
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorylated chitin and cellulose nanocrystals as colloidal bio-templates towards mesoporous aluminophosphates†
Sara
Blilid
abc,
Nadia
Katir
 b,
Abdelkrim
El Kadib
b,
Mohammed
Lahcini
de,
Valérie
Flaud
a,
Bruno
Alonso
*a and
Emmanuel
Belamie
*ac
b,
Abdelkrim
El Kadib
b,
Mohammed
Lahcini
de,
Valérie
Flaud
a,
Bruno
Alonso
*a and
Emmanuel
Belamie
*ac
aICGM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: emmanuel.belamie@enscm.fr; bruno.alonso@enscm.fr
bEuromed University of Fes (UEMF), BP 51, Fes, Morocco
cEcole Pratique des Hautes Etudes, PSL Research University, 75014 Paris, France
dIMED-Lab. Faculty of Sciences and Techniques, Cadi-Ayyad University, Av. A. Khattabi. BP 549, 40000 Marrakech, Morocco
eMohammed VI Polytechnic University Lot 660, Hay Moulay Rachid Ben Guerir 43150 Morocco, Marrakech, Morocco
First published on 21st March 2025
Abstract
This work demonstrates the interest of phosphorylated chitin and cellulose nanocrystals as colloidal bio-templates towards the formation of mesoporous aluminophosphates. Chitin and cellulose nanocrystals (ChitNC and CellNC, respectively) were phosphorylated using POCl3. This procedure does not affect the internal structure of the nanoparticles while it opens new opportunities for materials’ design. From the analysis of dimensions and zeta potentials of the phosphorylated nanoparticles (ChitNC-P and CellNC-P), we demonstrate notably the enhanced stability of chitin suspensions across a pH range of 4 to 13. Further we investigated the interactions between the polysaccharide nanocrystals and aluminium oxo–hydroxo clusters ε-Al13O4(OH)24(H2O)127+ to form mesoporous alumina and aluminophosphate materials through spray-drying and calcination in air. The spatial proximity between phosphorous and aluminum sites in the final materials was probed by solid-state NMR studies using notably 27Al{31P} REDOR experiments.
1. Introduction
Crystalline polysaccharide nanoparticles (NPs) are efficient bio-based precursors for the formation of textured materials (e.g. mesostructured organic–inorganic hybrids or mesoporous oxides).1–8 Compared to conventional colloids (micelles, non-crystalline polymers, …), these NPs are not only interesting for their biomass origin (allowing waste recycling and limiting pollution and toxicity) but also for their physicochemical characteristics. Among the latter, the nanoparticles possess a rigid shape that remains almost unaffected during the templating processes. The calibrated dimensions of the polysaccharide NPs favor the design of mesoporous materials with known pore dimensions. In addition, their defined surface chemistry opens potentially to a high variety of functional groups and final compositions.The polysaccharide NPs are manipulated in suspension. Above a critical concentration, they self-assemble into nematic mesophases.9–13 This property is beneficial for the formation of textured materials. Two main strategies have been developed for this purpose: (i) the infiltration of the nematic mesophases by inorganic precursors,14–16 and (ii) the cooperative self-assembly between the polysaccharide NPs and inorganic or organometallic colloidal precursors (siloxane oligomers, metal oxide–hydroxide clusters).17,18 In both cases, bio-sourced polysaccharide NPs can act as colloidal bio-templates since they form stable colloidal suspensions, and they can serve as a template for the organization of inorganic phases and generate porosity after removal. In previous studies, we have shown how combining this cooperative self-assembly with sol–gel processes results in textured materials with defined morphology, like microparticles by spray-drying,17,19,20 thin films by spin-coating,21,22 and fibers by electrospinning.23 This approach follows principles similar to those of self-assembly methods such as evaporation-induced self-assembly (EISA).24
Under certain conditions, the texture of the final material replicates that of the nematic mesophase of polysaccharide NPs, paving the way to the design of hierarchical and/or optically active materials.25–29 The NPs can also be manipulated with external fields (shearing, electrical, magnetic)30 in the suspension before solidification, to impart the resulting hybrid material with long-range orientation.17,23,31 Using these strategies, several textured materials with various compositions have been formed among which mesoporous silica,17,18,20,23,25,27,32,33 titania,20,34–36 carbon that can be nitrogen-doped using chitin,37–39etc. As mentioned above, an important property of the polysaccharide NPs is their surface chemistry. For instance, we have favored the presence of TiO4 tetrahedral sites at the mesopores’ surface of silica thanks to the presence of de-acetylated amino groups at the chitin surface, allowing to increase the catalytic efficiency of the final silica–titania microspheres.20,36
Hitherto, only downsized but non-functionalized polysaccharide NPs were used as building-blocks for assembling inorganic nanostructures, leveraging on interactions of hydroxyl and sulfonate in the case of nanocellulose, amine and acetamide when using chitin nanocrystals. Owing to the pivotal role played by interfacial interactions in designing organic–inorganic hybrid materials, there is a need to investigate such assembly by harnessing new functionalities. In this direction, the surface functionalization of polysaccharide NPs is able to widen the diversity of materials formed. Among the possible strategies, phosphorylation provides many advantages considering the highest reactivity of such derivatives (uptake of pollutants, easy dispersion in recalcitrant medium, stabilizing nanoparticles, etc.)40,41 and the well-established stability of P–O–M bridges for templating metal oxides.42
The aim of the research presented here is to evaluate the opportunities offered by phosphorylated chitin and cellulose nanocrystals as colloidal bio-templates towards the formation of mesoporous aluminophosphates. To this end, we characterize the suspensions of phosphorylated cellulose and chitin NPs and their interactions with Al clusters in view of their use in a colloidal self-assembly approach. After the phosphorylation of NPs, we have determined the hydrodynamic radii and zeta potentials of the functionalized NPs in aqueous suspensions and compared the results to those of non-phosphorylated NPs over a wide range of pH. This study shows notably how the phosphorylation of chitin NPs ensures a high colloidal stability towards pH variations. Furthermore, by co-assembling the polysaccharide NPs (phosphorylated or not) with aluminium oxo–hydroxo clusters (tridecamer ε-Al13O4(OH)24(H2O)127+, hereafter referred to as Al13), we have synthesized textured microparticles through spray-drying. The calcination of the resulting hybrids leads to mesoporous aluminophosphate materials. A thorough multinuclear solid-state NMR study allowed for a better understanding of the composition and structure of the final materials and their intermediates.
2. Results and discussion
2.1. Nanocrystals’ characterization
Chitin and cellulose nanocrystals (ChitNC and CellNC) were prepared and purified according to previously published methods9,11,43,44 and then submitted to phosphorylation with POCl3 as described in previous works by our group and others42,45,46 (see Experimental), forming the functionalized polysaccharides ChitNC-P and CellNC-P.2.2. Preserved polysaccharide structures
X-ray diffraction (XRD) analysis on powdered samples (Fig. S1, ESI†) indicates that the phosphorylation treatment did not alter the crystalline structure of cellulose or chitin. The crystallinity index was found to be 88% and 89% for CellNC before and after phosphorylation, and 92% and 90% for ChitNC before and after phosphorylation. The chemical structure of the polymers was preserved during phosphoryl chloride grafting, as confirmed by 13C solid-state nuclear magnetic resonance (NMR) (Fig. S2, ESI†).2.3. Phosphorous incorporation
Polysaccharides functionalized with phosphoryl oxychloride (POCl3) exhibit 31P NMR signals at ca. −0.3 ppm. These signals are different from those of the starting POCl3 at ca. 2 ppm,47 indicating the consumption of the starting phosphoryl chloride and the formation of new species on the cellulose and chitin surfaces (Fig. S3, ESI†), consistent with the large decrease in Cl/P ratio from element analyses.The overall composition of the pristine and phosphorylated nanocrystals was analyzed using energy-dispersive X-ray spectroscopy (EDX) and compared to expected atomic proportions based on the chemical structure of the polysaccharides (Table 1). For the pristine ChitNC and CellNC, the experimental values closely matched predicted ones, considering an acetylation degree of 80% for ChitNC (see Experimental and Table S1, ESI†). After phosphorylation, both ChitNC-P and CellNC-P displayed slightly different compositions, with lower C contents, as well as lower N content for ChitNC-P. In both cases, the phosphorous atomic concentration reaches ca. 2.5% by X-ray photoelectron spectroscopy (XPS) and EDX, except the EDX measurement for CellNC-P, which gives a lower value of about 1.0%.
| Element atom% | EDX | XPS | EDX | XPS | ||
|---|---|---|---|---|---|---|
| Mean ± SD | SD | Mean ± SD | SD | |||
| ChitNC | ChitNC-P | |||||
| C | 64.7 | 1.3 | 60.2 | 61.6 | 4.2 | 54.9 |
| N | 10.4 | 0.6 | 6.5 | 8.1 | 1.8 | 6.2 |
| O | 24.9 | 1.1 | 33.0 | 28.1 | 2.9 | 35.1 |
| O/N | 2.4 | 0.2 | 5.0 | 3.6 | 1.0 | 5.6 |
| P | 2.2 | 1.4 | 2.4 | |||
| P/C | 0.04 | 0.02 | 0.04 | |||
| CellNC | CellNC-P | |||||
| C | 70.1 | 1.5 | 57.9 | 64.8 | 4.7 | 55.0 |
| O | 29.4 | 1.6 | 41.8 | 34.0 | 4.4 | 40.9 |
| S | 0.45 | 0.05 | 0.35 | 0.2 | 0.04 | 0.4 |
| C/O | 2.4 | 0.2 | 1.4 | 1.9 | 0.4 | 1.3 |
| P | 0.95 | 0.53 | 2.8 | |||
| P/C | 0.02 | 0.01 | 0.05 | |||
The corresponding phosphorous EDX weight concentrations amount to 4.5 ± 0.7 wt% and 2.2 ± 0.2 wt% for ChitNC-P and CellNC-P respectively, hence a ratio of more than two between chitin and cellulose. This relatively large difference in phosphorylation rate is in line with the global phosphorous content measured by inductive coupled plasma (ICP), which amounts to more than 3.18 ± 0.84 wt% for chitin nanocrystals and 1.2 ± 0.15 wt% for cellulose nanocrystals.
2.4. Possible association modes
For ChitNC-P, the ratio O/N should be indicative of the mode of association of phosphorous species with the nanocrystals (NC). The addition of one P atom brings along three O atoms in the case of a N–P covalent bond, and four O atoms if a phosphate ion is associated with the surface. Interestingly for ChitNC-P, setting the P/N atomic ratio to 0.35–0.40, values determined by EDX and XPS respectively, yields a predicted O/N ratio between 5.9 and 6.0 in the covalent case (3 oxygen atoms per phosphorous), which is slightly above but close to the value measured by XPS (5.6) (Table 1). In the non-covalent hypothesis, this O/N ratio is predicted to be in the 6.2–6.4 range, hence much higher than the experimental value.In addition, since the P/C atomic ratios measured by XPS are approximately 0.04–0.05, and considering that each saccharide unit contains 6 carbons (8 in the case of N-acetyl-glucosamine), there would be roughly 0.25–0.4 phosphorous moiety per sugar ring. Moreover, because of the nanometric lateral dimensions of the particles, about 1/3rd of the polysaccharide chains are located on the nanocrystals’ surface (see for instance44 for cotton cellulose NC). If we assume that only the surface chains are accessible to phosphorylation by POCl3 (no structure modification observed by XRD), it follows that 0.8–1.2 phosphorus moiety is associated with every glucose (CellNC) or glucosamine (ChitNC) unit on the NC surface.
Comparing P/C ratios obtained by XPS and EDX can provide insights into the aggregation state of the nanoparticles, as the first technique is more influenced by surface composition, while the second has a penetration depth of up to several microns. In fact, the P/C ratio measured for CellNC-P by EDX (0.02) is much lower than that obtained by XPS (0.05), while the values for ChitNC-P are consistent across both methods. It can be speculated that the location of CellNC-P inside larger aggregates might result in the underestimation of phosphorous moieties by EDX as compared to XPS, indicating a higher aggregation state than that of ChitNC-P. This hypothesis aligns well with size measurements obtained by dynamic light scattering (DLS) and observations by transmission electron microscopy (TEM) following the reaction with POCl3 (vide infra, Fig. 3 and Fig. S4, ESI†).
In addition to providing information on the elemental composition of the materials, deconvolution of XPS spectra (see survey spectra for all samples in Fig. S5, ESI†) gives access to the contribution of different atom environments, i.e. their valence and possibly covalently bound neighbors. It is noteworthy that the deconvolution of the N 1s spectra for ChitNC after reaction with POCl3 (Fig. 1B) reveals an additional contribution as compared to untreated ChitNC (Fig. 1A), visible as a shoulder at higher binding energy near 402 eV. Although it is difficult to unequivocally attribute this additional contribution to the formation of a N–P bond, it agrees well with similar observations of Wang and Liu with phosphorylated chitosan.48 The shift to higher binding energy in the N 1s spectrum would be consistent with the substitution of one hydrogen by one phosphorus atom bearing strongly electro-attractive OH groups.49
 | ||
| Fig. 1 N1s XPS spectra deconvolution for chitin nanocrystals before (A) (ChitNC) and after (B) (ChitNCP) reaction with POCl3. | ||
The spectra for the other elements (C 1s, O 1s) are very similar in all samples, except for a slight difference in the Full Width at Half Maximum (FWHM) of the phosphorous 2p spectrum (P 2p) between CellNC-P and ChitNC-P. This could also suggest an additional contribution for ChitNC-P at a lower binding energy (Fig. S6, ESI†) possibly related to the formation of a P–N bond.
Overall, these analyses demonstrate that the compositions of both cellulose and chitin nanocrystals are modified by the reaction with POCl3, most likely through an association with surface polysaccharide chains, preserving the internal structure, with 0.75 to 1.2 phosphorous moiety per saccharide unit. Although these interactions are difficult to assess, our data suggest the formation of a P–N phosphamide bond in the particular case of ChitNC.
3. Colloidal suspensions
3.1. Colloidal stability by visual inspection
Pristine and phosphorylated chitin and cellulose nanocrystals were tested for their ability to form stable dispersions in aqueous media as a function of pH (Fig. 2). Cellulose nanocrystals obtained by acid hydrolysis in H2SO4 can be dispersed over a wide pH range from 2 to 13 without visible change in turbidity. After phosphorylation with POCl3, although the turbidity appears slightly increased, the colloidal suspensions remain stable at all pH values. In contrast, unmodified chitin nanocrystals obtained by hydrolysis with HCl form stable suspensions in acidic water, only at pH = 5 or below (Fig. 2 top of right panel). Beyond this threshold, close to and beyond the pKa of chitosan amine groups, turbid suspensions are observed, which behave like a soft gel. The presence of free amine groups on the surface of chitin nanocrystals results from partial de-acetylation during the hydrolysis step of the ChitNC preparation. The ionization of these amine groups at pH below ca. 6 responsible for the colloidal stability of ChitNC suspensions is cancelled at higher pH. Interestingly, the phosphorylated chitin nanoparticles form stable dispersions over the whole pH range investigated, from 4 to 13. | ||
| Fig. 2 Visual inspection of aqueous dispersions of cellulose and chitin nanocrystals (0.1 wt%) and their phosphorylated analogues as a function of pH. | ||
3.2. Suspension stability and colloids’ size evolution by DLS
Dynamic light scattering (DLS) measurements were performed to assess the dispersion state of the particles in water as a function of pH. The hydrodynamic diameter inferred from measuring the diffusion coefficient of the rodlike dispersed particles, corresponds roughly to the diameter of an equivalent sphere diffusing in the liquid, hence close to the length of the elongated particles. This equivalent diameter scales as the inverse of the diffusion coefficient as drawn in Fig. 3. As observed visually with chitin nanocrystals, the diffusion coefficient is stable for pH values below 6 and then rapidly decreases (a large increase of 1/D) indicating aggregation as the pH of the solution approaches the pKa of the amine groups present on the surface of ChitNC (Fig. 3). In contrast, ChitNC-P exhibit a nearly constant diffusion coefficient over the whole pH range, which again agrees well with visual observations in Fig. 2. Globally, the values for the modified nanocrystals are slightly larger than for pristine ones at low pH, thus indicating larger objects. This suggests some marginal aggregation of initial ChitNC particles during phosphorylation. The same observation holds for cellulose nanocrystals, although in this case, both modified and unmodified samples exhibit a nearly constant diffusion coefficient, irrespective of pH. The larger size difference between pristine and phosphorylated CellNC (×5.8) compared to ChitNC (×1.6) at low pH suggests a higher aggregation state for the cellulose nanocrystals upon phosphorylation. This observation is in line with differences in P distribution suggested by discrepancies between EDX and XPS measurements (vide supra, Section 1.3). | ||
| Fig. 3 Dimensions (equivalent sphere diameter) and inverse diffusion coefficient (1/D) for polysaccharide cellulose and chitin nanocrystals dispersed in aqueous medium at different pH: (a) pristine and phosphorylated cellulose nanocrystals (CellNC, CellNC-P); (b) pristine and phosphorylated chitin nanocrystals (ChitNC, ChitNC-P). For most samples, the error bars displayed in the figure are smaller than the symbols size. | ||
3.3. Zeta potential
According to zeta potential measurements reported in Fig. 4, the overall negative charge of cellulose nanocrystals is apparently not modified upon reaction with POCl3 and stays roughly comprised between −30 and −40 mV. Therefore, the modification of the particles surface chemistry has no visible effect on the colloidal behavior and stability of cellulose nanocrystals, most likely because the initial particles already bear sulfate groups, negatively charged over the pH range investigated (pKa ∼ 1.9–2.5).50 In contrast, the behavior of chitin nanocrystals is strongly affected by POCl3 treatment. Pristine ChitNC exhibits a positive charge of about 40 mV in the 2 to 4 pH range. The zeta potential then steeply decreases as the pH approaches neutrality, in accordance with the neutralization of charged surface amine groups (pKa ≈ 6). Beyond this pH range, the suspension precipitates, and the zeta potential is no longer measurable. Interestingly, after phosphorylation, the contribution of phosphate groups has a striking effect on the colloidal properties of chitin nanocrystals with a switch of the zeta potential to negative charge (−20 to −40 mV) over a wide pH range (2–13).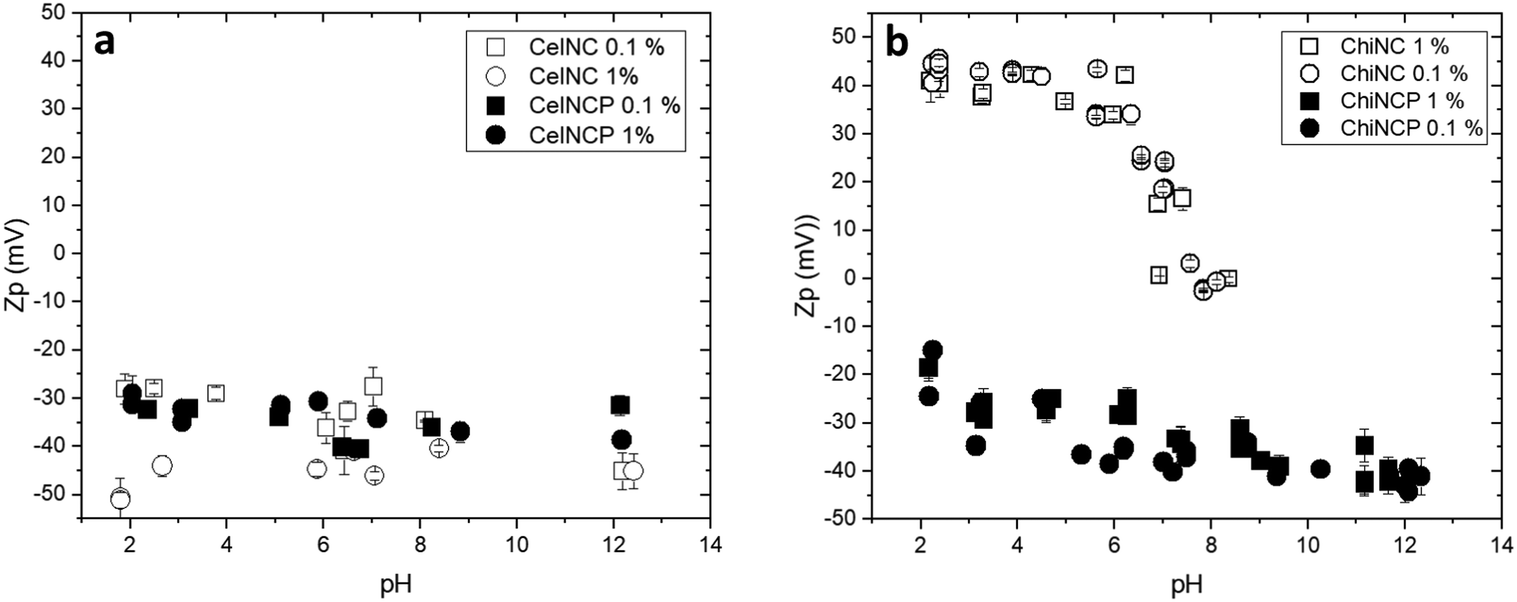 | ||
| Fig. 4 Zeta potential for polysaccharide cellulose and chitin nanocrystals dispersed in aqueous medium as a function of pH: (a) pristine (CellNC, open symbols) and phosphorylated (CellNC-P, closed symbols) cellulose nanocrystals; (b) pristine and phosphorylated chitin nanocrystals (ChitNC, ChitNC-P). In both figures, squares and circles correspond to measurements performed at respectively 0.1 wt% and 1 wt%. | ||
In Fig. 5, we plotted the fractional charge calculated as a function of pH for amine groups as those present on the surface of ChitNC particles, combined with the negative charges possibly brought by the association of phosphate moieties. The evolution of the charge estimated for pristine ChitNC as a function of pH agrees well with the experimental data. In the case of phosphorylated chitin nanocrystals assuming all amine groups remain unaltered and ionizable (open black circles), the fractional charge should be positive at low pH, even in the presence of phosphate groups. This does not agree with the experimental data as shown in the blow-up view of Fig. 5b. As discussed above, based on composition analyses, we can hypothesize that phosphorylation resulted in the introduction of phosphamide groups by reaction with the surface amine groups. In this situation, these substituted amines would not be ionizable in the pH range investigated and would not contribute positive charges at acidic pH. In fact, the predicted evolution of the net charge as a function of pH (Fig. 5d) better matches that of the experimental measurements (Fig. 4b). This indicates that the phosphorylation of chitin nanocrystals with POCl3 results in the introduction of additional negative charges and hampers the ionization of surface amine groups.
 | ||
| Fig. 5 Zeta potential evolution for (a) pristine (ChitNC, 1 wt%) and (b) phosphorylated chitin nanocrystals (ChitNC-P, 1 wt%) as a function pH (as in Fig. 3); (c) calculated fractional charges of ionizable groups on chitin nanocrystals before and after treatment with POCl3 as a function of pH. [i] Open blue squares are for pristine ChitNC with fully ionizable amine groups [NH2]. The two other conditions are for chitin nanocrystals after modification with POCl3, considering the association with phosphate moieties (0.5 phosphate per amine group based on EDX and XPS data): [ii] open black circles with the addition of negative charges from phosphate ions (three acidic functions) [NH2 + 3P] and [iii] open red circles considering only two negative charges from bound phosphate [2P]. The calculated data for the situation corresponding to the [2P] case are reproduced in figure (d) to emphasize the agreement with experimental data shown in (b) for ChitNC-P. Lines are guides for the eyes. | ||
4. CellNC and ChitNC interactions with Al13 clusters
With the aim of preparing hybrid or porous alumina and aluminophosphate materials, we investigated the colloidal association of Keggin-type aluminium oxo–hydroxo clusters (ε-Al13O4(OH)24(H2O)12)7+, hereafter named Al13, with polysaccharide nanocrystals. In a previous work, we demonstrated the opportunities of synthesizing mesoporous alumina microparticles by co-assembling ChitNC and Al13 clusters and further spray-drying.51 Herein, we study how the phosphorylation of the polysaccharide particles would affect their colloidal interactions with the positively charged Al13 clusters.4.1. Hydrodynamic diameter of mixtures
DLS provides a rough evaluation of these interactions through the measurement of the hydrodynamic diameter of the resulting species in suspension. The compositions of all mixtures (Al13 clusters/polysaccharide nanocrystals) were adjusted to result in a ratio of one Al atom per saccharide unit. The pH was set to 4.5 to ensure the colloidal and chemical stability of all samples. In all cases, the addition of Al13 clusters resulted in significant aggregation as indicated by a large increase of the measured diffusion coefficient, hence of the deduced Z-average hydrodynamic diameters reported in Fig. 6. | ||
| Fig. 6 Z-averaged hydrodynamic diameter measured by DLS for hybrid suspensions of (a) chitin and (b) cellulose nanocrystals after phosphorylation or not, in the presence or absence of Al13 clusters. | ||
4.2. Zeta-potential of mixtures
The evolution of the zeta potential (Zp) upon mixing also gives insight into the association of the Al13 clusters with the polysaccharide nanoparticles. The zeta potential of ChitNC is positive (ca. +33.4 ± 0.2 mV) and stays positive after mixing with Al13 clusters. In contrast, the surface charge of ChitNC-P is negative (ca. −25.1 ± 0.8 mV) and takes a positive value after mixing with these clusters (Fig. 7). These data strongly indicate a close association of the Al13 clusters with the polysaccharide nanocrystals surface, the particles charge being reverted from negative to positive upon mixing with the positively charged clusters. CellNC and CellNC-P both have negative zeta potentials and the mixtures with Al13 clusters also exhibit positive charges, respectively +22.0 ± 3.7 mV and +28.2 ± 2.9 mV. | ||
| Fig. 7 Zeta potential measurements of hybrid suspensions à pH 4.5 for Al13 clusters alone, polysaccharide nanocrystals phosphorylated (ChitNC-P, CellNC-P) or not (ChitNC, CellNC) alone, and in the presence of Al13 (ChitNC@Al13, ChitNC-P@Al13, CellNC@Al13, CellNC-P@Al13). | ||
From these results, it appears that Al13 clusters are associated to all polysaccharide nanocrystals, irrespective of their surface charge, leading to larger aggregated particles. The mechanistic association mode (complexation by hydroxyl groups at polysaccharide surfaces, attraction by surface charges, or solvated ionic layers) is difficult to assess at this stage. What we do prove is the formation of positively charged colloids in the presence of Al13, notably when interacting with phosphorylated polysaccharides that are negatively charged. In these latter cases, we hypothesize a close proximity between phosphate-like groups and Al13.
5. Mesoporous aluminophosphate materials by spray-drying
The co-suspensions containing the pristine or phosphorylated polysaccharide nanocrystals and the Al13 clusters were processed by spray-drying. The resulting solids were notably analyzed by EDX and by solid-state NMR to assess their composition and probe the environment of phosphorous and aluminum atoms obtained with our synthetic approach.5.1. Chemical and textural analysis of spray-dried microparticles
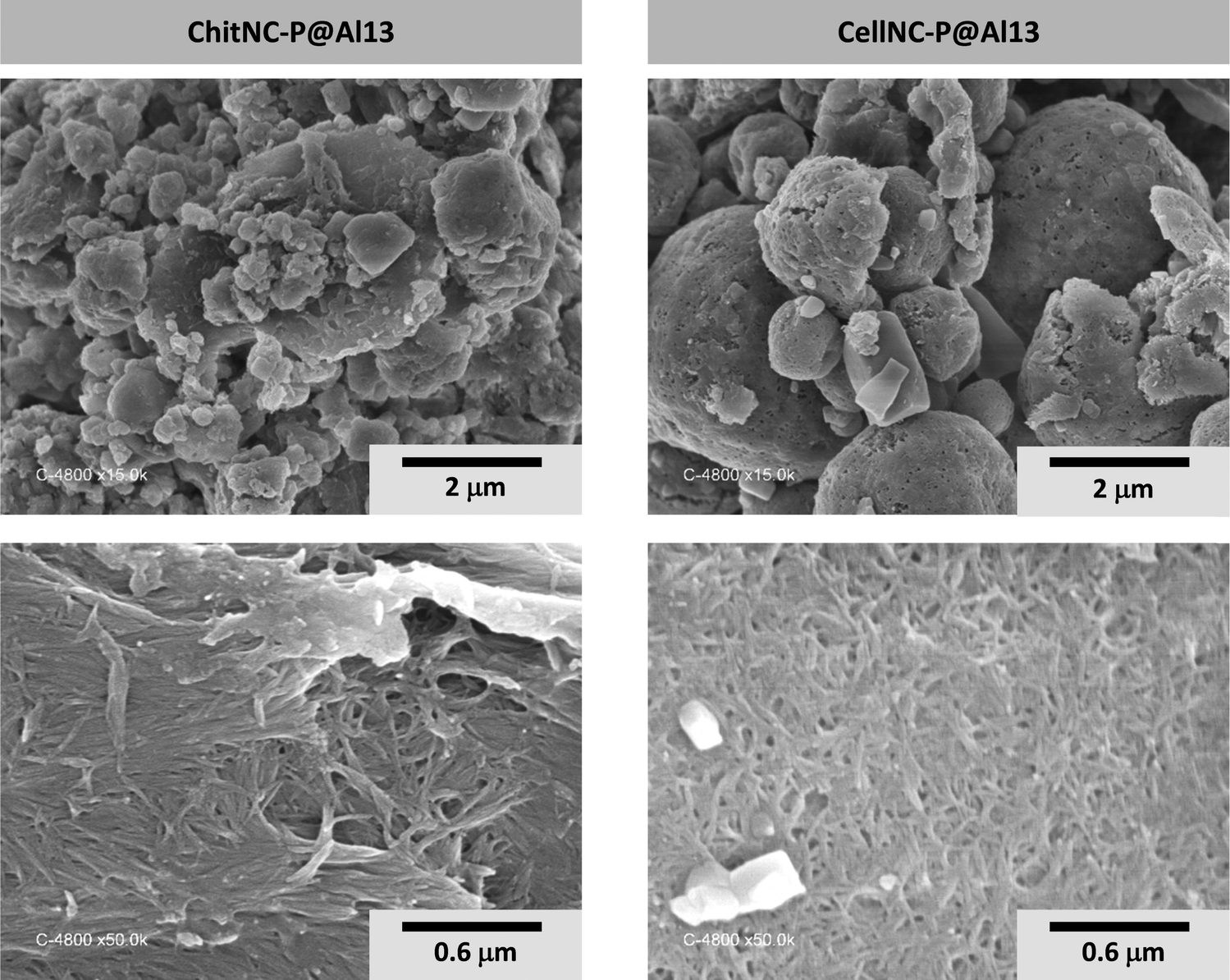 | ||
| Fig. 8 SEM images of hybrid spray-dried microparticles obtained with phosphorylated polysaccharide NPs and Al13 clusters. | ||
After calcination of the hybrid materials at high temperature (823 K in air), the porosity of the resulting solids was analyzed by N2 sorption (Fig. 9 and Table 2). All samples exhibit type IV isotherms with hysteresis loops between H2 and H3 types,52 characteristic of mesoporous solids with ill-defined interconnected porosity. As observed in an earlier study, the estimated pore diameters (11 to 17 nm range) reveal that the polysaccharide NPs are acting as templates.51 It appears however that the modification of the polysaccharide nanocrystals with POCl3 (dotted lines) has a strong impact on the porosity of the calcined materials as compared to non-phosphorylated ones (full lines). This is also consistent with the observations of spray-dried microparticles’ surfaces analyzed in parallel by scanning electron microscopy (SEM) (Fig. 10). With chitin nanocrystals, the BET (Brunauer, Emmett and Teller) mesoporous surface area (SBET) drops by a factor of 3, and the isotherm is shifted to the right indicating a larger pore diameter (Table 2). The same effect is visible with cellulose nanocrystals although to a lesser extent. This again is consistent with a higher content of incorporated phosphorous with chitin NC as compared to cellulose (Table 1). Although the mechanism leading to reduced SBET values for phosphorylated nanocrystals remains unclear, it might be related to the formation of a denser oxide network thanks to the thermally-induced expansion of polyphosphates and/or the stronger interactions between the phosphorylated NPs and the Al13 clusters.
 | ||
| Fig. 9 N2 adsorption—desorption isotherms (77 K) for spray-dried and calcined materials obtained with both polysaccharide nanocrystals (chitin and cellulose), phosphorylated or not, and reacted with Al13 clusters. | ||
| Initial hybrid sample | ChitNC@Al13 | ChitNC-P@Al13 | CelNC@Al13 | CellNC-P@Al13 |
|---|---|---|---|---|
| BET surface area SBET (m2 g−1) | 44.3 | 12.5 | 26.2 | 20.1 |
| Maximum adsorbed volume (cm3 g−1) | 137 | 42 | 103 | 70 |
| Pore volume (cm3 g−1) | 0.21 | 0.06 | 0.16 | 0.11 |
| Pore diameter DBdB from adsorption branch (nm)53 | 15 | 17 | 12 | 17 |
| Pore diameter DBdB from desorption branch (nm)54 | 14 | 17 | 11 | 13 |
 | ||
| Fig. 10 Surfaces of calcined spray-microparticles analyzed by SEM. The observed porosity is formed by the imprints of the polysaccharide NPs (removed during calcination in air). The crystalline shapes observed in some of the micrographs correspond to residual NaCl crystals formed by the presence of chlorine and sodium ions in the NCs and Al13 suspensions respectively.51 The magnification is the same for all samples, and the scale bar represents 600 nm. | ||
5.2. Solid-state NMR characterization of interfaces
 | ||
| Fig. 11 13C{1H} CP-MAS spectra of spray-dried microparticles obtained from different polysaccharide colloids: pristine chitin nanocrystals (ChitNC), phosphorylated ChitNC (ChitNC-P), and ChitNC-P in interaction with Al13 (ChitNC-P@Al13). The spectra were recorded at ν0{13C} ≡ 75.4 MHz and νMAS = 12 kHz (see Experimental), acquiring 4096 scans. | ||
 | ||
| Fig. 12 31P{1H} CP-MAS spectra recorded at ν0{31P} ≡ 242.8 MHz and νMAS = 12 kHz (τc = 2 ms), acquiring 64 scans for the ChitNC-P precursor, and 256 for the hybrid and calcined materials. The spectrum of the hybrid (middle) was fitted using the peak's characteristics of the ChitNC-P precursor and two other peak functions freely optimized. | ||
 | ||
| Fig. 13 27Al single pulse spectra recorded at ν0{27Al} ≡ 156.3 MHz and νMAS = 15 to 20 kHz. The scheme of the Al13 cluster highlights the central AlO4 tetrahedra (in red) surrounded by the 12 AlO6 octahedra (in blue). | ||
After calcination in air, 27Al NMR spectra change drastically and present ill-defined peaks for four-, five- and six-coordinated Al sites (Fig. 13B). The peaks have a dissymmetric shape characteristic of a distribution in quadrupolar coupling constants. These spectra indeed resemble those of amorphous61,62 or some transition alumina.63
 | ||
| Fig. 14 27Al{31P} REDOR experiments on ChitNC-P@Al13 samples. (A) and (B) Typical 27Al NMR REDOR spectra obtained with (green lines) and without (blue lines) the application of 31P pulses for the hybrid ChitNC-P@Al13 sample (A) and its calcined counterpart (B). (C) 27Al{31P} REDOR curves for the AlVI sites of the hybrid sample (orange squares) and all Al sites of the calcined sample (blue disks). (D) 27Al{31P} REDOR curves for the four 27Al contributions of the calcined sample obtained from fitting 27Al spectra (δiso values associated to each curve are indicated in the legend). | ||
For the non-calcined hybrid ChitNC-P@Al13 sample, only the 27Al peaks of AlVI sites are significantly affected by the 31P pulses (Fig. 14A). These octahedral sites, at the periphery of the Al13 cluster, are therefore spatially close to P moieties as confirmed by the efficient REDOR effect (Fig. 14C).
In the case of the calcined Al13@ChitNC-P sample (Fig. 14B), peak overlapping renders difficult a direct analysis. In the first approach, we integrate the whole 27Al spectra not considering the individual peak contributions. The resulting REDOR curve shows a gradual increase with N·Tr (Fig. 14C) demonstrating the presence of P sites in the vicinity of Al sites. By extrapolating the curve, we can infer that the dephasing ratio ΔS/S0 at infinite time will roughly be around 0.8. Further, we observe that all 27Al peaks seem affected by the application of 31P pulses (Fig. 14B). Therefore, we can consider that a majority of Al sites are close to P sites (distances below 1 nm).
In a second approach, we fitted the 27Al spectra with four contributions of fixed parameters, using Czjzeck functions65,66 only varying the amplitude. The fixed parameters (isotropic chemical shifts, quadrupolar coupling constant distribution, chemical shift distribution) were estimated by preliminary fits (see Fig. S8 in ESI† for details on the fitting procedure). The REDOR curves for the four contributions are presented in Fig. 14D. It is interesting to note that three of these curves possess a similar build-up, also similar to that of the whole 27Al spectra (Fig. 14C), and one curve drastically differs. This latter curve, corresponding to a small contribution located at δiso = 51.7 ppm, increases faster and reaches ΔS/S0 values that can be extrapolated to 1. Fitting the initial build-up of the REDOR curves allows measuring the heterodipolar second moment M2Al–P and the dipolar coupling in the case of a single Al–P pair.56,67 A rough analysis of the initial build-up of the more dephasing curve allows estimating M2Al–P of ca. 0.6 kHz2. This value is smaller than that obtained for model aluminophosphates with Al(OP)4 or Al(OP)6 moieties (typically between 5 and 7 kHz2)68 and for aluminophosphate glasses (between 1.5 and 7.2 kHz2)56,69 for which Al sites are bonded to several P sites. Nonetheless, it might correspond to an Al site directly bonded to one P site. Considering a single 27Al–31P pair, the initial REDOR build-up leads to an approximate 27Al–31P dipolar coupling of ca. 320 ± 20 Hz, and hence to an Al–P distance of about 3.4 ± 0.1 Å. For this Al site, such approximate distance, possibly overestimated for the closer Al–P pair, might be compatible with one direct Al–O–P bond and more distant interactions with other P. Indeed, in AlPO4 crystalline compounds, the Al–P distances for bonded AlO4 and PO4 tetrahedra fall between 2.9 and 3.3 Å.70–73 In amorphous aluminophosphates the shortest Al–P distances have also been estimated to ≈3.1 Å.74 Besides, the 27Al chemical shift of this site (51.7 ppm) is consistent with that of tetrahedral units Al(OAl)n(OP)4−n.75 This 27Al peak could then be assigned to AlIV sites bonded to one P site. For the other three 27Al peaks, the rough analysis of the initial REDOR build-up considering single 27Al–31P pairs leads to Al–P distances slightly above 4 Å, compatible with the presence of more distant P units.76
From these solid-state NMR analyses, we, therefore, conclude that P moieties and Al sites are spatially close in the hybrid and calcined samples thanks to favored interactions in the colloidal suspensions.
6. Experimental
6.1. Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 20 min), dialyzed to neutrality against distilled water, and sonicated with an ultrasound homogenizer (Omni Ruptor 250 W) operated at 50% power in the pulse mode for 20 min effective sonication time. The resulting CellNC suspension was finally freeze-dried and stored at 277 K.
000 rpm, 20 min), dialyzed to neutrality against distilled water, and sonicated with an ultrasound homogenizer (Omni Ruptor 250 W) operated at 50% power in the pulse mode for 20 min effective sonication time. The resulting CellNC suspension was finally freeze-dried and stored at 277 K.
In the case of CellNC, the atomic sulfur contents (S%ato) determined by EDX and XPS (Table S1, ESI†) are 0.45 and 0.35 respectively, in line with literature values.50,77–79 Calculated atomic contents for CellNC were thus obtained considering an average sulfur content S%ato of 0.4. Note that the carbon contribution is overestimated by EDX, which is rather usual and possibly due to contaminations. In turn, the oxygen and nitrogen contents appear underestimated. The values obtained by XPS agree relatively well with the predicted ones.
000 rpm, 4 h) centrifugation cycles. Under these conditions, chitin deacetylation was estimated to be approximately 20% (by conductivity and IR measurements). The aqueous ChitNC suspension was finally freeze-dried and stored at 277 K.
In the case of chitin nanocrystals, the nitrogen content calculated assuming a DA of 80% based on previously published experimental determination11 is bracketed by the EDX and XPS measurements (Table S1, ESI†).
To prepare hybrid suspensions, polysaccharide nanocrystals (cellulose: CellNC, CellNC-P or chitin: ChitNC, ChitNC-P) were dispersed in water with the pH set to 4.5. The Al13 cluster suspension was then simply added under stirring at room temperature for at least 30 minutes before analysis or further processing. The hybrid suspensions were prepared with a final Al concentration set at 0.025 M, a molar ratio Al/saccharide unit set to 1 and a pH between 4 and 5.
6.2. Characterization
NMR spectrum fitting was undertaken using the Dmfit software,82 and mathematical models for Gaussian/Lorentzian lines and quadrupolar lineshapes (Central Transition for nuclei with I = n/2 (n > 1) considering an infinite MAS frequency) described in the dedicated webpage.83 For 27Al spectra of calcined samples, the fitting uses a mathematical function that describes the distribution in the quadrupolar coupling parameters and the related electric field gradient through the Czjzeck model (Gaussian Isotropic Model).65,66 Details are given in ESI† S10.
XRD patterns were recorded using a Bruker D8 diffractometer equipped with a Bruker Lynx Eye detector and using the Kα line of copper at λ = 1.5405 Å. The diffractograms were acquired between 4 and 50° in 2θ steps of 0.02° with a duration of 2 s per step.
N 2 sorption isotherms at 77 K were recorded using a Micromeritics Tristar apparatus. Each calcined sample was outgassed at 553 K until a stable static vacuum of 0.13 Pa was reached. Specific surface areas of SBET were calculated using the BET method.84
EDX spectroscopy was performed by analyzing back-scattered electrons (5 independent measurements) with a SEM FEI Quanta 200F operated at 15 kV under low vacuum (40 Pa).
The XPS measurements were performed using a Thermoelectron ESCALAB 250 device. The photoelectron emission spectra were recorded using Al-Kα radiation (hν = 1486.6 eV) from a monochromatized source. The analyzed area was approximately 0.5 mm2. The pass energy was fixed at 20 eV. The spectrometer energy calibration was made using the Au 4f7/2 (83.9 ± 0.1 eV) and Cu 2p3/2 (932.8 ± 0.1 eV) photoelectron lines. All spectra were recorded using electron charge neutralizer guns to minimize the surface charging effect that may occur at the insulating powder surface during the photoemission process. All spectra were calibrated using the adventitious carbon C 1s emission at 284.8 eV. XPS spectra were treated using AVANTAGE software. The background signal was removed using the Shirley method.85 The atomic concentrations were determined with an accuracy less than 10% from photoelectron peak areas using the atomic sensitivity factors reported by Scofield.86
Elemental analyses used inductive coupled plasma (ICP) and were done at the CNRS “Central Analysis Facility” (Vernaison, France).
Dynamic Light Scattering (DLS) and zeta potential measurements were performed with a Malvern Zetasizer Nano ZS. Each data represented in the figures is the mean value from 3 repetitions, each being the average over 10 individual measurements. Error bars represented in the figure correspond to standard deviation.
Scanning electron microscopy (SEM) images were collected with a HITACHI S4800 microscope using platinum coating on the samples (<50 Å) prior to the observation.
7. Conclusions
Phosphorylation of chitin and cellulose nanocrystals (ChitNC and CellNC respectively) has been successfully achieved using POCl3 as a reagent. This process results in stable phosphorylated polysaccharide nanoparticles with preserved internal structures (XRD, 13C NMR). The amount of phosphorous at the particles’ surface is between 0.75 and 1.2 per polysaccharide cycle (XPS, EDX). The cellulose nanocrystals become more aggregated during the phosphorylation process (DLS of suspensions) leading to a lower global weight percentage of P in the final CellNC-P particles (1 wt% compared to 3 wt% and more for ChitNC-P as measured by ICP). On the contrary, no additional particle aggregation is observed for ChitNC in suspension at low pH (below 6). This observation, along with XPS data, suggests that phosphorylation may have led to the formation of a P–N phosphamide bond.The resulting phosphorylated nanoparticles, CellNC-P and ChitNC-P, form stable dispersions over the whole pH ranges investigated (from 2 to 12 for CellNC-P and from 4 to 13 for ChitNC-P). Interestingly, the phosphorylation of ChitNC largely extends the pH stability range of the particles’ suspensions, which is usually limited to pH < 6 for pristine ChitNC. This increased stability is explained by the presence of phosphate functions at the polysaccharide's surface that maintain an overall negative zeta potential. In the case of ChitNC-P, the zeta potential values are consistent with the presence of a P–N bond at the nanoparticles’ surface.
The interactions between pristine and phosphorylated polysaccharide nanocrystals with Keggin-type aluminum oxo–hydroxo clusters (ε-Al13O4(OH)24(H2O)12)7+ (Al13) systematically lead to aggregated particles with hydrodynamic radii between 1 and 10 μm (DLS). It is noteworthy that the zeta potential values are all positive in the presence of Al13, with an Al/(saccharide unit) molar ratio adjusted to one. Al13 clusters are therefore associated with the pristine and phosphorylated polysaccharide nanocrystals to form positively charged colloids.
The suspensions prepared by mixing both types of colloids, polysaccharide nanocrystals and Al13 clusters were spray-dried to form hybrid organic–inorganic materials (SEM) with preserved polysaccharide and Al13 structures (NMR). After calcination in air, these hybrid solids are transformed into mesoporous alumina and aluminophosphate materials (N2 sorption isotherms). From solid-state NMR studies (31P, 27Al and 27Al{31P} REDOR experiments), it appears that P and Al sites are spatially close in hybrid and calcined samples. This is due to favored interactions in the precursors’ suspensions, a result of our approach combining phosphorylation of polysaccharide nanocrystals with colloidal self-assembly.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from PHC Toubkal CNRST-CNRS (N° TBK//19/87, Campus France N°41517TF).Notes and references
- Q. Song, Z. Wang, D. Xu, S. Liu, H. Liu and K. Zhang, Prog. Polym. Sci., 2024, 148, 101768 CAS.
- E. Lizundia, T.-D. Nguyen, R. J. Winnick and M. J. MacLachlan, J. Mater. Chem. C, 2021, 9, 796–817 CAS.
- Y. Wang, Z. Chen, J. Tang and N. Lin, in Advanced Functional Materials from Nanopolysaccharides, ed.N. Lin, J. Tang, A. Dufresne and M. K. C. Tam, Springer, Singapore, 2019, pp. 87–136 Search PubMed.
- G. R. Meseck, A. S. Terpstra and M. J. MacLachlan, Curr. Opin. Colloid Interface Sci., 2017, 29, 9–20 CAS.
- M. Giese, L. K. Blusch, M. K. Khan and M. J. MacLachlan, Angew. Chem., Int. Ed., 2015, 54, 2888–2910 CAS.
- E. Belamie and B. Alonso, in Handbook of Sol-Gel Science and Technology, ed. L. Klein, M. Aparicio and A. Jitianu, Springer International Publishing, Cham, 2016, pp. 1–39 Search PubMed.
- H. Liu, Z. Wang, H. Xin, J. Liu, Q. Wang, B. Pang and K. Zhang, ACS Nano, 2024, 18, 22675–22708 CAS.
- J. Tang, J. Sisler, N. Grishkewich and K. C. Tam, J. Colloid Interface Sci., 2017, 494, 397–409 CAS.
- J. F. Revol and R. H. Marchessault, Int. J. Biol. Macromol., 1993, 15, 329–335 CAS.
- J. F. Revol, L. Godbout, X. M. Dong, D. G. Gray, H. Chanzy and G. Maret, Liq. Cryst., 1994, 16, 127–134 CAS.
- E. Belamie, P. Davidson and M. M. Giraud-Guille, J. Phys. Chem. B, 2004, 108, 14991–15000 CAS.
- L. Heux, G. Chauve and C. Bonini, Langmuir, 2000, 16, 8210–8212 CAS.
- R. M. Parker, T. G. Parton, C. L. C. Chan, M. M. Bay, B. Frka-Petesic and S. Vignolini, Acc. Mater. Res., 2023, 4, 522–535 CAS.
- K. E. Shopsowitz, W. Y. Hamad and M. J. MacLachlan, J. Am. Chem. Soc., 2012, 134, 867–870 CAS.
- T. D. Nguyen, W. Y. Hamad and M. J. MacLachlan, Chem. Commun., 2013, 49, 11296–11298 RSC.
- J. A. Kelly, K. E. Shopsowitz, J. M. Ahn, W. Y. Hamad and M. J. MacLachlan, Langmuir, 2012, 28, 17256–17262 CrossRef CAS PubMed.
- B. Alonso and E. Belamie, Angew. Chem., Int. Ed., 2010, 49, 8201–8204 CrossRef CAS PubMed.
- E. Belamie, M. Y. Boltoeva, K. Yang, T. Cacciaguerra and B. Alonso, J. Mater. Chem., 2011, 21, 16997–17006 RSC.
- L. Cardoso, T. Cacciaguerra, P. Gaveau, L. Heux, E. Belamie and B. Alonso, New J. Chem., 2017, 41, 6014–6024 RSC.
- A. Sachse, V. Hulea, K. L. Kostov, N. Marcotte, M. Y. Boltoeva, E. Belamie and B. Alonso, Chem. Commun., 2012, 48, 10648–10650 RSC.
- G. Smolyakov, S. Pruvost, L. Cardoso, B. Alonso, E. Belamie and J. Duchet-Rumeau, Carbohydr. Polym., 2016, 151, 373–380 CrossRef CAS PubMed.
- G. Smolyakov, S. Pruvost, L. Cardoso, B. Alonso, E. Belamie and J. Duchet-Rumeau, Carbohydr. Polym., 2017, 166, 139–145 CrossRef CAS PubMed.
- B. Alonso, N. Witczak, C. Vallicari, B. Lecourt-Capdeville, M. Guiraud, L. Vachoud, K. L. Kostov, S. Spirk, G. Trimmel and E. Belamie, Micropor. Mesopor. Mater., 2022, 341, 112057 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- K. E. Shopsowitz, H. Qi, W. Y. Hamad and M. J. MacLachlan, Nature, 2010, 468, 422–425 CrossRef CAS PubMed.
- K. E. Shopsowitz, A. Stahl, W. Y. Hamad and M. J. MacLachlan, Angew. Chem., Int. Ed., 2012, 51, 6886–6890 CrossRef CAS PubMed.
- M. Schlesinger, W. Y. Hamad and M. J. MacLachlan, Soft Matter, 2015, 11, 4686–4694 RSC.
- G. Chu, X. S. Wang, H. Yin, Y. Shi, H. J. Jiang, T. R. Chen, J. X. Gao, D. Qu, Y. Xu and D. J. Ding, ACS Appl. Mater. Interfaces, 2015, 7, 21797–21806 CrossRef CAS PubMed.
- D. Qu, J. N. Zhang, G. Chu, H. J. Jiang, C. F. Wu and Y. Xu, J. Mater. Chem. C, 2016, 4, 1764–1768 CAS.
- B. Frka-Petesic, T. G. Parton, C. Honorato-Rios, A. Narkevicius, K. Ballu, Q. Shen, Z. Lu, Y. Ogawa, J. S. Haataja, B. E. Droguet, R. M. Parker and S. Vignolini, Chem. Rev., 2023, 123, 12595–12756 CAS.
- M. Y. Boltoeva, I. Dozov, P. Davidson, K. Antonova, L. Cardoso, B. Alonso and E. Belamie, Langmuir, 2013, 29, 8208–8212 CAS.
- J. A. Kelly, C. P. K. Manchee, S. S. Cheng, J. M. Ahn, K. E. Shopsowitz, W. Y. Hamad and M. J. MacLachlan, J. Mater. Chem. C, 2014, 2, 5093–5097 CAS.
- K. E. Shopsowitz, J. A. Kelly, W. Y. Hamad and M. J. MacLachlan, Adv. Funct. Mater., 2014, 24, 327–338 CrossRef CAS.
- A. Ivanova, D. Fattakhova-Rohlfing, B. E. Kayaalp, J. Rathousky and T. Bein, J. Am. Chem. Soc., 2014, 136, 5930–5937 CrossRef CAS PubMed.
- A. Ivanova, M. C. Fravventura, D. Fattakhova-Rohlfing, J. Rathousky, L. Movsesyan, P. Ganter, T. J. Savenije and T. Bein, Chem. Mater., 2015, 27, 6205–6212 CAS.
- A. Sachse, V. Hulea, K. L. Kostov, E. Belamie and B. Alonso, Catal. Sci. Technol., 2015, 5, 415–427 RSC.
- K. E. Shopsowitz, W. Y. Hamad and M. J. MacLachlan, Angew. Chem., Int. Ed., 2011, 50, 10991–10995 CAS.
- L. Yao, W. Zhong, L. Qiu and L. Deng, Int. J. Electrochem. Sci., 2018, 13, 5798–5809 CAS.
- J. Zhou, L. Bao, S. Wu, W. Yang and H. Wang, Carbohydr. Polym., 2017, 173, 321–329 CAS.
- A. Anouar, N. Katir, A.-S. Mamede, A. Aboulaich, K. Draoui, S. Royer and A. E. Kadib, Mater. Chem. Front., 2019, 3, 242–250 CAS.
- F.-E. Zirar, A. Anouar, N. Katir, I. A. Ichou and A. E. Kadib, RSC Adv., 2021, 11, 28116–28125 CAS.
- N. Illy, M. Fache, R. Ménard, C. Negrell, S. Caillol and G. David, Polym. Chem., 2015, 6, 6257–6291 RSC.
- J. F. Revol, H. Bradford, J. Giasson, R. H. Marchessault and D. G. Gray, Int. J. Biol. Macromol., 1992, 14, 170–172 CrossRef CAS PubMed.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J.-L. Putaux, L. Heux, F. Dubreuil and C. Rochas, Biomacromolecules, 2008, 9, 57–65 CrossRef CAS PubMed.
- S. Blilid, N. Katir, J. E. Haskouri, M. Lahcini, S. Royer and A. E. Kadib, New J. Chem., 2019, 43, 15555–15562 RSC.
- D. J. Suchyta, R. J. Soto and M. H. Schoenfisch, Polym. Chem., 2017, 8, 2552–2558 RSC.
- K. R. Dixon, Phosphorous to bismuth in Multinuclear NMR, ed. J. Mason, Plenum Press, New York, 1987 Search PubMed.
- K. Wang and Q. Liu, Carbohydr. Res., 2014, 386, 48–56 CrossRef CAS PubMed.
- P. Vassileva, V. Krastev, L. Lakov and O. Peshev, J. Mater. Sci., 2004, 39, 3201–3202 CrossRef CAS.
- H. Wang, C. Qian and M. Roman, Biomacromolecules, 2011, 12, 3708–3714 CrossRef CAS PubMed.
- A. Sachse, L. Cardoso, K. L. Kostov, C. Gerardin, E. Belamie and B. Alonso, Chem. Eur. J., 2015, 21, 3206–3210 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- J. C. P. Broekhoff and J. H. de Boer, J. Catal., 1967, 9, 15–27 CrossRef CAS.
- J. C. P. Broekhoff and J. H. de Boer, J. Catal., 1968, 10, 377–390 CrossRef CAS.
- H. Kono, Biopolymers, 2004, 75, 255–263 CrossRef CAS PubMed.
- L. Zhang and H. Eckert, J. Mater. Chem., 2004, 14, 1605–1615 RSC.
- J. W. Akitt, N. N. Greenwood, B. L. Khandelwal and G. D. Lester, J. Chem. Soc., Dalton Trans., 1972, 604–610 RSC.
- G. Fu, L. F. Nazar and A. D. Bain, Chem. Mater., 1991, 3, 602–610 CrossRef CAS.
- A. C. Kunwar, A. R. Thompson, H. S. Gutowsky and E. Oldfield, J. Magn. Reson., 1984, 60, 467–472 CAS.
- L. Allouche, C. Huguenard and F. Taulelle, J. Phys. Chem. Solids, 2001, 62, 1525–1531 CrossRef CAS.
- S. Acosta, R. J. P. Corriu, D. Leclercq, P. Lefèvre, P. H. Mutin and A. Vioux, J. Non-Cryst. Solids, 1994, 170, 234–242 CrossRef CAS.
- G. Kunath-Fandrei, T. J. Bastow, J. S. Hall, C. Jaeger and M. E. Smith, J. Phys. Chem., 1995, 99, 15138–15141 CrossRef CAS.
- C. V. Chandran, C. E. A. Kirschhock, S. Radhakrishnan, F. Taulelle, J. A. Martens and E. Breynaert, Chem. Soc. Rev., 2019, 48, 134–156 RSC.
- T. Gullion and J. Schaefer, J. Magn. Reson., 1989, 81, 196–200 CAS.
- J.-B. d’Espinose de Lacaillerie, C. Fretigny and D. Massiot, J. Magn. Reson., 2008, 192, 244–251 CrossRef PubMed.
- G. Le Caër and R. A. Brand, J. Phys.: Condens. Matter, 1998, 10, 10715 CrossRef.
- M. Bertmer and H. Eckert, Solid State Nucl. Magn. Reson., 1999, 15, 139–152 CrossRef CAS PubMed.
- J. C. C. Chan and H. Eckert, J. Magn. Reson., 2000, 147, 170–178 CrossRef CAS PubMed.
- L. Zhang and H. Eckert, Solid State Nucl. Magn. Reson., 2004, 26, 132–146 CrossRef CAS PubMed.
- A. Goiffon, J.-C. Jumas, M. Maurin and E. Philippot, J. Solid State Chem., 1986, 61, 384–396 CrossRef CAS.
- Y. Muraoka and K. Kihara, Phys. Chem. Miner., 1997, 24, 243–253 CrossRef CAS.
- H. Graetsch, Acta Crystallogr., Sect. C:Struct. Chem., 2000, 56, 401–403 CrossRef PubMed.
- S. N. Achary, O. D. Jayakumar, A. K. Tyagi and S. K. Kulshresththa, J. Solid State Chem., 2003, 176, 37–46 CrossRef CAS.
- G. D. Wignall, R. N. Rothon, G. W. Longman and G. R. Woodward, J. Mater. Sci., 1977, 12, 1039–1049 CAS.
- T. T. P. Cheung, K. W. Willcox, M. P. McDaniel, M. M. Johnson, C. Bronnimann and J. Frye, J. Catal., 1986, 102, 10–20 CAS.
- G. D. Wignall, R. N. Rothon, G. W. Longman and G. R. Woodward, J. Mater. Sci., 1977, 12, 1039–1049 CAS.
- S. Beck-Candanedo, M. Roman and D. G. Gray, Biomacromolecules, 2005, 6, 1048–1054 CAS.
- X. M. Dong, J. F. Revol and D. G. Gray, Cellulose, 1998, 5, 19–32 CAS.
- S. Beck, M. Méthot and J. Bouchard, Cellulose, 2015, 22, 101–116 CAS.
- N. Sanson, F. Bouyer, M. Destarac, M. In and C. Gérardin, Langmuir, 2012, 28, 3773–3782 CAS.
- T. Gullion, D. B. Baker and M. S. Conradi, J. Magn. Reson., 1990, 89, 479–484 CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J. O. Durand, B. Bujoli, Z. H. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CAS.
- Dmfit software, https://nmr.cemhti.cnrs-orleans.fr/Dmfit/help/Models/Default.aspx, last accessed March 17 2025.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CAS.
- D. A. Shirley, Phys. Rev. B:Condens. Matter Mater. Phys., 1972, 5, 4709–4714 Search PubMed.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nj00413f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |