
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConfigurational assignments of type-I polyketide synthase (PKS)-derived natural products based on spectroscopic and chemical analysis: methodologies and case studies
Jinsheng
Cui†
a,
Prima F.
Hillman†
b,
Geum Jin
Kim†
 c,
Thinh T. M.
Bui†
d,
Kyuho
Moon
*d,
Sang-Jip
Nam
*e,
Hyukjae
Choi
*f and
Dong-Chan
Oh
*a
c,
Thinh T. M.
Bui†
d,
Kyuho
Moon
*d,
Sang-Jip
Nam
*e,
Hyukjae
Choi
*f and
Dong-Chan
Oh
*a
aNatural Products Research Institute, College of Pharmacy, Seoul National University, Seoul, 08826, Republic of Korea. E-mail: dongchanoh@snu.ac.kr
bDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Andalas, Kampus Limau Manis, Padang, 25163, Indonesia
cDepartment of Pharmacology, School of Medicine, Dongguk University, Gyeongju, Gyeongsangbukdo 38066, Republic of Korea
dCollege of Pharmacy, Kyung Hee University, Seoul, 02447, Republic of Korea. E-mail: kmoon@khu.ac.kr
eDepartment of Chemistry and Nanoscience, Ewha Womans University, Seoul, 03760, Republic of Korea. E-mail: sjnam@ewha.ac.kr
fCollege of Pharmacy and Research Institute of Cell Culture, Yeungnam University, Gyeongsan, Gyeongsangbukdo 38541, Republic of Korea. E-mail: h5choi@yu.ac.kr
First published on 23rd April 2025
Abstract
Covering: 1992 to 2024
Type-I polyketide synthase (PKS)-derived metabolites are structurally diverse bioactive natural products containing multiple stereogenic centres. This review focuses on the configurational analysis of type-I PKS-derived natural products, emphasizing the methodologies and challenges associated with determining their stereochemistry due to their complex structures with multiple chiral centres. Key strategies include J-based configuration analysis (JBCA), chemical derivatizations with chiral reagents, degradation methods, NMR spectroscopic analysis, and the exploitation of chiroptical properties. Case studies demonstrate the practical applications of these methods in elucidating the stereochemistry of type-I polyketide natural products.
 Kyuho Moon | Kyuho Moon is an assistant professor at the College of Pharmacy, Kyung Hee University, since 2024. He received his PhD degree in Seoul National University under Prof. Dong-Chan Oh's guidance in 2016. In the same year, he joined Prof. Mohammad Seyedsayamdost's group in the Department of Chemistry, Princeton University, as a postdoctoral researcher. His research is focused on microbial natural products, chemical biology, specifically discovering cryptic microbial secondary metabolites and investigation of bacteria in unreported habitats. |
 Sang-Jip Nam | Sang-Jip Nam is currently a professor at Chemistry and Nanoscience Department, Ewha Womans University, Korea, since 2013. He received his MS degree (2001) and PhD degree (2006) from Seoul National University, Korea. In 2007, he joined as a postdoc in the Prof. William Fenical Lab., Scripps Institution of Oceanography, University of California, San Diego. In 2012, he was appointed as an assistant professor at the College of Pharmacy, Sunchon National University. His research interests are in the discovery and development of natural products as potential treatments for diseases. |
 Hyukjae Choi | Hyukjae Choi earned his BS in oceanography at Seoul National University. He received his MS (2001) and PhD (2009) in marine natural products chemistry at Seoul National University under the supervision of Prof. Heonjoong Kang. After a postdoctoral fellowship (2009–2012) with Prof. William H. Gerwick at Scripps Institution of Oceanography, he joined Yeungnam University's College of Pharmacy as a faculty member in 2012. His research focuses on the discovery and structure elucidation of bioactive compounds from diverse natural sources. |
 Dong-Chan Oh | Dong-Chan Oh earned a BS degree in Oceanography as well as an MS degree in Marine Chemistry, from Seoul National University, Republic of Korea. He completed his doctoral studies in Marine Natural Products Chemistry in 2006 under the mentorship of Professor William Fenical at the Scripps Institution of Oceanography, University of California, San Diego. Following this, he pursued postdoctoral research in the laboratory of Professor Jon Clardy at Harvard Medical School. In 2009, he commenced his independent academic career at the College of Pharmacy, Seoul National University, where his research focuses on the discovery, structural elucidation, and stereochemical analysis of new bioactive natural products from microbial sources. From 2012 to 2017, he was recognized as a Howard Hughes Medical Institute International Early Career Scientist. Dong-Chan Oh formerly held the position of Director of the Natural Products Research Institute, Seoul National University from 2019 to 2025. Currently, he is a Full Professor in the College of Pharmacy at Seoul National University. |
1. Introduction
Polyketides represent a diverse and structurally complex class of secondary metabolites produced by a wide range of microorganisms (actinobacteria, cyanobacteria, and fungi), plants, and marine organisms (sponges and dinoflagellates). These compounds are synthesized through polyketide synthases (PKSs), a family of multifunctional enzymes that catalyse the stepwise polymerization of acyl-CoA precursors. Polyketides exhibit remarkable structural diversity owing to the flexibility of their biosynthetic pathways and the remarkable diversity of post-modifications mediated by tailoring enzymes.1,2 The significance of polyketides extends not only to their structural complexity but also to their potent biological activities and their pivotal roles in medicine and biotechnology. Their utility also spans agricultural applications, where they function as natural pesticides and growth regulators.Among the various classes of polyketides, those synthesized by type-I polyketide synthases (PKSs) have garnered considerable attention owing to their complex structures and intricate biosynthetic pathways. These type-I PKS-derived polyketides often contain multiple chiral centres, which result from their biosynthesis through the stepwise assembly of polyketide chains via elongation and modification processes mediated by PKSs. These PKSs are multi-enzyme complexes that utilize a combination of starter and extender units, typically acetyl-CoA and malonyl-CoA, to introduce chiral centres into the growing polyketide chain.3 Furthermore, the actions of tailoring enzymes, such as oxidases, reductases, methyltransferases, and cyclases, further modify the polyketide backbone, resulting in the formation of multiple stereogenic centres. These enzymatic modifications contribute to the remarkable structural diversity and chirality of type-I PKS-derived polyketides.4
While the configurational diversity of type-I PKS-derived polyketides enhances their therapeutic potential, it introduces notable challenges in their structural elucidation.3,4 This stereochemical complexity often arises from several factors, including the presence of multiple chiral centres, existence of remote stereogenic centres, unanticipated conformational changes, stereochemistry derived from hybrid biosynthetic pathways, and instability.
Consequently, analysing the absolute configurations of type-I PKS-derived polyketides is an essential but daunting task. For instance, during derivatization, eliminating alcohol groups can lead to misinterpretations, while generating multiple methoxy(trifluoromethyl)phenylacetyl (MTPA, Mosher's reagent) derivatives can complicate the analysis and yield incorrect stereochemical predictions.5 A notable example of this complexity is gibbosol, a super carbon-chained polyketide, which features multiple hydroxy chiral centres along its extended carbon chain, making the determination of its absolute configuration particularly difficult (Fig. 1).6 Polyketides that are unstable typically have specific structural or chemical features, making them reactive or susceptible to environmental factors. The instability of these compounds is often attributed to the presence of functional groups or molecular configurations that are highly reactive or sensitive to pH, temperature, light, or enzymatic degradation. Common features of unstable and easily degradable polyketides include epoxides, unsaturated conjugations, lactones, glycosylated polyketides, halogenated polyketides, etc. Hence, acquiring a comprehensive understanding of these challenges is crucial for developing reliable methods to characterize these compounds, harness their bioactive properties for pharmaceutical applications, and establish accurate synthetic targets for total synthesis.
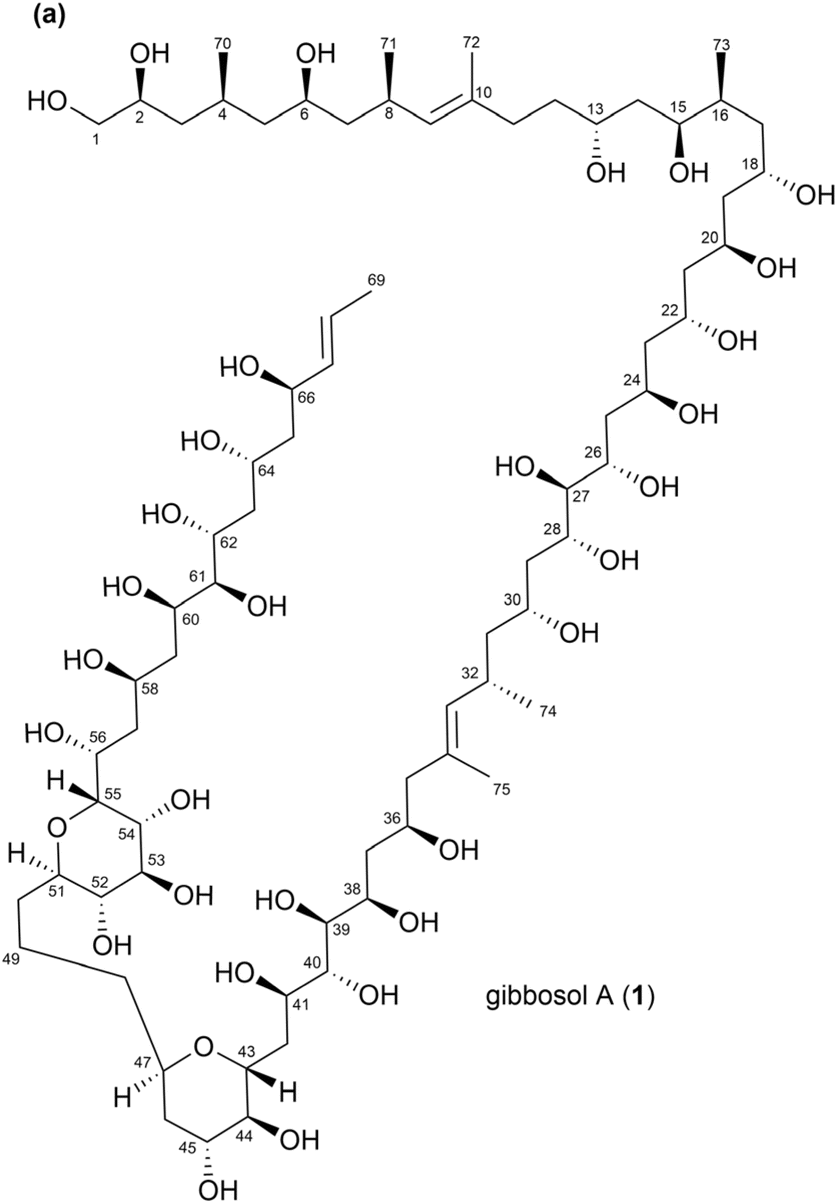 | ||
| Fig. 1 (a) Structure of gibbosol A (1). | ||
This review aims to offer insights into the complexities and methodologies involved in the analysis of type-I PKS-derived natural products, underscoring the critical need for robust analytical techniques. We introduce several prominent methods employed by researchers in recent decades to examine the stereochemistry of type-I PKS-derived polyketides, including J-based configuration analysis, chiral derivatizations, chemical degradation, NMR shift comparisons with universal databases, and chiroptical analysis paired with quantum mechanical calculations. Through several case studies, we demonstrate the practical application of these techniques in resolving relative and absolute configurations, offering valuable insights into stereochemical structural elucidation.
2. Methods and strategies for configurational analysis of type-I PKS-derived natural products
2.1. J-based configuration analysis (JBCA)
 | ||
| Fig. 2 Murata's JBCA strategy: (a) dihedral angle dependence of spin-coupling constants, 3JH,H and 2,3JC,H (values in parentheses correspond to the 1,2-dioxygenated systems. R represents H, alkyl, or acyl groups). (b) Dependence of 3JH,H and 2,3JC,H on the dihedral angles between vicinal methine carbons in 2,3-disubstituted butane systems. (c) Dependence of 3JH,H, and 2,3JC,H on the dihedral angles between vicinal methine carbons in alternating butane systems. (d) Dependence of 3JH,H and 2,3JC,H on the dihedral angles between methine and methylene carbons in 2-substituted butane systems. (e) Dependence of 3JH,H and 2,3JC,H on the dihedral angles between methine and methylene carbons in alternating butane systems. | ||
Notably, the value of 3JH,H depends on the dihedral angles of protons, as described by the Karplus equation.8 Similarly, 3JH,C values also exhibit a Karplus-like dependency on dihedral angles. Although 2JH,C values are typically independent of dihedral angles, this dependence becomes relevant when the α-carbon is bonded to an electronegative atom, such as O, N, or a halogen. In such cases, the 2JH,C value depends on the dihedral angle between the proton and the electronegative atom on the α-carbon.9,10
Notably, Murata's JBCA strategy was originally designed to determine the relative configurations of two adjacent (1,2) or alternately positioned (1,3) stereogenic carbons substituted with hydroxy, alkoxy, or methyl groups in acyclic compounds.7 In 1,2-methine systems, as illustrated in Fig. 2b, six possible staggered rotamers exist for the 2,3-disubstituted butane stereoisomers. Among these, the two rotamers in the threo configuration (A-1 and A-2) and the two others in the erythro configuration (B-1 and B-2) can be distinctly identified based on the 3JH,H and 2,3JC,H values. Although rotamers A-3 (in threo) and B-3 (in erythro) cannot be uniquely assigned using only the 3JH,H and 2,3JC,H values, they can be differentiated based on the NOE (or rotating-frame Overhauser effect (ROE)) correlations among the protons located at the C1, X, Y, and C4 positions. A moderate 3JH,H value suggests that two protons are distributed between two predominant rotamers with anti and gauche orientations. As depicted in Fig. 2c, four pairs of rotamers (A-2/A-3, A-3/A-1, B-2/B-3, and B-3/B-1) can be clearly distinguished based on 3JH,H and 2,3JC,H values. However, for the rotameric pairs A-1/A-2 and B-1/B-2, which feature a gauche conformation between H2 and H3, the relative configurations cannot be resolved using 3JH,H and 2,3JC,H values. Notably, these unresolved rotamer pairs are thermodynamically less favored because their substituents at C2 and C3 lie on the same side. For 1,3-methine systems, the JBCA approach is applicable if the two methylene protons are magnetically distinct and thus can be stereospecifically assigned. In these cases, the relative positions of each diastereotopic methylene proton between the two adjacent methines can be distinguished among the six possible conformers using 3JH,H and 2,3JC,H values and NOE/ROE correlations, as illustrated in Fig. 2d. Similar to 1,2-methine systems, moderate 3JH,H values in 1,3-methine systems suggest the presence of multiple rotamers (Fig. 2e). Rotameric exchange is commonly observed between C-1/C-3 or D-1/D-3, while other pairs, such as C-2 and D-2, are thermodynamically less stable and hence less frequently encountered. The JBCA method is also applicable to 1,4-methine systems, provided an ethylene linker is present between the two methine carbons and all relevant proton signals in the 1H NMR spectrum are stereospecifically assigned without overlapping.11 Furthermore, previous studies have extended the application scope of the JBCA to stereogenic carbons bonded to alkyl groups or electronegative atoms such as N, S, and Cl.10–15
Notably, selective irradiation pulse techniques—including 1D TOCSY, 1D NOESY, and 1D ROESY—are effective in eliminating the multiplicity associated with the irradiated proton signals,16,17 thereby resolving overlapping signals and simplifying the 1H NMR spectra for precise J value measurements.
Meanwhile, homonuclear J-resolved NMR spectroscopy separates chemical shifts (δ) along the f2 axis and coupling constants (J) along the f1 axis.18 This separation eliminates overlapping multiplet patterns along the f2 axis, transforming all proton signals into singlets at their corresponding chemical shifts. In this scenario, multiplicity information is retained along the f1 axis, allowing the measurements of J values. Nevertheless, homonuclear J-resolved NMR spectroscopy often exhibits tilting patterns caused by proton couplings along both the f1 and f2 axes.19 To address this issue, post-processing techniques, such as t1-noise reduction and tilting correction at an angle of 45°, are required.
Notably, phase-sensitive DQF-COSY is a powerful technique for measuring J values owing to its ability to generate high-resolution crosspeak multiplets with a pure absorption lineshape.20 Interestingly, the diagonal peaks in DQF-COSY also display an anti-phase absorption lineshape, suppressing the tailing commonly observed in standard COSY spectra. This suppression enhances spectral clarity near the diagonal peaks.21
Compared to DQF-COSY, E.COSY offers simpler crosspeak multiplet patterns. By employing E.COSY, both the magnitudes and signs of JH,H values can be accurately determined in complex spin systems.22–24
Among these, HETLOC yields a 2D 1H–1H spectrum that reveals nJC,H values through E.COSY-type splitting. This experiment enables the determination of both the magnitudes and signs of 1JC,H and 2,3JC,H based on signal displacements along the f1 and f2 dimensions, respectively.25 HETLOC is relatively sensitive and can produce spectra at millimolar concentrations. Consequently, HETLOC is a popular choice for measuring 2,3JC,H values in configurational studies owing to its high sensitivity and relatively simple data analysis. However, HETLOC relies on TOCSY transfer, which limits its applicability to measuring nJC,H values only between protons and protonated carbons. Consequently, HETLOC is incapable of measuring 2,3JC,H values for non-protonated carbons. Furthermore, frequent signal overlap in HETLOC spectra can interfere with precise J value measurements. Moreover, if the 1H–1H vicinal coupling constant (3JH,H) is small, the crosspeak intensity in HETLOC may be substantially reduced.
HSQC–HECADE spectra display E.COSY-type crosspeaks, making the HSQC–HECADE technique inherently similar to HETLOC. However, in HECADE, the f1 dimension represents the 13C NMR spectrum, allowing signals to disperse over a wider spectral range compared to HETLOC,26 thereby mitigating spectral crowding and accidental overlaps. Nevertheless, similar to HETLOC, this technique employs TOCSY transfer, which prevents the measurement of 2,3JC,H values for non-protonated carbons or heteroatoms. Furthermore, the heteronuclear coupling magnitude in HECADE depends on the 1H–1H vicinal coupling constant (3JH,H), similar to that in HETLOC, which is a limitation of this approach.
In contrast, J-HMBC generates a 2D 1H–13C spectrum, where the crosspeaks are split along the f1 dimension, with the distance being proportional to the nJC,H value. This distance can be adjusted by modifying J-scaling factors in experimental parameters, enabling the measurement of nJC,H values without requiring high resolution in the f1 dimension.27J-HMBC does not rely on TOCSY transfer and allows the measurement of nJC,H values between specific protons and both protonated and non-protonated carbons not linked by TOCSY transfers. Additionally, J-HMBC can provide information on the signs of nJC,H values in a single experiment. However, while determining nJC,H values based on the distance between two split crosspeaks is straightforward, identifying their signs is still challenging. Other drawbacks of this technique include accidental signal overlaps owing to peak doubling and relatively lower sensitivity compared to HETLOC.
The IPAP–HSQMBC technique involves acquiring two separate 2D 1H–13C spectra—one with IP data and the other with AP data.28 Once recorded, these datasets are subsequently added and subtracted to produce α- and β-HSQMBC spectra, respectively. The displacement of crosspeaks between the α/β-HSQMBC spectra along the f2 dimension provides both the magnitudes and the signs of nJC,H constants.28 By examining crosspeaks from separate spectra, IPAP–HSQMBC reduces the likelihood of accidental overlaps, leading to more accurate measurements of nJC,H. Similar to J-HMBC, this technique can measure nJC,H values independent of carbon protonation. However, it requires selective or band-selective excitation and multiple experiments, increasing the complexity of the measurement.
 | ||
| Fig. 3 Application of JBCA for the relative configuration assessment of formicolides: (a) C19–C21 of formicolide A and (b) C19–C22 of formicolide B. Newman projections were adapted from the original report and re-drawn as indicated in the JBCA models. | ||
Strasseriolides A–D (4–7) are 18-membered macrolides isolated from the endophytic fungus Strasseria geniculate CF-247251. These macrolides exhibit antimalarial activity against Plasmodium falciparum without cytotoxic effects on the HepG2 cell line (Fig. 4a).37 The planar structures of compounds 4–7 were elucidated through an integrative analysis of mass spectrometry (MS) and NMR (1H, 13C, HSQC, COSY, HMBC, and NOESY) data. Notably, the absolute configuration of compound 5 was determined using X-ray crystallography, while those of 4 and 6 were inferred based on similarities in NMR data, including chemical shifts and NOESY correlations. Strasseriolide D (7) comprises an additional stereogenic centre at C13. The relative configurations of C13 and C14 were determined using the JBCA strategy. The 3JH,H values of overlapping multiplets in compound 7 were estimated using J-resolved NMR spectroscopy and 1D TOCSY, while the 2,3JH,C values were accurately determined by J-HMBC with a scaling factor of 60. Notably, the measured 3JH,H and 2,3JH,C values between relevant nuclei indicated that the relative orientation between C13 and C14 can adopt either the A-3 or B-3 rotameric conformation. A NOESY correlation between H12a and H15 confirmed the presence of the A-3 rotameric conformation. Based on this, the absolute configurations of compound 7 were determined to be 2R, 3S, 6S, 8R, 11R, 13S, 14S, and 18S.
 | ||
| Fig. 4 Application of JBCA for the relative configuration assessment of strasseriolide D: (a) structures of strasseriolides A–D (4–7). (b) JBCA-based assessment of the relative configuration of C13–C14 in strasseriolide D (7). | ||
The authors attempted to extend the relative configuration assignment from C11 to C13 in compound 7 using a 1,3-methine system. However, this approach was unsuccessful owing to discrepancies in the 3JH,H and 2,3JH,C values around the C12–C13 bond (3JH12b,H13 = 8.0 Hz for anti-orientation between H12b and H13; 3JC11,H13 = 6.4 Hz for anti-orientation between C11 and H13). These inconsistencies were attributed to a partially eclipsed relative orientation between H13 and C11, highlighting the limitations of JBCA in dealing with cyclic compounds featuring fixed conformations that include non-staggered rotamers.
Neaumycin B (8), a cytotoxin isolated from the marine-derived Micromonospora sp. CNY-010, is a polycyclic polyketide possessing a complex structure comprising an epoxide, two tetrahydropyran rings connected by a spiroketal, and a 28-membered macrolide, as depicted in Fig. 5a.38,39 This polyketide has a highly intricate architecture with 19 chiral centres. One of its congeners, neaumycin A, was first reported in 2012,40 and its planar structure was substantially revised in 2015.39 Along with the structural revision of neaumycin A, the structures of neaumycins B and C were also reported in 2015. However, the absolute configurations of these neaumycins were not comprehensively assessed at that time. In 2018, Fenical and co-workers comprehensively determined the absolute configurations of neaumycin B by integrating the stereospecificities of biosynthetic enzymes with extensive NMR data analysis.38 In particular, the relative configurations at C4–C7, C10–C14, C22–C25, and C37–C38 were separately assigned based on the JBCA and ROESY correlations. The authors measured 2,3JH,C values using HETLOC and 3JH,H values from 1H NMR data and homonuclear J-resolved NMR spectroscopy. In the C3–C4–C5–C6 fragment, 3JH4,H5 and 2JH2,C3 values were determined to be 2.0 and 8.6 Hz, respectively. In a 1,2-methine system, these coupling values indicated two possible configurations at C4–C5: the threo (A-2 rotamer) and erythro (B-1 rotamer) forms. The presence of a ROESY correlation between H5 and H42 confirmed the A-2 rotamer as the correct conformation, as depicted in Fig. 5b. Through further analyses of ROESY correlations, the relative configurations at C4, C5, and C7 were assigned as syn/anti. The relative configurations at C10–C11–C12–C13–C14 were assigned using JBCA with a 1,3-methine system, along with ROESY analyses. The 3JH11,H12h and 3JH11,H12l values were determined to be 2.3 and 10.5 Hz, respectively, while the 2JC11,H12h value was observed to be below 2.0 Hz. These observations supported the presence of a C-1 conformer at the C11–C12 position. At the C13–C14 positions, the relatively small 3JH13,H14, 2JC13,H14, and 2JH13,C14 values indicated an A-1 rotameric configuration.
 | ||
| Fig. 5 Application of JBCA for the relative configuration assessment of neaumycin B: (a) Chemical structure of neaumycin B. Application of JBCA for the configurational assessment of the (b) C4–C7, (c) C10–C14, (d) C21–C23, and (e) C37–C38 positions. | ||
Through additional ROESY correlation analyses, the relative configurations at C10, C11, C13, and C14 were assigned as anti/syn/syn orientations (Fig. 5c). At the C22–C23–C25 positions, the small 3JH22,H23 and 2JH22,C23 values suggested two possible rotamers: A-1 and B-2. However, the observed ROESY correlations between H23 and H44 ruled out the A-1 rotameric orientation. Through further analyses of the ROESY correlations between H20/H22, H21/H24l, and H23/H25, the relative configuration at C22, C23, and C25 was determined as anti/syn (Fig. 5d). Next, the large values of vicinal 1H–1H coupling constants (3JH25,H26 and 3JH26,H27) in the pyran ring allowed the extension of relative configurations from C25 to C26 and C27. ROESY correlations then confirmed the configurations within the bicyclic spiroketal structure.38 The relative configurations at the C34–C38 positions were determined through meticulous ROESY correlation analysis and JBCA. Specifically, a small 3JH37,H38 value (2.3 Hz) combined with a large 2JC37,H38 value (7.2 Hz) suggested two possible rotamers: A-2 in the threo form and B-1 in the erythro form. However, the observed ROESY correlations between H37 and H47 ruled out the possibility of the B-1 rotameric configuration. Consequently, the relative configuration at the C37–C38 position was determined to be a threo configuration, as depicted in Fig. 5d. Additionally, the ROESY correlations between H34/H37, H36/H46, and H36/H47 facilitated the extension of relative configurations to the C34–C38 positions, which encompassed a trans epoxide at the C36–C37 position.
Bahamaolide A (9) is a polyene-polyol-type 36-membered macrocyclic lactone produced by the marine actinomycete Streptomyces sp. CNQ343 (Fig. 6a).41 The relative configurations of its repeating 1,3-diol units were assigned through acetonide derivatization followed by NMR data analysis. The relative configurations of the isolated stereogenic centres at C34 and C35 were further determined using JBCA. A large 3JH35,C39 value indicated two possible rotameric configurations: A-1 in the threo form and B-1 in the erythro form, both characterized by an anti orientation between H35 and C39. However, the observed ROESY correlations between H36 and H39 supported the existence of only the A-1 rotamer. Consequently, the relative configurations at C34 and C35 were both assigned as S* (Fig. 6b). The absolute configurations of bahamaolide A were determined using Mosher's method, which involved analysing a tetra-acetonide derivative and its methanolysis product. Details of this analysis are provided in Section 2.3.
 | ||
| Fig. 6 Application of JBCA for relative configuration assessment at C34–C35 in bahamaolide A: (a) chemical structure of bahamaolide A. (b) Newman projection of C33–C36. | ||
Pulvomycins are 22-membered macrolactones isolated from the bacterium Streptomyces sp. These macrolactones feature polyene fragments and the sugar unit labilose. Although the first compound of this class, pulvomycin (also known as labilomycin), was discovered in 1957, its planar structure was only fully established in 1985. The absolute configuration was later confirmed by X-ray crystallography in 2006. Pulvomycins B–D were subsequently identified as additional members of this structural class.42 Among these, pulvomycin B (10) contains 13 stereogenic centres, with five located in the labilose sugar unit and eight within the polyketide moiety (Fig. 7a). The sugar unit was identified as labilose based on the 1JC35,H35 and 3JH,H values, along with ROESY correlations. The relative configurations of the consecutive stereogenic centres at C21–C24 were successfully determined using JBCA. Relatively large 3JH21,H22 and 2JH22,C21 values, along with the small 3JC20,H22, 3JH21,C23, and 2JH21,C44 values, indicated two possible rotamers: A-3 or B-3. The observed ROESY correlation between H20 and H23 confirmed the relative configuration at C21–C22 as the A-3 rotamer in the threo form, as depicted in Fig. 7b. Similarly, the relative configurations at C22–C24 were successfully identified as the threo form at C22–C23 and the erythro form at C23–C24 (Fig. 7c and d). At the C32–C33 position, JBCA analysis indicated a configuration similar to the A-3 rotamer in a 1,2-methine system. However, given that the 2JH32,C33 value was moderate, the configuration was further confirmed through density functional theory (DFT) modeling and additional ROESY observations. Ultimately, the relative configuration at C32–C33 was identified as an A-3 rotamer in the threo form (Fig. 7e). Finally, the absolute configurations at C3, C13, C32, and C37 were determined using the modified Mosher's method. Meanwhile, the absolute configuration at C23, which was sterically hindered and inactive in Mosher's esterification, was determined using Kishi's bidentate chiral solvation technique, as will be detailed in Sections 2.2 and 2.4.
 | ||
| Fig. 7 Application of JBCA for the relative configuration assessment in pulvomycin B: (a) chemical structure of pulvomycin B. Application of JBCA for the configuration assessment of the (b) C21–C22, (c) C22–C23 (d) C23–C24, and (e) C32–C33 positions. | ||
2.2. Derivatization with auxiliary chiral reagents
In the traditional Mosher's method, determination of the absolute configurations of chiral carbinol carbons through the analysis of Δδ values obtained from (R)- and (S)-MTPA ester derivatives was applied. However, this method has limitations when the substituents on the stereogenic centre, R1 and R2, are similar in size or electronic properties, resulting in ambiguous Δδ patterns that complicate accurate configuration assignment. Additionally, the traditional method does not systematically account for steric and electronic effects, which can lead to errors in more complex molecular environments (Fig. 8).50
 | ||
| Fig. 8 Synthesis of (R)- and (S)-MTPA esters from a secondary alcohol. | ||
The modified Mosher's method emerged as an evolution of the traditional approach to address its limitations, particularly when dealing with sterically hindered alcohols or molecules with complex stereochemistry. This method emphasizes the use of high-resolution 1H NMR and integrates advanced multi-dimensional NMR techniques such as COSY, NOESY, and HSQC to enhance the precision of proton signal assignments. The modified Mosher's method also incorporates the use of alternative solvents like deutero-chloroform (CDCl3), which improve its effectiveness for sterically hindered systems. Furthermore, it incorporates complementary validation methods, such as X-ray crystallography, to confirm the configuration of chiral centres in cases where NMR results may be ambiguous. These improvements allow the modified Mosher's method to overcome challenges associated with steric-hindrance, solvent compatibility, and the limitations of one-dimensional NMR spectroscopy. As a result, the method has become a gold standard in stereochemical analysis, offering superior versatility and accuracy compared to the traditional approach.47,49
In Mosher's method, MTPA is the most commonly used CDA for assigning the absolute configurations of secondary alcohols. In a previous study, deplelides A (11) and B (12) (Fig. 9), which represent 36-membered polyol macrolides, were isolated from Streptomyces MM581-NF15. The absolute configuration of deplelide A (11)51 was determined through a degradation process followed by the application of a modified Mosher ester method. In particular, compound 11 was subjected to a four-step degradation process, involving the following steps: (1) reduction using NaBH4, (2) methanolysis in the presence of NaOMe, (3) oxidative cleavage of vicinal diols using NaIO4, and (4) reduction of the resulting aldehydes using NaBH4 (Fig. 9). This degradation process yielded alcohols 13 (C1–C15) and 14 (C32–C43 and C1′–C6′) as the products (Fig. 10). These alcohols, 13 and 14, were then transformed into the (S)- and (R)-MTPA esters 13a/13b and 14a/14b, respectively. The differences in chemical shifts (ΔδS–R) derived from the 1H NMR data of 13a and 13b assigned the configurations at C5, C7, and C11 in 13 as R, R, and R, respectively (Fig. 11a). Similarly, the configurations of C35, C3′, and C5′ in 14 were determined to be R, R, and R, respectively (Fig. 11c). Although the ΔδS–R values for 14a and 14b at C47 around C35 were inconsistent, the relative configuration between C3′ (3′R*) and C35 (35R*) confirmed that C35 in 14 featured the same configuration as 35R. Hence, the absolute configurations of the C1 to C15, C32 to C43, and C1′ to C7′ fragments in compound 11 were comprehensively determined (Fig. 11). Meanwhile, the absolute configuration of deplelide B (12) was inferred to be identical to that of deplelide A (11) given the similarity in their planar structures, except for the substitution of the methyl group at C14′′ in 11 with an ethyl group in 12.51
 | ||
| Fig. 9 Structures of deplelides A (11) and B (12). | ||
 | ||
| Fig. 10 Degradation of deplelide A (11). | ||
 | ||
| Fig. 11 (a and c) ΔδS–R values of MTPA esters 13a and 13b (a) and 14a and 14b (c). (b) Structure of 14. | ||
Menisporopsin A (15),52 a macrocyclic polylactone isolated from the fungus Menisporopsis theobromae, exhibits antimalarial activity. To determine the absolute configuration of each acid residue in compound 15, its methylated derivative (16) was hydrolysed in 1 M NaOH and purified by high performance liquid chromatography (HPLC), yielding compounds (17) and (18) (Fig. 12). The absolute configuration of 18 was then determined using Mosher's method. Both the (S)- and (R)-MTPA esters of 18 (18a and 18b) were prepared separately and analysed using 1H NMR spectroscopy (Fig. 13). The obtained ΔδS–R values indicated that the absolute configuration at C2′ in 18 was R. Consequently, the configuration at C39 in menisporopsin A (15) was also established as R. Further, the absolute configuration at C37 was determined as S based on spatial arrangements, in which the methyl groups at H37 and H40 of 15 are positioned in the same plane. The configuration at C3 in 3-hydroxybutyric acid was determined as R using chiral HPLC analysis. However, owing to the lack of a chromophore, the direct analysis of the 3-hydroxybutyric acid using chiral HPLC offered limited information regarding its identity. To address this limitation, a derivative was formed using Mosher's reagent ((S)-MTPA-Cl), which generated diastereomers with an aromatic chromophore. The resulting Mosher's esters, (R)- and (S)-3-hydroxybutyric acid, were successfully distinguished using chiral HPLC. Based on this analysis, the 3-hydroxybutyric acid residue in 15 was determined to be (R)-3-hydroxybutyric acid. Consequently, the absolute stereochemistry of all six chiral centres in menisporopsin A (15) was assigned as 9R, 13R, 23R, 27R, 37S, and 39R.52
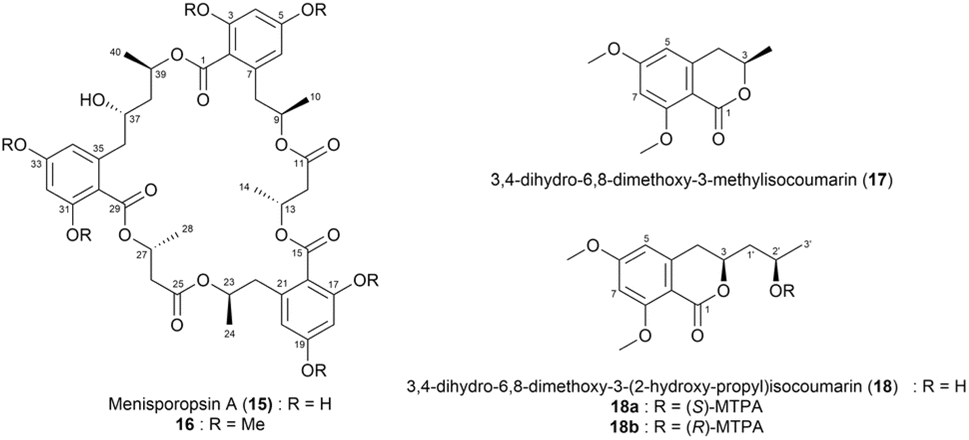 | ||
| Fig. 12 Structures of menisporopsin A (15), its methylated derivative (16), 3,4-dihydro-6,8-dimethoxy-3-methylisocoumarin (17), 3,4-dihydro-6,8-dimethoxy-3-(2-hydroxy-propyl)isocoumarin (18), and the (S)- and (R)- MTPA esters of 18 (18a and 18b). | ||
 | ||
| Fig. 13 ΔδS–R values of the MTPA esters (18a and 18b) of 18. | ||
Modiolides D-G (19–22)53 were isolated from Paraconiothyrium sp. VK-13 (Fig. 14a). The absolute configuration at C7 in compounds 19–22 was assigned using the modified Mosher's method. Based on the ΔδS–R values at C7, the configurations were determined as S for 19 and R for both 20 and 21 (Fig. 14b). For modiolide G (22), the absolute configurations at C7 and C9 were found to be inverted compared to those in modiolide A owing to the assignment of C4 as R using the modified Mosher's method (Fig. 14b).53
 | ||
| Fig. 14 (a) Structures of 19–22 and (b) ΔδS–R values of the MTPA esters of compounds 19–22. | ||
A previous study isolated a new tetronate-class polyketide, maklamicin (23),54 from the culture extract of Micromonospora sp. GMKU326 (Fig. 15a). Maklamicin exhibited strong to moderate antimicrobial activity against Gram-positive bacteria. The absolute stereochemistry of the secondary hydroxy group at C31 in 23 was assigned using the modified Mosher's method. During this test, the enolic hydroxy group at C24 was protected by treating 23 with TMSCHN2 in CHCl3/MeOH, forming a methyl ether. The resulting methylated derivative, 24, was then treated with (S)- and (R)-MTPA-Cl to yield the bis-(R)- and (S)-MTPA esters (24a and 24b), respectively. Notably, the ΔδS–R values for H332 were positive in the 1H NMR spectra of 24a and 24b, while those for H30, H21, H22, H29, H19, H28, and H17 were negative (Fig. 15b). Based on these data, the absolute configuration of C31 was determined as R.54
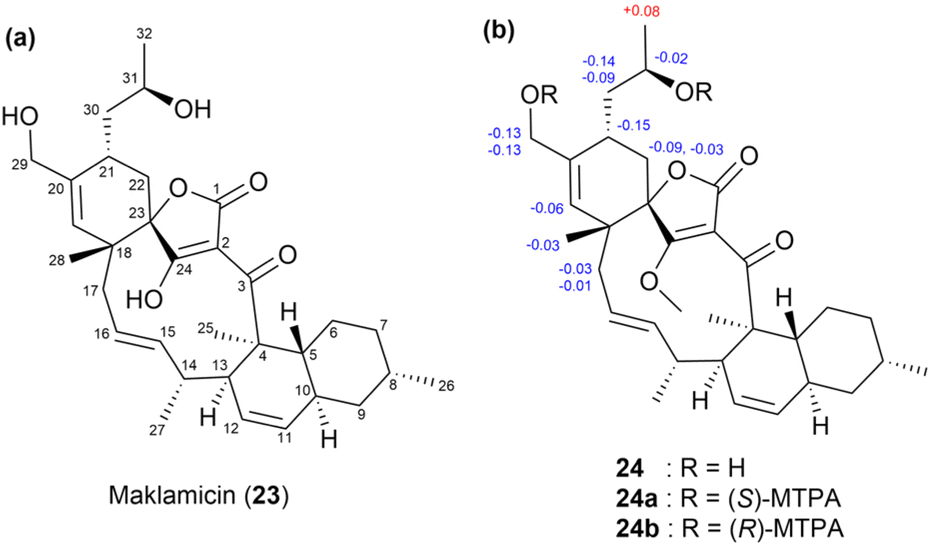 | ||
| Fig. 15 (a) Structure of maklamicin (23) and (b) the ΔδS–R values of the MTPA esters (24a and 24b) of 24. | ||
Another successful application of Mosher's method for determining the absolute stereochemistry of chiral carbons was demonstrated in pulvomycins B–D (10, 25, and 26) (Fig. 16).42 The absolute configurations at C3, C13, C32, and C37 were determined using the modified Mosher's method. For this, the hydroxy groups at these positions were esterified with (R)- and (S)-MTPA-Cl to yield tetra-(S)- and (R)-MTPA esters, 10a and 10b, respectively. The ΔδS–R values confirmed the absolute configurations at C3, C13, C32, and C37 as 3S, 13S, 32S, and 37S, respectively (Fig. 17). Pulvomycin C (25) was identified as a geometric isomer of compound 10 with a 6Z configuration. The absolute configurations of pulvomycin D (26) were confirmed by analysing the 1H chemical shifts of the bis-(S)- and (R)-MTPA esters (26a and 26b) (Fig. 17).42
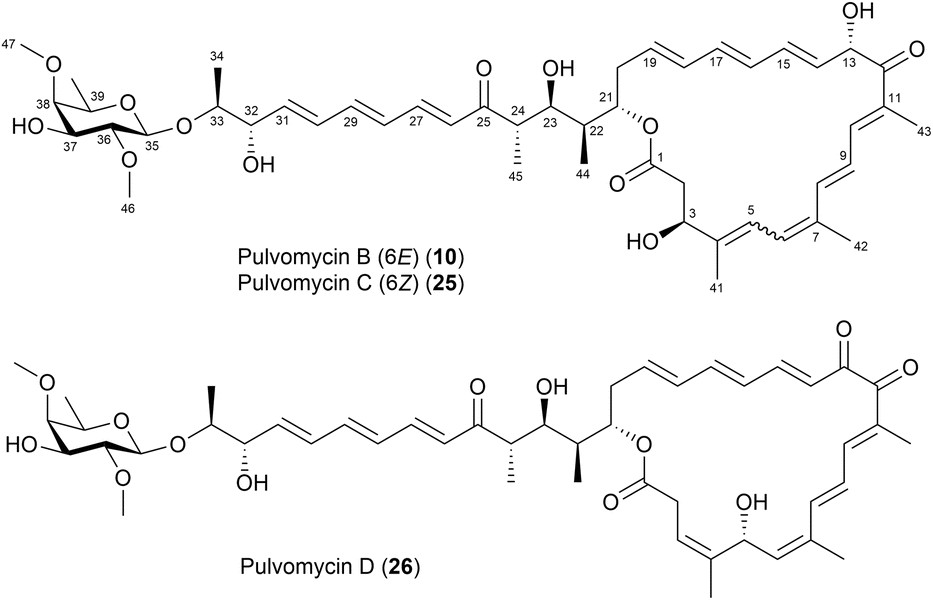 | ||
| Fig. 16 Structures of pulvomycins B–D (10, 25 and 26). | ||
 | ||
| Fig. 17 (a) ΔδS–R values of the tetra-(S)- and (R)-MTPA esters (10a and 10b) of 10 and (b) ΔδS–R values of di-(S)- and (R)-MTPA esters (26a and 26b) of 26. | ||
In a recent study, Park et al. described the use of Mosher's method to determine the absolute configurations of lenzioxazole (27), tenebriazine (28), and methyl-oxazolomycin A (29) (Fig. 18). The absolute configurations of compound 27 at positions C11, C21, C23, C24, and C25 were identified as 11R, 21S, 23S, 24R, and 25S, respectively, using the modified Mosher's method and NMR spectroscopic data analysis (Fig. 19a). Meanwhile, the absolute configuration of the chiral centre at C11 in compound 28 was analysed using the modified Mosher's method with (S)- and (R)-MTPA reagents. By evaluating the chemical shift differences between the (S)- and (R)-MTPA esters, the C11 configuration was identified as R (Fig. 19b). Moreover, the absolute configurations of compounds 29 were identified as 11R and 19R based on the differences in chemical shifts (ΔδS–R) of (S)- and (R)-MTPA esters from the 1H NMR data (Fig. 19c).55
 | ||
| Fig. 18 Structures of lenzioxazole (27), tenebriazine (28), and methyl-oxazolomycin A (29). | ||
 | ||
| Fig. 19 (a) ΔδS–R values of the (S)- and (R)-MTPA esters (27a and 27b) of 27; (b) ΔδS–R values of (S)- and (R)-MTPA esters (28a and 28b) of 28; and (c) ΔδS–R values of (S)- and (R)-MTPA esters (29a and 29b) of 29. | ||
The PGME method is another highly advanced approach for determining absolute configurations using CDAs. This approach is capable of determining the absolute configuration of a methine carbon adjacent to a carboxylic moiety. When dealing with carboxylic acids, the methine proton promotes an NOE interaction with the amide NH, indicating that the PGME moiety adopts a conformation that takes advantage of its anisotropic effect.43
In a previous study, pectenotoxins (PTXs, Fig. 20),57 a family of polyether macrolide toxins, were isolated from the dinoflagellate genus Dinophysis.58 Among the members of this family, PTX1 (30), PTX2 (31), PTX3 (32), and PTX6 (33) share the same skeleton, except for variations at the C43 position. The absolute configuration of PTX6 (33) was assigned using PGME-based NMR spectroscopy. Although PTX6 (33) lacks a methine proton at C18, the electrostatic repulsion between the lone pair electrons on the ether oxygen (O18) and the carbonyl oxygen at C43 causes the PGME moiety in PTX6-(R)- or PTX6-(S)-PGME amide to adopt a consistent conformation. This conformation enables the phenyl group of PGME to effectively extend its diamagnetic field to the protons near the chiral carbon at C18. NOEs from the amide proton to H17β, H19β, and H22 were clearly detected from the NOESY spectra of both (R)- and (S)-PGME amides acquired in C5D5N (Fig. 21). The calculated ΔδS–R values for H42 (12-Me) to H45 (27-Me) are presented in Fig. 22. Notably, the signs of ΔδS–R values were negative for protons from 12-Me to H17 and positive for protons from H19 to 27-Me in both C5D5N and CDCl3 (Fig. 22). Based on these observations, the absolute configuration at C18 was determined as S.57
 | ||
| Fig. 20 Structures of PTXs. | ||
 | ||
| Fig. 21 PGME plane and NOE data. Dashed arrows indicate NOEs in a C5D5N solution at 5 °C. | ||
 | ||
| Fig. 22 Partial structures of PTX6, with ΔδS–R values in (a) C5D5N and (b) CDCl3 at 20 °C. | ||
In a recent study, Yu et al. determined the absolute configuration of penifellutin A (34) (Fig. 23a) at C18 using the PGME method. The calculated ΔδS–R values derived from the (S)- and (R)-PGME amides (Fig. 23b) indicated that C18 has an S configuration.59
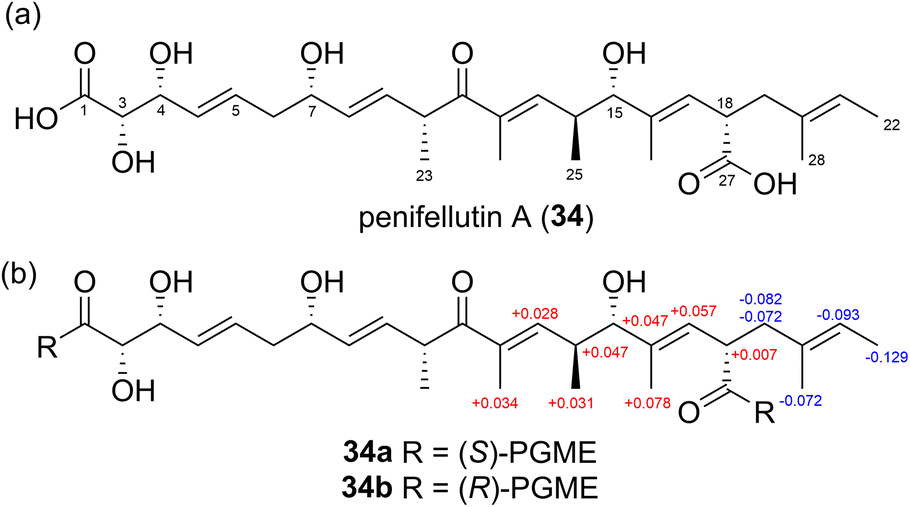 | ||
| Fig. 23 (a) Structure of penifellutin A (34) and (b) ΔδS–R values of the PGME derivatives (34a and 34b) of 34. | ||
 | ||
| Fig. 24 (a) Structure of 5-hydroxy-migrastatin and (b) the ΔδS–R values of the (S)- and (R)-MTPA esters of 35. | ||
Notably, the absolute stereochemistry of a carbinol group or an amine centre can be inferred by comparing the chemical shifts observed in the 19F-NMR spectra of (R)-MTPA and (S)-MTPA esters. This is based on a subtle conformation change caused by the interaction between the bulky phenyl group and the substituents at the stereogenic centre. When the phenyl group in a MTPA ester is ipsilateral with the relatively bulkier substituent (L2 or L3 in Fig. 25), the conformation is more destabilized than the most stable state and thus the CF3 group chemical shifts in (S)-MTPA and (R)-MTPA esters can be affected. Kakisawa and Kashman's latest modification of the Mosher approach emphasizes the benefits of using more accurate proton NMR chemical shifts while highlighting the limitations of relying solely on fluorine chemical shift data for configuration assignment. The aforementioned fluorine chemical shift measurement offers only a single-point comparison. Furthermore, accurately identifying the diastereoisomer that predominates in the most stable conformation, where the CF3 group is positioned in the carbonyl's deshielding plane, is essential (Fig. 25).50
 | ||
| Fig. 25 Most stable conformation of (S)-MTPA and (R)-MTPA esters. L2 and L3 are substituents at the stereogenic centre. | ||
Based on this, to determine the absolute stereochemistry at C5 and C9, basic Mosher-ester 19F chemical shift analysis was employed. Notably, the ΔδS–R values for 19F were positive for both trifluoromethyl groups in 35a and 35b, suggesting that the bulky phenyl groups in the (R)-MTPA esters were positioned adjacent to a bulky region in 35 (specifically the carbon bearing the methoxy group), resulting in destabilization. The trifluoromethyl groups in the bis-(R)-MTPA esters (35b) were shifted upfield compared to the bis-(S)-MTPA esters (35a) owing to their shorter residence time in the MTPA carbonyl's deshielding cone. Based on this analysis, the 5S and 9S configurations were assigned, forming an anti-1,5 diol system. Seco et al.61 established a sign distribution pattern for the ΔδS–R values of diols, which validated these assignments based on the 1H values of 35a and 35b.
 | ||
| Fig. 26 Conformational equilibrium in MPA esters. | ||
In a recent study, phytohabitols A–C (36–38), a novel group of linear polyketides featuring terminal δ-lactone rings, were identified in the extract of a rare actinomycete belonging to the genus Phytohabitans in 2022.64 Mosher's method by applying MPA was utilized to determine the absolute configuration of the multiple secondary hydroxy groups at C7, C19 and C21 of compound 36. Esterification of compound 36 with both (R)- and (S)-α-methoxyphenylacetic acid (MPA) in the presence of N,N′-diisopropylcarbodiimide (DIC) and N,N-dimethyl-4-aminopyridine (DMAP) yielded tri (R)- and (S)-MPA esters (36a and 36b). The ΔδR–S values were calculated, showing negative values for protons on the left side of C7 and positive values for protons on the right, confirming a 7S-configuration. Similarly, negative ΔδR–S values were observed for protons between C19 and C21, while positive values were recorded for protons on the left side of C19 and the right side of C21, establishing a 19R,21S-configuration (Fig. 27).
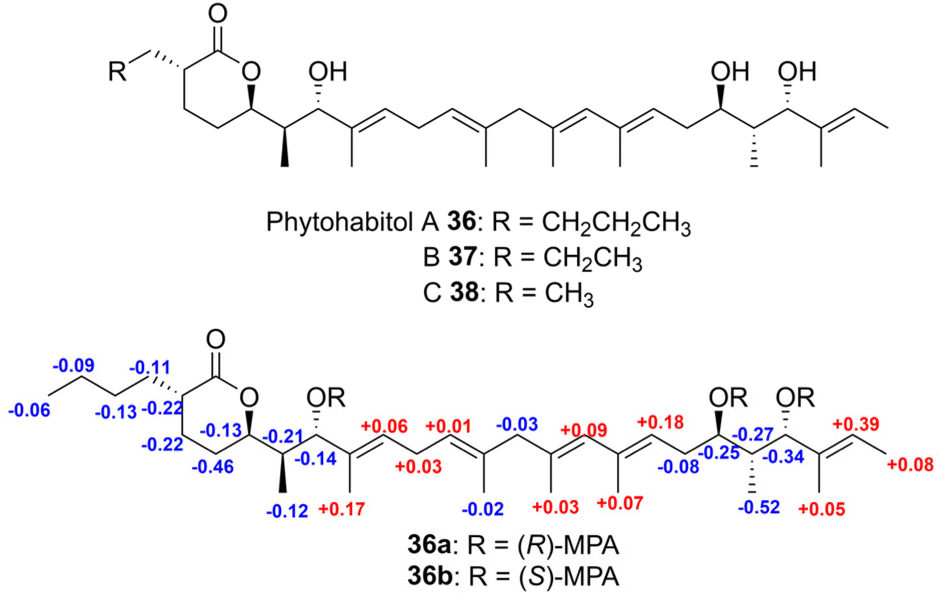 | ||
| Fig. 27 Structures of phytohabitols A–C (36–38) and 1H NMR ΔδR–S values of tri (R)- and (S)-MPA esters 36a and 36b. | ||
Notably, when researchers utilize these traditional CDA approaches, they often need to conduct multiple steps, including reaction, purification, and separation, which can be time-consuming and lead to sample loss, particularly in double derivatization methods. To overcome these limitations, Riguera's research group developed a method that utilizes solid matrix-bound CDAs, deuterated NMR solvents as reaction media, and direct high-yield derivatization within NMR tubes, eliminating the need for purification or separation. In this method, the chiral substrate reacts with the CDA-resin inside the NMR tube, forming the desired derivative in solution while the resin remains out of solution.65 This approach significantly reduces experimental time and complexity, allowing for rapid configurational analysis. Additionally, HR-MAS (High Resolution-Magic Angle Spinning) NMR was used to characterize the new CDA-resins and assess their stability and regioselectivity.
However, the application of CDA is challenging due to multiple asymmetric carbons and overlapping anisotropic effects from chiral auxiliaries. This issue is worsened by the incorrect choice of chiral auxiliaries, such as MTPA, whose high conformational flexibility leads to unpredictable results. Moreover, the chiral auxiliaries sometimes interact with each other, distorting the chemical shift patterns. As a solution, conformational studies enable the prediction of shielding effects and ΔδS–R signs, leading to new assignment methods that utilize alternative proton chemical shifts and novel NMR parameters, including 13C chemical shifts, for improved absolute configurational analysis.66
2.3. Degradation and chemical modification
 | ||
| Fig. 28 (a) Structures of gibbosols A (1) and C (39). (b) ΔδS–R values of the (S)- and (R)-MTPA esters of 39. | ||
 | ||
| Fig. 29 (a) Structures of the ozonolysed fragments of gibbosol A (1a–c). (b) Structures of the ozonolysed fragments of gibbosol C (39a′–c′). | ||
The absolute stereochemistry of C2, C7, and C9 in fragment 39a′ was determined using the modified Mosher's method. By comparing the ΔδS–R values of the MTPA esters of fragments 39a and 39b, the absolute configurations at C2 and C7 in fragment 39a′ were determined as S and R (Fig. 29b), respectively. Meanwhile, the configuration at C9 was confirmed to be R based on the widely separated H210 signals of fragment 39b (δH 4.29, 4.18) compared to those of fragment 39a (δH 4.18, 4.13). Furthermore, the configuration of C5 in fragment 39a′ was determined to be R based on the syn relationship between Me-71 and 7-OH.
Therefore, the absolute configurations of fragment 39a′ were determined to be 2S, 5R, 7R, and 9R (Fig. 29b), resembling those of fragment 1a. The absolute configurations at the C17–C25 and C37–C42 positions of 39 were determined as 17R, 19S, 21S, 23R, and 25S and 37S, 39R, 40S, 41R, and 42R through comparisons with gibbosol A. Meanwhile, the configurations at C16 in 39b′ and at C37 in 39c′ were confirmed to be R and S, respectively. Overall, the complete absolute configurations of gibbosol C (39) were determined to be 2S, 5R, 7R, 9R, 16R, 17R, 19S, 21S, 23R, 25S, 26R, 27R, 29R, 31S, 36S, 37S, 39R, 40S, 41R, 42R, 44R, 45S, 46R, 48R, 52R, 53S, 54S, 55R, 56R, 57R, 59S, 61R, 62R, 63R, 65R, and 67R.6
In another study, kalimantacin A (40)69 (Fig. 30a) was isolated from Alcaligenes sp. YL-02632S. This compound exhibited activity against Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus. Its absolute configuration was determined using a multidisciplinary approach, involving natural product isolation and chemical degradation procedures such as ozonolysis, hydrolysis, and methanolysis. In particular, the absolute configurations at C15, C17, C19, C26, and C27 were determined by comparing the NMR correlations of kalimantacin A (40) with those of the natural diol (41). Notably, the natural diol (41) was degraded, and its NMR data were compared with those of a synthetic standard (Fig. 31). The absolute configurations of diol 41 were determined to be 17S, 19S, 26R, and 27R. Ozonolysis of diol 41, followed by treatment with AcOH/H2O2, resulted in the cleavage of the 12,13-double bond to yield a dihydroxy acid, which was subsequently cyclized to form lactone (42).
 | ||
| Fig. 30 (a) Structures of kalimantacin A (40) and (b) diol (41). | ||
 | ||
| Fig. 31 Oxidative cleavage of diol (41). | ||
The absolute configuration at C15 was determined by comparing the NMR data of the synthetic lactone with those of compound 42. This synthetic lactone was obtained through a five-step reaction sequence involving (2R,4S,6S)-2,4,8-tris((tert-butyldimethylsilyl)oxy)-6-methyloctan-1-amine and ethyl-(2R,3R)-3-hydroxy-2-methylbutanoate (Fig. 32). Simultaneously, degradation procedures and chemical syntheses were employed to determine the absolute configuration at C5. Diol 41 was methylated to obtain (−)-43, which exhibited an optical rotation value of [α]D −6.8 (Fig. 33). The NMR spectra of the degradation product (−)-43 resembled those of a synthetic unsaturated methyl ester (+)-43; however, the positive optical rotation value ([α]D + 5.0) of (+)-43 suggested that it was an enantiomer of the degradation product. Hence, the absolute configuration at C5 in kalimantacin A (40) was determined to be R. Overall, the absolute configurations of diol 41 were assigned as 5R, 15S, 17S, 19R, 26R, and 27R (Fig. 30b).69
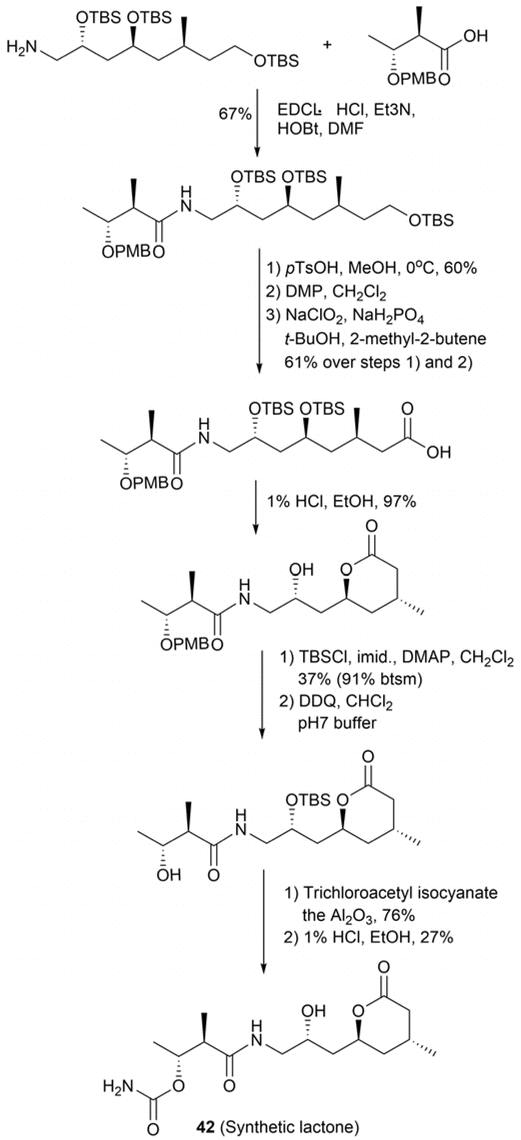 | ||
| Fig. 32 Synthesis of lactone (42). | ||
 | ||
| Fig. 33 Methylation of diol (41) followed by degradation to ester (−)-43. | ||
In a previous study, phaeospelide A (44),70 a novel 34-membered polyene macrolide, was isolated from Aspergillus oryzae. Chemical modifications were employed to determine its absolute configurations (Fig. 34a). Phaeospelide A (44) was first acetylated to produce compound 45 and subsequently decomposed via ozonolysis to yield fragments 46a–c. Among these, fragment 46b was further derivatized to yield triacetate (48), as depicted in Fig. 34b. The vibrational circular dichroism spectrum of compound 48 was compared to those of (S)- or (R)-1,2,4-butanetriols, revealing a 25S configuration for compound 44. Meanwhile, fragment 46a was subjected to a three-step reaction sequence to produce acetonides 47a and 47b (Fig. 34b). NOESY spectral analysis revealed that both compounds 47a and 47b displayed a chair conformation, with all oxygen atoms oriented along the same direction. Moreover, fragment 46c underwent similar reactions to produce compounds 47c and 47d. NOESY spectral data analyses of compounds 47c and 47d revealed that compound 47d adopted a chair conformation, while compound 47c exhibited a twist-boat conformation. The oxygen atoms at C29 and C31 in compound 47d were determined to have cis configurations, while that at C33 in compound 47c was oriented in the opposite direction relative to C31. Finally, the absolute configurations at C17 and C29 were determined as S for both compounds 47a and 47c based on the ΔδS–R values of their MTPA esters (47a-1, 47a-2, 47c-1, and 47c-2) (Fig. 34c). Ultimately, the absolute configurations of 44 were identified as 17S, 19S, 21S, 25S, 29S, 31S, and 33S.70
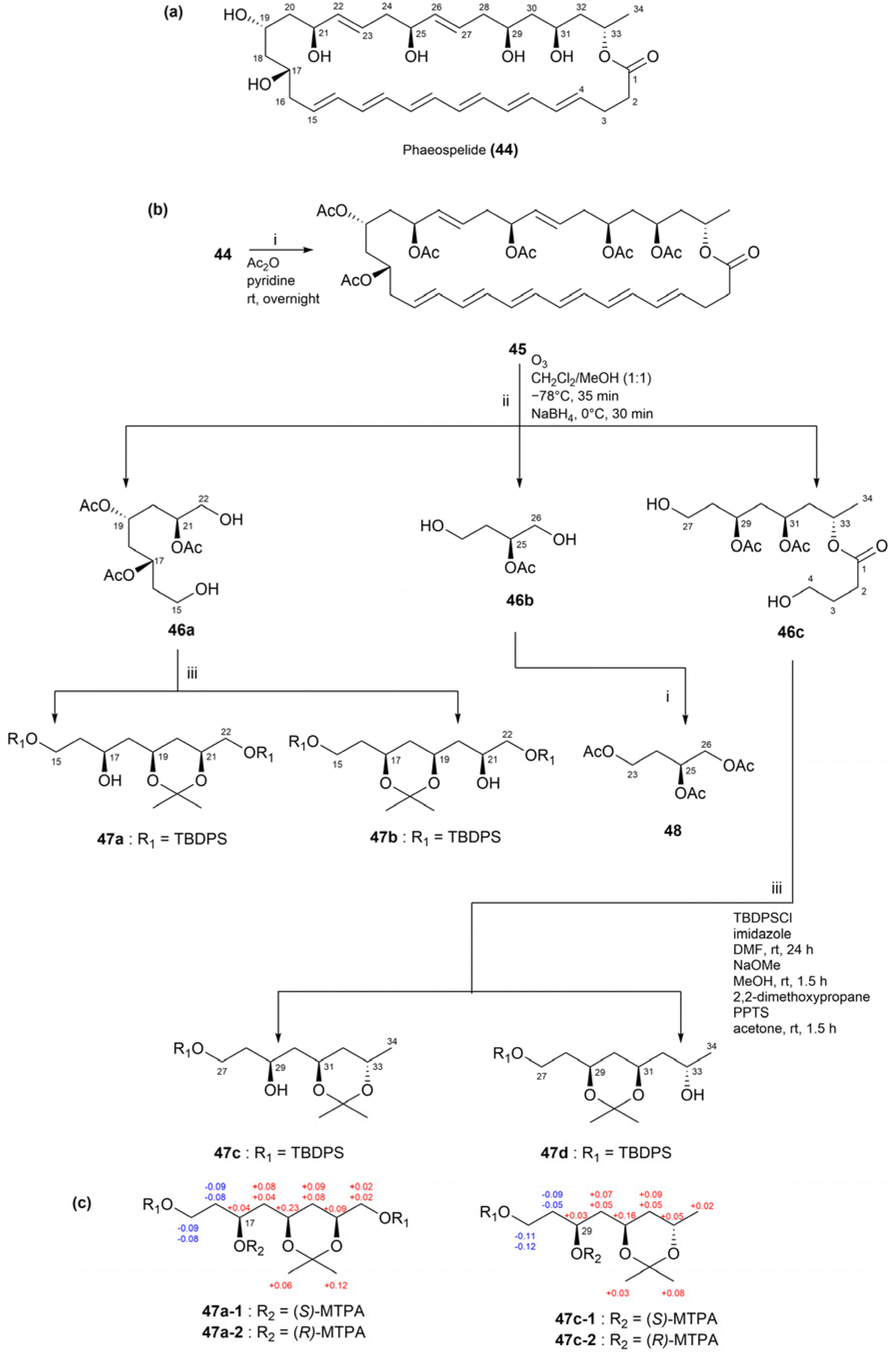 | ||
| Fig. 34 (a) Structure of phaeospelide A (44). (b) Scheme of fragmentation and derivatization of 44. (c) ΔδS–R values of the MTPA derivatives. | ||
In another study, four cytotoxic macrocyclic lactams, bombyxamycins A–C (49–51) and piceamycin (52) (Fig. 35), were isolated from the intestinal Streptomyces sp. SD53 of the silkworm Bombyx mori.71 Among the bombyxamycins, compound 49 was subjected to ozonolysis and acid hydrolysis to determine the stereochemistry at C24. During this process, following step-by-step reaction sequences, the resulting β-amino acid, 3-amino-2-methylpropanoic acid, was derivatized using Sanger's reagent to yield 3-(2,4-dinitrophenylamino)-2-methylpropanoic acid (53). This derivative was further reacted with (S)- and (R)-PGME to produce (S)- and (R)-PGME amides (53a and 53b), respectively. Analyses of the ΔδS−R values in the 1H NMR spectra of these PGME derivatives revealed that the absolute configuration of compound 53 was S (Fig. 36). Consequently, the absolute configuration at C24 in compound 49 was established as 24R. Subsequently, the secondary alcohol at C11 was derivatized with (R)- and (S)-MTPA-Cl, yielding (S)- and (R)-MTPA esters. The ΔδS–R values of these esters were determined by analysing the 1H and COSY NMR spectra of the MTPA esters. Consequently, the absolute configuration at C11 was determined as 11R (Fig. 37). Adopting the same procedure, the absolute configuration of 50 was determined as 8R, 9S, 10S, and 24R.71
 | ||
| Fig. 35 Structures of bombyxamycins A–C (49–51) and piceamycin (52). | ||
 | ||
| Fig. 36 Schemes for the ozonolysis, hydrolysis, and PGME treatment of compound 49. | ||
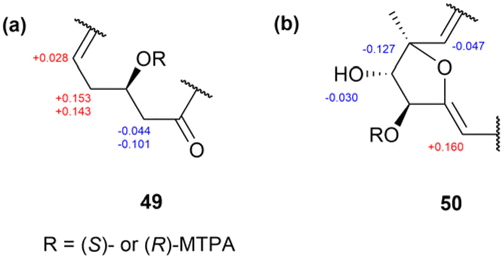 | ||
| Fig. 37 ΔδS–R values of the (S)- and (R)-MTPA esters of compounds 49 and 50. (a) Partial assignment of C9–C13 in 49. (b) Partial assignment of C7–C12 in 50. | ||
Meanwhile, for piceamycin (52), the authors refined the entire process by introducing a liquid chromatography-mass spectrometry (LC-MS) analysis step to compare the retention times of the (S)- and (R)-PGME amides derived from 52 with those derived from the authentic (2S)- and (2R)-3-amino-2-methylpropanoic acids (Fig. 38).72 This approach eliminated the need for lengthy chemical derivatization, purification, and 1H NMR spectroscopic analysis of the products.
 | ||
| Fig. 38 Improved chemical reactions for compound 52. | ||
Another study isolated four polycyclic macrolactams, cyclamenols A–D (54–57), from the Antarctic Streptomyces sp. OUCMDZ-4348 (Fig. 39).73 To determine the stereochemistry at C18 in cyclamenol A (54), the authors performed ozonolysis followed by acid hydrolysis, which yielded 3-amino-2-methylpropanoic acid (Fig. 40). This compound was then derivatized using Sanger's reagent to yield derivative 58. Subsequently, compound 58 was reacted with (S)- and (R)-PGME to form (S)- and (R)-PGME amides (58a and 58b), respectively. The observed chemical shift differences (ΔδS–R) in the NMR spectra of these amides confirmed the R configuration at the stereogenic centre C18 of compound 58 (Fig. 40). Based on these results, the absolute configuration at C18 in cyclamenol A (54) was determined to be S. Furthermore, the absolute configuration at C9 in 54 was established as R based on the modified Mosher's method (Fig. 41). Subsequently, the absolute configurations of compounds 55–57 were also determined by adopting the same procedures.73
 | ||
| Fig. 39 Structures of cyclamenols A–D (54–57). | ||
 | ||
| Fig. 40 Schemes for the ozonolysis, hydrolysis, and PGME treatment of compound 54. | ||
 | ||
| Fig. 41 ΔδS–R values of the (S)- and (R)-MTPA esters of 54–57. | ||
In another study, arenicolides A–C (59–61), a series of 26-membered macrolides, were isolated from the saline fermentation broth of the marine actinomycete Salinispora arenicola (Fig. 42).74 To confirm whether compound 59 contained a 26- or 27-membered macrocycle, the authors performed degradation via olefin cross metathesis, inspired by a previous study on a simpler polyacetylenic oxylipid. The authors then mixed compound 59 with a second-generation Grubbs catalyst in dichloromethane under a 5 atm pressure of ethylene gas. Although the rate of the reaction was slow, LC-MS analysis detected a small amount of a macrocyclic product with a 27-carbon backbone (Fig. 43). Simultaneously, the structure of compound 61 was elucidated, providing clear NMR spectroscopic data to confirm its carbon backbone. Based on this information, the researchers concluded that compound 59 was a 26-membered macrolide. Consequently, they opted not to pursue further analysis of the metathesis reaction products.
 | ||
| Fig. 42 Structures of arenicolides A–C (59–61). | ||
 | ||
| Fig. 43 Olefin cross-metathesis reaction of compound 59 and its products. | ||
The configurations of the stereogenic centres in compound 59, a poly-hydroxy macrolide, were primarily determined using the modified Mosher's method, revealing arrangements of 7R, 17R, 23S, 29S, and 33S (Fig. 44). To determine the configuration at C25, compound 59 was initially subjected to a methanolysis reaction, yielding compound 62. Acetonide formation produced compound 63. Based on this, the 1,3-dioxane ring was determined to exhibit a twist-boat conformation, given the identical chemical shifts of the gem-dimethyl carbons (25.2 ppm and 24.8 ppm). The anti-configuration between H23 and H25 in compound 63 was confirmed by analysing the proton–proton coupling constants, specifically 3JH23,H24 = 2.4 Hz and 3JH24,H25 = 5.8 Hz, within the 1,3-dioxane ring. This deduction was further supported by comparisons with models of compound 63 established by previous studies. The findings confirmed the anti and syn stereochemical relationships in compound 63, with H24/H25 exhibiting an anti relationship and H23/H24 showing a syn relationship.74 However, the absolute configurations of the epoxide group in the side chain and the C12 chiral centre remained unidentified.
 | ||
| Fig. 44 Methanolysis, acetonide formation, and Mosher's reaction of compound 59. | ||
In another study, a pair of trisoxazole macrolides, miuramides A and B (64 and 65), were isolated from a marine sponge, Mycale sp.75 Both compounds 64 and 65 exhibited potent cytotoxicity against 3Y1 cells. To determine the absolute configurations at the C25–C35 segment, the authors leveraged the similarity between miuramide A (64) and kabiramide C (66), both of which contain a C32-methoxy group (Fig. 45a). Miuramide A (64) was subjected to oxidation with RuO4, followed by reduction using a borane dimethyl sulfide complex and alkaline hydrolysis, yielding compound 67 (Fig. 45b). Further, a comparable fragment was synthesized from kabiramide C (66) through the following steps: NaBH4 reduction, acetylation of the resulting C30 alcohol, oxidation with RuO4, and reduction of the resulting dicarboxylic acid, yielding compounds 68 and 69, which are epimeric at C30. The relative configurations at C30 in compounds 68 and 69 were determined based on selective decoupling NMR experiments. The 1H NMR spectrum of compound 67 was superimposable with that of compound 68, confirming that both compounds 67 and 68 shared the same total relative configuration. Given the established (22S, 23R, 24S) absolute configurations of compound 64, the authors inferred that the absolute configurations of the remaining stereogenic centres in compounds 67 and 68 were also identical.75
 | ||
| Fig. 45 (a) Structures of miuramides A and B (64 and 65) and kabiramide C (66). (b) Chemical reactions of compounds 64 and 66 to yield fragments 67–69. | ||
 | ||
| Fig. 46 NMR chemical shift correlations of anti and syn 1,3-diol acetonides (trans- and cis-4,6 dialkyl-2,2-dimethyl-1,3-dioxanes). | ||
Macrolactin B (70)76 was the first natural product whose absolute configuration was determined using the [13C]acetonide method. Here, macrolactin B was subjected to hydrogenation and hydrolysis in HCl–methanol before the formation of the acetonide derivative. The 13C chemical shift observed at 24.8 ppm for the acetonide methyl groups in compound 71 indicated an anti-configuration for the C13–C15 diols (Fig. 47).76 The configuration of macrolactin B was further confirmed through degradation and Mosher's analysis, as illustrated in Fig. 47.
 | ||
| Fig. 47 Relative configurations of C13–C15 diols in macrolactin B, obtained from the [13C]acetonide analysis. | ||
Roflamycoin (72)76 is a polyene macrolide derived from Streptomyces roseoflavus. Its stereochemistry was determined by synthesizing several polyacetonide derivatives and evaluating their relative configurations (Fig. 48).76 Notably, roflamycoin, a cyclic hemiacetal, readily undergoes cyclization to form a spiroacetal upon acid treatment. The NMR data analysis of the configuration at C15–C19 was assigned as syn. The primary acetonide derivative obtained from the spiroacetal (compound 73) contained acetonide groups at C23–C25 and C29–C31, both of which were identified as syn through [13C]acetonide analysis. Degrading compound 72 yielded tetrahydropyran 76, which subsequently formed the diacetonide derivative 78. The 13C NMR spectral data analysis of compound 78 indicated that its C23–C25 and C27–C29 segments exhibited syn conformations. Meanwhile, the 13C NMR data analysis of pentaacetonide 74 revealed the presence of four anti rings and one syn ring. Despite five possible configurations for the arrangement of one syn and four anti acetonides, the [13C]acetonide analysis was unable to pinpoint which acetonide ring adopted the syn conformation, necessitating additional analysis. Further assessments revealed that the C29–C31 acetonide in compound 73 exhibited a syn conformation, suggesting that compound 74 must also adopt this same syn configuration. The remaining acetonides, including the C21–C23 and C25–C27 rings, exhibited anti conformations. The stereochemical assignment of roflamycoin was successfully completed by confirming the relative stereochemical assignments of C21–C23 and C25–C27, and the absolute configuration of the C35 alcohol was established using Mosher's analysis.
 | ||
| Fig. 48 (a) Structures of roflamycoin (72). (b) Polyacetonide derivatives of 72. | ||
In another study, filipin III (80),76 a polyene macrolide isolated from Streptomyces filipinensis, was structurally analysed using the [13C]acetonide method. Its polyacetonide derivatives of compound 80 were obtained by reacting it with acetone, 2,2-dimethoxypropane and pyridinium p-toluenesulfonate (PPTS) (see Fig. 49). [13C]acetonide analysis of polyacetonides 82, 83 and 84 provided key stereochemical details essential for the structural determination of filipin III. The analysis revealed that tetraacetonide 82 possessed three syn acetonide rings and one anti acetonide ring, triacetonide 83 comprised three syn acetonides, and triacetonide 84 contained two syn acetonide rings and one anti acetonide ring. Given that compound 83 only contained syn acetonide rings, the stereochemical assignments at C3–C5, C7–C9, and C11–C13 were confirmed as syn. Both 82 and 84 contained one anti acetonide ring, and because the C3–C5 region was assigned as syn based on compound 83, the anti acetonide ring common to both 82 and 84 must be located at either C9–C11 or C13–C15. The remaining acetonide rings in 82, specifically at C1′–C3 and C5–C7, were confirmed to have a syn configuration. This narrowed the possible configurations to two locations for the single anti relationship in the polyol chain: C13–C15 or C9–C11.
 | ||
| Fig. 49 (a) Polyacetonide derivatives of filipin III (80). (b) Degradation of compounds 82 and 84. (c) Overview of the configurational assignment of filipin III. | ||
To confirm the location of the anti-diol in filipin III at C13–C15, compounds 82 and 84 were degraded as illustrated in Fig. 49b, yielding compounds 85 and 86, respectively. The methyl peaks observed at 25.69 and 24.78 ppm in 85 and at 25.94 and 24.32 ppm in 86 clearly indicated the presence of an anti-1,3-diol acetonide. This confirmed that the anti acetonide ring in both compounds 82 and 84 is positioned at C13–C15. The relative configurations of the stereogenic centres in the C1′–C15 region are illustrated in Fig. 49c. Additionally, derivatization of compounds 87 and 88 using MTPA, followed by the calculation of ΔδS–R values for the resulting MTPA derivatives (87a, 87b, 88a, and 88b), revealed that the absolute configurations at C1′ and C27 were both R (Fig. 50b). Consequently, the absolute stereochemistry of filipin III was determined to be 1′R, 2R, 3S, 5S, 7S, 9R, 11R, 13R, 15S, 26S, and 27R.76
 | ||
| Fig. 50 (a) Degradation products (87 and 88) of filipin III. (b) ΔδS–R values of the MTPA derivatives. | ||
Marinisporolides A and B (89 and 90), 34-membered conjugated pentaene macrolides, were isolated from the saline culture of the marine actinomycete strain Marinispora CNQ-140.77 However, the presence of multiple pairs of 1,3-dihydroxy groups in the marinisporolides complicated their absolute configuration assignment.
Methanolysis of compound 89 produced the methyl ester 91 (Fig. 51). Based on the Kishi database, the relative configurations of the hydroxy groups at C25, C27, C29, and C31 were determined as either syn/anti/syn or anti/syn/anti. Fig. 51 illustrates the synthesis of bis-acetonide 92 from compound 89 through acetonide formation, resulting in the formation of ketals on the C25, C27, C29, and C31 hydroxy groups. Based on the chemical shifts (δC 20.2, 30.6, 20.5 and 30.6), the configurations of the hydroxy groups at C25 and C27 and at C29 and C31 were determined as syn. Further characterization using NOE correlations revealed the relative configurations of other chiral centres. Specifically, the relative configurations of the 1,3-diols were obtained as 25,27-syn, 27,29-anti, 29,31-syn, 31,32-anti, and 32,33-syn. The absolute stereochemistry of compound 89 was established using the modified Mosher's method with the methanolysis product 93 utilized in this analysis.
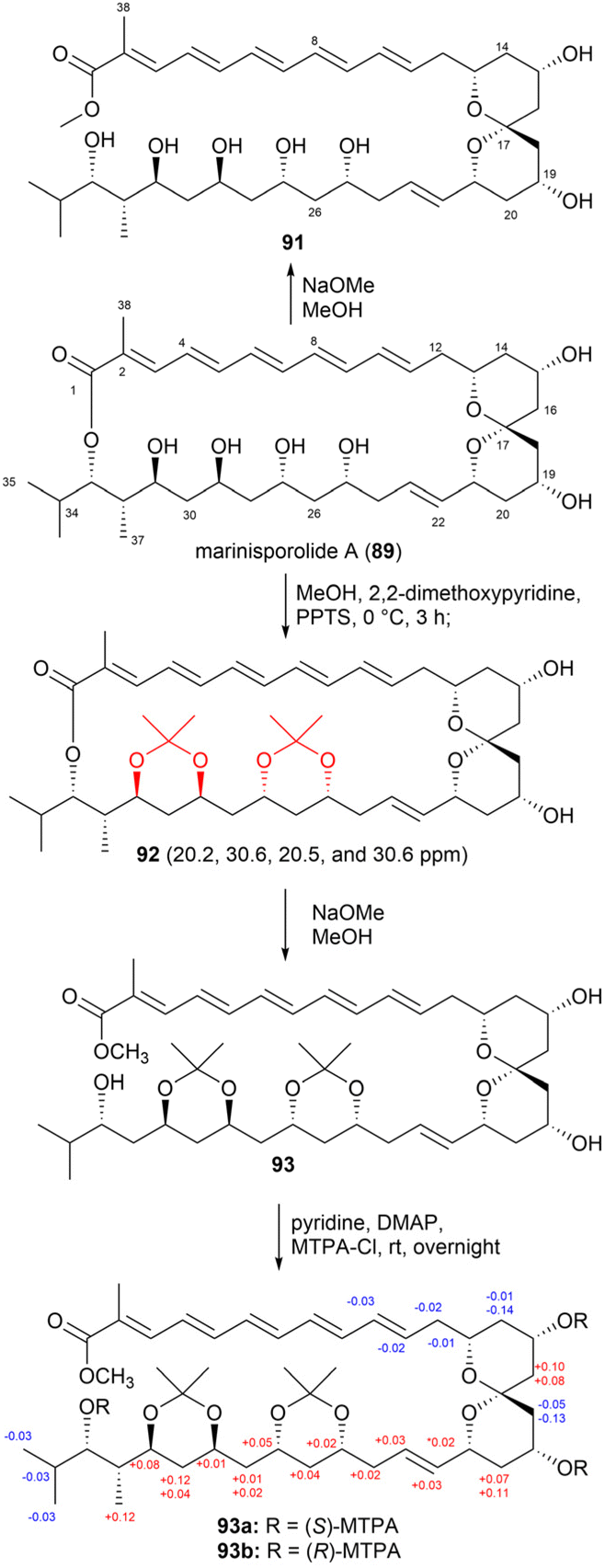 | ||
| Fig. 51 Chemical reactions of marinisporolide A (89). | ||
Methanolysis of compound 90 with NaOMe in MeOH yielded a methyl ester (Fig. 52). HMQC NMR spectroscopy analysis of the resulting compound 94 enabled the assignment of the relative configurations at C19, C21, C25, C27, C29, C31, C32, and C33 through comparisons of the observed carbon chemical shifts with those of the methanolysis product 91. Notably, the carbon chemical shifts at C25, C27, C29, C31, C32, and C33 in 94 aligned with those observed in compound 91, indicating identical relative configurations at C25–C33 in both compounds. To confirm these assignments, compound 90 was treated with Dowex X2-100 resin in MeOH for 10 min, which yielded marinisporolide A (89) as the major product. The 1H and CD spectra of compound 89, derived from 90, matched those of compound 89, confirming their identical absolute configurations.77
 | ||
| Fig. 52 Methanolysis of marinisporolide B (90). | ||
To determine the relative and absolute configurations of bahamaolides A (9) and B (95, with a 13Z configuration), the authors of a previous study subjected the 1,3-diols to acetonide formation.41 The resulting tetraacetonide products were analysed using various NMR techniques to determine the stereochemistry of the hydroxy groups. The absolute configuration was further confirmed using the modified Mosher's method and chemical derivatizations (Fig. 53).
 | ||
| Fig. 53 Acetonide formation, hydrolysis, methylation, and Mosher's reaction of bahamaolide A (9). | ||
Tetraacetonide 96 featured a free alcohol at C15 and displayed characteristic methyl group chemical shifts, indicating the presence of two syn and two anti 1,3-diols. ROESY correlations revealed that the hydroxy groups at C17 and C19 exhibited a syn configuration, while those at C21 and C23 displayed an anti configuration. Tetraacetonide 97 comprised a free hydroxy group at C31. In this compound, the configuration at C19 and C21 was syn, while that at C23 and C25 was anti. Notably, the configurations at C25, C27, and C29 were classified as syn or anti based on the appearance of distinct NMR peaks. JBCA revealed that the stereogenic centres at C34 and C35 exhibited an anti relationship. The application of the modified Mosher's method to tetraacetonide 97 and its linear methyl ester 99 revealed that the absolute configuration at C31 was S. Ultimately, the polyol configuration was confirmed to be 15R, 17S, 19S, 21S, 23S, 25R, 27S, 29S, and 31S, with the stereogenic centres at C34 and C35 being assigned as S and S, respectively.41
Iriomoteolide-3a (100), a 15-membered macrolide featuring an allyl epoxide, was isolated from a marine benthic dinoflagellate-derived strain, Amphidinium sp. HYA024.78 Determining the stereochemical configuration of compound 100 was particularly complex in the C6–C9 region, where overlapping signals from H7 and H8 complicated the assignment of the relative configuration.
To overcome these challenges, iriomoteolide-3a (100) was converted into the 7,8-O-isopropylidene derivative (101) through acetonide formation (Fig. 54). ROESY correlations confirmed a trans configuration between H7 and H8 (Fig. 55). Furthermore, the values of coupling constants (3JH6,H7, 3JH7,H8, and 3JH8,H9) for derivative 101, all measured at 8.6 Hz, suggested anti relationships among these hydrogen atoms. The absolute configuration of compound 100 was ultimately determined using the modified Mosher's method.
 | ||
| Fig. 54 Acetonide formation of iriomoteolide-3a (100) and its MTPA ester products (100a and 100b). | ||
 | ||
| Fig. 55 Key ROESY correlations for the C4–C11 region in the derivative (101). | ||
Niizalactams A and B (102 and 103, Fig. 56) belong to the same structural family as niizalactam C, but both compounds 102 and 103 possess bicyclic skeletons. These compounds were isolated from a terrestrial bacterium, Streptomyces sp. NZ-6, and co-cultured with the mycolic acid-containing bacterium Tsukamurella pulmonis TP-B0596.79
 | ||
| Fig. 56 Structures of niizalactams A and B (102 and 103). | ||
The relative stereochemistries of the pyrrolidinol rings (C22 to C25) in compounds 102 and 103 were determined as (22S*, 23R*, and 24S*) based on NOESY correlations. However, the NOESY data did not clearly resolve the stereochemistries of the 1,2,3-triol (in 102) and 1,2-diol (in 103) moieties. To address this, acetonide derivatives 104 and 107 were synthesized from compounds 102 and 103 using 2,2-dimethoxypropane and PPTS (Fig. 57).
 | ||
| Fig. 57 Chemical modifications of niizalactams A and B (102 and 103). | ||
The similar chemical shifts of the isopropylidene methyl groups and a large value of the coupling constant (3JH10,H11 = 9.0 Hz) indicated an anti configuration for the 10,11-diol in compound 102. Meanwhile, dissimilar chemical shifts (ΔδH = 0.16) and a small value of the coupling constant (3JH11,H12 = 4.5 Hz) confirmed a syn configuration for the 11,12-diol in compound 103. Consequently, the relative configurations of the triol moiety in compound 102 were assigned as (10R*, 11S*, and 12S*), and the corresponding moiety in compound 103 was inferred to share the same configuration.
The absolute configurations of compound 102 were further determined using the modified Mosher's method and Trost's method (Fig. 57). First, derivative 104 was treated with MTPA-Cl to yield MTPA esters (105a and 105b), which established the pyrrolidinol configurations as (22S, 23R, 24S). Next, the absolute configurations of the 1,2,3-triol moiety were determined using bis-MPA derivatives (106a and 106b), resulting in the assignments (10R, 11S, 12S). Structural comparisons between compounds 102 and 103 allowed the absolute configuration of compound 103 to be identified as (11S, 12S, 22S, 23R, and 24S).79
In another study, marinomycins were isolated from the saline culture of a newly discovered group of marine actinomycetes called Marinispora.80 Structural elucidation revealed that marinomycins are unusual macrodiolides composed of dimeric 2-hydroxy-6-alkenyl-benzoic acid lactones, featuring conjugated tetraene-pentahydroxy polyketide chains (Fig. 58). Biological evaluation demonstrated that these compounds exhibit potent antimicrobial activity against drug-resistant bacterial pathogens and impressive selective cytotoxicity.
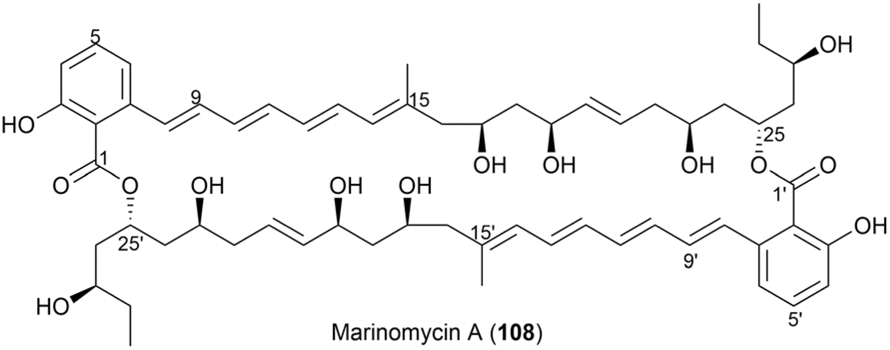 | ||
| Fig. 58 Structure of marinomycin A (108). | ||
Methanolysis of marinomycin A (108) resulted in the cleavage of both lactone linkages, yielding the monomeric methyl ester (compound 109). This product was characterized using LC-MS and NMR spectroscopic analyses, confirming compound 108 to be a dimeric macrodiolide with a unique 44-membered ring structure (Fig. 58). NMR spectral analysis of 109 enabled the assignment of the relative stereochemistry of the polyol functionalities at C17, C19, C23, C25, and C27 in compound 108.
Acetonide formation of 108 yielded a bis-acetone ketal (110, Fig. 59). NMR data analysis of this compound revealed chemical shifts of the acetonide methyl groups at δC 19.6 and 30.1, indicating that the 1,3-dioxane ring adopts a chair conformation. Based on these shifts, the authors assigned the hydroxy groups at C17 and C19 (and their symmetry equivalents, C17′ and C19′) to a syn configuration. Methanolysis product 109 was subjected to acetonide formation, resulting in two diacetonides, labeled compounds 111 and 112. NMR data analysis of these bis-ketals confirmed the relative stereochemistry of the three 1,3-diols in 108 as follows: 17,19-syn (and 17′,19′-syn), 23,25-anti (and 23′,25′-anti), and 25,27-anti (and 25′,27′-anti).
 | ||
| Fig. 59 Methanolysis and acetonide formation of marinomycin A (108). | ||
To determine the absolute stereochemistry of marinomycin A, the modified Mosher's method was employed, using acetonides 111 and 112 (Fig. 61). Analysis of the 1H NMR chemical shift differences (ΔδS–R) between the (S)- and (R)-MTPA esters (111a and 111b) revealed that the absolute stereochemistry at C23 is S. Similarly, the preparation of (S)-MTPA ester (112a) and (R)-MTPA ester (112b) from compound 112, followed by NMR data analysis, confirmed that the absolute stereochemistry at C27 is R.
To resolve the absolute configuration at C19, various derivatives were synthesized. For instance, hydrogenation of the olefinic bonds in compound 108 produced compound 113, which was further modified through acetonide formation, acetylation, and subsequent acetonide deprotection, yielding derivative 115 with six acetyl groups (Fig. 60). Treating compound 115 with (R)-MTPA-Cl and (S)-MTPA-Cl under standard acylation conditions yielded tetra-(S)-MTPA ester (115a) and tetra-(R)-MTPA ester (115b), respectively. Using both of the conventional Mosher ester 19F NMR approach and the modified Mosher's analysis, the absolute configurations of C17, C17′, C19, and C19′ were determined. The negative ΔδS–R values observed for the four fluorine signals of compounds 115a and 115b (Fig. 61) indicated that the absolute configurations at C17, C17′, C19, and C19′ are all S.
 | ||
| Fig. 60 Hydrogenation of olefinic bonds in 108, acetonide formation, acetylation, and acetonide deprotection to form 115. | ||
 | ||
| Fig. 61 Chemical shift differences (ΔδS–R) between (S)- and (R)-MTPA esters. (a) Partial assignment of (S)- and (R)-MTPA esters (111a/111b). (b) Partial assignment of (S)- and (R)-MTPA esters (112a/112b). (c) Assignment of (S)- and (R)-MTPA esters (115a/115b). | ||
Consequently, the absolute configurations at C17, C17′, C19, C19′, C23, and C25 were assigned as S, while those at C27 and C27′ were determined as R.80
For the phytohabitols A–C (36–38) mentioned in Section 2.2.5, the absolute configuration at C5 was determined using [13C] acetonide analysis.64 Methanolysis of the lactone with NaOMe in methanol produced methyl ester 116, which was subsequently reacted with 2,2-dimethoxypropane in the presence of PPTS as a catalyst, yielding bisacetonide 117 (Fig. 62). Analysis of the HSQC and HMBC spectra enabled the assignment of the 13C NMR chemical shifts for the acetonide methyl groups and acetal carbons: 24.2 ppm (×2) and 100.7 ppm for the 5,7-acetonide, and 28.6, 28.9, and 100.9 ppm for the 19,21-acetonide. These chemical shifts were consistent with a six-membered 1,3-dioxane ring in a twist-boat conformation, confirming anti relationships for the 5,7- and 19,21-diols. Consequently, the R-configuration at C5 was established.
 | ||
| Fig. 62 Preparation of bisacetonide (117). | ||
2.4. Comparisons of chemical shifts without chemical derivatization
Kishi's NMR database was constructed using small model molecules containing partial structures commonly found in natural products, such as α,γ,ε-triol motifs84 and contiguous propionate units.85 All Kishi's model molecules share an α,γ-diol core structure. This is attributed to not only the frequent occurrence of the α,γ-diol motif in natural products but also its distinctive spectroscopic features. The 13C NMR chemical shifts of the α,γ-diol motif recorded in DMSO-d6 revealed shifts of approximately 68 ppm and 66 ppm for the syn isomer and anti isomer, respectively. These results indicated a consistent trend: the 13C NMR chemical shift of the syn diol was approximately 2 ppm higher than that of the anti diol (Fig. 63).84 Additional 13C NMR data from an expanded structural motif, the α,γ,ε-triol motif (model 5), confirmed this trend. A comparison between the NMR data of α,γ-diol and allylic α,γ-diol demonstrated that functional groups attached to the motif could influence chemical shifts; however, the 2 ppm difference between the syn and anti diol isomers remained unaffected. Thus, monomethylene-spaced polyols exhibit a unique characteristic: the chemical shifts of internal carbons and their corresponding hydrogen atoms are independent of the functional groups outside the motif but depend on the relative configurations at Cα/Cγ and Cγ/Cε. This behavior remains consistent even for higher polyol systems, such as tetraols and pentaols (Fig. 64).84
 | ||
| Fig. 63 Kishi's NMR database for common motifs found in natural products, including two contiguous propionate units (model 1); α,β,γ,ε-tetraol peracetate (model 2); α,γ-diol (model 3); and allylic α,γ-diol (model 4). (a) Recorded in DMSO-d6, (b) recorded in CD3OD, and (c) recorded in CDCl3. | ||
 | ||
| Fig. 64 Kishi's NMR database for additional motifs found in natural products, including α,γ,ε-triol (model 5), along with the empirical rule for monomethylene-bridged polyols. (a) recorded in DMSO-d6 and (b) recorded in CD3OD. | ||
As detailed in Section 2.3, to determine the configuration of marinomycin A (108), methanolysis of 108 was performed to obtain the ester derivative (109), and the 13C NMR spectrum of this derivative (109) was recorded in DMSO-d6 (Fig. 59).80 Notably, the alkenyl chain in (109) comprises two stereoclusters: the first from C16 to C20 and the second from C22 to C28 (Fig. 66). The relative configurations of these stereoclusters (stereocluster 1 and stereocluster 2) were determined by comparing the experimental 13C NMR data with those of model 4 and model 5, respectively. Consequently, stereocluster 1 was determined to have a syn orientation, while the three hydroxy groups at C23, C25, and C27 were determined to exhibit anti/anti orientations.
To confirm the configurations assigned by the Kishi's database, acetonide derivatives of 108 and 109 were prepared. The 13C NMR spectra of these derivatives were analysed following the method described by Rychnovsky, enabling the assignment of the relative configurations of the two stereoclusters. The results of Rychnovsky's method demonstrated good agreement with the configurations predicted by Kishi's NMR database. Finally, the absolute configuration of marinomycin A was assigned as illustrated in Fig. 58. This assignment was achieved by determining the S configurations at C17, C19, and C23 through a combination of chemical modifications and both classical and modified Mosher's methods. Marinomycins B (118) and C (119), geometric isomers of marinomycin A, were identified as photoconversion products of 108. Hence, the stereochemistry of 118 and 119 was assumed to be identical to that of 108 (Fig. 65).80
 | ||
| Fig. 65 Structures of marinomycins B and C (118 and 119). | ||
 | ||
| Fig. 66 Comparison of the experimental 13C NMR data of stereocluster A in the degradation product (109) with the corresponding carbons in model 1. Data recorded in DMSO-d6. | ||
Oasomycins and desertomycins are 42-membered macrolactones isolated from Streptoverticillium baldacii subsp. netropse (Fig. 67).86 These compounds typically undergo natural structural modifications on the side chains at C22 and C50, while the macrolactone core remains preserved. Given that these compounds share the same macro-ring skeleton, oasomycin A (120) was selected as a representative compound for the stereochemical analysis of the macrolactone moiety. Notably, the ring structure consists of two stereoclusters: stereocluster A (from C5 to C10) and stereocluster B (from C21 to C38). The relative configuration of stereocluster A was determined by comparing the 13C NMR chemical shifts of the C5–C10 fragment with those of model 1, which contains a two-contiguous-propionate-unit motif. As illustrated in Fig. 68, C6, C8, C48, and C49 in stereocluster A corresponded to C8, C6, C12, and C11 in model 1, respectively. The differences in the 13C NMR chemical shifts between the corresponding carbons revealed that stereocluster A exhibits anti/anti/syn configurations (Fig. 68).81
 | ||
| Fig. 67 (a) Structures of oasomycins A (120) and B (121). (b) Relative configurations of stereocluster A (from C5 to C10). (c) Relative configurations of stereocluster B (from C21 to C38). | ||
 | ||
| Fig. 68 Comparison of the experimental 13C NMR data of stereocluster A in compound 120 with the data of corresponding carbons in model 1. NMR data were recorded in DMSO-d6. | ||
Next, stereocluster B (Fig. 67a) of compound 120 was divided into four parts: part B1 (red box, from C22 to C23), part B2 (green box, from C23 to C29), part B3 (blue box, from C29 to C33), and part B4 (orange box, from C33 to C37) (Fig. 67c). Parts B2 and B4 were compared with model molecules 5 and 5A owing to the presence of monomethylene-spaced polyol motifs. Notably, the 13C NMR chemical shifts of C25 and C27 in part B2 were similar to those of the internal carbons in model 5A, suggesting that the relative configurations at C23/C25/C27/C29 are anti/anti/syn (Fig. 69). Similarly, the triol in part B4 was assigned an anti/anti configuration, as the chemical shift of C25 matched that of the internal carbon of the anti/anti isomer in model 5 (Fig. 69).87
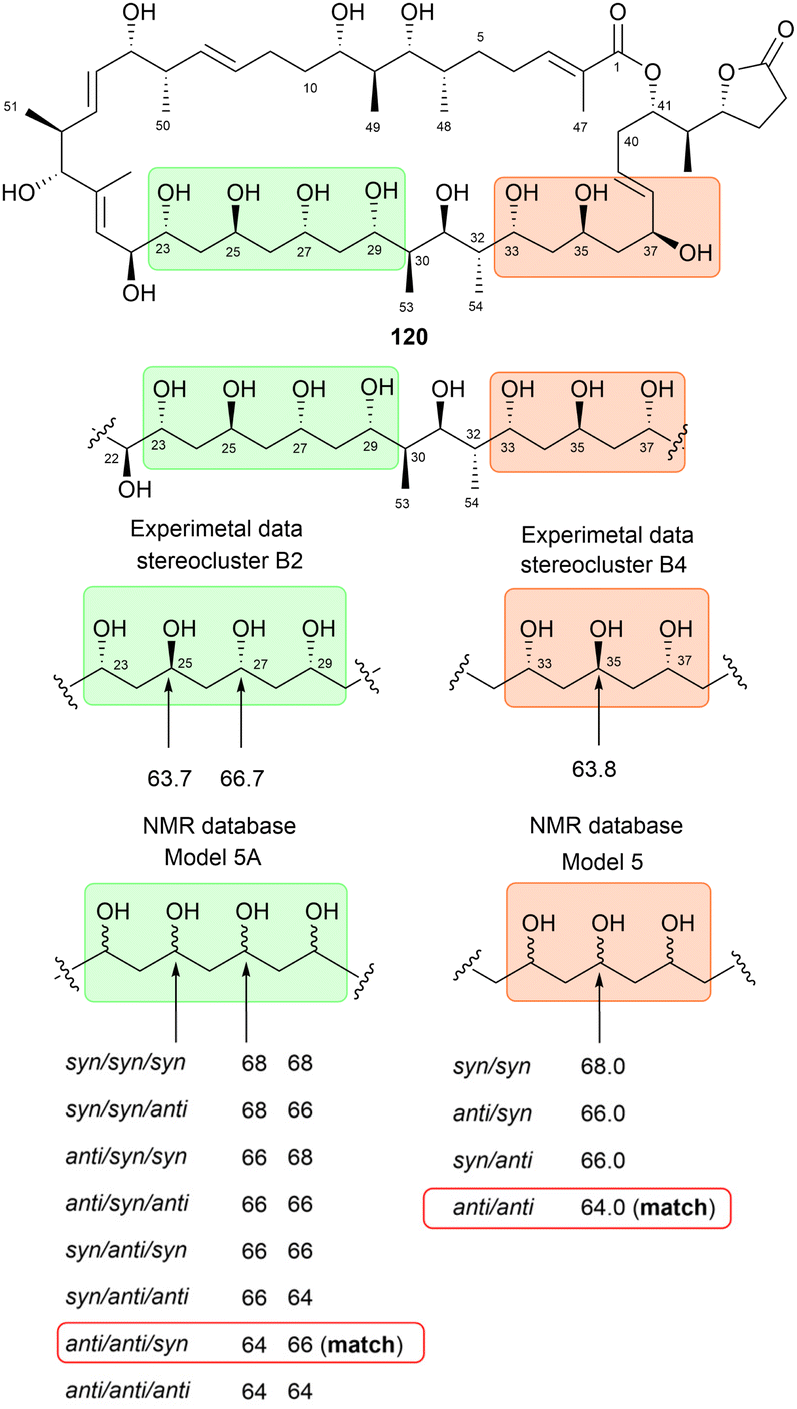 | ||
| Fig. 69 Determination of the relative configurations of stereoclusters B2 (green box, from C23 to C29) and B4 (orange box, from C33 to C37) of 120. NMR data were recorded in DMSO-d6. | ||
For part B3, which contains a two-contiguous-propionate-unit motif, comparison with model 1 revealed two correlation modes. The first mode (C29–C31) and the second mode (C31–C33) corresponded to the C5–C7 fragment of model 1. The relative configurations of C29/C30/C31/C32 were assigned as anti/syn/anti based on the similarities in the 13C NMR chemical shifts of C30 and C53 and their corresponding carbons, C6 and C11, in model 1. However, determining the configuration of the substituents in the C31–C33 fragment proved more challenging. This was because comparisons between the 13C NMR data of C32 and C54 and the data of the corresponding carbons from Kishi's database suggested three possible relative configurations: syn/anti/syn, syn/syn/anti and anti/syn/syn (Fig. 70). Given that the configuration from C30 to C32 must be consistent across both correlation modes, only the syn/anti/syn isomer of the second mode met this requirement. Therefore, the relative configuration of part B3 was successfully assigned as anti/syn/anti/syn (Fig. 70).87
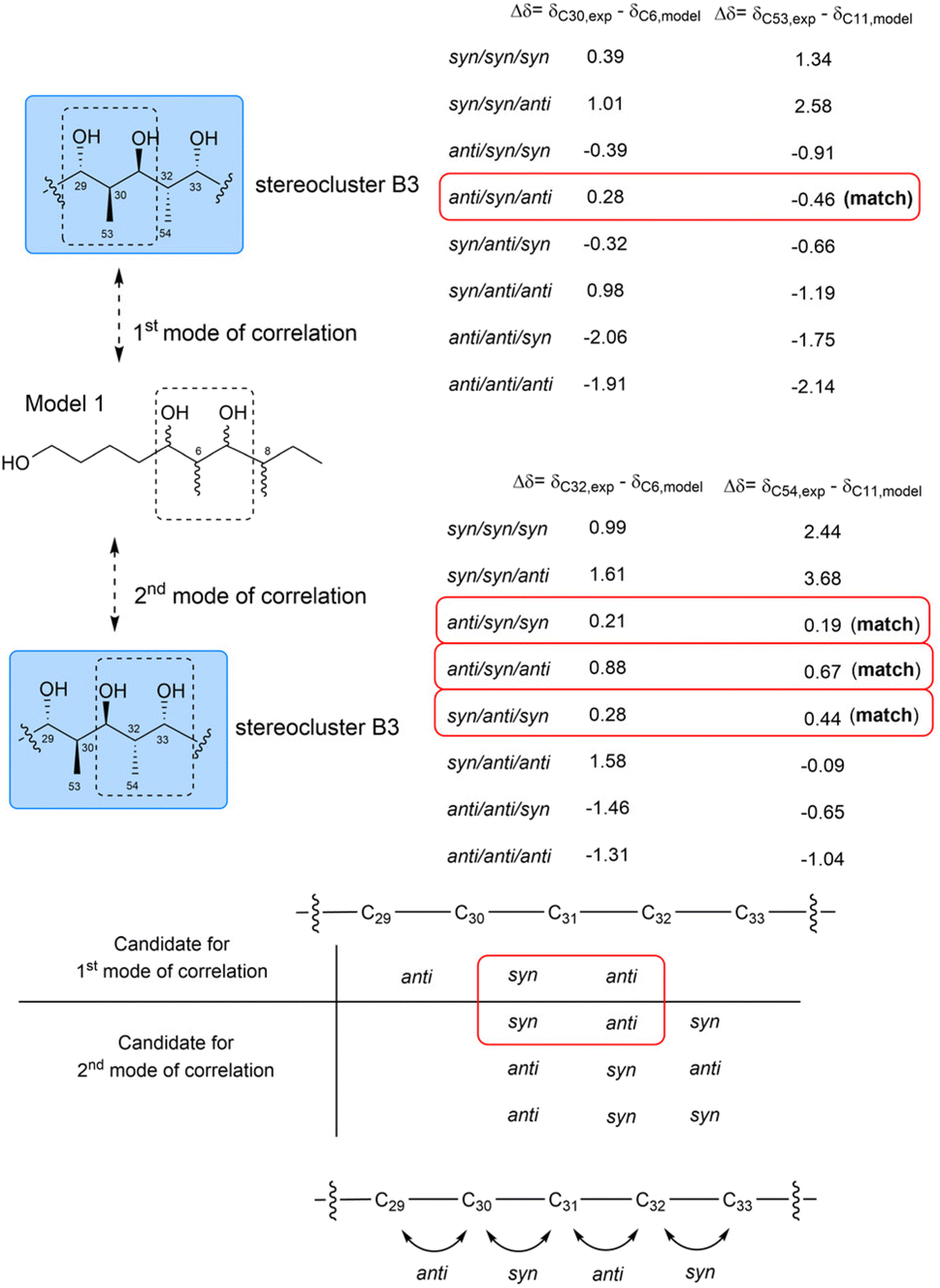 | ||
| Fig. 70 Determination of the relative configurations of part B3 (blue box, C29 to C33). NMR data were recorded in DMSO-d6. | ||
To determine the configuration of part B1, compound 122 was prepared through multiple steps of chemical degradation and modification of 120. Model 4, which shares the α,β,γ,ε-tetraol peracetate partial structure of compound 122, was also prepared to expand the database for assigning the relative configuration of C22 and C23 in part B1. The 13C NMR chemical shifts of C21, C22, and C23 in compound 122 were similar to those of C1, C2, and C3, respectively, confirming the anti/anti orientation of substituents attached to C22, C23, and C25 (Fig. 71). Interestingly, the anti configuration of the two hydroxy groups at C23 and C25, determined through the analysis of model 2, aligns with the relative configuration of C23/C25 determined using model 5A (Fig. 69). This consistency across analyses utilizing Kishi's NMR database demonstrates the reliability and robustness of the method.87
 | ||
| Fig. 71 Determination of the relative configurations of part B1 (red box, from C21 to C23). NMR data were recorded in CDCl3. | ||
The combined analysis of parts B1, B2, B3, and B4 enabled the researchers to determine the relative configuration of stereocluster B, as depicted in Fig. 67c. The 1H NMR spectrum of compound 122 was compared with that of the degradation product derived from natural oasomycin A, demonstrating excellent agreement with the configuration predicted using Kishi's NMR database.88
These successful case studies demonstrate that Kishi's NMR database is a powerful and straightforward tool for determining the relative configurations of polyketides, particularly polyols. Additionally, Kishi's database serves as a useful alternative to JBCA, especially when J-coupling values are altered or cannot be observed. However, this method has several limitations. First, it is applicable only to specific structural motifs, such as monomethylene-spaced polyols and allylic diols. Second, in numerous cases, Kishi's database cannot reliably distinguish between syn/anti and anti/syn isomers.84 For instance, in the case of α,γ,ε-triols, the chemical shift of the internal carbon is approximately 66 ppm in both the syn/anti and anti/syn isomers. Finally, given that the NMR data included in Kishi's database have only been recorded in DMSO-d6 and CD3OD, this approach is not applicable to compounds that are insoluble in these solvents.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of BMBA-p-Me and an achiral solvent (such as CDCl3, DMSO-d6, pyridine-d5, or CD3OD) was used in the NMR experiments.89 The absolute configurations of alcohols were assigned by analysing the chemical shift behaviors of the adjacent carbons in (R,R)- and (S,S)-BMBA-p-Me, following the empirical rule shown in Fig. 72c.90 Notably, this method allows natural product chemists to determine the absolute configuration of hydroxy groups without requiring chemical derivatization, making Kishi's bidentate solvents a valuable alternative when the Mosher's method fails. Furthermore, each hydroxymethine unit can be analysed independently and simultaneously, simplifying the process of absolute configuration determination. This technique also has broad applications in determining the stereochemistry of a variety of alcohol types, including secondary, benzyl, biaryl, and tertiary alcohols.89,90
:2 mixture of BMBA-p-Me and an achiral solvent (such as CDCl3, DMSO-d6, pyridine-d5, or CD3OD) was used in the NMR experiments.89 The absolute configurations of alcohols were assigned by analysing the chemical shift behaviors of the adjacent carbons in (R,R)- and (S,S)-BMBA-p-Me, following the empirical rule shown in Fig. 72c.90 Notably, this method allows natural product chemists to determine the absolute configuration of hydroxy groups without requiring chemical derivatization, making Kishi's bidentate solvents a valuable alternative when the Mosher's method fails. Furthermore, each hydroxymethine unit can be analysed independently and simultaneously, simplifying the process of absolute configuration determination. This technique also has broad applications in determining the stereochemistry of a variety of alcohol types, including secondary, benzyl, biaryl, and tertiary alcohols.89,90
 | ||
| Fig. 72 (a) Structure of BMBA-p-Me. (b) Recognition mode between BMBA-p-Me and an isolated hydroxy group. (c) Empirical rule for determining the absolute configurations of alcohols using Kishi's bidentate chiral solvent. | ||
The absolute configuration of the hydroxy group at C23 in pulvomycin B (10) could not be determined using Mosher's method owing to the failure of MTPA derivatization by steric-hindrance. To address this problem, the 13C NMR behavior of adjacent carbons in the presence of (R,R)- and (S,S)- BMBA-p-Me solvents was analysed, leading to the successful assignment of the C23 hydroxy group as an S-configuration (Fig. 73).42
 | ||
| Fig. 73 Chemical shift differences of the carbon adjacent to C23 in pulvomycin B (10) observed in a 5:2 mixture of (R,R)- and (S,S)-BMBA-p-Me and CDCl3. | ||
Mohangic acids A–E (123–127), a series of p-amino acetophenoic acids, were isolated from Streptomyces sp. SNM31, a mudflat-derived strain. The 13C NMR spectrum of mohangic acid A (123) was recorded in a 5:2 mixture of BMBA-p-Me and pyridine-d5. Using Kishi's empirical rules, the three secondary alcohols in the alkyl chain of mohangic acid A were quickly assigned as 3R, 11S, and 15R without requiring chemical derivatization or degradation (Fig. 74). Owing to their shared biosynthetic origin and the similarities in the NMR data of the side chains, the absolute configurations of mohangic acids B–E (124–127) were also assigned as 3R, 11S, and 15R, consistent with those of mohangic acid A (123).91
 | ||
| Fig. 74 (a) Structures of mohangic acids A–E (123–127). Chemical shift differences of the carbons adjacent to tertiary alcohols in mohangic acid A (123): (b) C15, (c) C11, and (d) C3, observed in a 5:2 mixture of (R,R)- and (S,S) BMBA-p-Me and pyridine-d5. | ||
Another successful application of Kishi's bidentate solvent was demonstrated with glisoprenin A (128). Glisoprenin A (128), a member of the polyprenol subclass isolated from the fungus Gliocladium sp. FO-1513, contains four stereogenic tertiary alcohols.92 The initial attempt to assign the absolute configurations of the stereogenic centres was unsuccessful owing to overlapping 13C NMR signals of adjacent carbons, recorded in (R,R)- and (S,S)-BMBA-p-Me, making it impossible to analyse their chemical shift behavior. This challenge was overcome by adding chiral shift reagents, (R)- and (S)-Pr(tfc)3 (Fig. 75a), to the NMR mixture at a concentration of 25 mol% per hydroxy group in the structure of 128. The addition of these reagents successfully spread out the 13C resonances of all eight carbons adjacent to the tertiary alcohols, allowing for the detection of chemical shift differences between the α-carbons in (R,R)- and (S,S)-BMBA-p-Me. This analysis revealed that the absolute configurations of all four stereogenic centres were S (Fig. 75b). The assignment was further confirmed by comparing the 1H NMR spectra of natural and fully synthetic glisoprenin A (128).93,94
 | ||
| Fig. 75 (a) Structures of (R)- and (S)-Pr(tfc)3, chiral shift reagents. (b) Structures and chemical shift differences of the carbons adjacent to the tertiary alcohols of glisoprenin A (128). | ||
Leiodermatolide (129), a 16-membered macrolactone connected to a δ-lactone via an alkenyl chain, was first isolated from the marine sponge Leiodermatium sp. by Amy E. Wright's research group. Intensive NMR data analysis determined the relative configuration of 129 (Fig. 76). However, Mosher's method failed to assign its absolute configuration. To address this, the DP4 methodology was applied. Model structures 129a (corresponding to the marcolactone part) and 129b (corresponding to the δ-lactone part) were used for the calculations. The experimental 1H and 13C NMR data of 129 and the DP4-predicted 1H and 13C NMR data of 622 conformers of 129a, generated from 32 possible diastereoisomers, and 99 conformers of 129b, generated from four possible diastereoisomers, were analysed. This analysis assigned the absolute configurations of the macrolactone core as (6R, 7S, 8S, 9S, 14S, and 15S) and the δ-lactone part as (21S, 22S, and 23S), with >99% probability. Given that Mosher's method failed to assign the absolute configurations, the final confirmation of these assignments required comparisons of NMR data and optical rotation between synthetic and natural leiodermatolide (129).97 The macrolactone core of 129 and a non-natural isomer of 129 (6S, 7S, 8R, 9S, 14S, 15S) were synthesized independently by the research groups of Stephen M. Dalby and Martin E. Maier, respectively, confirming the DP4-predicted configuration for the macrolactone.98,99 Finally, the proposed stereochemistry of the δ-lactone part (21S, 22S, 23S) was validated by Alois Fürstner through total synthesis.100
 | ||
| Fig. 76 (a) Structure of leiodermatolide (129). (b) Model structures 129a and 129b used in DP4 calculations for the macrolactone and δ-lactone parts, respectively. | ||
Neopeltolide (130), a 14-membered macrolide, was isolated from a marine sponge belonging to the family Neopeltidae. The planar structure and relative configuration of neopeltolide were initially determined by 2D NMR data analysis (Fig. 77). However, owing to the limited quantity of 130, its absolute configuration remained ambiguous.101 Application of the DP4 method revealed that the correct configuration of neopeltolide was 130b, the 11,13-epi-isomer of the originally proposed structure.
 | ||
| Fig. 77 Structures of the original (130) and modified neopeltolides (130a and 130b). | ||
The absolute configurations of formicolides A (2) and B (3) were determined through a combination of 2D NMR, genomic approaches, and DP4 calculations.36 For formicolide A (2), DP4 analysis was used to determine the absolute configurations at C5, C13, and C19. An analysis of the calculated 1H and 13C NMR data of the 50 conformers of two possible diastereomers (2a and 2b) of 2 with the experimental NMR data of 2 led to the stereochemical assignment of C5, C13, and C19 as 5R, 13S, and 19S, with 100.0% probability. Meanwhile, for formicolide B (3), DP4 calculations focused on the side chain (C20 to C28). Using 17 conformers of two possible diastereomers, 3a (19S, 20S, 21R, 22R, and 25R) and 3b (19S, 20S, 21R, 22R and 25S), the side-chain configuration was determined as 19S, 20S, 21R, 22R, and 25R (Fig. 78).36
 | ||
| Fig. 78 (a) Two diastereomers considered in DP4 calculations for the macrolactone core of formicolide (2). (b) Two diastereomers considered in DP4 calculations for the side chain of formicolide B (3). | ||
As mentioned in Section 2.3, the absolute configuration of C12 of arenicolide A (59) was unidentified although other chiral centers were assigned by combination of various methods such as JBCA, modified Mosher's method, and chemical modification. Recently, the stereocenter at C12 was assigned as 12-(R) with 100% probability by employing DP4. In total, 40 conformers of two C12-epimers of the macrolactone ring of 59 were considered for the calculation. Bioinformatic analysis revealed the absence of key tyrosine residue in the ER domain of module 12 of biosynthetic PKS genes, suggesting an R configuration of C12, which is consistent with the result from the DP4 calculation.102
Although DP4 is simple and robust aid for the determination of structure as well as configuration of a natural product, it has the common limitation of all comparison-based methods, where an incorrect structure may coincidentally match the experimental data more closely than the correct candidate.103 There are several reasons for this problem, such as energy miscalculation, subtle differences in chemical shift between isomers, and the uncertain correctness of proposed structure candidates. To overcome this, Jonathan M. Goodman's group developed DP5 probability. Generally, DP5 is similar to DP4 in giving the probability that a proposed structure candidate is correct. However, the advancement of DP5 is that the structure candidates are proposed without any assumption about their correctness, since DP5 compares each proposed isomer to the chemical space, while DP4 compares each isomer against each other.104 As of the drafting of this paper, DP5 has been available for only two years, so there are no case studies reporting the use of DP5 to determine the configuration of type-1 polyketide-derived natural products. In conclusion, both DP5 and DP4 each have their own unique advantages. DP5 shows higher reliability for cases where the proposed structures are of uncertain correctness, while DP4 is suitable for cases where the correctness of the structure candidates is guaranteed.103,104 By understanding the strengths and limitations of each method, natural product chemists can choose the most appropriate method for each individual case to obtain a result with the highest probability.
2.5. Exploitation of chiroptical properties
 | ||
| Fig. 79 Relationship between the steric configuration of the substrate and the sign of the O–C–C–O dihedral in the Cottonogenic derivative. | ||
This phenomenon, known as induced ECD, occurs when an achiral complex, such as [Mo2(OAc)4], interacts with a chiral environment, leading to the appearance of ECD signals. The presence of chiral substrates or solvents can induce ECD responses in the molybdenum complex, enabling stereo-specific interaction analysis. The ECD spectrum of the [Mo2(OAc)4] complex can be further analysed to identify the specific electronic transitions responsible for the observed circular dichroism. By comparing experimental ECD data with theoretical calculations, researchers can determine how the chiral ligand influences the complex's electronic structure. Specific peaks in the ECD spectrum correspond to different conformational states, providing insights into stereochemistry.
A practical application of this method was demonstrated in the study of dentigerumycin (131, Fig. 80), a type-I PKS-derived non-ribosomal peptide synthetase (NRPS) natural product produced by the bacterium Pseudonocardia associated with fungus-growing ants, such as Apterostigma dentigerum. These ants maintain a mutualistic relationship with the fungi they cultivate for food and actinobacteria that produce antibiotics, such as Pseudonocardia, which inhibit the parasitic fungus Escovopsis sp.106 Molybdenum acetate [Mo2(OAc)4] was used to determine the absolute configuration of dentigerumycin. The researchers determined the absolute configuration of the acyl side chain through the CD spectral analysis of the [Mo2(OAc)4] complex with the 1,2-diol at positions C29 and C30. Following the empirical rule, the dentigerumycin complex displayed a negative sign at 305 nm in its induced CD spectrum, indicating that the C29 and C30 stereogenic centres adopt the 29S and 30R configurations if the 1,2-diol remains in its expected low-energy conformation.
 | ||
| Fig. 80 Structure of dentigerumycin (131). | ||
The process begins by identifying the potential conformers of the compound. This step is typically performed using Monte Carlo methods in combination with molecular mechanics (such as MMFF94) or semi-empirical approaches (e.g., AM1) to estimate the relative energies of the conformers. Once identified, these conformers are further optimized using DFT techniques. In the next step, UV/ECD TDDFT calculations are performed on the optimized conformers using computational software such as Gaussian, TURBOMOLE, or NWChem.107–109 The UV/ECD spectra of each individual conformer are then averaged using Boltzmann statistics to generate the final calculated ECD spectrum. The accuracy of these calculations depends on the choice of basis sets and functionals. Larger basis sets typically provide more accurate results but require more computational time. The level of ECD calculation is expressed as a combination of the functional and basis set used, indicating the methods employed at each stage. The TDDFT calculations yield key parameters, including excitation energies, oscillator strengths, and rotatory strengths. Oscillator strengths simulate the UV spectrum, while rotatory strengths are used to generate the ECD spectrum. The calculated rotatory strength values are converted into ECD curves using either a Gaussian or Lorentzian distribution function.
 | ||
| Fig. 81 Structures of spirosnuolides A–D (132–135). | ||
 | ||
| Fig. 82 Experimental ECD and calculated ECD spectra of spirosnuolide B (133). | ||
In piceamycin (52) mentioned in Section 2.3.2,72 the stereochemistry of the unprotonated C8 carbon of the cyclopentenone remained undetermined. Due to the inability to chemically derivatize this chiral centre for stereochemical analysis, ECD calculations were employed. To determine the stereochemistry, three-dimensional models representing the two possible diastereomers (8S/24R and 8R/24R) were constructed, and ECD spectra were calculated for both. The experimental ECD spectrum of piceamycin revealed a positive Cotton effect at 300 nm and a negative Cotton effect at 455 nm. Notably, the calculated ECD spectrum for the 8S/24R diastereomer closely matched the experimental data (Fig. 83). This alignment suggests that the C8 configuration of piceamycin is S.72
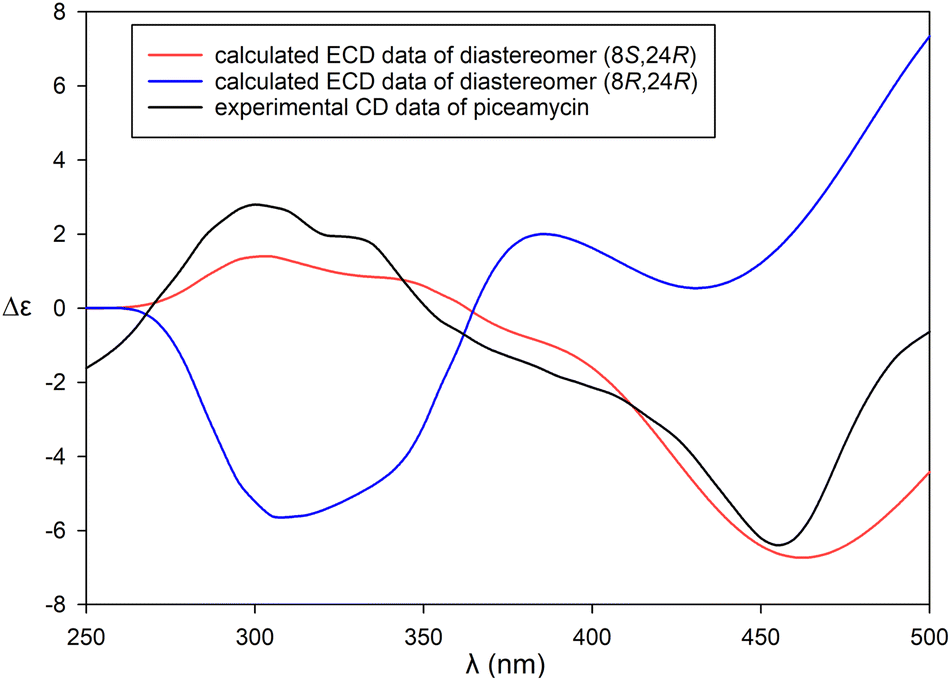 | ||
| Fig. 83 Experimental ECD of piceamycin (52) and calculated ECD spectra of two possible diastereomers (8S/24R and 8R/24R). | ||
(+)-Phorbasides A (136a) and B (136b) are two macrolides structurally related to the rare callipeltosides, such as (−)-callipeltoside A (137). These compounds were isolated from a sponge of the Phorbas genus in Western Australia (Fig. 84).111 The authors conducted a comprehensive configuration analysis of 136a and 136b using semi-quantitative CD analysis, leveraging the Cotton effect (CE) caused by hyperconjugation between the cyclopropyl ring's σ-like bonds and the extended ene-yne π system. This method revealed that the configuration of the trans-chlorocyclopropane ring in 136a and 136b is opposite to that in 137.
 | ||
| Fig. 84 Structures of (+)-phorbasides A and B (136a and 136b), (−)-callipeltoside A (137), and synthesized enantiomeric models ((+)-138a, (−)-138b, (+)-139, and (+)-140). | ||
The CD spectra of 136a and 136b are nearly identical, exhibiting a pronounced positive CE [λmax 232 (Δε +9.1), 241 (+8.1)] and vibronic fine structure associated with the asymmetrically perturbed ene-yne chromophore. A red shift in the UV spectrum of 136a, attributed to significant hyperconjugation with the chlorocyclopropane ring, supported these findings. The authors hypothesized that the sign of the CE would depend on the configurations of both the adjacent trans-chlorocyclopropane ring and the C13 acyloxy substituent.
To determine the configurations at C18 and C19 in 136a and 136b, the authors synthesized enantiomeric models (+)-138a and (−)-138b with defined chirality at the chlorocyclopropane rings (Fig. 84). CD spectral analysis confirmed that the observed CE arises from the asymmetry of the ene-yne chlorocyclopropane chromophore. The CE of 138a aligned with that of (+)-138a and opposed that of (−)-138b. The acyloxy substituent at C13 affected the magnitude but not the sign of the CE, as demonstrated using models (+)-139 and (+)-140. Consequently, the complete macrolide configurations of 136a and 136b were assigned as (2S, 3S, 5S, 6R, 7R, 8R, 9R, 13R, 18R, and 19S).
The stereochemical determination of long-chain natural product lipids is particularly challenging, especially for those with multiple hydroxy groups, such as caylobolide A, derived from the cyanobacterium Lyngbya majuscula.112 These lipids, common in polyketides and glycolipids, present significant difficulties in elucidating the relative configurations of isolated stereogenic centres. Polyketides often contain segments with different glycol architectures (1,3-, 1,5-, or even 1,7-glycols), adding complexity to stereochemical analysis.
Several methodologies have been employed to resolve the configurations of 1,2- and 1,3-glycols, including 13C NMR data analysis, exciton coupling circular dichroism (ECCD), J-based analysis, and the use of universal NMR databases. However, in acyclic chains or macrocyclic polyketides, hydroxy groups separated by four or more carbon–carbon (C–C) bonds often fail to provide meaningful configurational information through NMR or CD spectral properties. For example, the CD spectra of acyclic 1,5-glycol diarylcarboxylate esters in isotropic solutions typically show only baseline signals.
The authors proposed a solution involving the pre-alignment of long-chain lipids through partial ordering within the lipid bilayers. This approach allows for non-averaged orientations of chromophore charge-transfer dipole moments, facilitating the long-range transmission of stereochemical information within 1,n-glycol lipids (where n = 5, 7, and 9). The resulting data provide insights into the relative and absolute configurations of glycol molecules by analysing ECCD signals from their derived porphyrin carboxylate diesters encapsulated in submicrometer-sized liposomes.
A model study was conducted to validate the principles of ECCD. Enantiomeric C2-symmetric glycols, specifically (R, R)-141 and (S, S)-142, with over 99% enantiomeric excess, were synthesized from (R)- and (S)-1,2-epoxypentane (Fig. 85). The meso-isomer, 144, was separated from a by-product mixture through subsequent chemical reactions. These stereoisomers were acylated with carboxylic acids or N-acylimidazoles to yield purified diesters (141a–c, 142c, and 144c) (Fig. 85).
 | ||
| Fig. 85 Model glycols, esters, and chromophores. | ||
CD measurements revealed that (R, R)-141a–c in methanol or (R, R)-141a–b in liposomes produced baseline spectra. However, (R, R)-141c in liposomes displayed a pronounced positive bisignate signal owing to exciton coupling, with a λmax of 430 nm and an intensity of +52. In contrast, the enantiomer (S, S)-142c exhibited a bisignate CD spectrum with inverted signs and magnitudes relative to (R, R)-141c. Conversely, the meso-144c ester produced only a baseline signal.
The signs of the bisignate CD spectra correlated with the extended conformational helicity of the lipid chains, highlighting the relationship between the observed CD signals and the helicity of the electric transition dipole moments of the coupled chromophores. The ECCD of (R, R)-144c exhibited nonlinear concentration dependence, with optimal signal detection occurring around 1 μM and a detection sensitivity of approximately 40 pmol. The authors also explored the distance dependence of ECCD in TPP diesters of acyclic 1,n-glycols. They observed that diastereomeric glycols with hydroxy groups at positions n = 7 (compound 145) and n = 9 (compound 146) adopted anti (pseudo-C2 symmetric) configurations, whereas the glycol with n = 9 in compound 147 exhibited a syn (pseudomeso) arrangement. The CD spectra of (R, R)-145c and (R, R)-146c displayed strong positive bisignate signals with specific A values of 51 and 27, respectively. These signals decreased linearly with increasing n and the distance between ester oxygens, which was estimated to be around 10 Å for n = 9. In contrast, the pseudomeso form (147c) exhibited only a baseline signal. Based on linear extrapolation from their findings, the authors suggested that the ECCD detection limit could extend to n = 13, with an estimated interatomic distance of approximately 15 Å, as predicted by Chem3D modeling. In summary, the study demonstrated that ECCD signals in TPP diesters of acyclic 1,n-glycols are strongly dependent on the anti (pseudo-C2 symmetric) configuration of the glycols. The intensity of these signals decreases linearly with increasing interatomic distance, while the pseudomeso configuration produces only baseline signals.
The determination of the complete stereochemical structure of (–)-sagittamide A (148), a novel polyacetoxy long-chain α,ω-dicarboxylic acid extracted from a tropical didemnid tunicate, was challenging (Fig. 86).113 Initial analyses using conventional 2D NMR techniques allowed for partial stereochemistry assignments. While the configurations of the terminal amino acids, L-ornithine and L-valine, were straightforward to identify, the central hexol region (C5–C10) posed significant challenges due to complex stereochemistry and difficulties in interpreting J couplings.
 | ||
| Fig. 86 Structures of (−)-sagittamide A (148), stereo-structured models 149a–149h, and the per-benzoate ester derivatives (150, 151, and 152). | ||
To address these challenges, the researchers employed a progressive-convergent methodology that combined three advanced techniques for comprehensive stereochemical analysis. First, J-based analysis predicted an all-anti configuration for C6–C9, narrowing the possible diastereomers from 32 to 4. Next, Kishi's universal database was used to compare the 13C NMR chemical shifts of sagittamide A against predefined stereo-structured models (149a–149h) (Fig. 86).
The final step involved ECCD to determine the absolute stereochemistry. Molinsiki's group generated hexaol diastereomers corresponding to specific configurations (149e and 149f) and converted them into per-benzoate ester derivatives (150, 151, and 152) (Fig. 86). CD spectra of these derivatives revealed opposite Cotton effects for compounds 150 and 151, indicating that the absolute configuration of (–)-sagittamide A corresponds to the configuration of ent-148. Additionally, the authors validated their findings by comparing the CD spectra of ent-151 with that of L-ribose, which provided a reliable reference point for confirming the stereochemical assignments. The relationship established between ent-151 and the known structure of L-ribose helped validate their stereochemical assignments. Based on these analyses, they determined the complete configuration of (–)-sagittamide A as (5S, 6S, 7S, 8R, 9R, 10S).
3. Conclusions and perspectives
The exploration of configurational analysis methods for type-I PKS-derived natural products remains a fundamental area of research in natural product chemistry. This review highlights a variety of strategies for determining stereochemistry, including JBCA, chemical derivatizations using auxiliary chiral reagents, chemical degradations, NMR chemical shift comparisons with universal databases, and chiroptical properties coupled with quantum mechanics-based calculations applied to type-I polyketides. Case studies illustrate the practical applications of these methodologies, emphasizing their importance in accurately determining absolute and relative configurations, which provides critical insights into the stereochemical structures of type-I PKS-derived natural products.While current methodologies are highly effective, challenges persist. Complex natural products, particularly certain type-I PKS-derived polyketides, can pose significant difficulties, including low production yields, chemical instability, or the presence of unique functional groups for which established methods are unavailable. Looking forward, integrating chemical and spectroscopic techniques while accounting for the limitations of each method will provide more reliable and definitive stereochemical analysis. On the other hand, as bioinformatics analysis of polyketide synthases for configurational assignments, which can be complementary to chemical and spectroscopic analysis, continues to evolve,114,115 reviewing the recent analysis of type-I polyketide BGCs in respect to stereochemistry is required to address recent advancements and emerging challenges in this rapidly growing field. Complete stereochemical elucidation of bioactive type-I PKS-derived natural products will enable to provide accurate targets for total synthesis and enhance their potential applications in pharmaceutical development.
4. Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.5. Author contributions
D.-C. O., H. C., S.-J. N., and K. M. participated in conceptualization. J. C., P. F. H., G. J. K. and T. T. M. B. were responsible for manuscript writing and preparation of graphical content. All authors participated in manuscript revising and editing.6. Conflicts of interest
There are no conflicts to declare.7. Acknowledgements
This work was supported by the National Research Foundation of Korea grants funded by the Republic of Korean Government (Ministry of Science and ICT) (RS-2024-00352229, NRF-2020R1A6A1A03044512, 2021R1A2C1010727, 2021R1C1C1009609, 2022R1A2C1011848, and 2022R1A4A3022401).8. Notes and references
- A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2023, 40, 275–325 RSC.
- D. O'Hagan, Nat. Prod. Rep., 1993, 10, 593–624 RSC.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2012, 29, 1050–1073 RSC.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451–2458 CrossRef CAS PubMed.
- W.-S. Li, Z. Luo, Y.-L. Zhu, Y. Yu, J. Wu and L. Shen, Mar. Drugs, 2020, 18, 590–603 CrossRef CAS PubMed.
- N. Matsumori, D. Kaneno, M. Murata, H. Nakamura and K. Tachibana, J. Org. Chem., 1999, 64, 866–876 CrossRef CAS PubMed.
- M. Karplus, J. Chem. Phys., 1959, 30, 11–15 CrossRef CAS.
- G. Bifulco, P. Dambruoso, L. Gomez-Paloma and R. Riccio, Chem. Rev., 2007, 107, 3744–3779 CrossRef CAS PubMed.
- C. Nilewski, R. W. Geisser, M. O. Ebert and M. Erick, J. Am. Chem. Soc., 2009, 131, 15866–15876 CrossRef CAS PubMed.
- H. Choi, E. Mevers, T. Byrum, F. A. Valeriote and W. H. Gerwick, Eur. J. Org. Chem., 2012, 2012, 5141–5150 CrossRef CAS PubMed.
- C. Bassarello, G. Bifulco, A. Evidente, R. Riccio and L. Gomez-Paloma, Tetrahedron Lett., 2001, 42, 8611–8613 CrossRef CAS.
- C. Campagnuolo, C. Fattorusso, E. Fattorusso, A. Ianaro, B. Pisano and O. Taglialatela-Scafati, Org. Lett., 2003, 5, 673–676 CrossRef CAS PubMed.
- A. R. Pereira, T. Byrum, G. M. Shibuya, C. D. Vanderwal and W. H. Gerwick, J. Nat. Prod., 2010, 73, 279–283 CrossRef CAS PubMed.
- A. Aiello, E. Fattorusso, C. Imperatore, P. Luciano, M. Menna and R. Vitalone, Mar. Drugs, 2012, 10, 51–63 CrossRef CAS PubMed.
- C. Dalvit and G. Bovermann, Magn. Reson. Chem., 1995, 33, 156–159 CrossRef CAS.
- K. Scott, J. Keeler, Q. N. Van and A. J. Shaka, J. Magn. Reson., 1997, 125, 320–324 Search PubMed.
- W. P. Aue, J. Karhan and R. R. Ernst, J. Chem. Phys., 1976, 64, 4226–4227 CrossRef CAS.
- K. Nagayama, P. Bachmann, K. Wuthrich and R. R. Ernst, J. Magn. Reson., 1978, 31, 133–148 CAS.
- U. Piantani, O. W. Sørensen and R. R. Ernst, J. Am. Chem. Soc., 1982, 104, 6800–6801 CrossRef.
- A. L. Davis, E. D. Laue, J. Keeler, D. Moskau and J. Lohman, J. Magn. Reson., 1991, 94, 637–644 CAS.
- C. Griesinger, O. W. Sørensen and R. R. Ernst, J. Am. Chem. Soc., 1985, 107, 6394–6396 CrossRef CAS.
- C. Griesinger, O. W. Sørensen and R. R. Ernst, J. Chem. Phys., 1986, 85, 6837–6852 CrossRef CAS.
- C. Griesinger, O. W. Sørensen and R. R. Ernst, J. Magn. Reson., 1987, 75, 474–492 CAS.
- M. Kurz, P. Schmieder and H. Kessler, Angew. Chem., Int. Ed., 1991, 30, 1329–1331 CrossRef.
- W. Kozminski and D. Nanz, J. Magn. Reson., 1997, 124, 383–392 CrossRef CAS.
- A. Meissner and O. W. Sørensen, Magn. Reson. Chem., 2001, 39, 49–52 CrossRef CAS.
- S. Gil, J. F. Espinosa and T. Parella, J. Magn. Reson., 2011, 213, 145–150 CrossRef CAS PubMed.
- K. E. Kövér, V. J. Hruby and D. Uhrín, J. Magn. Reson., 1997, 129, 125–129 CrossRef PubMed.
- J. Boyd, N. Soffe, B. John, D. Plant and R. Hurd, J. Magn. Reson., 1992, 98, 660–664 CAS.
- A. Ross, M. Czisch, C. Ciselar and T. A. Holak, J. Biomol. NMR, 1993, 3, 215–224 CrossRef CAS.
- G. W. Vuister, J. Ruiz-Cabello and P. C. M. Van Zijl, J. Magn. Reson., 1993, 100, 215–220 Search PubMed.
- V. V. Krishnamurthy, J. Magn. Reson., Ser. A, 1996, 121, 33–41 CrossRef CAS.
- C. P. Butts, B. Heise and G. Tatolo, Org. Lett., 2012, 14, 3256–3259 CrossRef CAS PubMed.
- J. Sauri and T. Parella, Magn. Reson. Chem., 2012, 50, 717–721 CrossRef CAS PubMed.
- J. S. An, J. Y. Lee, E. Kim, H. Ahn, Y.-J. Jang, B. Shin, S. Hwang, J. Shin, Y. J. Yoon, S. K. Lee and D.-C. Oh, J. Nat. Prod., 2020, 83, 2776–2784 CrossRef CAS PubMed.
- F. Annang, G. Pérez-Moreno, V. González-Menéndez, R. Lacret, I. Pérez-Victoria, J. Martín, J. Cantizani, N. de Pedro, D. Choquesillo-Lazarte, L. M. Ruiz-Pérez, D. González-Pacanowska, O. Genilloud, F. Vicente and F. Reyes, Org. Lett., 2020, 22, 6709–6713 CrossRef CAS PubMed.
- M. C. Kim, H. Machado, K. H. Jang, L. Trzoss, P. R. Jensen and W. Fenical, J. Am. Chem. Soc., 2018, 140, 10775–10784 CrossRef CAS PubMed.
- M. Simone, S. Maffioli, A. Tocchetti, S. Tretter, M. Cattaneo, I. Biunno, E. Gaspari and S. Donadio, J. Antibiot., 2015, 68, 406–408 CrossRef CAS PubMed.
- S. X. Huang, X. J. Wang, Y. Yan, J. D. Wang, J. Zhang, C. X. Liu, W. S. Xiang and B. Shen, Org. Lett., 2012, 14, 1254–1257 CrossRef CAS PubMed.
- D.-G. Kim, K. Moon, S.-H. Kim, S.-H. Park, S. Park, S. K. Lee, K.-B. Oh, J. Shin and D.-C. Oh, J. Nat. Prod., 2012, 75, 959–967 CrossRef CAS PubMed.
- K. Moon, J. Cui, E. Kim, E. S. Riandi, S. H. Park, W. S. Byun, Y. Kal, J. Y. Park, S. Hwang, D. Shin, J. Sun, K.-B. Oh, S. Cha, J. Shin, S. K. Lee, Y. J. Yoon and D.-C. Oh, Org. Lett., 2020, 22, 5358–5362 CrossRef CAS PubMed.
- R. H. Contreras and J. E. Peralta, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 37, 321–425 CrossRef CAS.
- I. E. Ndukwe, A. Brunskill, D. R. Gauthier Jr, Y. L. Zhong, G. E. Martin, R. T. Williamson, M. Reibarkh and Y. Liu, Org. Lett., 2019, 21, 4072–4076 CrossRef CAS PubMed.
- P. Cimino, G. Bifulco, A. Evidente, M. Abouzeid, R. Riccio and L. Gomez-Paloma, Org. Lett., 2002, 4, 2779–2782 CrossRef CAS PubMed.
- M. Menna, C. Imperatore, A. Mangoni, G. D. Sala and T. S. Orazio, Nat. Prod. Rep., 2019, 36, 476–489 RSC.
- T. Kusumi, T. Ooi, Y. Ohkubo and T. Yabuuchi, Bull. Chem. Soc. Jpn., 2006, 79, 965–980 CrossRef CAS.
- J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1968, 90, 3732–3738 CrossRef CAS.
- J. M. Seco, E. Quiñoá and R. Riguera, Chem. Rev., 2004, 104, 17–118 CrossRef CAS.
- M. J. Rieser, Y. H. Hui, J. K. Rupprecht, J. F. Kozlowski, K. V. Wood, J. L. McLaughlin, P. R. Hanson, Z. Zhuang and T. R. Hoye, J. Am. Chem. Soc., 1992, 114, 10203–10213 CrossRef CAS.
- T. Takeuchi, M. Hatano, M. Umekita, C. Hayashi, S. Wada, M. Nagayoshi, R. Sawa, Y. Kubota, M. Kawada, M. Igarashi and M. Shibasaki, Org. Lett., 2017, 19, 4207–4210 CrossRef CAS PubMed.
- M. Chinworrungsee, P. Kittakoop, M. Isaka, P. Maithip, S. Supothina and Y. Thebtaranonth, J. Nat. Prod., 2004, 67, 689–692 CrossRef CAS PubMed.
- T. H. Quang, D. C. Kim, P. V. Kiem, C. V. Minh, N. X. Nhiem, B. H. Tai, P. H. Yen, N. Ngan, H. J. Kim and H. Oh, J. Anibiot., 2018, 71, 826–830 CrossRef CAS PubMed.
- Y. Igarashi, H. Ogura, K. Furihata, N. Oku, C. Indananda and A. Thamchaipenet, J. Nat. Prod., 2011, 74, 670–674 CrossRef CAS PubMed.
- J. Park, Y.-H. Shin, S. Hwang, J. Kim, D. H. Moon, I. Kang, Y.-J. Ko, B. Chung, H. Nam, S. Kim, K. Moon, K.-B. Oh, J.-C. Cho, S. K. Lee and D.-C. Oh, Angew. Chem., Int. Ed., 2024, 63, e202402465 CrossRef CAS PubMed.
- T. Yabuuchi and T. Kusumi, J. Org. Chem., 2000, 65, 397–406 CrossRef CAS PubMed.
- K. Sasaki, M. Satake and T. Yasumoto, Biosci., Biotechnol., Biochem., 1997, 61, 1783–1785 CrossRef CAS PubMed.
- B. Reguera, M. García-Portela, E. Velasco-Senovilla, P. Rial, L. Escalera, P. A. Díaz and F. Rodríguez, Front. Protistol., 2024, 1, 1328026 CrossRef.
- G. Yu, P. Sun, R. Aierken, C. Sun, Z. Zhang, Q. Che, G. Zhang, T. Zhu, Q. Gu, M. Li and D. Li, Mar. Life Sci. & Technol., 2021, 4, 237–244 Search PubMed.
- J. H. Im, S. Oh, E. S. Bae, S. Um, S. K. Lee, Y. H. Ban and D.-C. Oh, J. Ind. Microbiol. Biotechnol., 2023, 50, 1–10 CrossRef PubMed.
- J. M. Seco, E. Quiñoá and R. Riguera, Tetrahedron: Asymmetry, 2001, 12, 2915–2925 CrossRef CAS.
- K. M. Blazewska and T. Gajda, Tetrahedron: Asymmetry, 2009, 20, 1337–1361 CrossRef CAS.
- S. K. Latypov, J. M. Seco, E. Quinoa and R. Riguera, J. Org. Chem., 1995, 60, 504–515 CrossRef CAS.
- S. Saito, Y. Xiaohanyao, T. Zhou, J. Nakajima-Shimada, E. Tashiro, D. W. Triningsih, E. Harunari, N. Oku and Y. Igarashi, J. Nat. Prod., 2022, 85, 1697–1703 CrossRef CAS PubMed.
- S. Porto, J. M. Seco, J. F. Espinosa, E. Quiñoá and R. Riguera, J. Org. Chem., 2008, 73, 5714–5722 CrossRef CAS PubMed.
- J. M. Seco, E. Quiñoá and R. Riguera, Chem. Rev., 2012, 112, 4603–4641 CrossRef CAS PubMed.
- T. F. Molinski and B. I. Morinaka, Tetrahedron, 2012, 68, 9307–9343 CrossRef CAS PubMed.
- C. Imperatore, P. Luciano, A. Aiello, R. Vitalone, C. Irace, R. Santamaria, J. Li, Y.-W. Guo and M. Menna, J. Nat. Prod., 2016, 79, 1144–1148 CrossRef CAS PubMed.
- I. R. G. Thistlethwaite, F. M. Bull, C. Cui, P. D. Walker, S.-S. Gao, L. Wang, Z. Song, J. Masschelein, R. Lavigne, M. P. Crump, P. R. Race, T. J. Simpson and C. L. Willis, Chem. Sci., 2017, 8, 6196–6201 RSC.
- Y. Morishita, H. Zhang, T. Taniguchi, K. Mori and T. Asai, Org. Lett., 2019, 21, 4788–4792 CrossRef CAS PubMed.
- Y.-H. Shin, J. Y. Beom, B. Chung, Y. Shin, W. S. Byun, K. Moon, M. Bae, S. K. Lee, K.-B. Oh, J. Shin, Y. J. Yoon and D.-C. Oh, Org. Lett., 2019, 21, 1804–1808 CrossRef CAS PubMed.
- Y.-H. Shin, S. Kang, W. S. Byun, C.-W. Jeon, B. Chung, J. Y. Beom, S. Hong, J. Lee, J. Shin, Y.-S. Kwak, S. K. Lee, K.-B. Oh, Y. J. Yoon and D.-C. Oh, J. Nat. Prod., 2020, 83, 277–285 CrossRef CAS PubMed.
- J. Shen, Y. Fan, G. Zhu, H. Chen, W. Zhu and P. Fu, Org. Lett., 2019, 21, 4816–4820 CrossRef CAS PubMed.
- P. G. Williams, E. D. Miller, R. N. Asolkar, P. R. Jensen and W. Fenical, J. Org. Chem., 2007, 72, 5025–5034 CrossRef CAS PubMed.
- R. Suo, K. Takada, H. Kohtsuka, Y. Ise, S. Okada and S. Matsunaga, J. Nat. Prod., 2018, 81, 1108–1112 CrossRef CAS PubMed.
- S. Rychnovsky, B. Rogers and T. Richardson, Acc. Chem. Res., 1998, 31, 9–17 CrossRef CAS.
- H. C. Kwon, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Org. Chem., 2009, 74, 675–684 CrossRef CAS PubMed.
- K. Oguchi, M. Tsuda, R. Iwamoto, Y. Okamoto, J. Kobaysashi, E. Fukushi, J. Kawabata, T. Ozawa, A. Masuda, Y. Kitaya and K. Omasa, J. Org. Chem., 2008, 73, 1567–1570 CrossRef CAS PubMed.
- S. Hoshino, M. Okada, T. Wakimoto, H. Zhang, F. Hayashi, H. Onaka and I. Abe, J. Nat. Prod., 2015, 78, 3011–3017 CrossRef CAS PubMed.
- H. C. Kwon, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Am. Chem. Soc., 2006, 128, 1622–1632 CrossRef CAS PubMed.
- J. Lee, Y. Kobayashi, K. Tezuka and Y. Kishi, Org. Lett., 1999, 1, 2181–2184 CrossRef CAS PubMed.
- D. Menche, Nat. Prod. Rep., 2008, 25, 905–918 RSC.
- H. Seike, I. Ghosh and Y. Kishi, Org. Lett., 2006, 8, 3861–3864 CrossRef CAS PubMed.
- Y. Kobayashi, C. H. Tan and Y. Kishi, Helv. Chim. Acta, 2000, 83, 2562–2571 CrossRef CAS.
- Y. Kobayashi, J. Lee, K. Tezuka and Y. Kishi, Org. Lett., 1999, 1, 2177–2180 CrossRef CAS PubMed.
- G. Kretzschmar, M. Krause and R. Lajos, Tetrahedron, 1997, 53, 971–986 CrossRef CAS.
- Y. Kobayashi, C. H. Tan and Y. Kishi, Angew. Chem., Int. Ed., 2000, 39, 4279–4281 CrossRef CAS PubMed.
- C. H. Tan, Y. Kobayashi and Y. Kishi, Angew. Chem., Int. Ed., 2000, 39, 4282–4284 CrossRef CAS PubMed.
- Y. Kobayashi, N. Hayashi and Y. Kishi, Org. Lett., 2002, 4, 411–414 CrossRef CAS PubMed.
- Y. Kobayashi, N. Hayashi and Y. Kishi, Tetrahedron Lett., 2003, 44, 7489–7491 CrossRef CAS.
- M. Bae, K. Moon, J. Kim, H. J. Park, S. K. Lee, J. Shin and D.-C. Oh, J. Nat. Prod., 2016, 79, 332–339 CrossRef CAS PubMed.
- H. Nishida, X. H. Huang, H. Tomoda and S. Omura, J. Antibiot., 1992, 45, 1669–1676 CrossRef CAS PubMed.
- C. M. Adams, I. Ghosh and Y. Kishi, Org. Lett., 2004, 6, 4723–4726 CrossRef CAS PubMed.
- I. Ghosh, Y. Kishi, H. Tomoda and S. Omura, Org. Lett., 2004, 6, 4719–4722 CrossRef CAS PubMed.
- D. K. Nielsen, L. L. Nielsen, S. B. Jones, L. Toll, M. C. Asplund and S. L. Castle, J. Org. Chem., 2009, 74, 1187–1199 CrossRef CAS PubMed.
- S. G. Smith and J. M. Goodman, J. Am. Chem. Soc., 2010, 132, 12946–12959 CrossRef CAS PubMed.
- I. Paterson, S. M. Dalby, J. C. Roberts, G. J. Naylor, E. A. Guzmán, R. Isbrucker, T. P. Pitts, P. Linley, D. Divlianska, J. K. Reed and A. E. Wright, Angew. Chem., Int. Ed., 2011, 50, 3219–3223 CrossRef CAS PubMed.
- I. Paterson, K. K. H. Ng, S. Williams, D. C. Millican and S. M. Dalby, Angew. Chem., Int. Ed., 2014, 53, 2692–2695 CrossRef CAS PubMed.
- C. Rink, V. Navickas and M. E. Maier, Org. Lett., 2011, 13, 2334–2337 CrossRef CAS PubMed.
- J. Willwacher, N. Kausch-Busies and A. Fürstner, Angew. Chem., Int. Ed., 2012, 51, 12041–12046 CrossRef CAS PubMed.
- A. E. Wright, J. C. Botelho, E. Guzman, D. Harmody, P. Linley, P. J. McCarthy, T. P. Pitts, S. A. Pomponi and J. K. Reed, J. Nat. Prod., 2007, 70, 412–416 CrossRef CAS PubMed.
- S. Hwang, B. E. Heo, T. Q. Nguyen, Y. J. Kim, S. G. Lee, T. H. Huynh, E. Kim, S. I. Jo, M. J. Baek, E. K. Shin, J. Oh, C. Park, Y. J. Yoon, E. J. Park, K. T. Kim, S. Ryoo, D. G. Lee, C. Wood, M. Woo, D. D. Kim, S. Paik, E. K. Jo, J. Jang and D.-C. Oh, Angew. Chem., Int. Ed., 2025, 64, e202412994 CrossRef CAS PubMed.
- M. O. Marcarino, S. Cicetti, M. M. Zanardi and A. M. Sarotti, Nat. Prod. Rep., 2022, 39, 58–76 RSC.
- A. Howarth and J. M. Goodman, Chem. Sci., 2022, 13, 3507–3518 RSC.
- L. D. Bari, G. Pescitelli, C. Pratelli, D. Pini and P. Salvadori, J. Org. Chem., 2001, 66, 4819–4825 CrossRef PubMed.
- D.-C. Oh, M. Poulsen, C. R. Currie and J. Clardy, Nat. Chem. Biol., 2009, 5, 391–393 CrossRef CAS PubMed.
- A. E. Nugroho and H. Morita, J. Nat. Med., 2014, 68, 1–10 CrossRef CAS PubMed.
- X.-C. Li, D. Ferreira and Y. Ding, Curr. Org. Chem., 2010, 14, 1678–1697 CrossRef CAS PubMed.
- G. Bringmann, T. Bruhn, K. Maksimenka and Y. Hemberger, Eur. J. Org Chem., 2009, 17, 2717–2727 CrossRef.
- T.-H. Huynh, S. C. Jang, Y. H. Ban, E.-Y. Lee, T. Kim, I. Kang, J. S. An, S. Kang, J. Han, Y. Kwon, D. Oh, H.-G. Park, J.-C. Cho, J. Jang, K.-B. Oh, S.-J. Nam, S. K. Lee and D.-C. Oh, JACS Au, 2024, 4, 4821–4832 CrossRef CAS PubMed.
- C. K. Skepper, J. B. Macmillan, G.-X. Zhou, M. N. Masuno and T. F. Molinski, J. Am. Chem. Soc., 2007, 129, 4150–4151 CrossRef CAS PubMed.
- J. B. MacMillan and T. F. Molinski, J. Am. Chem. Soc., 2004, 126, 9944–9945 CrossRef CAS PubMed.
- S. C. Lievens and T. F. Molinski, J. Am. Chem. Soc., 2006, 128, 11764–11765 CrossRef CAS PubMed.
- D. H. Kwan and F. Schulz, Molecules, 2011, 16, 6092–6115 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2016, 33, 141–149 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |