
An efficient hydrogen evolution catalyst constructed using Pt-modified Ni3S2/MoS2 with optimized kinetics across the full pH range†
Maoyuan
Li‡
a,
Zhongrui
Yu‡
b,
Zulin
Sun‡
a,
Yuchen
Liu
c,
Simiao
Sha
a,
Jiancheng
Li
 a,
Riyue
Ge
d,
Liming
Dai
ef,
Bin
Liu
*a,
Qingqiao
Fu
*a and
Wenxian
Li
*ef
a,
Riyue
Ge
d,
Liming
Dai
ef,
Bin
Liu
*a,
Qingqiao
Fu
*a and
Wenxian
Li
*ef
aSchool of Materials Science and Engineering, Shanghai University, Shanghai 200444, China. E-mail: binliu@shu.edu.cn; fuqingqiao@shu.edu.cn
bShanghai Electric Hency Solar Technology Co., Ltd, Shanghai Electric Power Generation Group, Shanghai, 201199, China
cCollege of Science, Nanjing Agricultural University, Nanjing 210095, China
dSchool of Fashion and Textiles, The Hong Kong Polytechnic University, Hung Hom, Kowloon 999077, Hong Kong
eSchool of Chemical Engineering, The University of New South Wales, Sydney, New South Wales 2052, Australia. E-mail: wenxian.li1@unsw.edu.au
fAustralian Research Council Centre of Excellence for Carbon Science and Innovation, The University of New South Wales, Sydney, New South Wales 2052, Australia
First published on 17th December 2024
Abstract
Electrocatalyst materials play a crucial role in determining the efficiency of the hydrogen evolution reaction (HER), directly influencing the overall effectiveness of energy conversion technologies. Ni3S2/MoS2 heterostructures hold substantial promise as bifunctional catalysts, owing to their synergistic electronic characteristics and plentiful active sites. However, their catalytic efficacy is impeded by the relatively elevated chemisorption energy of hydrogen-containing intermediates, which constrains their functionality in different pH environments. In order to mitigate this limitation, trace amounts of Pt are introduced into the heterostructure, intending to enhance electronic transport and refining chemisorption energies, thereby facilitating significant enhancements in both HER and oxygen evolution reaction (OER) activities over a wide pH range. It is revealed that the Pt-modified catalyst achieves exceptional HER performance, requiring merely 64 mV and 83 mV overpotentials to attain a current density of 100 mA cm−2 in acidic and alkaline media, respectively. Furthermore, theoretical simulations corroborate that Pt modification optimizes local electronic configurations and augments electronic transfer, contributing to its superior catalytic performance. This investigation underscores the pivotal role of Pt modification in propelling the practical application of Ni3S2/MoS2 heterostructures as highly efficient and pH-universal bifunctional catalysts.
1. Introduction
As global energy demands and environmental challenges intensify, the development of renewable green energy has become increasingly crucial.1,2 Hydrogen, with its high energy density and zero emissions, stands out as a promising alternative to fossil fuels.3,4 Water electrolysis, driven by renewable energy sources such as solar or wind power, decomposes water into hydrogen and oxygen. However, the efficiency of water electrolysis for hydrogen production is limited by high reaction barriers and substantial energy consumption.5–8 The entire process largely depends on the performance of electrocatalysts used for the HER and OER.9,10 In recent years, significant progress has been made in enhancing the performance of electrocatalysts for the HER and OER under varying pH conditions, particularly in the field of transition metal-based catalysts. For instance, Gopalakrishnan et al. reported the development of a bifunctional catalyst with exceptional HER and OER performance through interface engineering, which significantly reduces the overpotential.11 Additionally, the dual doping of Mo and S notably improves the HER kinetics and durability of NiSe2.12 Meanwhile, the strategy of bimetallic doping to enhance the OER activity of nickel-based hydroxides also provides new insights for the design of HER catalysts.13 These advancements highlight the critical impact of doping on the electronic structure of catalysts, particularly in the potential to enhance HER and OER performance to address global energy and environmental challenges.Transition metal dichalcogenides, such as molybdenum disulfide (MoS2), have garnered significant attention as potential substitutes for noble metals in HER catalysis.14–16 MoS2, with its layered structure and abundant active sites, particularly when engineered to expose more edge sites where the HER occurs, has demonstrated promising catalytic activity.17 However, the inherent low conductivity and sluggish reaction kinetics of MoS2 limit its catalytic efficiency, especially in practical applications requiring high current densities where OER performance is suboptimal.18 In comparison, nickel sulfides, such as Ni3S2, have been employed as attractive candidates due to their good conductivity and high stability in both acidic and alkaline electrolytes, and complementary catalytic performance.19–21 The limitation of Ni3S2 lies in its suboptimal HER performance, which is attributed to a deficiency of active sites necessary for efficient proton adsorption and HO–H bond dissociation.22–24 By forming a heterojunction between MoS2 and Ni3S2, the composite structure effectively modulates the electronic configuration, thereby increasing the exchange current density and significantly boosting the catalytic activity of Ni3S2.25–27 Similarly, Ni3S2 can improve the charge transfer and durability of MoS2. The interface between MoS2 and Ni3S2 plays a pivotal role in determining the overall catalytic performance because the formation of heterojunctions at this interface can enhance charge separation and transfer, creating new active sites more favorable for catalytic reactions.28,29 In the design and optimization of electrocatalysts, rational interface structure design and precise electronic structure modulation are key strategies for enhancing catalytic performance. Zhang et al. developed a novel MoS2/Ni3S2 heterostructure catalyst through interface engineering, which exhibited excellent performance in redox reactions. The MoS2/Ni3S2 heterostructure showed a low overpotential of 218 mV in the OER, outperforming the then-state-of-the-art catalysts. In bifunctional catalyst applications, this catalyst achieved a current density of 10 mA cm−2 at a low cell voltage of approximately 1.56 V in an alkaline electrolyser. Combined with DFT calculations, it was shown that these interfaces facilitate the chemical adsorption of hydrogen and oxygen intermediates, thereby accelerating the overall water electrolysis.30 Yang et al. successfully synthesized MoS2/Ni3S2 heterojunction nanorods (HNRs/NF) via a simple hydrothermal method, growing these nanorods on nickel foam, which exhibited excellent electrocatalytic performance. The MoS2/Ni3S2 HNRs/NF showed low overpotentials of 98 mV and 249 mV for the HER and OER, respectively, and only required a cell voltage of 1.50 V for overall water splitting to achieve a current density of 10 mA cm−2. These outstanding performances are attributed to the favorable chemical adsorption at the MoS2/Ni3S2 interface, effective active site exposure, and electron transport along the 1D Ni3S2.31 Liu et al. successfully prepared MoS2/Ni3S2 nanospheres with a heterostructure and loaded them onto three-dimensional nanoporous nickel. This catalyst exhibited excellent catalytic performance in the HER and OER, with a HER overpotential of 109 mV and a Tafel slope of 71 mV dec−1 and an OER overpotential of 130 mV and a Tafel slope of 81 mV dec−1. For overall water splitting, a cell voltage of 1.49 V was required to achieve a current density of 10 mA cm−2.32 This superior performance is attributed to the fast electron transfer channels provided by the MoS2 and Ni3S2 interface, as well as the large specific surface area provided by the nanosphere structure.
Heterostructure engineering transcends the mere combination of two distinct materials. Specifically, Ni3S2/MoS2 heterostructures face challenges due to the elevated chemisorption energies associated with hydrogen-containing intermediates, which hinder their catalytic efficiency across varying pH conditions.33 Furthermore, the interfacial instability and suboptimal charge transfer between MoS2 and Ni3S2 can result in diminished catalytic performance.34 The lattice mismatch between the two materials is another critical issue, potentially inducing strain and defects that further undermine the heterostructure's overall efficiency.35 To address these limitations, the introduction of noble metal modifications, such as Pt or Pd, offers a promising strategy. These modifications can significantly enhance the electronic properties of the heterostructure, thereby improving catalytic activity. For instance, Pt or Au decoration has been shown to lower activation barriers and facilitate more efficient charge transfer, resulting in superior HER and OER performance.36 Xie et al. highlight the impact of Pt loading on heterojunction electronic structures. Their study on Pt–Au/graphene (Pt–Au/GNs) catalysts with varying Pt ratios showed that a Pt ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1 improves electrocatalytic performance for methanol oxidation and oxygen reduction. This enhancement is due to the optimized Pt–Au dispersion, which fine-tunes the electronic structure of the heterojunction. Excessive Au causes aggregation, negatively affecting catalytic efficiency, thereby underscoring the importance of optimal Pt loading.37 Kumar et al. demonstrated that the incorporation of noble metals such as Pt, Pd, Au, and Ag into three-dimensional graphene (3D-G) nanocomposites significantly alters the electronic structure of the material, leading to enhanced catalytic performance. Their work shows that the Pt/3D-G composite, in particular, outperforms traditional Pt/C catalysts in the HER, highlighting the critical role of 3D-G modification in optimizing the electronic structure and boosting catalytic activity.38 Ye et al. successfully developed a new method to synthesize single-atom Pt atoms anchored on aniline stacked graphene (Pt SASs/AG), which significantly improved the electronic structure and catalytic performance. The study coordinated the atomically isolated Pt with the nitrogen of aniline, optimized the electronic structure of Pt, increased its hydrogen adsorption energy, and thus improved the HER activity. This work highlights the key role of noble metal modification in regulating electronic properties to achieve excellent electrocatalytic performance.39 An et al. found that precious metal Pt has good adsorption and desorption ability of H* intermediates under acidic conditions. Studies have proved that the introduction of trace Pt can regulate the active sites of sulfide, and the electron shift between Pt and S sites reduces the D-band central position of Pt, weakens the binding ability of Pt and H*, which is conducive to the desorption of H*, and improves the kinetics of the HER.40 The addition of noble metal modifiers effectively addresses the limitations of Ni3S2/MoS2 heterostructures, significantly enhancing their electronic properties and catalytic efficiency.
:0.1 improves electrocatalytic performance for methanol oxidation and oxygen reduction. This enhancement is due to the optimized Pt–Au dispersion, which fine-tunes the electronic structure of the heterojunction. Excessive Au causes aggregation, negatively affecting catalytic efficiency, thereby underscoring the importance of optimal Pt loading.37 Kumar et al. demonstrated that the incorporation of noble metals such as Pt, Pd, Au, and Ag into three-dimensional graphene (3D-G) nanocomposites significantly alters the electronic structure of the material, leading to enhanced catalytic performance. Their work shows that the Pt/3D-G composite, in particular, outperforms traditional Pt/C catalysts in the HER, highlighting the critical role of 3D-G modification in optimizing the electronic structure and boosting catalytic activity.38 Ye et al. successfully developed a new method to synthesize single-atom Pt atoms anchored on aniline stacked graphene (Pt SASs/AG), which significantly improved the electronic structure and catalytic performance. The study coordinated the atomically isolated Pt with the nitrogen of aniline, optimized the electronic structure of Pt, increased its hydrogen adsorption energy, and thus improved the HER activity. This work highlights the key role of noble metal modification in regulating electronic properties to achieve excellent electrocatalytic performance.39 An et al. found that precious metal Pt has good adsorption and desorption ability of H* intermediates under acidic conditions. Studies have proved that the introduction of trace Pt can regulate the active sites of sulfide, and the electron shift between Pt and S sites reduces the D-band central position of Pt, weakens the binding ability of Pt and H*, which is conducive to the desorption of H*, and improves the kinetics of the HER.40 The addition of noble metal modifiers effectively addresses the limitations of Ni3S2/MoS2 heterostructures, significantly enhancing their electronic properties and catalytic efficiency.
In this study, we perform Pt cluster modification onto an array of Ni3S2/MoS2 nanorods grown on nickel foam to form a multistage structure that exposes more active sites. With the advantage of the high activity and conductivity of Pt and the stability of the Ni3S2/MoS2 heterojunction, more efficient and durable electrocatalysts could be obtained. The choice of nickel foam as a substrate not only provides mechanical support but also enhances the overall conductivity of the electrode. DFT calculations show that Pt cluster modification on the heterojunction interface effectively regulates the local electronic arrangement of Ni, S and Mo near the heterojunction interface, resulting in a decrease in the adsorption energy of the intermediates, which is beneficial for the HER. We found that the Pt5-NMS@NF catalyst has high HER and OER activity and reaction kinetics under both alkaline and acidic conditions. This research provides valuable insights into designing high-performance bifunctional catalysts for holistic water decomposition and addresses the challenges posed by integrating different materials into an effective electrocatalyst.
2. Experimental section
2.1. Chemicals and materials
Ammonium heptamolybdate tetrahydrate ((NH4)6Mo7O24·4H2O), hexachloroplatinic acid hexahydrate (H2PtCl6·6H2O), nickel foam, ethanol, distilled water, potassium hydroxide, and sulfuric acid were procured from China National Pharmaceutical Group Chemical Reagents Co., Ltd. Thiourea (CH3CSNH2) and sulfur powder were obtained from Shanghai Aladdin Biochemical Technology Co., Ltd.2.2. Synthesis of MoS2/Ni3S2
A Ni3S2/MoS2 nanorod array was grown on nickel foam (NF) by a simple hydrothermal method. The resulting sample was denoted as Ni3S2/MoS2 (NMS@NF) as described in the materials synthesis section of the ESI.†2.3. Characterization studies and DFT calculations
Details on X-ray diffraction (XRD), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), Brunauer–Emmett–Teller (BET) surface area analysis, electrochemical testing methods, and density functional theory (DFT) calculations are provided in the ESI.†3. Results and discussion
3.1. Synthesis and structural characterization of Pt-modified Ni3S2/MoS2 catalysts
After the Ni3S2/MoS2 nanorods array was grown on nickel foam, the sample size of nickel foam was changed (2 × 3 cm2), and then the sample of nickel foam supported nanoarray was placed in 10 mL of 0.3 mM chloroplatinic acid solution by the photoreduction method (Fig. 1a). The amount of Pt supported was controlled by changing the illumination time. A xenon lamp was irradiated for 30 min, 50 min and 70 min, and the obtained samples were named Pt3-NMS@NF, Pt5-NMS@NF and Pt7-NMS@NF, respectively. | ||
| Fig. 1 Synthesis and characterization of Pt-modified Ni3S2/MoS2 catalysts. (a) Schematic of Pt5-NMS@NF synthesis via photoreduction, with Pt loading adjusted by varying illumination time. (b) XRD patterns of NMS@NF and Pt-modified samples, highlighting Ni3S2 peaks (yellow) and indicating no new phase formation. (c–f) SEM images: (c) unmodified NMS@NF shows nanorods with surface nanosheets, (d) Pt3-NMS@NF maintains similar morphology, (e) Pt5-NMS@NF displays uniform Pt nanoparticles (∼2 nm) on nanorods, and (f) Pt7-NMS@NF shows a transition from nanosheets to nanoparticles. | ||
The XRD patterns of NMS@NF, Pt3-NMS@NF, Pt5-NMS@NF, and Pt7-NMS@NF samples are shown in Fig. 1b. Distinct Ni3S2 peaks are evident (marked in yellow in Fig. 1b), but no obvious MoS2 peaks are observed due to MoS2's poor crystallinity. After Pt modification, the XRD peaks of the catalyst samples show no significant changes, indicating no new phase formation.
SEM analysis reveals the morphological changes in the NMS@NF catalyst before and after Pt modification. The morphology of the NMS@NF catalyst without Pt modification is a nanorod structure with nanosheets on the surface (Fig. 1c), similar to the catalyst morphology with slight Pt modification (Fig. 1d). As shown in Fig. 1e, with Pt content increment, small nanoparticles start to appear on the nanorod surface. These nanoparticles, approximately 2 nm in size, are relatively uniform on the nanorod (Fig. 2a and b). When the reduction time is increased to 70 minutes, as shown in Fig. 1f, the number of nanosheets on the Pt7-NMS@NF nanorod surface decreases, transforming the morphology from nanosheets to nanoparticles, which reduces the catalyst's surface area and affects the exposure of active sites.
 | ||
| Fig. 2 TEM and HAADF-STEM analysis of Pt5-NMS@NF. (a and b) TEM images showing the nanorod structure. (c) HRTEM image with 0.62 nm MoS2 (002) lattice fringes. (d) Pt nanoparticles and Ni3S2/MoS2 interfaces. (e and f) Enlarged views showing lattice spacings of MoS2 and Ni3S2 planes. (g) HAADF-STEM image with EDX confirming Ni, Mo, S, O, and Pt distribution. | ||
TEM results for Pt5-NMS@NF are shown in Fig. 2. Fig. 2c demonstrates the nanorod structure of Pt5-NMS@NF, consistent with SEM observations. Fig. 2d shows the HRTEM image of the sample, with lattice fringes spaced at 0.62 nm corresponding to the (0 0 2) plane of MoS2. Rich Ni3S2 and MoS2 heterointerfaces are present at the edges. Fig. 2e shows a magnified image of interface 1 in Fig. 2d, with lattice fringes spaced at 0.267 nm corresponding to the (1 0 1) plane of MoS2 and 0.204 nm to the (2 0 2) plane of Ni3S2. Fig. 2f shows a magnified image of interface 2 in Fig. 2d, with lattice fringes of 0.163 nm corresponding to the (1 0 6) plane of MoS2 and 0.204 nm to the (2 0 2) plane of Ni3S2. Additionally, lattice fringes of Pt are observed in Fig. 2d, indicating successful Pt nanoparticle modification on the heterostructure surface. Fig. 2g shows the HAADF-STEM image and EDX spectrum, confirming the presence of Ni, Mo, S, O, and Pt elements in Pt5-NMS@NF, with Pt predominantly distributed on the nanorod surface.
3.2. Surface chemical states of Pt-modified catalysts
X-ray photoelectron spectroscopy was employed to characterize the elemental chemical states of NMS@NF and Pt5-NMS@NF catalysts. As shown in Fig. 3a, all surfaces contain Ni, Mo, S, and O, with Pt additionally present on Pt5-NMS@NF, indicating that Pt modification is successful. The composition percentages of each element are listed in Table S3,† where the percentage of Pt is found to be 0.21%. The C 1s XPS spectra of NMS@NF and Pt5-NMS@NF (Fig. 3b) show peaks corresponding to C–C, O–C–O, and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. In the O 1s XPS spectra (Fig. 3c), peaks for metal–O, OH−, SO42−, and H2O are observed at 529.1 eV, 530.7 eV, 531.7 eV, and 533.1 eV, respectively. Since XPS detected more H2O water molecules on Pt5-NMS@NF, the peak area of H2O at Pt5-NMS@NF was significantly larger, indicating that the adsorption of water on the Pt-modified surface was enhanced.
O. In the O 1s XPS spectra (Fig. 3c), peaks for metal–O, OH−, SO42−, and H2O are observed at 529.1 eV, 530.7 eV, 531.7 eV, and 533.1 eV, respectively. Since XPS detected more H2O water molecules on Pt5-NMS@NF, the peak area of H2O at Pt5-NMS@NF was significantly larger, indicating that the adsorption of water on the Pt-modified surface was enhanced.
 | ||
| Fig. 3 XPS analysis of NMS@NF and Pt5-NMS@NF catalysts. (a) Full XPS spectrum confirming the presence of Ni, Mo, S, O, and Pt on the surfaces, indicating successful Pt modification. (b) High-resolution C 1s XPS spectra showing peaks for C–C, O–C–O, and O–CO. (c) O 1s XPS spectra displaying peaks for metal–O, OH−, SO42−, and H2O. | ||
Fig. S1 of the ESI† shows a detailed XPS spectrum of each element. In Ni 2p, compared with NMS@NF, the Ni–S peak of the Pt5-NMS@NF sample shifted by −0.13 eV, indicating that the electrons of Ni–S shifted, the outer layer of Ni atoms gained electrons, and the electron cloud density increased. The Mo 3d XPS spectrum shows that the Mo–S peak of Pt5-NMS@NF shifted by 0.13 eV, indicating that Mo lost electrons and moved toward the high valence state. The S 2p XPS spectrum shows that the Mo–S and Ni–S peaks of Pt5-NMS@NF shifted by 0.33 eV, indicating that the outermost electrons of S decreased and the electron cloud density decreased. The electron shifts on the Mo, S, and Ni orbits highlight the effect of Pt modification on the electronic structure in the heterojunction. The detailed XPS analysis of the specific elements can be found in the ESI.†
3.3. Effect of Pt modification on HER performance under alkaline conditions
The HER performance of NF, NMS@NF, and Pt-modified samples (Pt-NF, Pt3-NMS@NF, Pt5-NMS@NF, Pt7-NMS@NF, and Pt/C@NF) was tested under alkaline conditions (1.0 M KOH solution) in a three-electrode system. As shown in Fig. 4a, Pt5-NMS@NF exhibits the best performance, reaching a current density of 100 mA cm−2 at an overpotential of 64 mV, surpassing commercial Pt/C electrodes and other catalysts. Fig. 4b shows that Pt5-NMS@NF maintains excellent catalytic performance at both low (100 mA cm−2) and high (500 mA cm−2) current densities, with overpotentials of 64 mV and 187 mV, respectively, outperforming NF (323 mV, 492 mV), Pt-NF (239 mV, 491 mV), NMS@NF (185 mV, 293 mV), Pt3-NMS@NF (112 mV, 229 mV), Pt7-NMS@NF (65 mV, 227 mV), and Pt/C@NF (110 mV, 392 mV). Pt5-NMS@NF exhibits remarkable catalytic activity, particularly at low overpotentials, where its efficiency surpasses that of other catalysts, maintaining stable performance across various current densities. This indicates that Pt modification improves the HER performance of NMS@NF catalysts. The excellent performance can be attributed to the coexistence of nanoparticles and nanosheets in the Pt-modified Ni3S2/MoS2 heterojunction structure, which provides abundant active sites and the good conductivity of Pt itself, which promotes the changes in the electronic structure on the surface of the heterostructure and accelerates the electron transfer rate.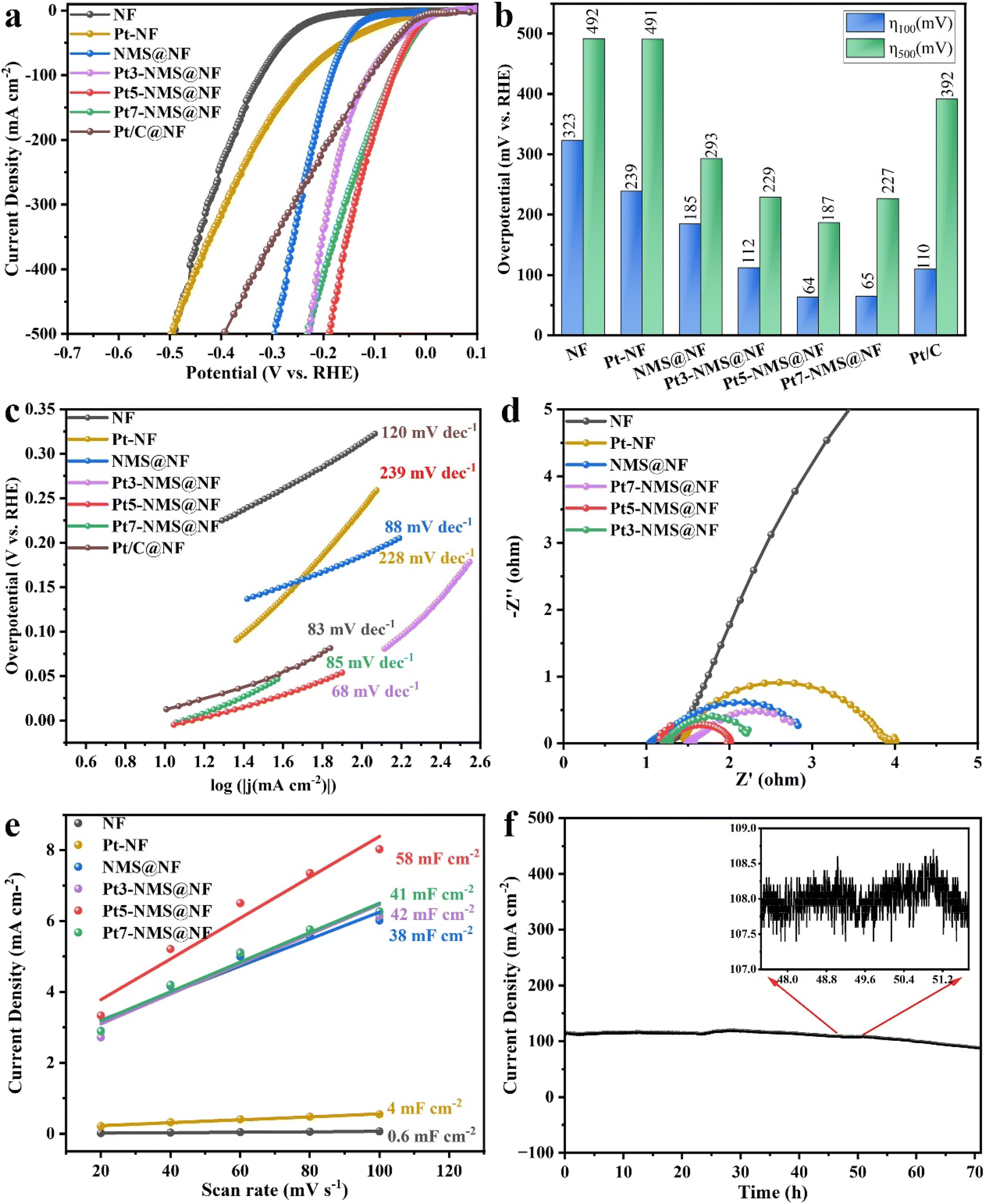 | ||
| Fig. 4 HER performance of various catalysts under alkaline conditions. (a) LSV curves of NF, NMS@NF, Pt-NF, Pt3-NMS@NF, Pt5-NMS@NF, Pt7-NMS@NF, and Pt/C@NF under 1.0 M KOH solution. (b) Comparison of overpotential at current densities of 100 mA cm−2 and 500 mA cm−2 for each catalyst. (c) Tafel slope analysis derived from polarization curves. (d) EIS spectrum demonstrating the charge transfer resistance of the catalysts. (e) Cdl values of the catalysts. (f) Results of the Pt5-NMS@NF stability test over 75 hours at 1.0 M KOH. | ||
The LSV curve in Fig. 4a shows that although the onset potential of Pt7-NMS@NF is lower than that of Pt5-NMS@NF, Pt5-NMS@NF finally exhibits excellent catalytic performance because of the optimal balance between its active surface area and Pt modification amount. A detailed explanation of this phenomenon is provided in the ESI (Fig. S2†).
The Tafel slope extrapolated from the polarization curve can be used to evaluate the reaction kinetics. As shown in Fig. 4c, the Tafel slopes of the synthesized catalysts Pt-NF, NMS@NF, Pt3-NMS@NF, Pt5-NMS@NF, Pt7-NMS@NF and Pt/C@NF are 239 mV dec−1, 88 mV dec−1, 228 mV dec−1, 68 mV dec−1, 85 mV dec−1 and 83 mV dec−1, respectively. From the above results, it can be seen that Pt5-NMS@NF exhibits an excellent performance with a lower overpotential than that of Pt/C@NF and has faster HER catalytic kinetics. The reaction mechanism and rate-limiting step of electrocatalytic hydrogen evolution can be judged by the Tafel slope. The Tafel slopes of the Pt-modified catalysts are all between 40 and 120 mV dec−1, which can determine that the HER process is the Volmer–Heyrovsky mechanism and the rate-limiting step is the Heyrovsky reaction. In addition, it can be seen that compared with the catalyst without Pt loading, Pt5-NMS@NF has the smallest Tafel slope, indicating that its HER catalytic kinetics under alkaline conditions is faster. The acceleration of the reaction kinetics is because the modification of Pt regulates the electronic structure of the heterojunction and promotes the electron transfer rate. In addition, due to the introduction of Pt, the adsorption and dissociation of hydrogen on the heterojunction are optimized, thereby improving the overall efficiency of the HER process. DFT calculations will be used to further describe the adsorption of hydrogen on the catalyst surface. In addition, the nanorod morphology in which an appropriate amount of nanoparticles and nanosheets coexist has a larger material transmission channel, which is more conducive to the transmission of materials and the overflow of hydrogen.
Electrochemical impedance spectroscopy (EIS) was employed to characterize the electrochemical impedance and evaluate the interfacial charge transfer process during catalysis. As depicted in Fig. 4d, among the various Pt-modified samples, Pt5-NMS@NF exhibits the lowest charge transfer resistance, indicating superior conductivity and rapid electron transport during the reaction. This improvement should be attributed to the formation of heterointerfaces that accelerate electron transfer rates. The electrochemical surface area (ECSA) is used to evaluate the number of active sites that actually participate in the reaction. The catalyst materials were tested with CV at different scan rates (Fig. S3†) and the double layer capacitance (Cdl) of each material was calculated. As shown in Fig. 4e, Pt5-NMS@NF (58 mF cm−2) has a larger Cdl value, which is better than Pt-NF (4 mF cm−2), NMS@NF (38 mF cm−2), Pt3-NMS@NF (42 mF cm−2) and Pt7-NMS@NF (41 mF cm−2). This shows that the modification of an appropriate amount of Pt introduces nanoparticles so that the catalyst has a larger electrochemical surface area, and the catalyst can have more active sites, which is conducive to the Volmer and Heyrovsky reactions.
The stability of a catalyst is a critical factor for its practical application. In order to further assess the stability of the catalyst under practical conditions, long-term stability tests were conducted using chronoamperometry in an alkaline environment. As shown in Fig. 4f, Pt5-NMS@NF was operated in a 1.0 M KOH electrolyte for about 75 h. Throughout the stable operation, dense hydrogen bubbles continuously emerged from the cathode and quickly detached from the catalyst surface into the electrolyte, indicating that mass transport was not hindered. After prolonged operation, the current density showed only a slight decrease, indicating the catalyst's excellent stability. To further verify whether corrosion occurred on the surface after the HER stability test, we examined the post-reaction SEM images and XRD patterns of Pt5-NMS@NF in an alkaline environment, as shown in Fig. S4.† After the HER stability test under alkaline conditions, the XRD pattern showed no new phases, with only slight decreases in peak intensity. SEM observations confirmed that the surface structure remained intact, with minimal morphological changes, indicating high corrosion resistance of the material under alkaline conditions. To further verify the corrosion resistance of Pt5-NMS@NF after HER stability testing in an alkaline environment, XPS measurements were performed, and the results are shown in Fig. S13.† Compared to the spectra before the reaction, the Ni–S peak shifted by 0.73 eV to a higher binding energy, and the Ni–Ni signal weakened. Additionally, significant changes were observed in the S 2p peaks, attributed to stability testing in an alkaline environment, where parts of S and Ni were oxidized.47,48 In the Mo 3d region, the attenuation of both Mo–O and Mo–S signals further confirmed the surface oxidation of Pt5-NMS@NF during the HER process. The Pt0+ peak shifted by 0.63 eV to a higher binding energy, and the Ptδ+ peak shifted by 1.53 eV, with a decrease in peak areas. These energy shifts indicate a rearrangement of the electronic structure of Pt, playing a crucial role in the electron transfer process during the HER. Although changes in peak intensity were observed, the coordination environments of all elements confirm that the surface composition of Pt5-NMS@NF remains essentially stable after prolonged HER operation. Combined with the structural stability demonstrated by SEM and XRD, this highlights the catalyst's remarkable durability in alkaline environments. This good stability may be due to the coexistence of nanoparticles and nanosheets of the Pt-modified Ni3S2/MoS2 heterojunction, which is conducive to the transport of substances. Moreover, the modification of Pt forms Pt–S bonds and Pt–Ni bonds with S atoms and Ni atoms, which accelerates the electron transfer between S, Mo and Ni atoms and optimizes the adsorption and desorption processes of the intermediates.
Benefiting from the regulation of the Pt-Ni3S2/MoS2 heterostructure, Pt5-NMS@NF demonstrates excellent HER activity under alkaline conditions. However, excessive Pt loading leads to performance degradation. The origin is attributed to the formation of a large number of nanoparticles on the surface, which cover the active Pt–S sites, limit the contact between the reactants and the active sites, and finally reduce the reaction rate. According to the XPS results, the binding energy of the S site is negatively shifted, and more vacancies are generated in the outer electron orbit of S, which is conducive to the adsorption of proton source H2O by the S site.41 As shown in Fig. S5,† the HER mechanism of the Pt5-NMS@NF catalyst under alkaline conditions is mainly to split water from the S site to produce H* and OH− intermediates, while the Ni site near the Pt cluster has a good adsorption effect on H*. The OH− generated at the interface will be adsorbed by the metal site, the separation rate of the reactants will be accelerated, and the Volmer reaction will be promoted. The Pt cluster will facilitate the Heyrovsky reaction and promote the adsorption and desorption of H*, which will be confirmed in the subsequent DFT calculation.
3.4. Effect of Pt modification on the HER performance of catalysts under acidic and neutral conditions
To meet the requirements for catalysts that are effective across the entire pH range, the catalytic performance of the catalysts under acidic conditions was evaluated. HER performance of various catalysts was tested in a 0.5 M H2SO4 electrolyte. Initially, HER activity for NF, Pt-NF, NMS@NF, Pt3-NMS@NF, Pt5-NMS@NF and Pt7-NMS@NF was assessed through iR-corrected LSV curves, as shown in Fig. 5a. At an overpotential of 200 mV, NMS@NF achieved a current density of 100 mA cm−2. However, the performance of Pt-modified catalysts exhibited substantial enhancement, with Pt5-NMS@NF achieving a current density of 100 mA cm−2 at an overpotential of 83 mV. The results indicate that Pt modification enhances the HER performance of NMS@NF catalysts under acidic conditions. This improvement can be attributed to the synergistic effect of Pt clusters and the underlying Ni3S2/MoS2 structure, which facilitates efficient electron transfer and increases active site availability. The fluctuations in the LSV curves may be attributed to current variations caused by bubble evolution. Fig. 5b illustrates the overpotentials required for different catalysts to reach current densities of 50 mA cm−2 and 100 mA cm−2. When varying the Pt loading, the Pt5-NMS@NF catalyst showed superior overpotential performance compared to Pt7-NMS@NF (85 mV) and Pt3-NMS@NF (124 mV). Similar to the principle under alkaline conditions, the performance degradation caused by the addition of a large amount of Pt may be related to the coverage of active sites on the catalyst surface due to excessive Pt loading. Compared with recent studies on transition metal-based HER catalysts under alkaline conditions, the Pt5-NMS@NF catalyst demonstrates superior performance, positioning it as one of the best catalysts reported to date (Table S1†). | ||
| Fig. 5 HER performance of various catalysts under acidic conditions. (a) iR-corrected LSV curves of NF, Pt-NF, NMS@NF, Pt3-NMS@NF, Pt5-NMS@NF, and Pt7-NMS@NF in 0.5 M H2SO4 electrolyte. (b) Overpotentials required for different catalysts to achieve current densities of 50 mA cm−2 and 100 mA cm−2. (c) Tafel slope analysis from the polarization curves. (d) EIS spectra showing charge transfer resistance for each catalyst. (e) Cdl values obtained from CV measurements at varying scan rates. (f) Stability test results of Pt5-NMS@NF after 90 hours of continuous operation at a current density of 100 mA cm−2. | ||
The reaction kinetics and mechanism of the electrochemical process were characterized by the Tafel slope, as depicted in Fig. 5c. Pt5-NMS@NF exhibited a lower Tafel slope (43 mV dec−1), outperforming Pt3-NMS@NF and Pt7-NMS@NF. This suggests superior reaction kinetics for Pt5-NMS@NF. Based on the Tafel slope value, the HER mechanism under acidic conditions can be identified as the Volmer–Heyrovsky mechanism. Unlike the HER mechanism under alkaline conditions, which involves water adsorption, the acidic HER mechanism does not include this step. Instead, H+ ions from the electrolyte are directly adsorbed at S sites during the Volmer process to form H*, followed by the Heyrovsky process at Pt sites to complete the reaction. Further analysis of the kinetics was conducted through EIS testing to evaluate conductivity. As shown in Fig. 5d, Pt5-NMS@NF exhibits the smallest semicircle diameter in acidic media, which indicates that the loading of an appropriate amount of Pt under acidic conditions can also accelerate the electron transfer rate of the catalyst. The above results show that the HER of Pt5-NMS@NF under acidic conditions has a smaller Tafel slope and charge transfer resistance and has the same excellent HER kinetics as under alkaline conditions.
Moreover, the ECSA of the catalyst for the HER under acidic conditions was characterized. The Cdl values were obtained by CV measurements at varying scan rates (Fig. S6†), as shown in Fig. 5e. Notably, the Pt5-NMS@NF sample exhibited the highest Cdl value, indicative of a larger ECSA. This underscores that Pt modification enhances the ECSA of the Ni3S2/MoS2 heterostructure, while excessive Pt modification leads to a reduction in ECSA. Lastly, the stability of Pt5-NMS@NF under acidic conditions was evaluated over extended operation periods. As shown in Fig. 5f, minimal activity decay was observed after 90 hours of continuous operation at a current density of 100 mA cm−2. This demonstrates excellent HER stability, which is attributed to the stable nanorod morphology of the catalyst (Fig. S7†). Post-reaction XRD analysis of Pt5-NMS@NF reveals that the phase remains the same as before the reaction, with only a slight decrease in intensity. SEM images show that the microstructure on the surface after testing in the acidic environment exhibits minimal change compared to the pre-reaction microstructure (Fig. 1e). Even after prolonged stability testing, the structure remains intact and undamaged. To evaluate the surface stability, XPS measurements were performed after HER testing under acidic conditions, as shown in Fig. S15.† The XPS spectra reveal that in the Ni 2p region, the Ni–Ni signal is enhanced, while the Ni–S signal is weakened. This may be due to changes in the local chemical environment during the HER process under acidic conditions, making the Ni–Ni sites more prominent and reducing the contribution of Ni–S sites. In the Mo 3d region, the binding energy of the Mo–O signal increases, and its peak area decreases, while the Mo–S signal shows significant enhancement. This could be attributed to the higher stability and activity of Mo–S under acidic conditions, resulting in a stronger signal.
Pt5-NMS@NF exhibited promising catalytic activity under both alkaline and acidic conditions. Subsequently, its HER performance under neutral conditions was assessed. As illustrated in Fig. S8a,† during the HER in neutral media, Pt5-NMS@NF required an overpotential of 429 mV to achieve a current density of 100 mA cm−2, which, albeit significant, still lags behind Pt/C. However, at a current density of 167 mA cm−2, Pt5-NMS@NF attained a comparable overpotential to Pt/C. As the current density further increased, the superiority of Pt5-NMS@NF became increasingly evident. Furthermore, the HER kinetics of Pt5-NMS@NF and Pt/C under neutral conditions were evaluated through Tafel slope analysis. Pt5-NMS@NF demonstrated a lower Tafel slope than Pt/C (Fig. S8b†), suggesting faster reaction kinetics. Under neutral conditions, the HER activity of the Pt-modified NMS@NF catalyst also shows a significant improvement. The presence of Pt clusters provides a favorable electronic environment for the adsorption of reaction intermediates and enhances charge transfer dynamics. The Cdl of Pt5-NMS@NF was probed via cyclic voltammetry curves at varying scan rates (Fig. S8c†). The smaller area corresponds to a relatively lower abundance of active sites under alkaline conditions, yielding a calculated Cdl value of 4.03 mF cm−2 (Fig. S8d†). As with the alkaline and acidic environments, the XRD pattern and SEM images after HER testing in a neutral environment are shown in Fig. S12.† The XRD pattern after HER testing in a neutral environment also shows no new phase formation, with minimal changes in peak intensity. SEM images reveal no significant morphological changes on the material's surface, indicating good corrosion resistance under neutral conditions. To gain further insights into the surface chemical stability, XPS measurements were conducted post-HER testing, as shown in Fig. S16.† The XPS spectra after HER testing in a neutral environment show that the changes in Ni, Mo, S, and Pt signal peaks are consistent with those observed under alkaline conditions. This indicates that the chemical environment changes on the catalyst surface during the HER process are similar under both neutral and alkaline conditions. Furthermore, the electronic structure rearrangement and chemical shifts of Pt also exhibit consistency under neutral and alkaline conditions, further highlighting the structural stability and electron transfer behavior of the catalyst across different environments. These results underscore the exceptional HER performance of Pt5-NMS@NF under both acidic and alkaline conditions, with superior performance compared to Pt/C, particularly in alkaline media. However, there remains room for optimization in enhancing the HER performance of Pt5-NMS@NF under neutral conditions.
3.5. Simulation of hydrogen adsorption on Pt-loaded Ni3S2/MoS2 heterostructures using density functional theory
We investigated the influence of Pt cluster deposition on the HER mechanism of the Ni3S2/MoS2 heterojunction. DFT calculations were performed to simulate hydrogen adsorption on the heterojunction. The crystal structure information of bulk Ni3S2 and MoS2 was obtained from the ICDD database. As shown in Fig. 2b, the Ni3S2 (1 0 1) surface and the MoS2 (0 0 2) surface were selected based on the TEM images to construct the heterostructure. The heterostructure we constructed is similar to the structure model used in the DFT theoretical calculations in ref. 43. To minimize lattice mismatch, the surfaces were expanded in 1 × 2 and 3 × 4 configurations, respectively, resulting in a supercell containing 96 atoms. To prevent non-physical interactions between the surface and its periodic images, a 12 Å vacuum layer was introduced in the cleaved surface model, yielding a final heterojunction structure denoted as NMS@NF (21.3096, 12.0510, and 13.0632), as shown in Fig. S9a.† Based on XPS spectra and electrochemical tests, the interaction between Pt and S was found to favor the Volmer step. Therefore, a tetrahedral Pt cluster was anchored at the interface of the NMS@NF heterojunction.42 As depicted in Fig. S9b,† the Pt cluster forms bonds with atoms near the Ni3S2 surface, with Pt–S and Pt–Ni bond lengths measuring 2.35 Å and 2.6 Å, respectively. This structure is referred to as Pt-NMS@NF.Li discovered that H* preferentially adsorbs at the Ni sites on the Ni3S2/MoS2 heterojunction interface.43 Based on this finding, we constructed the structures shown in Fig. S10,† where H atoms are adsorbed near the Ni sites on the Ni3S2 side of the NMS@NF and Pt-NMS@NF interfaces. Typically, the Gibbs free energy of hydrogen adsorption (ΔGH*) on the catalyst surface is used to evaluate the HER performance in electrochemical experiments. The closer this value is to zero, the better the HER efficiency.44,45 The definition of Gibbs free energy is provided in the ESI.† As shown in Fig. 6c, the adsorption free energy of H* (ΔGH*) at the Ni sites of the NMS@NF heterojunction is 0.868 eV. Remarkably, the ΔGH* at the Ni sites of the Pt-NMS@NF heterojunction is much closer to zero (−0.432 eV), indicating that it is more favorable for the HER.
 | ||
| Fig. 6 Charge density differences and hydrogen adsorption free energy of the heterostructures. (a) Differential charge density distribution of the NMS@NF heterostructure, where orange regions indicate charge depletion and yellow regions represent charge accumulation. (b) Differential charge density distribution of the Pt-NMS@NF heterostructure. (c) Gibbs free energy of hydrogen adsorption (ΔGH*) for NMS@NF and Pt-NMS@NF, presented in a ladder diagram. | ||
To further investigate the effect of Pt clusters on the electronic distribution of the heterojunction, the differential charge densities of NMS@NF and Pt-NMS@NF were plotted, as shown in Fig. 6a and b. In Fig. 6a, the orange regions indicate charge depletion, where electron density decreases. The yellow regions represent charge accumulation, corresponding to areas of increased electron density. The accumulation of charge enhances electronic interactions at the heterojunction interface, promoting more efficient charge transfer during the HER. It is evident that the modification by Pt clusters significantly affects the spatial distribution of electron density within the heterojunction, especially at the interface and around the atoms bonded to the Pt clusters. The charge is primarily concentrated around the Pt clusters, leading to an accumulation of electrons that accelerates the rate of electron transfer.
Chen et al. research demonstrated that Pt3-SnS2 exhibits strong chemical interactions with gases such as SO2 and H2S, whereby Pt clusters optimize the electronic structure and enhance electron transfer processes, thereby improving the material's sensing performance.46 This observation is consistent with our findings on the Pt-NMS@NF catalyst, where the modification by Pt clusters significantly improved the spatial distribution of electron density, facilitating charge transfer during the HER.
This explains the faster mass transport observed in Pt-NMS@NF after Pt modification. Moreover, after Pt modification, the Ni sites that adsorb H* (shown in Fig. S10†) accumulate more electrons. This suggests that the electron density around the Ni atoms increases, making H* adsorption more favorable. Consequently, the ΔGH* decreases, which enhances the HER performance. This observation is consistent with the findings from the high-resolution Ni 2p XPS spectra.
3.6. Effects of Pt modification on OER performance under alkaline conditions
In order to further evaluate the OER activity of the Pt-modified catalyst in an alkaline environment, electrochemical tests were performed in 1.0 M KOH solution in a three-electrode system. As shown in Fig. 7, the overpotential of the NMS@NF catalyst with an appropriate amount of Pt modified on the surface was reduced. Pt3-NMS@NF and Pt5-NMS@NF can reach a current density of 100 mA cm−2 at an overpotential of 290 mV and 275 mV, respectively. The overpotential increased after excessive Pt modification, which is related to the excessive Pt causing the nanoparticles to cover the surface active sites. The Tafel slopes of Pt3-NMS@NF and Pt5-NMS@NF catalysts are 41 mV dec−1 and 35 mV dec−1, respectively, which are smaller than the Tafel slope of NMS@NF (Fig. 7b). Compared with recent studies on transition metal-based OER catalysts under alkaline conditions, the Pt5-NMS@NF catalyst exhibits outstanding performance, ranking among the best OER catalysts reported to date (Table S2†). The modification of the catalyst surface with an appropriate amount of Pt and the generation of Pt–S coordination affect the surface chemical state of the catalyst and accelerate the OER kinetics of Pt5-NMS@NF. The EIS in Fig. 7c shows that the Pt-modified catalyst has a smaller semicircle diameter. This suggests enhanced electron transfer rates during the OER. As a result, the kinetics of the OER are improved due to the Pt modification. Fig. 7d shows the OER stability results. After ∼24 h of testing by chronoamperometry, the current density of the catalyst can still be maintained at ∼90%, and the surface catalyst also exhibits good OER stability. After prolonged operation, the current density showed only a slight decrease, indicating the catalyst's excellent stability. To verify the material's stability under alkaline OER conditions, we collected the post-reaction XRD pattern and SEM images of Pt5-NMS@NF. As shown in Fig. S11,† no new phases appeared in the XRD pattern, with only minor decreases in peak intensity observed. Furthermore, SEM images revealed no significant changes in surface morphology after testing, indicating that the material exhibits excellent structural stability under OER conditions without evident reconstruction. The XPS analysis shown in Fig. S14a† indicates a significant change in the chemical state of Ni after OER testing under alkaline conditions. The Ni–Ni peak of Pt5-NMS@NF disappears, while the Ni–S peak shifts by 1.33 eV to a lower binding energy, suggesting an increase in the electron density around Ni. This may be related to surface chemical restructuring leading to the formation of NiOOH.49 Such behavior is similar to the surface reconstruction of Ni described in the La-NMS@NF catalyst.50 Additionally, the Mo–O and Pt0+ peaks shift by 1.23 eV and 1.93 eV, respectively to lower binding energies, indicating an increase in the electron density around Mo and Pt during the OER process. | ||
| Fig. 7 Electrochemical performance of catalysts for the OER under alkaline conditions. (a) LSV curves for the OER of NMS@NF, Pt3-NMS@NF, and Pt5-NMS@NF catalysts. (b) Tafel slopes for the catalysts. (c) EIS spectra showing the impedance characteristics of the catalysts. (d) Chronoamperometric stability test results for Pt5-NMS@NF. | ||
3.7. Impact of Pt modification on the overall water splitting performance of the catalyst
The Pt5-NMS@NF catalyst was evaluated for overall water splitting performance in a dual-electrode system within a 1.0 M KOH electrolyte solution. As shown in Fig. 8a, the overall water splitting overpotential was assessed, with the cell voltage reaching 1.62 V at which point the current density achieved 100 mA cm−2. Fig. 8b illustrates the stability test for overall water splitting of the catalyst, conducted at a cell voltage of 1.62 V. After approximately ∼23 hours of testing, a slight loss in current density was observed, indicating that Pt5-NMS@NF retains excellent stability as a catalyst for overall water splitting. | ||
| Fig. 8 Performance of the Pt5-NMS@NF catalyst for overall water splitting. (a) LSV curves of Pt5-NMS@NF as an overall water splitting catalyst. (b) i–t stability curves during long-term operation at 1.62 V. | ||
4. Conclusion
Pt cluster decoration of the Ni3S2/MoS2 heterostructure nanorod array can optimize the electronic structure of active interface atoms, favor the adsorption of proton sources, and provide electron transport pathways during catalysis. Meanwhile, the Pt–S interaction facilitates the separation of Volmer process products, accelerating the overall reaction kinetics. The introduction of Pt clusters increases the number of electrons in the outermost layer of Ni atoms, reduces the adsorption energy of H* intermediates, and promotes the desorption of H2. Loading Pt onto nanorods made of nanosheets can form a hierarchical structure where nanoparticles and nanosheets coexist, in which the nanorods coated with an appropriate amount of Pt have a larger surface area, thereby exposing more active sites. This open configuration also enhances mass transport. In the calculation of free energy adsorption, Pt-NMS@NF shows the ΔGH* value closest to 0, and the electron density of Ni sites adsorbed with H in the differential charge density map increases significantly, indicating that Pt cluster loading improves the HER activity by changing the electronic structure of the interface. The optimized sample exhibits remarkable HER performance, requiring overpotentials of merely 83 and 64 mV, respectively, to achieve a current density of 100 mA cm−2. The Tafel slopes of 43 and 68 mV dec−1, respectively, underscore the catalyst's high activity and favorable reaction kinetics across varying pH electrolytes. Stability tests under acidic and alkaline conditions reveal minimal current loss over 90 hours in acidic media and ∼70 hours in alkaline media, demonstrating the catalyst's robust catalytic stability across these conditions.Author contributions
Maoyuan Li: investigation, DFT calculations, data curation, and writing – original draft. Zhongrui Yu: data visualization, writing – review & editing, and validation. Zulin Sun: electrochemical experiments, investigation, and writing – review & editing. Yuchen Liu, Simiao Sha, and Jiancheng Li: writing – review & editing, computational analysis, and data curation. Riyue Ge and Liming Dai: experimental supervision, project supervision, and writing – review & editing. Bin Liu: methodology, funding acquisition, and writing – review & editing (corresponding author). Qingqiao Fu: methodology, and writing – review & editing (corresponding author). Wenxian Li: conceptualization, formal analysis, and writing – review & editing (corresponding author).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council Future Fellowship (FT230100436), Australian Research Council Centre of Excellence for Carbon Science and Innovation (CE230100032), BAJC R&D project (BA23011), ARC Hub (IH230100010) -BAJC Project (SPDC-P09), and UNSW Science Translational Impact Seed Funding. B. Liu acknowledges research support from the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning. R. Ge acknowledges the financial support from the China Postdoctoral Science Foundation (No. 2021M702073). Q.Q. Fu acknowledges research support from the Postdoctoral Fellowship Program of CPSF (grant No. GZC20240969).References
- J. Staszak-Jirkovský, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef PubMed.
- M. Cabán-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H. Chang, M. Tsai, J. He and S Jin, Nat. Mater., 2015, 14, 1245–1251 CrossRef PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- I. H. Kwak, J. Y. Kim, G. M. Zewdie, J. Yang, K. Lee, S. J. Yoo, I. S. Kwon, J. Park and H. S. Kang, Adv. Mater., 2024, 36, 2310769 CrossRef CAS PubMed.
- L. C. Miao, W. Q. Jia, X. J. Cao and L. F. Jiao, Chem. Soc. Rev., 2024, 53, 2771–2807 RSC.
- G. Wu, X. Han, J. Y. Cai, P. Q. Cui, X. S. Zheng, H. Li, C. Chen, G. M. Wang and X. Hong, Nat. Commun., 2022, 13, 4200 CrossRef CAS PubMed.
- K. Sharma, A. Kumar, T. Ahamad, Q. V. Le, P. Raizada, A. Singh, L. H. Nguyen, S. Thakur, V. Nguyen and P. Singh, J. Mater. Sci. Technol., 2023, 152, 50–64 CrossRef CAS.
- S. M. Sha, R. Ge, Y. Li, J. M. Cairney, R. K. Zheng, S. Li, B. Liu, J. J. Zhang and W. X. Li, Front. Energy, 2024, 18, 265–290 CrossRef.
- F. Wang, B. Dong, J. Wang, N. Ke, C. Tan, A. Huang, Y. Wu, L. Hao, L. Yin, X. Xu, Y. Xian and S. Agathopoulos, J. Adv. Ceram., 2022, 11, 1208–1221 CrossRef CAS.
- W. X. Li, Y. Liu, A. Azam, Y. Liu, J. Yang, D. Wang, C. C. Sorrel, C. Zhao and S. Li, Adv. Mater., 2024, 36, 2404658 CrossRef CAS PubMed.
- S. Gopalakrishnan, H. S. Krishnan, S. K. Eswaran and N. Mani, ACS Appl. Nano Mater., 2024, 7, 22674–22683 CrossRef CAS.
- M. K. Sahoo, H. K. Lee, W. S. Yang, D. S. Kim and H. K. Cho, Int. J. Hydrogen Energy, 2024, 49, 25–36 CrossRef CAS.
- R. Li, P. Ren, P. Yang, Y. Li, H. Zhang, A. Liu, S. Wen, J. Zhang and M. An, J. Colloid Interface Sci., 2023, 631, 173–181 CrossRef CAS PubMed.
- J. Y. Loh, F. M. Yap and W. Ong, J. Mater. Sci. Technol., 2024, 179, 86–97 CrossRef CAS.
- L. Zhang, Z. Hu, J. Huang, Z. Chen, X. Li, Z. Feng, H. Yang, S. Huang and R. Luo, J. Adv. Ceram., 2022, 11, 1294–1306 CrossRef CAS.
- B. Liu, Y. Zhu, S. Sha, R. Ge, C. Cheng, J. Yin, Z. Huang, L. Dai, S. Li and W. Li, Adv. Funct. Mater., 2024, 34, 2408613 CrossRef CAS.
- X. Li, X. Sun, H. Yu, H. Li, X. Sun, X. Tao and Y. Zheng, Appl. Catal., B, 2022, 307, 121156 CrossRef CAS.
- Y. Xu, J. Cheng, L. Ding, H. Lv, K. Zhang, A. Hu, X. Yang, W. Sun and Y. Mao, Int. J. Hydrogen Energy, 2024, 49, 897–906 CrossRef CAS.
- H. Xu, Y. Liao, Z. Gao, Y. Qing, Y. Wu and L. Xia, J. Mater. Chem. A, 2021, 9, 3418–3426 RSC.
- R. Zhang, L. Cheng, Z. Wang, F. Kong, Y. Tsegazab, W. Lv and W. Wang, Appl. Surf. Sci., 2020, 526, 146753 CrossRef CAS.
- M. Zhu, L. Yu, S. Sha, R. Ge, C. Cheng, L. Dai, S. Li, B. Liu, Z. Qu and W. Li, Sustainable Mater. Technol., 2024, 41, e01090 CrossRef CAS.
- X. Wang, X. Yu, S. Wu, P. He, F. Qin, Y. Yao, J. Bai, G. Yuan and L. Ren, ACS Appl. Mater. Interfaces, 2023, 15, 15533–15544 CrossRef CAS PubMed.
- J. Cao, R. Zhao, L. Bai, Y. Wang, Z. Zhang, L. Wu, X. Du and J. Li, Appl. Surf. Sci., 2023, 627, 157287 CrossRef CAS.
- X. Gu, S. Zheng, X. Huang, H. Yuan, J. Li, M. Kundu and X. Wang, Chem. Commun., 2020, 56, 2471–2474 RSC.
- N. Zhang, Y. Li, R. Zhang, S. Huang, F. Wang, M. Tang and J. Liu, J. Colloid Interface Sci., 2023, 642, 479–487 CrossRef CAS PubMed.
- Y. Xu, R. Ge, J. Yang, J. Li, S. Li, Y. Li, J. Zhang, J. Feng, B. Liu and W. Li, J. Energy Chem., 2022, 74, 45–71 CrossRef CAS.
- X. Teng, Z. Wang, Y. Wu, Y. Zhang, B. Yuan, Y. Xu, R. Wang and A. Shan, Nano Energy, 2024, 122, 109299 CrossRef CAS.
- Y. Ren, C. Wang, W. Duan, L. Zhou, X. Pang, D. Wang, Y. Zhen, C. Yang and Z. Gao, J. Colloid Interface Sci., 2022, 628, 446–455 CrossRef CAS PubMed.
- X. Tong, Y. Li, Q. Ruan, N. Pang, Y. Zhou, D. Wu, D. Xiong, S. Xu, L. Wang and P. K. Chu, Adv. Sci., 2022, 9, 2104774 CrossRef CAS PubMed.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., 2016, 128, 6814–6819 CrossRef.
- Y. Yang, K. Zhang, H. Lin, X. Li, H. C. Chan, L. Yang and Q. Gao, ACS Catal., 2017, 7, 2357–2366 CrossRef CAS.
- H. Liu, D. Ouyang, Q. Zhou and C. Feng, J. Alloys Compd., 2022, 920, 165243 CrossRef CAS.
- Y. Yao, J. He, X. Zhu, L. Mu, J. Li, K. Li and M. Qu, Int. J. Hydrogen Energy, 2024, 51, 207–221 CrossRef CAS.
- M. Bhosale, N. Baby, S. S. Magdum, N. Murugan, Y. A. Kim, S. Thangarasu and T. Oh, J. Energy Storage, 2024, 80, 110301 CrossRef.
- M. He, S. Hu, C. Feng, H. Wu, H. Liu and H. Mei, Int. J. Hydrogen Energy, 2020, 45, 23–35 CrossRef CAS.
- H. Liu, J. Cheng, W. He, Y. Li, J. Mao, X. Zheng, C. Chen, C. Cui and Q. Hao, Appl. Catal., B, 2022, 304, 120935 CrossRef CAS.
- Y. Xie, Z. Li, Y. Liu, Y. Ye, X. Zou and S. Lin, Appl. Surf. Sci., 2020, 508, 145161 CrossRef CAS.
- S. Kumar, P. K. Sahoo and A. K. Satpati, Electrochim. Acta, 2020, 333, 135467 CrossRef CAS.
- S. Ye, F. Luo, Q. Zhang, P. Zhang, T. Xu, Q. Wang, D. He, L. Guo, Y. Zhang, C. He, X. Ouyang, M. Gu, J. Liu and X. Sun, Energy Environ. Sci., 2019, 12, 1000–1007 RSC.
- C. H. An, W. Kang, Q. B. Deng and N. Hu, Rare Met., 2022, 41, 378–384 CrossRef CAS.
- A. Fan, P. Zheng, C. Qin, X. Zhang, X. Dai, D. Ren, X. Fang, C. Luan and J. Yang, Electrochim. Acta, 2020, 358, 136927 CrossRef CAS.
- A. Sihag, Z. L. Xie, H. V. Thang, C. Kuo, F. Tseng, M. S. Dyer and H. T. Chen, J. Phys. Chem. C, 2019, 123, 25618–25627 CrossRef CAS.
- W. Li, X. Xing, R. Ge, Y. Zhang, S. Sha, Y. Li, J. Cairney, R. Zheng, S. Li and B. Liu, Sustainable Mater. Technol., 2023, 36, e00743 CrossRef.
- Y. Xu, R. Ge, J. Yang, J. Li, S. Li, Y. Li, J. Zhang, J. Feng, B. Liu and W. Li, J. Energy Chem., 2022, 74, 45–71 CrossRef CAS.
- H. Liu, Y. Zhang, J. Li, R. Ge, J. M. Cairney, R. Zheng, S. Li, B. Liu, L. Dai, T. Liao and W. Li, J. Mater. Chem. A, 2024, 12, 5100–5114 RSC.
- J. Chen, Q. Zhou, L. Jia, X. Cui and W. Zeng, Appl. Surf. Sci., 2022, 597, 153693 CrossRef CAS.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351–9355 CrossRef CAS PubMed.
- Z. Dai, H. Geng, J. Wang, Y. Luo, B. Li, Y. Zong, J. Yang, Y. Guo, Y. Zheng, X. Wang and Q. Yan, ACS Nano, 2017, 11, 11031–11040 CrossRef CAS PubMed.
- S. Wang, X. Ning, Y. Cao, R. Chen, Z. Lu, J. Hu, J. Xie and A. Hao, Inorg. Chem., 2023, 62, 6428–6438 CrossRef CAS PubMed.
- W. Li, Z. Sun, R. Ge, J. Li, Y. Li, J. M. Cairney, R. Zheng, Y. Li, S. Li, Q. Li and B. Liu, Small Struct., 2023, 4, 2300175 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03811h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |