other rich in Na, Al, and Si. Further, it can be a saponite-type mineral, which can precipitate under these given experimental conditions, as observed in.11 Similar types of microstructure elemental compositions were observed for samples R-30 d (Fig. 8e) and R-60 d (Fig. 8f). The elemental mapping of sample L-5 h-60 d (Fig. 8g) showed that the rod-like structures separated or loosely adhered to the GBFS surface were rich in Mg, Al, Si, Na, and C and had depleted Ca and Ti. The TEM images of this precipitate (Fig. S4†) under high magnification revealed the formation of nano-smectite or LDH-like precipitate covering the rod–like structure. However, the morphology and XRD result could not confirm the presence of hydrotalcite as they did for the reference system, even though they were rich in similar elements, which indicated that this precipitate could be amorphous. This indicated that the ligand affected the morphology of the smectite or LDH-like precipitate that was originally observed in the reference system.
It was not possible to analyze the micro sized pyramidal-shaped crystals (Fig. 5e & 6c) using TEM since the crystals were too big to be collected in a TEM grid. However, SEM elemental mapping (Fig. 9) and averaged EDS data revealed that the micro sized pyramidal-shaped structures with varying sizes were rich in Na, Ca, and C, which indicated that these crystals belonged to the gaylussite phase identified by XRD analysis.
 | ||
| Fig. 9 Representation of SEM-EDS elemental mapping of gaylussite crystals (left) and averaged SEM-EDS elemental composition of gaylussite crystals (right). ± values in the table represent standard deviation. | ||
4. Discussion
In both systems, the dissolution of the Ca-rich aluminosilicate glass (GBFS) showed an increasing dissolution rate, which slowed after 1 d and had a residual rate regime from 1–30 d (Fig. 3). The ligand influenced the system by increasing the extent of slag dissolution, thereby accelerating the precipitation of phases. This is evidenced by the dissolution experiment results where the Si dissolution rate at the initial stages (for the first 2.5 h) was almost 24 times higher in the ligand system than in reference. Further, the residual dissolution rate (1–30 d) of the slag with the ligand was relatively high. The presence of the residual dissolution rate in both systems indicated the saturation of the solution with respect to the precipitation products, as discussed later in this section.The increased slag dissolution with the ligand is likely due to the complexation of elements by DHNP. During the initial hours (Fig. 2), where the pH was approximately 11.3 in the ligand system, NLSi was greater than that in the reference system, where the pH was approximately 11.8. It is commonly acknowledged10,11,36 that high pH accelerates glass dissolution. However, the results presented here show that pH, particularly in the initial hours, did not play a significant role for the increased dissolution; instead, it indicates the complexation of Si with the ligand. Additionally, Ti concentration increased significantly during 1 d (Fig. 3b), indicating complexation with Ti, which is plausible as dihydroxy aromatic ligands have been observed to complex with Ti at alkaline pH.39 Previous works11,36,40 have reported the formation of an amorphous Ti-rich layer and Ti incorporation with LDH structures on the surface of glasses with similar compositions and under similar experimental conditions as those used here. These Ti-rich precipitates may inhibit the dissolution of the slag, as considered in.40 Thus, the complexation of Ti by DHNP and the consequent prevention of the formation of Ti-rich precipitates may explain the increased forward dissolution rate of the slag with the ligand. After 1 d, the Ti concentration in the solution gradually decreased. This may be due to the instability of the metal–ligand complex under these experimental conditions (particularly varying pH), causing Ti to participate again in the precipitation reactions. Based on the TOC results (Fig. 4b), the ligand concentration fluctuated during the reaction, supporting the reversible behavior of the ligand. This shows the importance of understanding metal–ligand complex formation and identifying their stability properties.
Based on the SEM/TEM-EDS and XRD results (section 3.2), the precipitation of Mg–Al–Si-smectite or LDH-like and well-crystallized gaylussite and calcite was accelerated with the ligand. Additionally, the quantitative XRD analysis results (Fig. 7) showed high quantities of gaylussite and calcite in the ligand system. This correlates with the TIC results (Fig. 4a), where the carbonate concentration in the ligand system was much lower than that in the reference system. Thus, calcium concentration is controlled by the precipitation of gaylussite (Na2Ca(CO3)2·5H2O) in the reference system and by gaylussite and calcite (CaCO3) precipitation in the ligand system. The extensive formation of well-crystallized calcite in the ligand system is likely due to the increased extent of dissolution of the slag, which increases the quantity of available Ca for calcite precipitation. Additionally, the surface chemistry of the slag is different in the presence of the ligand compared to reference system, which may favor the surface precipitation of calcite. From the XRD result, peaks corresponding to the formation of C–A–S–H or tobermorite were not observed even when the reaction pH stayed at approximately 12.8–12.9 (Fig. 2) from 1 to 60 d of dissolution. This is expected under the given L/S conditions of the experiment, where the high carbonate ion concentration drives the favor toward carbonate salt precipitation reactions.
Mg, Al, Si, Ti, C, and Na concentrations were controlled by the formation of smectite or LDH-like reaction products completely covering the surface of the BFS in the reference system. In contrast, in the ligand system, rods covered with smectite or LDH-like structures loosely bound to the BFS surface and consisting of Mg, Al, Si, C, and Na were detected. The morphology of the precipitate was modified with the ligand because the ion activity product of the phase was affected, which altered the composition of the precipitate (Table 2), or because the ligand was incorporated into the precipitates.41,42
As observed from the results, the significant increase in the precipitation of phases observed in the ligand system compared with that in the reference system evidences the statement that the increased dissolution caused in the presence of the ligand accelerated the precipitation reactions. This could be one of the main reasons for the acceleration of the hydration kinetics of the sodium carbonate-activated BFS observed with the DHNP ligand.
5. Conclusion
This study aimed to understand the mechanisms controlling the early-stage dissolution kinetics of GBFS in Na2CO3-solution with and without DHNP ligand at 25 mM concentration. Detailed investigation of the dissolution and precipitation reactions is crucial for understanding the acceleration mechanism of Na2CO3-activated slag hydration and the effect of the ligand. Overall, this work provides insights into developing new admixtures for low-CO2 cements for sustainable concrete.The DHNP ligand increased the extent of the dissolution of the slag, which accelerated the precipitation kinetics and quantity of phases. The precipitation products in the reference system were (1) gaylussite (Na2Ca(CO3)2·5H2O), depicting a pyramidal morphology with sharp edges, and (2) smectite or LDH-like precipitate formed on the GBFS surface, consisting of Mg, Si, Al, Ti, and Na. In contrast, in the ligand system, three types of precipitates were observed: (1) the extensive formation of rhombohedral calcite (CaCO3) on the BFS surface during the early stages of the reaction, (2) gaylussite, and (3) loosely bound amorphous rods covered with smectite or LDH-like structures rich in Mg, Si, Al, and Na. The DHNP ligand affected the solution chemistry by having initially low and later high pH and by increasing the concentration of soluble elemental species, particularly Si and Ti. Additionally, the ligand exhibited reversible characteristics by participating in the dissolution–precipitation reactions and by being able to be released back into the solution. The ligand–metal ion interactions, i.e., complexation reactions and stability properties, should be explored further to understand the role of ligands in detail.
Author contributions
Rajeswari Ramaswamy: conceptualization, data curation, formal analysis, methodology, software, investigation, validation, visualization, writing – original draft, writing – review & editing. Juho Yliniemi: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing, visualization.Data availability
Research data associated with this article can be accessed at [https://doi.org/10.23729/fd-f1a643da-540b-3e49-8ccd-f5218706e369] in the national Finnish Etsin Fairdata services (https://etsin.fairdata.fi/) with persistent identifier (DOI). In addition, some of the data supporting this article have been included as part of the ESI.†Fairdata IDA (ida.fairdata.fi) is a continuous service for safe research data storage, offered free of charge to Finnish universities, universities of applied sciences and state research institutes. IDA provides storage space for data that is described as a research dataset with the Fairdata Qvain tool. The described data can be set openly available for anyone to download or its availability can be restricted. The data owner decides on the openness and usage policies for their data. The research dataset published with Qvain gets a persistent identifier (DOI) and a landing page in https://etsin.fairdata.fi/. This makes the dataset findable for others, and enables re-use of the data and creating a scientific reference.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Research Council of Finland (#322786 and #354263). Characterization of materials were conducted with the support of the Centre of Material Analysis and lab technicians from Fibre and Particle Engineering unit, University of Oulu, Finland.References
- R. M. Andrew, Earth Syst. Sci. Data, 2018, 10, 195–217 CrossRef
.
- D. N. Huntzinger and T. D. Eatmon, J. Cleaner Prod., 2009, 17, 668–675 CrossRef CAS
- L. K. Turner and F. G. Collins, Constr. Build. Mater., 2013, 43, 125–130 CrossRef
- J. L. Provis, Cem. Concr. Res., 2018, 114, 40–48 CrossRef CAS
- E. Gartner, Cem. Concr. Res., 2004, 34, 1489–1498 CrossRef CAS
- K. L. Scrivener, V. M. John and E. M. Gartner, Cem. Concr. Res., 2018, 114, 2–26 CrossRef CAS
- R. Snellings, P. Suraneni and J. Skibsted, Cem. Concr. Res., 2023, 171 DOI:10.1016/j.cemconres.2023.107199
-
S. A. Bernal, J. L. Provis, A. Fernández-jiménez, P. V. Krivenko, E. Kavalerova, M. Palacios and C. Shi, Alkali Activated Materials: state-of-the-art report, RILEM TC 224-AAM, Springer Dordrecht, 1st edn, 2014 Search PubMed
- A. Hajimohammadi, J. L. Provis and J. S. J. Van Deventer, Chem. Mater., 2010, 22, 5199–5208 CrossRef CAS
- H. Maraghechi, F. Rajabipour, C. G. Pantano and W. D. Burgos, Cem. Concr. Res., 2016, 87, 1–13 CrossRef CAS
- R. Ramaswamy, J. Yliniemi and M. Illikainen, Cem. Concr. Res., 2022, 160, 106922 CrossRef CAS
- G. Habert, J. B. D'Espinose De Lacaillerie and N. Roussel, J. Cleaner Prod., 2011, 19, 1229–1238 Search PubMed
- J. L. Provis and S. A. Bernal, Annu. Rev. Mater. Res., 2014, 44, 299–327 CrossRef CAS
- M. C. G. Juenger, R. Snellings and S. A. Bernal, Cem. Concr. Res., 2019, 122, 257–273 CrossRef CAS
- C. Jolicoeur and M. A. Simard, Cem. Concr. Compos., 1998, 20, 87–101 Search PubMed
-
Science and technology of concrete admixtures, ed. P. C. Aïtcin and R. J. Flatt, Woodhead Publishing Limited, 2016 Search PubMed
-
W. Kurdowski, Cement and Concrete Chemistry, Springer Dordrecht, 1st edn, 2016 Search PubMed
-
P. K. Mehta and P. Monteiro, Concrete: Microstructure, Properties, and Materials, 4th edn, 2001 Search PubMed
- X. Liu, S. Li, Y. Ding, Z. Lu, D. Stephan, Y. Chen, Z. Wang and S. Cui, J. Build. Eng., 2023, 64, 105694 Search PubMed
- T. Raju, K. P. Ramaswamy and B. Saraswathy, Mater. Today: Proc., 2022, 65, 846–851 CAS
- R. Ramaswamy, M. Illikainen and J. Yliniemi, Constr. Build. Mater., 2024, 422, 135753 Search PubMed
- J. Yliniemi, Langmuir, 2022, 38, 987–1000 Search PubMed
-
S. Bagheri, T. Luukkonen and J. Yliniemi, in Proceedings of the DuRSAAM 2023 Symposium ADVANCING ALKALI-ACTIVATED Alkali-Activated materials, ed. S. Matthys and A. Proia, 2023 Search PubMed
- B. Lina, L. Barbara, F. M. Alejandro and L. Christophe, Cem. Concr. Res., 2021, 149, 106563 Search PubMed
- L. Bouzouaid, B. Lothenbach, A. Fernandez-Martinez and C. Labbez, Cem. Concr. Res., 2022, 160, 106894 Search PubMed
- H. Xie, X. Liu, Y. Zheng, B. Chi, J. Guo, X. Dai, Z. Zhang, M. Sun, L. Duan, Z. Wang and S. Cui, Constr. Build. Mater., 2023, 393, 132105 CrossRef CAS
- S. Bagheri, O. Mankinen, S. Ojala, V. V. Telkki, T. Luukkonen and J. Yliniemi, J. Non-Cryst. Solids, 2024, 633, 122962 CrossRef CAS
- L. De Windt, A. Bertron, S. Larreur-Cayol and G. Escadeillas, Cem. Concr. Res., 2015, 69, 25–36 CrossRef CAS
- C. Nalet and A. Nonat, Cem. Concr. Res., 2017, 91, 87–96 CrossRef CAS
- R. J. Flatt, N. Roussel, H. Bessaies-Bey, L. Caneda-Martínez, M. Palacios and F. Zunino, Cem. Concr. Res., 2023, 172, 107243 CrossRef CAS
- A. Adesina, Resour. Environ. Sustain., 2021, 3, 100016 Search PubMed
- X. Deng, H. Guo, H. Tan, K. Nie, X. He, J. Yang, Y. Wang and J. Zhang, Ceram. Int., 2022, 48, 1611–1621 CrossRef CAS
- F. Winnefeld, M. Ben Haha, G. Le Saout, M. Costoya, S. C. Ko and B. Lothenbach, J. Sustainable Cem.-Based Mater., 2014, 4, 85–100 CrossRef
- S. Blotevogel, L. Steger, D. Hart, L. Doussang, J. Kaknics, M. Poirier, H. Bornhöft, J. Deubener, C. Patapy and M. Cyr, J. Am. Ceram. Soc., 2021, 104, 128–139 CrossRef CAS
- B. Traynor, H. Uvegi, E. Olivetti, B. Lothenbach and R. J. Myers, Cem. Concr. Res., 2020, 135, 106122 CrossRef CAS
- K. C. Newlands, M. Foss, T. Matschei, J. Skibsted and D. E. Macphee, J. Am. Ceram. Soc., 2017, 100, 1941–1955 CrossRef CAS
- E. Chibowski, L. Hołysz, A. Szcześ and M. Chibowski, Colloids Surf., A, 2003, 225, 63–73 CrossRef CAS
-
R. Egerton, Physical Principles of Electron Microscopy, Springer International Publishing, 2016, II Search PubMed
- M. J. Sever and J. J. Wilker, J. Chem. Soc., Dalton Trans., 2004, 4, 1061–1072 RSC
- S. Blotevogel, M. Poirier, D. Vantelon, E. Chesneau, C. E. Dutoit, V. Montouillout, F. Fayon, J. Kaknics, G. Landrot, G. D. Saldi, J. Schott, H. Vezin, C. Patapy and M. Cyr, J. Non-Cryst. Solids, 2022, 591 DOI:10.1016/j.jnoncrysol.2022.121708
- N. K. Ilango, H. Nguyen, A. German, F. Winnefeld and P. Kinnunen, Cem. Concr. Res., 2024, 175, 107357 CrossRef
- H. Nguyen, H. Santos, H. Sreenivasan, W. Kunther, V. Carvelli, M. Illikainen and P. Kinnunen, Cem. Concr. Res., 2022, 153, 106696 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04251d |
| This journal is © The Royal Society of Chemistry 2025 |