
A-site composition tuning in methylammonium-based metal halide perovskite colloidal nanocrystals†
Shekhar
Mondal
 ab,
Gauri
Sharma
bc,
Sunanda
Bhoi
a,
Sadashiv
Wadepalli
ab,
Pralay K.
Santra
bc,
Dipankar
Saha
d and
Abhijit
Hazarika
*ab
ab,
Gauri
Sharma
bc,
Sunanda
Bhoi
a,
Sadashiv
Wadepalli
ab,
Pralay K.
Santra
bc,
Dipankar
Saha
d and
Abhijit
Hazarika
*ab
aPolymers and Functional Materials Division, CSIR-Indian Institute of Chemical Technology, Uppal Road, Tarnaka, Hyderabad, 500007, India. E-mail: abhijit@iict.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
cCentre for Nano and Soft Matter Sciences (CeNS), Bengaluru, 562162, India
dDepartment of Chemistry, University of Oslo, Blindern 0315, Oslo, Norway
First published on 17th December 2024
Abstract
Utilizing the soft-lattice nature of metal halide perovskites, we employ post-synthetic cross-ion exchange to synthesize a series of narrow band-gap colloidal nanocrystals of methylammonium-based lead iodide solid solutions of composition FAxMA1−xPbI3, as well as those of triple-cation composition CsxFAyMA1−x−yPbI3 (TCPbI3). The ability to finely tune the compositions not only helps in tailoring the optical properties in the near-infrared region, but also improves the stability of these colloidal nanocrystals towards moisture, which has been demonstrated as compared to their bulk counterparts. The thermal stability of these solid solutions is also comparable to that of the bulk, as evidenced by thermogravimetric studies. This study helps in expanding the composition space of stable 3D lead halide perovskites with band gaps suitable and relevant for photovoltaic applications.
 Abhijit Hazarika | Dr Abhijit Hazarika received his PhD in chemical science from the Indian Institute of Science, Bangalore in 2015. He then moved to the University of Chicago to carry out research as a post-doctoral scholar (2015–2017). He also worked as a post-doctoral researcher at NREL, Colorado, during 2017–2019. He joined the CSIR-Indian Institute of Chemical Technology, Hyderabad as a scientist in September 2019. His research interests are in the area of colloidal inorganic semiconducting nanomaterials, their synthesis, surface chemistry and photophysics. His current research focuses are on metal halide perovskite solar cells and colloidal quantum materials for optoelectronic applications. |
Introduction
No other semiconductor offers the ease and range of composition tuning found in metal halide perovskites (MHPs). Owing to the soft and ionic nature of the lattice, all the elemental sites in the unit cell of the three-dimensional structure ABX3 (X = halide) can be easily varied even under ambient conditions through simple chemistry, which in turn helps in tuning all the important optoelectronic properties such as optical band gap, photoluminescence, carrier life-time, etc.1–3 However, the varying sizes of different elements puts a geometric constraint on ABX3 MHPs to retain their three-dimensional structure with a network of corner-shared BX6 octahedra. This geometric criterion, known as the Goldschmidt tolerance factor (GTF), provides a fair prediction of crystallographic arrangements in ABX3 compounds. Typically, a GTF value of 0.8–1.0 is considered favourable to form a 3D perovskite structure under ambient conditions with either ideal cubic geometry or with distorted corner-shared octahedra. Beyond this limit, ABX3 compounds crystallize in either the hexagonal phase or in the orthorhombic phase with face-shared or edge-shared octahedra, respectively. Although these MHP semiconductors came into the limelight after the realization of their potential in solar photovoltaics in 2009,4 such materials were investigated as early as 1958.5 This particular study introduced cesium plumbohalide (CsPbX3) semiconductor systems and reported their crystal structure and photoconductivity. Here, all the constituents in ABX3 structures are inorganic elements, and thus they can be termed all-inorganic lead halide perovskites. In 1978, it was first demonstrated that organic compounds with suitable size can be incorporated into the A-site in ABX3, forming a 3D perovskite structure.6 This study reported a series of methylammonium lead halides, CH3NH3PbX3 or MAPbI3 (MAPI), where methylammonium ions occupied the cages in the 3D network of corner-shared PbX6 octahedra, showing the possibility of hybrid metal halide perovskites. The pioneering work by Miyasaka's group in 2009 demonstrated the feasibility of CH3NH3PbI3 as the effective photosensitizer in a solar cell achieving a photoconversion efficiency of ∼3.8%.4 Since then, MHP material systems have been explored extensively, showing their potential not only in photovoltaics,7,8 but also in almost all areas of optoelectronics such as LEDs,9 lasing,10 spintronics,11 photodetectors,12etc. In photovoltaics, perovskite solar cells (PSCs) have already achieved a PCE of 26.1% for lab-scale devices within an unprecedented short period of time.13In their colloidal nanocrystalline forms, MHPs have gained interest since their synthesis via hot injection was reported in 2015 by Kovalenko's group.14 Since then, perovskite nanocrystals (PNCs) or perovskite quantum dots (PQDs) have taken the center stage of colloidal quantum dot (CQD) research, which was earlier solely dominated by metal chalcogenide and metal pnictide-based semiconductors. Like bulk MHPs, colloidal nanocrystalline MHPs have shown tremendous promise in various optoelectronic applications.15–19 PNCs offer certain advantages as compared to the corresponding thin film/bulk MHPs in terms of the range of composition tunability and solvent compatibility. Composition tuning in MHPs, in general, has been one of the major focuses, particularly in solar-cell research involving lead halide perovskites, to shift the band gap towards lower energy than that of MAPI. The introduction of a larger organic cation, formamidinium (FA, CH(NH2)2+), at the A-site of APbX3 has resulted in lowering the band gap, but its larger ionic radius makes it difficult to stabilize the band gap in the required 3D perovskite structure under ambient conditions. On the other hand, all-inorganic perovskites, such as CsPbI3, are ideally suited to achieve better thermal stability with a narrow band gap. However, the small ionic radius of Cs+ does not result in the favourable geometric factors required for crystallization in the 3D perovskite network. PNCs offer unique advantages here: surface-related strains in these small particles cause tilting of the PbX6 octahedra, thus compensating for the geometric constraints to remaining in 3D geometry.20,21 Furthermore, many other MHP semiconductors with tunable A-, B- and X-site compositions can easily be synthesized by simple ion-exchange reactions in their colloidal nanocrystalline forms, which remain stable in the 3D crystal phase at room temperature and under ambient pressure.1–3
Considering the effective radii of methylammonium, lead and iodide ions, the composition of MAPI has the near-ideal GTF (∼0.9) to be in the 3D cubic perovskite phase, and probably this is the reason why organic–inorganic hybrid perovskite research was started with this particular composition. Despite reports of numerous research works on bulk and thin films,4,7,20,22 not much research has been carried out on different aspects of MAPI in colloidal nanocrystalline form. One of the key challenges is the traditional hot-injection-based high-temperature synthesis of colloidally stable MAPI nanocrystals due to the lack of availability of suitably stable methylammonium salts. There have been reports demonstrating the synthesis of MAPI PNCs at room temperature using MA-THF in oleic acid (OA),23,24 an MA–benzoyl iodide mixture,25 and MAI.26
However, these syntheses resulted in the formation of MAPI PNCs with morphologies other than 3D cuboidal nanocrystals during the course of the reaction. At a slightly elevated temperature of 70 °C, MAPI PNCs have been synthesized using methylamine as the major precursor.23,25,27 However, due to the high volatility of methylamine, it is challenging to control the reaction. Due to the lack of enough literature reports on MAPI, A-site composition tuning with the methylammonium ion as one of the components has also not gained much attention, thus limiting the compositional parameter space in lead halide perovskite nanocrystals.
Herein, we report an in-depth study on the optical and structural properties of colloidal MAPI nanocrystals and their A-site solid solutions. The MAPI nanocrystals have been synthesized via the hot injection method under inert conditions using methylamine acetate. A series of compounds of chemical formula FAxMA1−xPbI3 (0 < x < 1) have been synthesized via post-synthetic A-site cross-ion exchange between the MAPI and FAPbI3 (FAPI) PNCs. In the case of thin-film lead halide perovskites, it has been shown that by incorporating three cations (Cs+, FA+ and MA+) at the A-site, it is possible to overcome the thermal and compositional stability issues. This so-called triple-cation perovskite, when used as an absorber, resulted in high-performance solar cells with enhanced stability.28,29 We utilized the colloidal MAPI PNCs to demonstrate that in their colloidal nanocrystalline form, such triple-cation compositions CsxFAyMA1−x−yPbI3 (TCPbI3) can be achieved via ion exchange. All the nanocrystals are stable in their 3D perovskite phase at room temperature within an ambient environment. The nanocrystal solid solutions are found to be homogeneous without the formation of any secondary phase. The MAPI nanocrystal films demonstrate better moisture stability than those of the bulk MAPI films. We have also tried to elucidate the crystal structure of the colloidal MAPI nanocrystals from powder XRD patterns, which have been found to be closely matched with the orthorhombic (Fmmm) space group.
Results and discussion
We employed methylamine acetate (MAOAc) salt as an MA precursor for the synthesis of colloidal MAPI PNCs. Unlike methylamine, which is a liquid with a boiling point of 40 °C, MAOAc is a solid at room temperature with a melting point of ∼75 °C. However, due to its hygroscopic nature, it needs to be handled under an inert atmosphere. MAOAc has been reacted with oleic acid (OA) to form MA-oleate, which has a relatively high boiling point suitable for the hot injection synthesis. Using this MA-oleate, we followed two synthetic routes to the colloidal MAPI PNCs: in route I, MA-oleate was injected into a solution containing PbI2, oleic acid (OA) and oleylamine (OlAm) in 1-octadecene (ODE) at a designated temperature, while route II involves three precursors where a solution of oleylammonium iodide (OlAmI) salt in toluene was injected into a mixture of Pb-oleate and MA-oleate precursors. In both these synthetic schemes, the reactions were quenched immediately after injection to control the growth of MAPI PNCs. The details of the synthesis procedure can be found in the Experimental section. The resulting PNCs exhibit good colloidal dispersion in nonpolar solvents as discussed in the following paragraphs.The UV-Vis absorption and steady-state photoluminescence (PL) emission spectra of the MAPI PNCs, as shown in Fig. 1a, indicate a signature of quantum confinement with an absorption edge of ∼750 nm and a PL emission maximum of ∼725 nm, which are slightly blue-shifted (∼0.14 eV) as compared to that of thin-film MAPbI3 (see Fig. S1 in the ESI† for comparison). Fig. S2† shows the powder XRD patterns of the MAPI PNCs, revealing that they crystallize in the 3D near-cubic perovskite structure where the MA cation occupies the body center of the cube with eight PbI6 octahedra occupying the eight corners of the cube. Previous studies have discussed and debated the space group of bulk MAPI under ambient temperature and pressure conditions. While most of the literature confirms the tetragonal structure with I4cm,30I4/mcm,31 and I4/m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 space groups, a detailed crystallographic study by Karunadasa's group recently reported that MAPI at ambient pressure crystallizes in the orthorhombic Fmmm space group.33 In the colloidal nanocrystalline form, the MAPI PNCs have been indexed with a tetragonal geometry.23 As shown in Fig. S2a in the ESI,† the XRD pattern matches closely with both orthorhombic (Fmmm) and tetragonal (I4cm) symmetries. Moreover, closer observation at 2θ = 24.3° (see Fig. S2b and S2c†) clearly reveals splitting of the cubic (1 1 1) reflection into (2 1 1) and (2 0 2) reflections despite small particle size-related peak broadening, indicating that the synthesized MAPI PNCs deviate from the highest symmetry cubic geometry. A tetragonal model (I4cm) was tested but this resulted in a poor fit (see Fig. S3 in the ESI†). While it accounts for the splitting, it fails to reproduce the intensity accurately. In contrast, an orthorhombic model (Fmmm) provided a reasonable fit, as shown in Fig. 1b. Based on these observations, we propose that the structure of the MAPI PNCs is likely to adopt an orthorhombic symmetry. We note here that it is not straightforward to determine the crystallographic structure accurately for nanomaterials with small-range atomic ordering using traditional crystallographic methods due to inherent problems such as defects, size distribution and disorder.34–36 Careful determination of the atomic structure for the PNCs requires total scattering approaches using synchrotron-based sources and neutron scattering, as well as local-to-average structure modelling based on a variety of other complementary techniques, such as atomically resolved high-resolution transmission emission microscopy (HRTEM) and extended X-ray absorption fine structure (EXAFS). Such approaches have been adopted for different nanomaterials for structure determination in different reports.37,38
32 space groups, a detailed crystallographic study by Karunadasa's group recently reported that MAPI at ambient pressure crystallizes in the orthorhombic Fmmm space group.33 In the colloidal nanocrystalline form, the MAPI PNCs have been indexed with a tetragonal geometry.23 As shown in Fig. S2a in the ESI,† the XRD pattern matches closely with both orthorhombic (Fmmm) and tetragonal (I4cm) symmetries. Moreover, closer observation at 2θ = 24.3° (see Fig. S2b and S2c†) clearly reveals splitting of the cubic (1 1 1) reflection into (2 1 1) and (2 0 2) reflections despite small particle size-related peak broadening, indicating that the synthesized MAPI PNCs deviate from the highest symmetry cubic geometry. A tetragonal model (I4cm) was tested but this resulted in a poor fit (see Fig. S3 in the ESI†). While it accounts for the splitting, it fails to reproduce the intensity accurately. In contrast, an orthorhombic model (Fmmm) provided a reasonable fit, as shown in Fig. 1b. Based on these observations, we propose that the structure of the MAPI PNCs is likely to adopt an orthorhombic symmetry. We note here that it is not straightforward to determine the crystallographic structure accurately for nanomaterials with small-range atomic ordering using traditional crystallographic methods due to inherent problems such as defects, size distribution and disorder.34–36 Careful determination of the atomic structure for the PNCs requires total scattering approaches using synchrotron-based sources and neutron scattering, as well as local-to-average structure modelling based on a variety of other complementary techniques, such as atomically resolved high-resolution transmission emission microscopy (HRTEM) and extended X-ray absorption fine structure (EXAFS). Such approaches have been adopted for different nanomaterials for structure determination in different reports.37,38
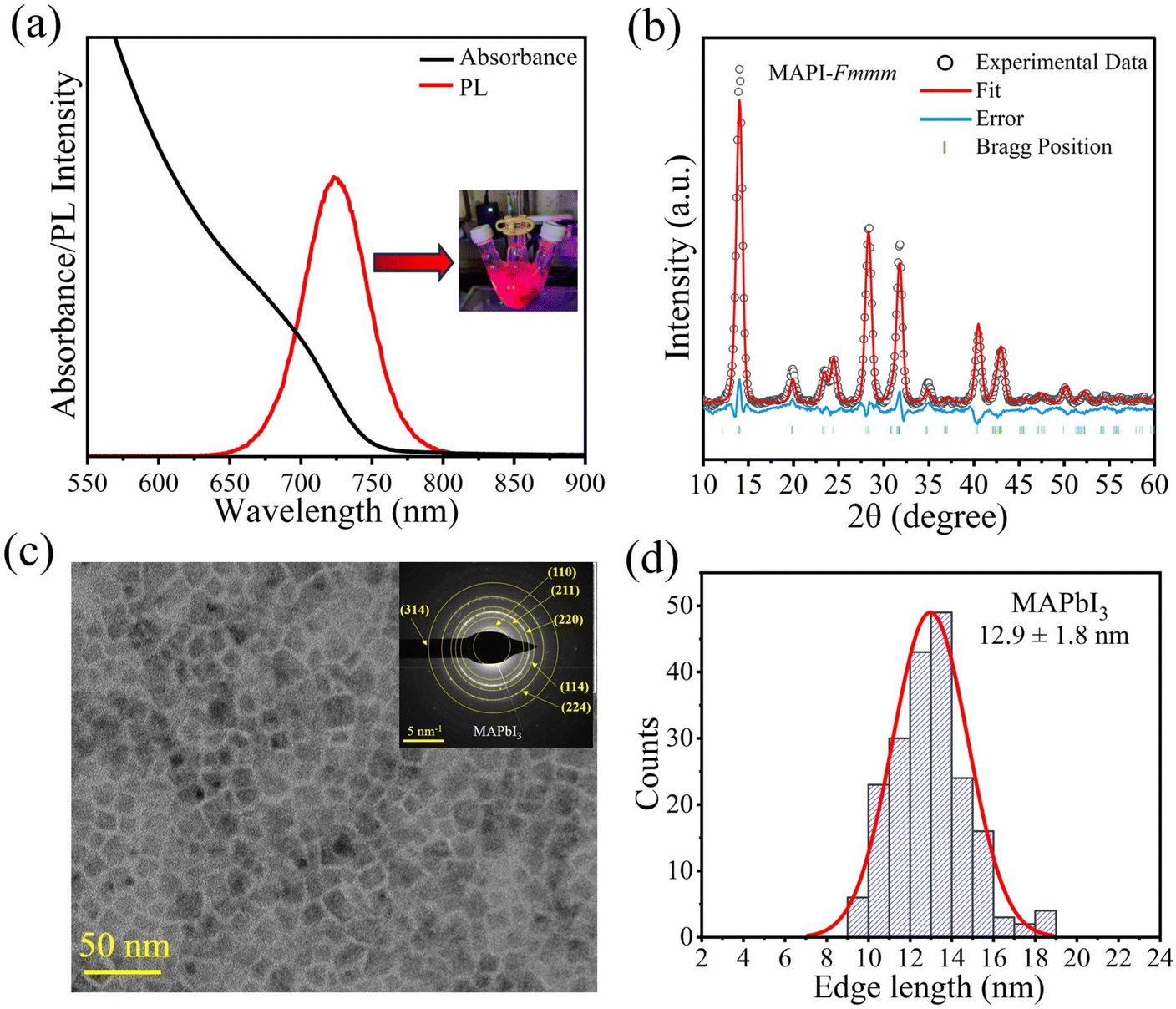 | ||
| Fig. 1 (a) UV-Vis absorption and PL emission spectra of the MAPI PNCs; the inset shows the MAPbI3 reaction mixture under UV light illumination. (b) Rietveld refinement of the powder XRD pattern of the MAPI PNCs with reported crystallographic information for Fmmm symmetry. (c) TEM image of the MAPI PNCs; the inset shows the SAED patterns of the MAPI PNCs; the planes were assigned according to the d-spacing value calculated from the powder XRD pattern. (d) Size distribution histogram calculated from the TEM image, indicating that the MAPI PNCs have an average edge length of 12.9 ± 1.8 nm. | ||
The transmission electron microscopy (TEM) image shows the cuboidal shape of the MAPI PNCs (Fig. 1c) with an average edge length of 12.9 ± 1.8 nm, while the SAED pattern reveals the crystalline nature of the MAPI NCs (Fig. 1d and the inset of Fig. 1c, respectively). The MAPI PNCs synthesized via route II exhibit similar optical and structural properties (see Fig. S4 in the ESI†).
One of the major issues with lead halide perovskite semiconductors, in general, has been their poor stability towards air and moisture. Several mechanisms have been proposed to explain the degradation pathways of hybrid perovskites. Unlike in all-inorganic perovskites, such as CsPbI3, where degradation takes place via crystallographic phase change to the optically inactive δ-phase,20,39 hybrid perovskites undergo compositional change during interaction with air and moisture. For instance, methylammonium lead iodide, when it comes into contact with water, decomposes into lead iodide and methylamine/hydrogen iodide/ammonia.40–47 We checked the stability of the MAPI PNC solid films under 50–70% relative humidity (RH), normal light and ambient conditions and compared their performance to that of MAPI polycrystalline (PC) thin films. As can be seen from Fig. 2c, the MAPI thin films start degrading from day 1 itself (appearance of yellow coloration). In contrast, the MAPI PNC films appear to become lighter with time without yellow coloration. This degradation can be better visualized by the extent of band gap bleaching in the corresponding absorption spectra, as shown in Fig. 2a and b; the relative absorbance at the exciton energy decreases faster in the MAPI thin film compared with that of the PNC film (Fig. 2d). The resistance of the MAPI PNC films towards moisture degradation can be attributed to the presence of hydrophobic surface ligands.
 | ||
| Fig. 2 UV-vis absorption spectra of (a) the MAPI PNC film and (b) the MAPI thin film recorded from day 1 to day 5 after preparation in 50–70% relative humidity (RH) under normal light and ambient conditions. (c) Photographs of both the MAPI PNC and thin films showing how the colour of the film changes with time. (d) Comparison of the change of relative absorption of both films with time; the PNC film shows better phase stability compared with the thin film. | ||
Post-synthetic ion exchange in colloidal PNCs has been shown to be an effective way to achieve compositional tunability that is beyond the reach of that in thin-film or bulk perovskites.1–3,48 Such control, in turn, has helped in tuning optoelectronic and photovoltaic properties.48–50 While X-site ion exchange is known to occur instantaneously at room temperature, for A-site ion-exchange reactions in PNCs, elevated temperature/longer time/excess amounts of ligands are required.48,50
Following a similar A-site cross-exchange reaction between CsPbI3 and FAPbI3,48,50,51 we synthesized a series of PNCs with composition FAxMA1−xPbI3 by mixing the MAPI PNCs with the colloidal FAPI PNCs at different ratios. As shown in Fig. 3a and b, controlled variation of “x” resulted in tuning optical absorption and photoluminescence (PL) in the range of ∼700 nm–800 nm, with larger values of “x” resulting in a red-shift in absorption and PL emission. We plotted the bandgap energy as well as the PL emission maximum energy of these solid solutions as a function of composition “x”, and both of these properties exhibited a linear relationship (Fig. S5†). Such a linear relationship of band gaps with composition “x” measured from diffuse reflectance has been reported in the case of bulk FAxMA1−xPbI3 solid solutions as well.52 Apart from the steady-state PL emission, the time-resolved PL (TRPL) decay kinetics also exhibits monotonic variation with the introduction of more MA ions, resulting in faster decay, as can be seen from Fig. S6 in the ESI.† The absence of bimodal absorption in the UV-Vis absorption spectra and the narrow symmetric PL emission peaks indicate the formation of nearly homogeneous solid solutions all throughout the FAxMA1−xPbI3 composition range. Photoluminescence excitation (PLE) spectroscopy is a commonly used technique to study inhomogeneous broadening of PL emission in colloidal quantum dots, which primarily occurs due to size distribution in these quantum-confined particles, which means that this technique indirectly provides information on sample inhomogeneity.53–55 In the weakly quantum-confined hybrid lead halide perovskite NC solid solutions, in the size range of 10 nm–15 nm, slight compositional variation is expected to result in inhomogeneous broadening of PL emission. We have collected PLE spectra of a few of the FAxMA1−xPbI3 compositions (Fig. 3c), and it has been observed that the PLE spectra at different emission energies overlap with each other in all the compositions, indicating negligible compositional inhomogeneity.
 | ||
| Fig. 3 (a) UV-Vis absorption and (b) steady-state PL emission spectra of the FAxMA1−xPbI3 compositions showing tunability over the range of 720 nm–770 nm; the inset in figure b shows the digital photograph of the FAxMA1−xPbI3 alloys under UV illumination. (c) Photoluminescence excitation (PLE) spectra of the FAxMA1−xPbI3 NCs at different emission energies as indicated by the colour-coded arrow in the emission spectra, showing the absence of substantial inhomogeneous broadening of the PL emission due to alloying; the dashed line represents the PL spectra of the corresponding alloy. (d) Powder XRD patterns of the as-synthesized alloy compositions of formula FAxMA1−xPbI3 NCs along with their parent NCs; all of these diffraction patterns indicate the formation of the 3D perovskite phase. | ||
Powder XRD patterns (Fig. 3d) of the FAxMA1−xPbI3 PNCs indicate that all compositions retain the 3D perovskite phase even after the ‘A’-site cross-ion exchange reaction between MAPI and FAPI PNCs. Detailed crystallographic analyses of these solid solutions were carried out by performing Rietveld refinement. The results from the refinement are shown in Fig. S7 and Table S4.† While the FAPI PNCs crystallize in the cubic Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, as also reported in the previous literature,56,57 it has been found that introduction of even a small amount of FAPI results in transformation of the orthorhombic phase of pure MAPI into the cubic phase, and this cubic phase is retained in all the FA-containing compositions (x = 0.25, 0.5 and 0.75). Similar phase transformation has also been observed in the case of bulk FAxMA1−xPbI3, although from the tetragonal phase to the cubic phase.52,58,59 However, in the case of the bulk, a higher FA content results in degradation of FAxMA1−xPbI3 from the cubic phase to the hexagonal phase with time.52 In the case of our FAxMA1−xPbI3 PNCs, the stability of the cubic phase remains stable for months. From the calculated crystallographic information as shown in Table S1,† we observed that with an increase in FA content, the lattice parameters and unit cell volumes increase linearly (Fig. S8 in the ESI†). Such variations can also be found in the case of bulk FAxMA1−xPbI3 solid solutions.52,58 Linear variation of the optical bandgap, PL emission energy maxima and lattice parameters as functions of “x”, all point towards the formation of homogeneous solids of FAxMA1−xPbI3 PNCs in the range of 0 ≤ x ≤ 1. The TEM images show the formation of cuboidal-shaped particles for all the FAxMA1−xPbI3 PNCs with an average edge length in the range of 13.2 ± 1.7–14.18 ± 1.9 nm, comparable to that of their parent MAPI (average edge length of 12.9 ± 1.8 nm) and FAPI (average edge length of 14.6 ± 2.3 nm, Fig. S9†) PNCs (see Fig. S10a–f† for complete TEM analysis of the FAxMA1−xPbI3 PNC alloys). Combining optical spectroscopic and structural data discussed above confirms the formation of homogeneous solid solutions of FAxMA1−xPbI3 PNCs in cuboidal 3D perovskite phases across all compositions.
m space group, as also reported in the previous literature,56,57 it has been found that introduction of even a small amount of FAPI results in transformation of the orthorhombic phase of pure MAPI into the cubic phase, and this cubic phase is retained in all the FA-containing compositions (x = 0.25, 0.5 and 0.75). Similar phase transformation has also been observed in the case of bulk FAxMA1−xPbI3, although from the tetragonal phase to the cubic phase.52,58,59 However, in the case of the bulk, a higher FA content results in degradation of FAxMA1−xPbI3 from the cubic phase to the hexagonal phase with time.52 In the case of our FAxMA1−xPbI3 PNCs, the stability of the cubic phase remains stable for months. From the calculated crystallographic information as shown in Table S1,† we observed that with an increase in FA content, the lattice parameters and unit cell volumes increase linearly (Fig. S8 in the ESI†). Such variations can also be found in the case of bulk FAxMA1−xPbI3 solid solutions.52,58 Linear variation of the optical bandgap, PL emission energy maxima and lattice parameters as functions of “x”, all point towards the formation of homogeneous solids of FAxMA1−xPbI3 PNCs in the range of 0 ≤ x ≤ 1. The TEM images show the formation of cuboidal-shaped particles for all the FAxMA1−xPbI3 PNCs with an average edge length in the range of 13.2 ± 1.7–14.18 ± 1.9 nm, comparable to that of their parent MAPI (average edge length of 12.9 ± 1.8 nm) and FAPI (average edge length of 14.6 ± 2.3 nm, Fig. S9†) PNCs (see Fig. S10a–f† for complete TEM analysis of the FAxMA1−xPbI3 PNC alloys). Combining optical spectroscopic and structural data discussed above confirms the formation of homogeneous solid solutions of FAxMA1−xPbI3 PNCs in cuboidal 3D perovskite phases across all compositions.
We further extended the ‘A’-site composition tuning to synthesize phase-stable hybrid triple-cation perovskite NCs by introducing the ‘Cs’ cation. For hybrid lead halide perovskite thin films, it has been reported that incorporation of both Br− and I− at the X-site is essential to simultaneously accommodate Cs, MA, and FA at the A-site, resulting in the formation of 3D triple-cation (TC) perovskites.29,60–62 Although triple-cation perovskites provide structural stability and desirable band gap in a thin film, it is well known that the presence of both Br− and I− leads to halide phase segregation under constant light illumination.63–66 In the colloidal nanocrystalline form, only a few reports are available for triple-cation PNCs. For instance, the ligand-assisted reprecipitation (LARP) approach to synthesize spherical-shaped CsFAMAPbBr3 triple-cation PNCs has been reported.67 Very recently, Manna's group demonstrated the formation of colloidal CsFAMAPbBr3 triple-cation perovskite NCs via post-synthetic ‘A’-site ion exchange reactions.51 Following the synthesis of FAxMA1−xPbI3, as discussed above, we applied A-site cation cross-exchange to form I-only triple-cation PNCs of chemical composition CsxFAyMA1−x−yPbI3, abbreviated as TCPbI3, which may act as a better alternative to CsxFAyMA1−x−yPb(BrzI1−z)3-based triple-cation perovskite thin films. 3D perovskite phase-stable triple-cation TCPbI3 PNCs can be obtained in a number of different post-synthetic A-site ion-exchange reaction pathways: for instance, by mixing CsPbI3, FAPbI3 and MAPbI3 PNCs, or by mixing the CsPbI3 PNCs with the FAxMA1−xPbI3 PNCs described previously. To demonstrate the feasibility of this pathway, we mixed CsPbI3 (see Fig. S11† for the details of CsPbI3), FAPbI3 and MAPbI3 PNCs in a 1:1:1 concentration ratio and allowed them to undergo A-site ion exchange until a thermodynamic equilibrium was reached, resulting in a single symmetric PL peak centered at ∼728 nm and a sharp absorption onset at ∼750 nm, as can be seen in Fig. 4a. The powder X-ray diffraction pattern of TCPbI3, as can be seen from Fig. 4b, suggests the formation of a 3D perovskite phase with cubic/pseudo-cubic symmetry, which is also proved from the TEM images that show the resulting TCPbI3 retains its cubic morphology after the extended ‘A’-site ion-exchange reaction of its parent counterpart (see Fig. S12b†).
 | ||
| Fig. 4 (a) UV-Vis absorption and PL emission spectra of the TCPbI3 NCs; the inset shows the complete absorption spectrum over the 300 nm to 900 nm range. (b) Powder XRD pattern of the TCPbI3 NCs along with their parent NCs, showing the diffraction pattern of the perovskite phase. (c) UV-Vis absorption spectra of the TCPbI3 NC film recorded from day 1 to day 7 after preparation in 50–70% relative humidity (RH) under normal light and ambient conditions. (d) Comparison of the change of relative band gap bleaching of the TCPbI3 NCs and the MAPI-PNC film with time; TCPbI3 shows comparatively better stability than the MAPI PNC film. | ||
A closer look at the XRD data can qualitatively inform us about the effective A-site cation mixing in the TCPbI3 PNCs. For instance, the diffraction peak of TCPbI3, at around 2θ = 14.29°, shifts towards a lower angle as compared to that of pure CsPbI3 (at 2θ = 14.53°), but when compared to that of pure FAPI (2θ = 14.13°), this peak has slightly higher 2θ value (see Fig. S13 in the ESI†). This can be correlated with the effective A-site ionic radius in TCPbI3. Considering the Shannon ionic radii68,69 of Cs, FA, and MA, the effective A-site ionic radius of TCPbI3, assuming a 1:1:1 ratio, can be calculated and is found to be 2.13 Å, which is larger than that of Cs but smaller than that of FA ions. This indicates lattice expansion in TCPbI3 as compared to CsPbI3 due to the incorporation of larger-sized FA and MA ions and lattice compression as compared to the unit cell of FAPbI3. Since the estimated A-site ionic radius of TCPbI3 is almost similar to that of the MA+ ion, there is no significant shift in that particular diffraction peak (2θ = 14.29°) of TCPbI3 as compared to that in MAPI. Moreover, the absence of the signature doublet peaks corresponding to the (211) and (022) diffraction planes of pure MAPI at around 25° in the case of the TCPbI3 PNCs further indicates that the crystallographic parameters between them are different, although the effective ionic radii are similar in both these compositions. We have further synthesized four different compositions of TCPbI3 NCs by mixing CsPbI3, FAPbI3 and MAPbI3 PNCs in different volume ratios. The optical and structural properties of these synthesized compositions are shown in Fig. S14.† All these compositions are formed in the black perovskite phase with linear variation of their optical properties.
The optical absorption and emission energy of the TCPbI3 PNCs is quite similar to that of MAPI, and the diffraction patterns are also very much similar, questioning the necessity of triple-cation perovskite compositions. However, if we compare the stability of these compositions under ambient conditions, the TCPbI3 PNCs are comparatively better than the MAPI PNCs. Fig. 4c shows the optical absorption spectra of solid films of TCPbI3 PNCs recorded for one week at 50–70% RH. If we compare their relative band gap bleaching with that of MAPI PNC solid films, we can clearly see that the TCPbI3 PNC composition still demonstrates good band-gap absorption performance at the same energy even after 7 days (Fig. 4d), which can also be visualized from the digital images of the films under normal light (see Fig. S15†).
Furthermore, such triple-cation PNC compositions can also be synthesized by cation-exchange reactions between CsPbI3 and FAxMA1−xPbI3 PNCs under ambient conditions. For example, by reacting the three FAxMA1−xPbI3 compositions discussed in the previous section with CsPbI3 at similar concentrations, stable homogeneous solid solutions of chemical formula CsxFAyMA1−x−yPbI3 can be obtained with tunable optical properties as shown in Fig. S16a–f.† All of these TCPbI3 PNCs retain their colloidal stability and structural integrity for over a month.
Apart from moisture stability, thermal stability is another important aspect of perovskite nanocrystals in their practical applications to achieve high-performing, repeatable, and stable optoelectronic devices. Several studies in the past highlighted the thermal decomposition pathways of lead halide perovskite thin films and single crystals using thermogravimetric analysis (TGA).70–73 For example, in the case of MAPI thin films, two weight-loss events have been described: the first one, between 250 °C and 380 °C, corresponds to the decomposition of the organic component (MAI) while the second one, until 450 °C, corresponds to the decomposition of PbI2. In the case of FAPI thin films, on the other hand, the first weight loss, corresponding to the decomposition of FAI, occurs at the much higher temperature of 420 °C, which has been ascribed to the stronger interaction of FA+ with the inorganic [PbI6]4− octahedral matrices as compared to that of MA+. As discussed earlier, the decomposition of FA/MA-based perovskites occurs via compositional change.
Ma et al. have shown, using the TGA-FTIR technique, that at lower temperatures, MAPI decomposed via the release of CH3I and NH3 gases, and at high temperatures, the decomposition of MAPI leads to the formation of CH4. However, the decomposition of FAPI leads to the formation of s-triazine (HCN)3 and NH3 at low temperatures, and HCN and NH3 at high temperatures.73 A few groups have reported the decomposition of MAPI via the formation of HI and CH3NH2.70,71
To check the thermal stability and decomposition temperatures of the PNCs, we carried out the TGA study by heating samples up to 800 °C under nitrogen at a ramp rate of 10 °C min−1. Unlike the thin films, all the PNC compositions discussed above display three-step weight-loss events, as can be seen from the TGA and their derivative plots shown in Fig. 5a: (a) initial loss (∼2–6%) below 200 °C, which may be attributed to the decomposition of organic surface ligands such as OA and OlAm; (b) the second step of mass loss, occurring above 300 °C, may be attributed to the decomposition of organic A-site cations and the remaining organic surface ligands; and (c) the final step, above 500 °C, where the major mass loss occurs due to decomposition of PbI2. A similar three-step degradation process has also recently been reported by Li's group for CsxFA1−xPbI3 PNCs.74 This noted that, unlike MAI and FAI, CsI decomposes at very high temperatures, above 600 °C, hence in the second step, only ∼15% of the total weight-loss event occurs in the case of pure CsPbI3 PNCs as opposed to ∼30% in the PNCs with organic-only A-site cations. As also observed in hybrid lead halide perovskite thin films, in the case of pure FAPI PNCs, the second weight-loss event occurs at a slightly higher temperature (378 °C) than that in pure MAPI PNCs (360 °C) due to the stronger interaction of FA cations with the [PbI6]4− octahedral units compared to the MA cation.73 All the solid solutions of FAxMA1−xPbI3 and TCPbI3 PNCs followed similar three-step weight-loss events. However, when we compare the thermal degradation of the TCPbI3 PNC composition with the MA- and FA-based PNC compositions, we can see that the percentage mass loss below 400 °C is ∼28% for TCPbI3, which is lower when compared to that of the FA- and MA-based PNCs where we observed 32–35% mass loss, indicating better thermal stability of TCPbI3 due to the incorporation of Cs into the A-site. When compared to the degradation of the TCPbI3 PNCs with the corresponding single crystals of similar compositions, we observed that both exhibit comparable thermal stability, although the second mass-loss event occurs at a slightly lower temperature in the case of the PNCs. However, in the case of bulk TCPbI3, there is substantial mass loss below 100 °C, which is not observed in the case of the nanocrystals. Such low-temperature decomposition may occur via the release of CH3NH2 and HI, which is reported for MAPI thin films.75 It is interesting to note here that in their thin-film form, both MAPI and FAPI undergo temperature-dependent crystallographic phase transformations, as reported in multiple literature reports.52,58,76 For instance, Ambra Pisanu et al. reported that the MAPI thin film undergoes a tetragonal-to-cubic phase transition at ∼57 °C, while the as-prepared yellow FAPI undergoes a hexagonal-to-cubic phase transition at ∼165 °C; moreover, the FAxMA1−xPbI3 compositions do not undergo such crystallographic phase transitions. Unlike in thin films, FAPI PNCs crystallize as the black cubic perovskite phase at room temperature, and temperature-dependent in situ XRD and thermogravimetric studies in the recent literature74 prove that FAPI PNCs directly decompose into PbI2 without undergoing any intermediate crystal phase transformation. However, similar studies on methylammonium-based PNCs have not been reported so far. Our TGA results do not reveal any signature of temperature-dependent crystallographic phase transformations in any of the PNC compositions including FAPI and MAPI.
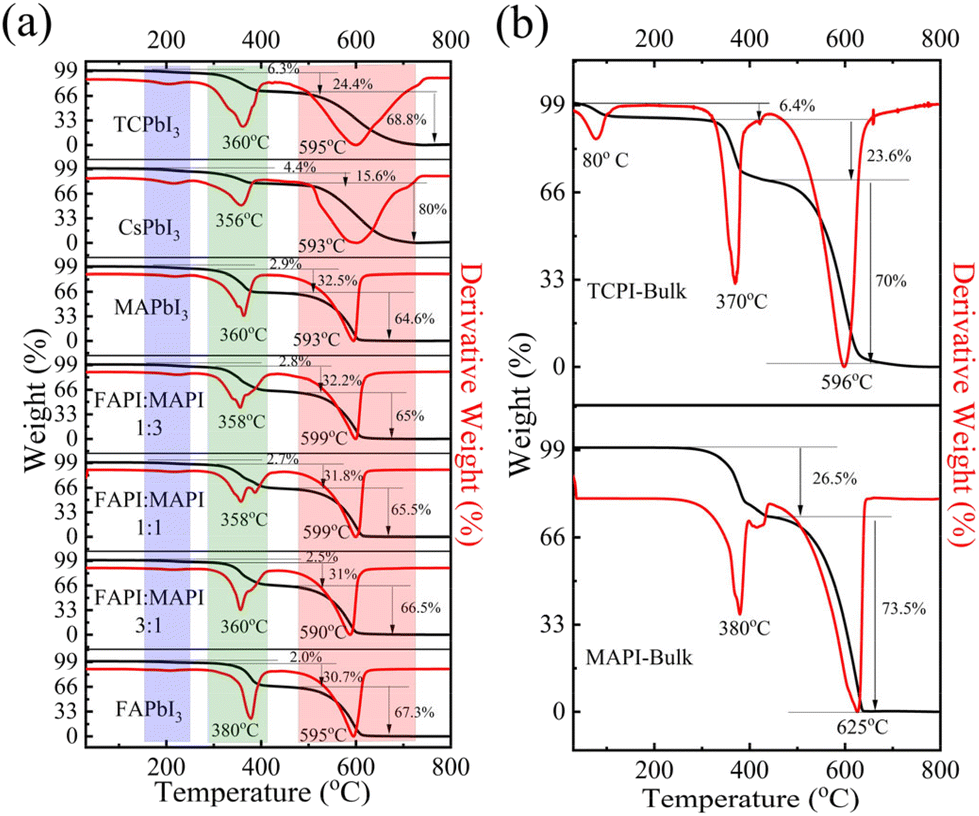 | ||
| Fig. 5 TG curves and their corresponding DTG plots of (a) TCPbI3, CsPbI3, FAPbI3, MAPbI3 and FAxMA1−xPbI3 PNCs, and (b) the MAPbI3 and TCPbI3 bulk thin-film perovskites. The percentage mass loss in the three different steps is shown by the arrow along with the corresponding value. | ||
The observed enhanced stability of the colloidal PNCs can be attributed to the hydrophobic surface ligands that resist the penetration of water molecules, which in thin-film lead halide perovskites initiates chemical reactions to disintegrate the chemical compositions forming individual components such as lead halides, methylamine, and/or ammonia. By incorporating multiple cations at the A-site, particularly the inorganic cation Cs, such moisture stability is further improved most likely due to reduced reactivity of the inorganic component towards moisture. Inclusion of these inorganic cations also helps in improving the thermal stability as compared to the simple MAPI composition where the organic A-site cation is known to be very volatile. Surface ligand binding strength with different A-site cations also plays a crucial role in the enhancement of moisture and thermal stability. However, further theoretical studies are required for a better understanding.
Conclusions
In conclusion, we have demonstrated the versatility of A-site composition tuning in colloidal lead halide perovskite nanocrystals by post-synthetic cross-ion exchange that involves methylammonium ions. Such ability has not only provided freedom to tune the optoelectronic properties, but also helped to achieve better moisture stability and thermal stability comparable to that of the bulk/thin-film lead halide perovskites. We have also demonstrated the synthesis of triple-cation iodide-only perovskite PNCs, expanding the compositional space of lead halide perovskite NCs. Such fine control over composition tuning may help to further strengthen perovskite research in general by developing efficient and stable perovskite optoelectronic devices. While in colloidal form, PNC inks are stable for months without losing phase stability, the long-term stability of their bare films under ambient conditions is still not good enough for practical purposes. However, it is to be noted that in real optoelectronic devices, the active layer deposited from the PNC inks is sandwiched between other charge transport layers and electrodes. There have been numerous reports on PNC-based encapsulated optoelectronic devices with operational stability up to hundreds of hours. So, the stability of the PNC films demonstrated in this study does not necessarily reflect their utilization in stable optoelectronic devices. We envisage that decorating the surface of the methylammonium-based PNCs with suitable ligands, such as zwitterionic ligands and amino acid ligands, as well as encapsulating these PNCs with suitable matrices, such as polymers and silica, can further enhance the stability of PNC inks leading to stable optoelectronic devices.Experimental section
Chemicals
Cesium carbonate (Cs2CO3; 99.9%), oleic acid (OA; technical grade, 90%), oleylamine (OlAm; technical grade, 70%), 1-octadecene (1-ODE; technical grade, 90%), formamidinium acetate (FA-acetate, 99%), methylamine acetate (MA-acetate; >97.0%), methyl acetate (MeOAc, anhydrous, 99.5%), PbI2 (99.99%, trace metal basis), Pb(acetate)2·3H2O, hexane (reagent grade, ≥95%), toluene (AR/ACS, 99.5%), ethanol (Sigma-Aldrich, used without further purification), hydroiodic acid (HI) (57% aq. solution), and diethyl ether (extrapure, 99%) were used in this study.Synthesis of CsPbI3 PNCs
CsPbI3 was synthesized by following a reported method where a mixture of 1.08 mmol (0.5 g) PbI2 and 25 ml of 1-ODE was degassed at 120 °C for 30 minutes. Then a mixture of 2.5 ml of OA and 2.5 ml of OlAm, which was preheated at 120 °C, was injected into the PbI2 precursor solution. The reaction vessel was further degassed to produce a clear solution of dissolved PbI2. Then it was heated to 180 °C under a continuous N2 flow. At a temperature of 175 °C, 2 ml of Cs-oleate stock solution (the synthesis procedure is mentioned in the ESI†) was swiftly injected into the PbI2 precursor solution and the reaction mixture was quenched in an ice bath within 5 seconds to slow down the growth steps. The crude reaction mixture was transferred to a centrifuge tube and washed with 70 ml of MeOAc, followed by centrifugation at 8000 rpm for 10 minutes. The resulting precipitate was dispersed in 3 ml of hexane and stored for further reaction.Synthesis of FAPbI3 PNCs
For FAPbI3 synthesis, 0.74 mmol (0.344 g) PbI2 and 20 ml of 1-ODE were loaded into a 3-necked round bottom flask and degassed at 120 °C for 30 minutes. Then, a mixture of 4 ml of OA and 2 ml of OlAm, preheated at 120 °C, was injected to dissolve the PbI2. The reaction mixture was further degassed to completely dissolve PbI2 and form a clear solution. Then the mixture was transferred into an N2 atmosphere and the temperature was reduced to 80 °C. At 80 °C, 5 ml of FA-oleate stock solution (see the ESI† for FA-oleate preparation) was swiftly injected into the PbI2 precursor and the reaction mixture was quenched in an ice water bath after 20 seconds. Then, 5 ml of MeOAc was added to the reaction mixture. Then it was centrifuged at 8000 rpm for 30 minutes. After that, the precipitate was dispersed in 3 ml of hexane and stored for the ion-exchange reaction.Synthesis of MAPbI3 PNCs via the two-precursor method using PbI2 (route I)
MAPbI3 NC was synthesized by transferring 0.37 mmol (0.172 g) PbI2 and 10 ml of 1-ODE into a 3-necked round bottom flask, which was then degassed at 120 °C for 30 minutes. Then a mixture of 2 ml of OA and 1 ml of OlAm, preheated to 120 °C, was injected into the PbI2 precursor solution. Then it was further degassed for complete dissolution of PbI2. The reaction mixture was transferred into an N2 atmosphere and the temperature was decreased to 80 °C. At 80 °C, 5 ml of MA-oleate stock solution (synthesis of MA-oleate is provided in the ESI†) was swiftly injected and the reaction mixture was quenched after 15 seconds in an ice water bath. The crude product was transferred to a centrifuge tube and 1 ml of hexane and 3 ml of MeOAc were added. Then it was centrifuged at 8000 rpm for 15 minutes and the resulting precipitate was dispersed in 2 ml of hexane and stored for further use.Synthesis of MAPbI3 PNCs via the three-precursor method using OAmI (route II)
Pb(acetate)2·3H2O (0.152 g, 0.4 mmol, Aldrich, 99.99%), MA-acetate (0.136 g, 1.5 mmol), ODE (16 mL, dried), and OA (4 mL, dried) were transferred into a 100 ml 3-necked round bottom flask and dried under vacuum for 30 min at 60 °C. Under these conditions, all the salts were dissolved in ODE. Under N2, the resulting mixture was heated to 80 °C, and subsequently into it 1.2 mmol OAmI in 4 ml of toluene was swiftly injected (synthesis of OAmI is described in the ESI†). The mixture was immediately placed in an ice water bath and the reaction was quenched within 20 s. The crude solution was divided into two equal parts in two centrifuge tubes and, to each part, 10 ml of MeOAc was added. Then each tube was centrifuged at 8000 rpm for 10 min, the resulting precipitate was dispersed in 3 ml of n-hexane and stored in a glass vial. After 1 hour, a small amount of precipitate was observed at the bottom of the glass vial. The upper part of the solution was isolated and centrifuged at 3000 rpm for 5 minutes. The supernatant was then stored for further use.Synthesis of PNC solid solution via A-site cation exchange
To synthesize the solid solution of NCs of FAPbI3 and MAPbI3, their concentration was first matched by adjusting their absorption value in the UV-Vis absorption spectra. Then the colloidal solution of FAPbI3 and MAPbI3 (synthesized via route I) was mixed at different volume ratios at room temperature to make an alloy of FAxMA1−xPbI3. The triple-cation PNCs were synthesized by reacting CsPbI3 with different compositions of FAxMA1−xPbI3. In this case also the absorption spectra of individual samples were recorded and their concentration was matched in a similar way to that mentioned before. Then the colloidal solution of CsPbI3 and FAxMA1−xPbI3 was mixed in a 1:1 ratio at room temperature. In an alternative method, all the three parent NCs, i.e., CsPbI3, FAPbI3 and MAPbI3, were mixed in different volume ratios at room temperature to form TCPbI3 NCs. In all of these ion exchange reactions, the parent NCs were cleaned only once with MeOAc after synthesis, and used for ‘A’ site composition tuning at room temperature under ambient conditions. The equilibrium of these reactions is reached within ∼2 minutes, as evidenced by the homogeneous PL emission spectra that do not change over time in peak energy and width.
Characterization
UV-Vis absorption spectra were recorded using a Cary 5000 UV-Vis-NIR spectrophotometer while steady-state PL emission and PLE spectra were recorded using a Cary Eclipse fluorescence spectrometer. Photoluminescence lifetime measurements were carried out using a Fluoro Log3-Triple Illuminator and an IBH Horiba Jobin Yvon instrument and the samples were excited with a 484 nm picosecond-light-emitting diode laser (NanoLED). Powder XRD measurements were performed on drop-cast QD films using a Bruker D-8 Advance powder X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å). TEM images were captured by using an FEI-Talos 200× electron microscope operated at 200 kV. All TGA measurements were carried out using a TGA Q500 instrument. The samples were heated in a temperature range of 25–800 °C at a heating rate of 10 °C min−1 under an inert atmosphere. For the TGA measurements, the samples were washed two times with MeOAc and 20–30 mg of each solid NC sample was used.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to be declared.Acknowledgements
AH and SM acknowledge financial support via DST/TMD/IC-MAP/2K20/03. GS and SW acknowledge the Department of Science and Technology, India for their INSPIRE fellowships (respectively, DST/INSPIRE Fellowship/2021/IF210674 and DST/INSPIRE Fellowship/2022/IF220292). AH, SM, SB and SW acknowledges CSIR-Indian Institute of Chemical Technology, Hyderabad, India (IICT/Pubs./2024/405).References
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15(8), 5635–5640 CrossRef
.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137(32), 10276–10281 CrossRef PubMed
- M. Suri, A. Hazarika, B. W. Larson, Q. Zhao, M. Vallés-Pelarda, T. D. Siegler, M. K. Abney, A. J. Ferguson, B. A. Korgel and J. M. Luther, ACS Energy Lett., 2019, 4, 1954–1960 CrossRef
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131(17), 6050–6051 CrossRef
- C. K. Møller, Nature, 1957, 180, 981–982 CrossRef
- D. Weber, Z. Naturforsch., B, 1978, 33(12), 1443–1445 CrossRef
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338(6107), 643–647 CrossRef PubMed
- K. Wang, Z. Jin, L. Liang, H. Bian, D. Bai, H. Wang, J. Zhang, Q. Wang and S. Liu, Nat. Commun., 2018, 9(1), 4544 CrossRef PubMed
- W. Xu, Q. Hu, S. Bai, C. Bao, Y. Miao, Z. Yuan, T. Borzda, A. J. Barker, E. Tyukalova and Z. Hu,
et al.
, Nat. Photonics, 2019, 13(6), 418–424 CrossRef
- M. Saliba, S. M. Wood, J. B. Patel, P. K. Nayak, J. Huang, J. A. Alexander-Webber, B. Wenger, S. D. Stranks, M. T. Hörantner and J. T.-W. Wang,
et al.
, Adv. Mater., 2016, 28(5), 923–929 CrossRef PubMed
- V. V. Belykh, D. R. Yakovlev, M. M. Glazov, P. S. Grigoryev, M. Hussain, J. Rautert, D. N. Dirin, M. V. Kovalenko and M. Bayer, Nat. Commun., 2019, 10(1), 673 CrossRef PubMed
- X. Geng, F. Wang, H. Tian, Q. Feng, H. Zhang, R. Liang, Y. Shen, Z. Ju, G.-Y. Gou and N. Deng,
et al.
, ACS Nano, 2020, 14(3), 2860–2868 CrossRef PubMed
- https://www.nrel.gov/pv/cell-efficiency.html (accessed 2024 May 25).
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15(6), 3692–3696 CrossRef
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27(44), 7162–7167 CrossRef
- Y. Wang, X. Li, J. Song, L. Xiao, H. Zeng and H. Sun, Adv. Mater., 2015, 27(44), 7101–7108 CrossRef
- J. Pan, S. P. Sarmah, B. Murali, I. Dursun, W. Peng, M. R. Parida, J. Liu, L. Sinatra, N. Alyami and C. Zhao,
et al.
, J. Phys. Chem. Lett., 2015, 6(24), 5027–5033 CrossRef PubMed
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami and J. Liu,
et al.
, Adv. Mater., 2016, 28(39), 8718–8725 CrossRef PubMed
- Q. Zhao, A. Hazarika, X. Chen, S. P. Harvey, B. W. Larson, G. R. Teeter, J. Liu, T. Song, C. Xiao and L. Shaw,
et al.
, Nat. Commun., 2019, 10(1), 2842 CrossRef PubMed
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354(6308), 92–95 CrossRef
- Q. Zeng, X. Zhang, Q. Bing, Y. Xiong, F. Yang, H. Liu, J.-Y. Liu, H. Zhang, W. Zheng and A. L. Rogach,
et al.
, ACS Energy Lett., 2022, 7(6), 1963–1970 CrossRef
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 RSC
- O. Vybornyi, S. Yakunin and M. V. Kovalenko, Nanoscale, 2016, 8(12), 6278–6283 RSC
- S. Das and A. Samanta, ACS Energy Lett., 2021, 6(11), 3780–3787 CrossRef
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140(7), 2656–2664 CrossRef
- F. Zhang, H. Zhong, C. Chen, X. G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9(4), 4533–4542 CrossRef PubMed
- Y. Zhang, T. D. Siegler, C. J. Thomas, M. K. Abney, T. Shah, A. De Gorostiza, R. M. Greene and B. A. Korgel, Chem. Mater., 2020, 32(13), 5410–5423 CrossRef
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate and A. Hagfeldt,
et al.
, Energy Environ. Sci., 2016, 9(6), 1989–1997 RSC
- Y. Sun, J. Peng, Y. Chen, Y. Yao and Z. Liang, Sci. Rep., 2017, 7(1), 46193 CrossRef PubMed
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52(15), 9019–9038 CrossRef
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13(4), 1764–1769 CrossRef PubMed
- M. I. Saidaminov, A. L. Abdelhady, B. Murali, E. Alarousu, V. M. Burlakov, W. Peng, I. Dursun, L. Wang, Y. He and G. Maculan,
et al.
, Nat.
Commun., 2015, 6(1), 7586 CrossRef PubMed
- A. Jaffe, Y. Lin, C. M. Beavers, J. Voss, W. L. Mao and H. I. Karunadasa, ACS Cent. Sci., 2016, 2(4), 201–209 CrossRef PubMed
- A. S. Masadeh, E. S. Božin, C. L. Farrow, G. Paglia, P. Juhas, S. J. L. Billinge, A. Karkamkar and M. G. Kanatzidis, Phys. Rev. B:Condens. Matter Mater. Phys., 2007, 76(11), 115413 CrossRef
- F. Bertolotti, D. Moscheni, A. Guagliardi and N. Masciocchi, Eur. J. Inorg. Chem., 2018, 34, 3789–3803 CrossRef
- S. J. L. Billinge and I. Levin, Science, 2007, 316(5824), 561–565 CrossRef
- S. Tao, J. Billet, J. De Roo and S. J. L. Billinge, Chem. Mater., 2024, 36(21), 10912–10921 CrossRef
- X. Yang, A. S. Masadeh, J. R. McBride, E. S. Božin, S. J. Rosenthal and S. J. L. Billinge, Phys. Chem. Chem. Phys., 2013, 15(22), 8480–8486 RSC
- T. Moot, D. R. Dikova, A. Hazarika, T. H. Schloemer, S. N. Habisreutinger, N. Leick, S. P. Dunfield, B. A. Rosales, S. P. Harvey and J. R. Pfeilsticker,
et al.
, Chem. Mater., 2020, 32(18), 7850–7860 CrossRef
- J. M. Frost, K. T. Butler, F. Brivio, C. H. Hendon, M. van Schilfgaarde and A. Walsh, Nano Lett., 2014, 14(5), 2584–2590 CrossRef PubMed
- B. Hailegnaw, S. Kirmayer, E. Edri, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2015, 6(9), 1543–1547 CrossRef PubMed
- W. Huang, J. S. Manser, P. V. Kamat and S. Ptasinska, Chem. Mater., 2016, 28(1), 303–311 CrossRef
- E. Smecca, Y. Numata, I. Deretzis, G. Pellegrino, S. Boninelli, T. Miyasaka, A. La Magna and A. Alberti, Phys. Chem. Chem. Phys., 2016, 18, 13413–13422 RSC
- N. Aristidou, C. Eames, M. S. Islam and S. A. Haque, J. Mater. Chem. A, 2017, 5, 25469–25475 RSC
- W. Wei and Y. H. Hu, Int. J. Energy Res., 2017, 41(7), 1063–1069 CrossRef
- J. A. McLeod and L. Liu, J. Phys. Chem. Lett., 2018, 9(9), 2411–2417 CrossRef
- C. Zheng and O. Rubel, J. Phys. Chem. C, 2019, 123(32), 19385–19394 CrossRef
- A. Hazarika, Q. Zhao, E. A. Gaulding, J. A. Christians, B. Dou, A. R. Marshall, T. Moot, J. J. Berry, J. C. Johnson and J. M. Luther, ACS Nano, 2018, 12(10), 10327–10337 CrossRef
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6(1), 8056 CrossRef PubMed
- M. Hao, Y. Bai, S. Zeiske, L. Ren, J. Liu, Y. Yuan, N. Zarrabi, N. Cheng, M. Ghasemi and P. Chen,
et al.
, Nat. Energy, 2020, 5(1), 79–88 CrossRef CAS
- C. Otero-Martínez, M. Imran, N. J. Schrenker, J. Ye, K. Ji, A. Rao, S. D. Stranks, R. L. Z. Hoye, S. Bals and L. Manna,
et al.
, Angew. Chem., Int. Ed., 2022, 61, e202205617 CrossRef
- A. Pisanu, C. Ferrara, P. Quadrelli, G. Guizzetti, M. Patrini, C. Milanese, C. Tealdi and L. Malavasi, J. Phys. Chem. C, 2017, 121(16), 8746–8751 CrossRef CAS
- D. J. Norris and M. G. Bawendi, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 53(24), 16338–16346 CrossRef CAS PubMed
- O. I. Micic, C. J. Curtis, K. M. Jones, J. R. Sprague and A. J. Nozik, J. Phys. Chem., 1994, 98(19), 4966–4969 CrossRef CAS
- S. Ithurria and D. V. Talapin, J. Am. Chem. Soc., 2012, 134(45), 18585–18590 CrossRef CAS PubMed
- J. A. Vigil, A. Hazarika, J. M. Luther and M. F. Toney, ACS Energy Lett., 2020, 5(8), 2475–2482 CrossRef CAS
- J. A. Vigil, B. M. Wieliczka, B. W. Larson, M. Abdelsamie, N. S. Dutta, A. M. Haque, A. Hazarika, J. M. Luther and M. F. Toney, Chem. Mater., 2023, 35(23), 9924–9934 CrossRef CAS
- O. J. Weber, B. Charles and M. T. Weller, J. Mater. Chem. A, 2016, 4, 15375–15382 RSC
- A. Francisco-López, B. Charles, M. I. Alonso, M. Garriga, M. Campoy-Quiles, M. T. Weller and A. R. Goñi, J. Phys. Chem. C, 2020, 124(6), 3448–3458 CrossRef
- M. Thambidurai, S. Foo, K. M. Muhammed Salim, P. C. Harikesh, A. Bruno, N. F. Jamaludin, S. Lie, N. Mathews and C. Dang, J. Mater. Chem. C, 2019, 7(17), 5028–5036 Search PubMed
- I. M. Pavlovetc, M. C. Brennan, S. Draguta, A. Ruth, T. Moot, J. A. Christians, K. Aleshire, S. P. Harvey, S. Toso and S. U. Nanayakkara,
et al.
, ACS Energy Lett., 2020, 5(9), 2802–2810 CrossRef
- N. Landi, E. Maurina, D. Marongiu, A. Simbula, S. Borsacchi, L. Calucci, M. Saba, E. Carignani and M. Geppi, J. Phys. Chem. Lett., 2022, 13(40), 9517–9525 CrossRef PubMed
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6(1), 613–617 RSC
- S. Draguta, O. Sharia, S. J. Yoon, M. C. Brennan, Y. V. Morozov, J. S. Manser, P. V. Kamat, W. F. Schneider and M. Kuno, Nat. Commun., 2017, 8(1), 200 CrossRef PubMed
- A. J. Barker, A. Sadhanala, F. Deschler, M. Gandini, S. P. Senanayak, P. M. Pearce, E. Mosconi, A. J. Pearson, Y. Wu and A. R. Srimath Kandada,
et al.
, ACS Energy Lett., 2017, 2(6), 1416–1424 CrossRef
- A. J. Knight, A. D. Wright, J. B. Patel, D. P. McMeekin, H. J. Snaith, M. B. Johnston and L. M. Herz, ACS Energy Lett., 2019, 4(1), 75–84 CrossRef
- P. Vashishtha, S. A. Veldhuis, S. S. H. Dintakurti, N. L. Kelly, B. E. Griffith, A. A. M. Brown, M. S. Ansari, A. Bruno, N. Mathews and Y. Fang,
et al.
, J. Mater. Chem. C, 2020, 8, 11805–11821 RSC
- R. Shannon, Acta Crystallogr., Sect. A, 1976, 32(5), 751–767 CrossRef
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2015, 6(6), 3430–3433 RSC
- A. Dualeh, P. Gao, S. I. Seok, M. K. Nazeeruddin and M. Grätzel, Chem. Mater., 2014, 26(21), 6160–6164 CrossRef
- A. E. Williams, P. J. Holliman, M. J. Carnie, M. L. Davies, D. A. Worsley and T. M. Watson, J. Mater. Chem. A, 2014, 2(45), 19338–19346 RSC
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9(11), 3406–3410 RSC
- L. Ma, D. Guo, M. Li, C. Wang, Z. Zhou, X. Zhao, F. Zhang, Z. Ao and Z. Nie, Chem. Mater., 2019, 31(20), 8515–8522 CrossRef
- S. Wang, Q. Zhao, A. Hazarika, S. Li, Y. Wu, Y. Zhai, X. Chen, J. M. Luther and G. Li, Nat. Commun., 2023, 14(1), 2216 CrossRef PubMed
- A. Ciccioli and A. Latini, J. Phys. Chem. Lett., 2018, 9(13), 3756–3765 CrossRef
- W. T. M. Van Gompel, R. Herckens, G. Reekmans, B. Ruttens, J. D'Haen, P. Adriaensens, L. Lutsen and D. Vanderzande, J. Phys. Chem. C, 2018, 122(8), 4117–4124 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Additional data on precursor synthesis, optical absorption, XRD, Rietveld refinement, time-resolved PL, HR-TEM of MAPI PNCs, MAPI thin films and different PNC compositions, and stability comparison. See DOI: https://doi.org/10.1039/d4nr03422h |
| This journal is © The Royal Society of Chemistry 2025 |