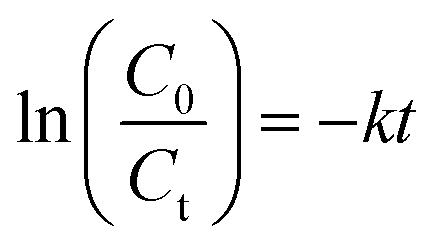DOI:
10.1039/D5NJ00407A
(Paper)
New J. Chem., 2025,
49, 8267-8278
Construction of Fe3O4/ZnO heterostructure photocatalysts derived from Fe-doped ZIF-8 for enhanced photocatalytic degradation of tetracycline and hydrogen peroxide production†
Received
30th January 2025
, Accepted 23rd April 2025
First published on 24th April 2025
Abstract
This study focuses on the development of Fe3O4/ZnO heterostructures derived from Fe-doped ZIF-8 through a precipitation and thermal decomposition approach. Fe3+ ions were incorporated into the ZIF-8 framework to partially replace Zn2+ ions, facilitating the formation of Fe3O4 during calcination under air. Optical and structural characterization, including ultraviolet-visible diffuse reflectance spectroscopy (UV-Vis DRS), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS), confirmed the successful formation of crystalline heterostructures. These heterostructures exhibited extended visible-light absorption, a narrowed bandgap, and the formation of defect states, which improved charge separation and suppressed electron–hole recombination. The Fe3O4/ZnO heterostructures demonstrate enhanced photocatalytic performance for two critical applications: tetracycline (TC) degradation and hydrogen peroxide (H2O2) production under visible light irradiation. Among the tested materials, Fe-ZIF-8 calcined at 500 °C showed the highest photocatalytic efficiency, achieving 74.8% TC degradation within 2 h, with a rate of 8.21 × 10−3 min−1. Additionally, it exhibited a significantly enhanced H2O2 production yield of 919.2 μM within 3 h of irradiation. Radical scavenging experiments identified superoxide radicals (˙O2−) as the primary reactive species driving the photocatalytic reaction. These findings highlight the potential of ZIF-8-derived photocatalysts as efficient and promising materials for sustainable wastewater treatment and green chemical production.
1. Introduction
The contamination of water resources with persistent organic pollutants, particularly antibiotics such as tetracycline (TC), poses a critical challenge to global sustainability. TC is frequently detected in water bodies due to its excessive use in agriculture, aquaculture, and healthcare, combined with inefficient wastewater treatment processes.1,2 Its presence disrupts aquatic ecosystems, promotes the proliferation of antibiotic-resistant bacteria, and poses severe health risks, including joint diseases and nephropathy, through bioaccumulation and prolonged exposure.2 Therefore, effective removal of TC from wastewater is critical for environmental remediation and public health. Concurrently, hydrogen peroxide (H2O2) has emerged as a green oxidant with applications in advanced oxidation processes, water disinfection, and eco-friendly chemical synthesis.3 The global market size for H2O2 was valued at USD 3.24 billion in 2023 and is projected to grow to USD 4.05 billion by 2027, reflecting its growing importance.4 Direct production of H2O2 offers a sustainable alternative to conventional energy-intensive, such as the anthraquinone process and electrocatalysis,3,5 making it a key focus for sustainable environmental technologies. However, the development of efficient, stable, and cost-effective methods capable of addressing both TC degradation and H2O2 production remains a significant scientific challenge.
Photocatalysis, driven by the activation of semiconductors under light irradiation, has gained attention as a promising approach for addressing environmental issues and advancing sustainable chemical processes. However, traditional photocatalysts like titanium dioxide (TiO2) and zinc oxide (ZnO) suffer from inherent limitations, including wide bandgaps that limit their activity to UV light and rapid recombination of photogenerated electron–hole pairs, which reduces their overall efficiency.6 To overcome these challenges, researchers have turned to innovative materials such as metal–organic frameworks (MOFs) for photocatalyst design.6–9 MOFs, particularly zeolitic imidazolate frameworks (ZIFs), offer unique advantages such as high surface area, tunable porosity, and chemical functionality.10,11 ZIF-8, a sodalite-type MOF composed of Zn2+ ions and 2-methylimidazole linkers, has emerged as a versatile precursor for functional materials due to its thermal stability and well-defined crystalline structure.12,13 Upon thermal decomposition, ZIF-8 transforms into ZnO while retaining its key structural features.14,15 Compared to other synthesis methods, MOF-derived metal oxides offer advantages such as high surface area, uniform porosity, abundant active sites, and efficient charge diffusion, capable of enhancing electrochemical and photochemical reactions.16–18 Viswanathan et al. reported a ZnO/C3N4 photocatalyst derived from ZIF-8 and C3N4 sheets, demonstrating improved photocatalytic performance due to interfacial coupling, which enhanced charge separation. The ZnO, derived from calcination of ZIF-8 at 600 °C, combined with C3N4 showed excellent sunlight-driven degradation of rhodamine B (RhB), achieving 95% degradation within 70 minutes.6 On the other hand, Su et al. developed a simple dipping-pyrolysis route to fabricate a novel 3D structure photocatalytic microreactor based on ZIF-8-derived porous ZnO nanocages supported on reduced graphene oxide-coated carbon sponge, which demonstrated excellent photocatalytic performance for RhB degradation and hydrogen production.19
Enhancing the performance of ZIF-8-derived materials can be achieved by incorporating transition metal dopants such as Ag,20 Cu,21 Co,22 and Fe.23 Metal doping introduces additional electronic states, reduces the bandgap, and enhances visible-light absorption, thereby improving the photocatalytic efficiency. Furthermore, the construction of heterostructures derived from ZIF-8, such as C3N4/ZnO,6 CuO/ZnO,8 Cu2O/ZnO,24 TiO2/ZnO,25 CuS/ZnO,26 SrTiO3/ZnO,27 and Co3O4/ZnO,28 offers a powerful strategy to further boost photocatalytic performance. Heterostructures provide enhanced charge separation through the formation of an internal electric field at the interface of the two phases, which suppresses electron–hole recombination.29,30 Previous studies have shown that FexOy/ZnO heterostructures exhibit superior photocatalytic performance due to their ability to facilitate efficient charge transfer between ZnO and iron oxide phases, resulting in prolonged lifetimes of charge carriers.31,32 Dien et al. synthesized ZnO nanoplates and Fe2O3 nanospindles using hydrothermal method and combined them in varying weight ratios to form Fe2O3/ZnO hybrids.31 The heterojunction formed between ZnO and Fe2O3 improved visible light absorption, facilitated enhanced electron–hole separation, and reduced recombination, contributing to its exceptional performance for methylene blue (MB) degradation under green LED illumination. On the other hand, Huong et al. demonstrated that Fe3O4/ZnO hetero-nanostructures synthesized via hydrothermal technique achieved significant enhancement in MB degradation efficiency, reaching up to 99.7% under UV light and 79.7% under visible light irradiation.32 This improvement was attributed to the transfer of electrons between the conduction bands of ZnO and Fe3O4, as well as the formation of defect states (e.g., Fe2+/Fe3+ levels) below the conduction band of ZnO, which promoted charge separation and prolonged carrier lifetimes. Despite these advancements, the development of FexOy/ZnO heterostructures derived from ZIF-8 remains both a challenge and an opportunity. Key challenges include achieving controlled phase transitions during calcination, retaining sufficient porosity after thermal decomposition, and optimizing the formation of heterostructures with efficient charge separation. Moreover, integrating transition metals like Fe into the ZIF-8 framework to introduce mixed-valence states can improve charge transfer and visible-light activity. Additionally, the practical application of FexOy/ZnO heterostructures for pollutant degradation and green chemical production remains underexplored.
This study addresses these challenges by synthesizing Fe-doped ZIF-8 and transforming it into Fe3O4/ZnO heterostructures through a precipitation method followed by thermal decomposition under air. During this process, Fe3+ ions partially replace Zn2+ within the ZIF-8 framework, enabling the formation of Fe3O4 during calcination. The resulting Fe3O4/ZnO heterostructures exhibit extended visible-light absorption, additional defect sites, and enhanced charge separation, significantly improving their photocatalytic performance for TC degradation and H2O2 production. By leveraging the unique properties of ZIF-8 and its derivatives, this work aims to address the challenges of inefficient photocatalysts and provide an environmentally friendly and sustainable solution for wastewater treatment and chemical production. Additionally, the structural and optical characteristics of these materials are investigated, correlating their physicochemical properties with their photocatalytic performance.
2. Experimental
2.1. Materials
The chemicals used in this study included zinc nitrate hexahydrate (Zn(NO3)2·6H2O, Fisher), 2-methylimidazole (C4H6N2, 99%, Acros) (denoted as meIm), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, R&M Chemicals), methanol (CH3OH, 99.9%, Fisher) (denoted as MeOH), tetracycline (C22H24N2O8, AR, 98%, Sigma-Aldrich) (denoted as TC), oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV) ([TiO(tpypH4)]4+, Tokyo Chemistry Industry Co., Ltd) (denoted as Ti-TPyP), hydrogen peroxide (H2O2, 30%, Nacalai Tesque), benzyl alcohol (C6H5CH2OH, >97%, Nacalai Tesque) (denoted as BA), acetonitrile (CH3CN, 99%, Nacalai Tesque) (denoted as ACN), perchloric acid (HClO4, 60%, Nacalai Tesque), hydrochloric acid (HCl, 16.9%, Nacalai Tesque), isopropyl alcohol ((CH3)2CHOH, 99.8%, QRec Chemicals) (denoted as IPA), 1,4-p-benzoquinone (C6H4O2, 99%, HmbG Chemical) (denoted as BQ) and ethylenediamine tetraacetic acid ([CH2N(CH2CO2H)2]2, 99.5%, R&M Chemicals) (denoted as EDTA). All chemicals used were of analytical grade and used without further purification.
2.2. Synthesis of Fe-ZIF-8 and Fe3O4/ZnO heterostructures
Fe-doped ZIF-8 (Fe-ZIF-8) was synthesized via a precipitation method, adapted from Du et al. with slight modification.33 First, 2.380 g of Zn(NO3)2·6H2O and 0.0238 g of Fe(NO3)3·9H2O were dissolved in 56 mL of MeOH (solution A). Separately, 5.287 g of meIm was dissolved in 56 mL of MeOH (solution B). Solution B was added dropwise to solution A, and the resulting mixture was stirred at 650 rpm for 6 h to facilitate the precipitation of Fe-ZIF-8. The precipitate was collected by centrifugation at 3000 rpm, washed three times with MeOH, and dried at 100 °C for 1 h. The dried Fe-ZIF-8 was calcined in a quartz tube furnace under an air atmosphere to form Fe3O4/ZnO heterostructures. Calcination was performed at 300, 500, 700, and 900 °C for 3 h with a heating rate of 5 °C min−1. The calcined samples were denoted as Fe-ZIF-8 300, Fe-ZIF-8 500, Fe-ZIF-8 700, and Fe-ZIF-8 900, respectively. For comparison, undoped ZIF-8 was prepared following the same procedure but without adding Fe(NO3)3·9H2O. The resulting undoped samples were similarly calcined and labeled accordingly. Fig. 1 shows the schematic representation of the synthesis of Fe3O4/ZnO heterostructures derived from Fe-ZIF-8 by thermal decomposition under an air atmosphere. The Fe-ZIF-8 precursor appeared as a fine beige powder, while calcination resulted in noticeable color changes due to thermal transformations. At 300 °C, the samples exhibited a brownish appearance, transitioning to an intense reddish-brown hue at 500 °C and above, reflecting the formation of Fe3O4/ZnO phases.
 |
| | Fig. 1 Schematic representation of the synthesis of Fe-ZIF-8, followed by thermal decomposition under an air atmosphere to produce Fe3O4/ZnO heterostructures. | |
2.3. Characterization
Various characterizations were conducted to investigate the optical, physical, and chemical properties of the photocatalytic materials. The optical absorption of the photocatalysts was assessed using solid-state ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS, Shimadzu UV-2450) from 200 to 800 nm with BaSO4 as the reference. The absorption spectra were analyzed using the Kubelka–Munk function to determine the bandgap energy of the photocatalysts.34 The crystalline phases and structural changes of the synthesized samples were recorded by powder X-ray diffraction (XRD) analysis (Rigaku Ultima IV) in the range of 5–60° with Cu Kα radiation (λ = 1.5405 Å). The thermal stability and decomposition behavior of the photocatalysts were evaluated using a PerkinElmer STA 6000 thermogravimetric analyzer (TGA). Samples were heated from room temperature to 900 °C at a rate of 5 °C min−1 under an air and nitrogen atmosphere. Surface morphology and elemental composition were analyzed using a scanning electron microscope coupled with an energy dispersive X-ray spectrometer (SEM-EDX, Model Quanta FEG 650, USA), operating at 0.9 kV and a magnification of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. The particle size has been calculated using the ImageJ software from the SEM images. The specific surface area and pore size distribution were determined using N2 physisorption measurements conducted on a BELSORP-max system (Microtrac BEL Corp., Inc.). Before the analysis, the samples were pretreated under vacuum at 120 °C for 5 h to eliminate moisture and surface impurities. Fourier-transform infrared (FTIR) spectroscopy (Model 2000, PerkinElmer, USA) was performed within the 4000–400 cm−1 range to identify functional groups and confirm the structural integrity of the photocatalysts. X-ray photoelectron spectroscopy (XPS) was carried out to analyze the surface composition and chemical states of the elements in the samples using an ESCA-3400 XPS system from Shimadzu equipped with a monochromatic Al Kα X-ray source. The binding energies were calibrated with reference to the C 1s peak at 284.6 eV.
000×. The particle size has been calculated using the ImageJ software from the SEM images. The specific surface area and pore size distribution were determined using N2 physisorption measurements conducted on a BELSORP-max system (Microtrac BEL Corp., Inc.). Before the analysis, the samples were pretreated under vacuum at 120 °C for 5 h to eliminate moisture and surface impurities. Fourier-transform infrared (FTIR) spectroscopy (Model 2000, PerkinElmer, USA) was performed within the 4000–400 cm−1 range to identify functional groups and confirm the structural integrity of the photocatalysts. X-ray photoelectron spectroscopy (XPS) was carried out to analyze the surface composition and chemical states of the elements in the samples using an ESCA-3400 XPS system from Shimadzu equipped with a monochromatic Al Kα X-ray source. The binding energies were calibrated with reference to the C 1s peak at 284.6 eV.
2.4. Evaluation of photocatalytic performance
2.4.1. Photocatalytic degradation of TC.
The photocatalytic degradation experiments were conducted in a custom-designed reactor equipped with a cooling fan to maintain room temperature and prevent thermal catalytic reactions. A 50 mg L−1 TC solution was used as the target pollutant, with 40 mg of the photocatalysts dispersed in 50 mL of the solution in a 100 mL beaker. Air was bubbled into the solution at a constant flow rate of 6 mL min−1, with continuous stirring using a magnetic stirrer to ensure proper mixing and maximum contact between the photocatalysts and the TC solution. A 120-W PAR38 warm light bulb (Philips), emitting visible light within 400–700 nm, was positioned 9 cm vertically above the beaker as the light source. The reaction was conducted in the dark for 1 h to reach adsorption–desorption equilibrium before being irradiated with light to initiate the photocatalytic reaction. At regular intervals, 5 mL aliquots were taken, centrifuged, and analyzed using a spectrophotometer (ThermoFisher Scientific, Genesys 30) at 370 nm. The degradation efficiency (R, %) of TC was calculated using the formula:| | 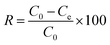 | (1) |
where C0 and Ct represent the initial concentration and the concentration of TC at a specific time interval, respectively, in mg L−1. The degradation rate (min−1), k, was derived by the Langmuir–Hinshelwood kinetic model given by:| |  | (2) |
2.4.2. Radical scavenging.
The radical scavenging experiment was conducted to determine the main active species involved in the photocatalytic degradation of TC. For this study, BQ, IPA, and EDTA were taken as scavengers for superoxide radicals (˙O2−), hydroxyl radicals (˙OH), and holes (h+), respectively.
2.4.3. Photocatalytic H2O2 production.
The photocatalytic production of H2O2 was assessed using a one-phase photocatalytic reaction system. Firstly, 5.0 mg of photocatalysts, 3.0 mL of ACN, and 2.0 mL of BA were added to a 30 mL reaction tube. The reactor was then sealed and sonicated for 5 minutes to ensure proper dispersion of the photocatalyst. Next, the mixture was bubbled with O2 at 20 mL min−1 for 30 minutes while magnetically stirred in the dark. The reaction tube was then irradiated with a 500 W Xe-lamp (Sanei Electric XEF-501S) through a 420 nm cut-off glass filter, maintaining a 5 cm distance between the tube and the lamp. The reaction was conducted at 25 °C and ambient pressure, with continuous stirring. Every hour, a 20 μL aliquot was withdrawn from the aqueous solution and diluted 100-fold with distilled water.
The concentration of H2O2 was determined using a [TiO(tpypH4)]4+ complex titration method.35 First, the Ti-TPyP reagent was prepared by dissolving 3.4 mg of the [TiO(tpypH4)]4+ complex in a 100 mL solution of 50 mM aqueous HCl. Then, 250 μL of the prepared Ti-TPyP reagent and 250 μL of 4.8 M HClO4 aqueous solution were mixed with 250 μL of the sample aliquot (diluted 100-fold with distilled water). After 5 minutes, the mixture was further diluted to a final volume of 2.5 mL with distilled water. The absorbance of the [TiO(tpypH4)]4+ complex at 434 nm decreased with the formation of H2O2, due to the formation of the peroxo[5, 10, 15, 20-tetra(4-pyridyl)porphyrinato]titanium(IV) complex. This decrease in absorbance (ΔA434) was directly proportional to the H2O2 concentration in the sample and was measured using a Shimadzu UV-2600 UV-vis spectrophotometer. The amount of H2O2 was calculated using the following equation:36
where
AB and
AS represent the absorbance of the blank solution and the sample solution, respectively. The H
2O
2 concentration was determined from the linear relationship between Δ
A434 and the molar concentration of H
2O
2, as established through a calibration curve.
3. Results and discussion
3.1. Characterization
The UV-vis diffuse reflectance spectra of Fe-ZIF-8, uncalcined and calcined at various temperatures (300, 500, 700, and 900 °C), provide valuable insights into the optical properties of the materials. These spectra are depicted as Kubelka–Munk functions in Fig. 2. The corresponding Tauc plots, shown in Fig. S1 (ESI†), illustrate the optical bandgap energies (Eg) of the samples, with the values summarized in Table 1. Uncalcined Fe-ZIF-8 exhibits minimal visible light absorption with an adsorption edge of approximately 250 nm, and a high bandgap energy of 5.26 eV. This high bandgap value is characteristic of MOFs like ZIF-8,23 limiting their photocatalytic activity to the UV region and restricting their potential for solar-driven applications. Calcination induces significant changes in the optical properties of the catalyst. At 300 °C, a significant red-shift in the absorption edge occurs, reducing the bandgap energy to 3.28 eV. This reduction is due to the degradation of organic linkers, the collapse of the MOF framework, and the formation of defect states, which extend light absorption into the visible region.7,37 At higher calcination temperatures (500–900 °C), the bandgap stabilizes slightly above 3.0 eV, consistent with the formation of thermally stable crystalline ZnO, whose bandgap energy is approximately 3.37 eV.37 Two key charge-transfer mechanisms that influence the optical and photocatalytic properties of these catalysts are intra-ligand charge transfer (ILCT) and ligand-to-metal charge transfer (LMCT).38,39 ILCT, dominant in uncalcined Fe-ZIF-8, involves electron transitions within the organic linker (meIm).39 As Fe-ZIF-8 was calcined at 300 °C, the partial degradation of the linker reduces ILCT, thus narrowing the bandgap. Between 500–900 °C, complete decomposition of organic linkers eliminates ILCT, resulting in metal oxide-like behavior. LMCT, which involves electron transfer from the linker to metal clusters,40 becomes dominant in calcined samples due to the formation of heterostructures. Compared to ZIF-8 (Fig. S2, ESI†), Fe-ZIF-8 demonstrates slightly enhanced visible light absorption due to the introduction of Fe3+ electronic states. The spectrum of Fe-ZIF-8 500 exhibits the most significant enhancement in visible light absorption and a slightly lower bandgap energy (Eg = 3.27 eV) compared to ZIF-8 500 (Eg = 3.29 eV). This improvement is attributed to the formation of Fe3O4/ZnO heterostructures, where the interaction between ZnO and Fe oxides promotes charge separation and creates defect states and interfacial pathways for efficient charge transfer.32 These effects result in broadened absorption and increased photocatalytic potential under visible light irradiation.
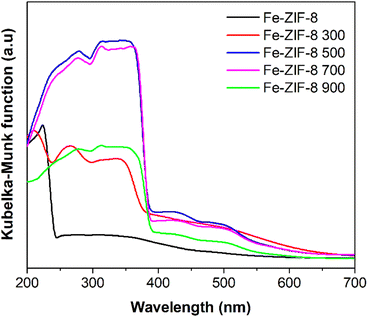 |
| | Fig. 2 UV-vis DRS spectra of uncalcined Fe-ZIF-8 and Fe-ZIF-8 calcined at different temperatures (300, 500, 700, and 900 °C). | |
Table 1 Bandgap energies and crystallite size of uncalcined Fe-ZIF-8 and Fe-ZIF-8 calcined at different temperatures (300, 500, 700, and 900 °C)
| Catalyst |
Bandgap energy (eV) |
Crystallite size, D (nm) |
| Fe-ZIF-8 |
5.26 |
30.6 ± 2.0 |
| Fe-ZIF-8 300 |
3.28 |
22.5 ± 0.6 |
| Fe-ZIF-8 500 |
3.27 |
23.1 ± 2.6 |
| Fe-ZIF-8 700 |
3.27 |
26.6 ± 2.4 |
| Fe-ZIF-8 900 |
3.23 |
31.2 ± 1.4 |
The phase transitions and crystal structures of Fe-ZIF-8 calcined at various temperatures were analyzed using XRD, as illustrated in Fig. 3(a). The diffraction patterns for uncalcined Fe-ZIF-8 confirm a sodalite (SOD)-type cubic crystal structure, similar to ZIF-8 (JCPDS 00-062-1030).41 With increasing calcination temperature, the XRD patterns demonstrate a gradual transformation from the MOF structure to a crystalline ZnO phase. This transition aligns with the thermal decomposition of organic linkers and the subsequent crystallization of metal oxides.7,15 At higher temperature of 500 °C and above, the diffraction peaks of Fe-ZIF-8 observed at 31.8°, 34.4°, 36.2°, 47.5°, and 56.5° correspond to the (100), (002), (101), (102), and (110) planes of wurtzite hexagonal-phase ZnO (JCPDS 36-1451).6 Additionally, new peaks appear corresponding to Fe3O4 magnetite (JCPDS 19-629),32,42 indicating the successful incorporation of Fe and the formation of Fe3O4/ZnO heterostructures. This differs from ZIF-8 500 (Fig. S3, ESI†), whose XRD pattern lacks Fe3O4-related peaks. Crystallite sizes calculated using the Debye–Scherrer equation,43 are summarized in Table 1. Calcination at 300 °C results in a significant reduction in crystallite size compared to the uncalcined sample, attributed to the structural degradation and collapse of the MOF framework. As the calcination temperature increases from 500 to 900 °C, the crystallite size grows from 23.1 ± 2.6 to 31.2 ± 1.4 nm. The values obtained at 500 °C are similar to the crystalline size of ZnO/α-Fe2O3 nanocomposites synthesized by the sol–gel method,44 indicating the development of well-crystallized FexOy/ZnO heterostructures.
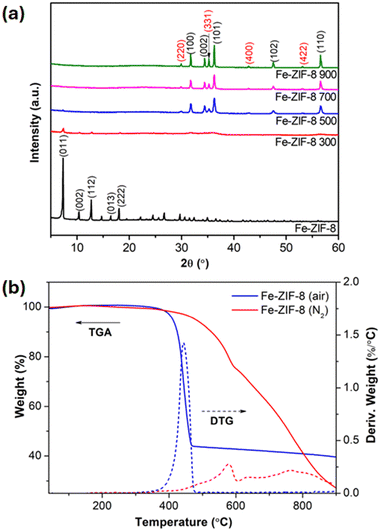 |
| | Fig. 3 (a) XRD patterns of uncalcined and calcined Fe-ZIF-8 at different temperatures (300, 500, 700, and 900 °C); (b) TGA and DTG of Fe-ZIF-8 under air and N2 atmosphere. | |
The TGA and DTG curves under air and N2 atmospheres, shown in Fig. 3(b), reveal key differences in the thermal stability and decomposition behavior of Fe-ZIF-8. Fe-ZIF-8 remains thermally stable up to 300 °C in both atmospheres, with minimal weight loss, indicating the absence of significant water or solvent desorption. A sharp weight loss occurs between 350 and 450 °C corresponding to the decomposition of the ZIF-8 framework. In air, this process is rapid due to the combustion of organic linkers in the oxidative environment, leading to the formation of stable residues beyond 500 °C. The final residue, primarily consists of ZnO and Fe3O4, derived from the complete combustion of organic components, as confirmed by the XRD analysis. In contrast, under N2, the decomposition is slower and less complete leading to partial oxidation of organic linkers. The residue includes ZnO, Fe3O4, and carbonaceous species, consistent with the formation of unburned carbon during the thermal decomposition of MOFs under inert conditions.7,45,46 The DTG curves further highlight the decomposition mechanisms. In air, a sharp peak at ∼440 °C signifies rapid, uniform oxidative degradation of the ZIF-8 framework. Under N2, a broad peak spanning 400–900 °C indicates a slower, stepwise decomposition process, characteristic of an inert atmosphere. Additional smaller peaks at higher temperatures in N2 suggest the degradation of residual carbonaceous species. The total weight loss also varies between the two atmospheres. In air, 60.4% weight loss is observed, reflecting efficient combustion and the removal of volatile organic compounds, leaving behind a higher proportion of inorganic oxides. In N2, 72.7% weight loss is observed, indicating a greater release of volatile products, but the residue includes unburned carbon, resulting in a lower proportion of crystalline oxides. These results signify that the calcination of Fe-ZIF-8 under air is preferred over inert conditions to promote the complete oxidation of the organic framework and the formation of well-crystallized Fe3O4/ZnO heterostructures. Additionally, it also implies that 500 °C is a sufficient temperature for the decomposition of Fe-ZIF-8 to achieve the desired heterostructures.
The SEM images in Fig. 4 illustrate the progressive morphological changes of Fe-ZIF-8 as it undergoes thermal treatment at various temperatures. The pristine Fe-ZIF-8 (Fig. 4(a)) exhibits a well-defined rhombic dodecahedral morphology with smooth surfaces and an average particle size of 815 ± 95 nm, reflecting its high crystallinity and intact SOD-type structure. Upon calcination at 300 °C (Fig. 4(b)), the structure largely retains its shape, though slight surface roughening is observed, and the average particle size reduces to 522 ± 81 nm, indicating the initial degradation of the organic linkers and partial collapse of the MOF framework. At 500 °C (Fig. 4(c)), the structure undergoes significant disintegration, transitioning into smaller, aggregated particles with increased porosity. The particle size significantly decreased to 81 ± 12 nm. This transformation corresponds to the decomposition of the organic framework and the formation of ZnO and Fe oxide phases. Calcination at 700 and 900 °C (Fig. 4(d) and (e)) leads to significant particle agglomeration, resulting in the formation of larger, irregularly shaped clumps with interconnected voids. This behavior is a characteristic of material sintering, occurs at elevated temperatures as thermal energy overcomes electrostatic repulsion and increased van der Waals forces, reducing surface energy and leading to particle agglomeration and the formation of compact, interconnected structures.44 The elemental analysis of Fe-ZIF-8 and Fe-ZIF-8 500, as shown in Fig. S4 (ESI†), highlights the compositional changes and elemental distribution resulting from the calcination process. The elemental maps of Fe-ZIF-8 reveal the uniform distribution of Zn, Fe, C, and N throughout the material. The presence of C and N corresponds to the organic linkers (meIm) within the ZIF-8 framework. The Fe mapping confirms the successful incorporation of Fe within the MOF, while Zn is uniformly dispersed as part of the framework's metal nodes. The EDX spectrum further supported these findings, showing significant peaks for Zn, Fe, C, and N. After calcination at 500 °C, the elemental composition undergoes significant changes. The elemental maps indicate the continued presence of Zn and Fe, which remain uniformly distributed. However, the C and N signals are no longer detectable, which is consistent with the thermal decomposition of the organic linkers and the collapse of the MOF framework. Instead, O appears prominently in the elemental maps, reflecting the formation of oxide phases such as ZnO and Fe oxides. The EDX spectrum shows strong peaks for Zn, Fe, and O, confirming the transition from an organic–inorganic hybrid framework to a primarily inorganic oxide structure.
 |
| | Fig. 4 SEM images of (a) Fe-ZIF-8, (b) Fe-ZIF-8 300, (c) Fe-ZIF-8 500, (d) Fe-ZIF-8 700, and (e) Fe-ZIF-8 900 at 20000× magnification. | |
The N2 adsorption–desorption isotherms of Fe-ZIF-8 and Fe-ZIF-8 500 (Fig. S5, ESI†) reveal key insights into the textural and structural changes, particularly porosity and surface area, resulting from calcination at 500 °C. The isotherm of Fe-ZIF-8 exhibits a Type I curve, characteristic of microporous materials, with a sharp increase in adsorption at low relative pressure (P/P0 < 0.1), indicating the dominance of micropores within its well-defined framework.47,48 This microporous structure is further supported by its high Brunauer–Emmett–Teller (BET) specific surface area of 1127 m2 g−1, which provides an abundance of active sites, making Fe-ZIF-8 a highly suitable precursor for functional photocatalytic applications. In contrast, Fe-ZIF-8 500 demonstrates a Type IV isotherm with a pronounced hysteresis loop at higher relative pressures (P/P0 > 0.5), indicative of mesoporous characteristics.41,47 This transition from microporosity to mesoporosity arises from the structural transformation induced by calcination at 500 °C. During this process, the organic linkers in the ZIF-8 framework decompose, leading to the collapse of the microporous structure and the formation of Fe3O4/ZnO heterostructures. Consequently, the BET surface area of Fe-ZIF-8 500 decreases significantly to 12.05 m2 g−1, reflecting a marked reduction in microporosity along with an increase in mesopore size or particle aggregation. Huang et al. obtained a comparable surface area for ZnO/ZnCo2O4 synthesized via the in situ growth on ZIF-8 templates followed by thermal annealing in air.49 Furthermore, mesoporous photocatalysts have been shown to improve accessibility for reactants to active sites, mitigating diffusion limitations, and enhance interaction between the photocatalyst and reactants, thereby improving overall catalytic performance.28,50,51
The FTIR spectra of Fe-ZIF-8 and its calcined forms at various temperatures (300, 500, 700, and 900 °C), shown in Fig. S6 (ESI†), provide insights into the structural changes and chemical transformations occurring during thermal decomposition. For uncalcined Fe-ZIF-8, distinct peaks are observed that are characteristic of the ZIF-8 framework.15,41 The bands observed in the 3000–2900 cm−1 region are attributed to the C–H stretching vibrations of the methyl group in the meIm linker. Additionally, peaks around 1600 and 1400 cm−1 correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching and C–N bending vibrations, respectively, which are indicative of the imidazole ring. In the fingerprint region (900–1350 cm−1), multiple sharp peaks appear due to the in-plane and out-of-plane bending of the imidazole ring. The bands in the range of ∼600–800 cm−1 are associated with the Zn–N bond, indicative of the metal–ligand coordination in the ZIF-8 structure. Upon calcination at 300 °C, the intensity of the C–H stretching bands decreases, indicating the partial decomposition of the organic linkers. Similarly, the CN stretching and C–N bending peaks weaken significantly. This reduction in intensity suggests the onset of thermal degradation of the ZIF-8 framework, although some organic components remain intact. At 500 °C, the spectra exhibit a complete disappearance of peaks associated with the ZIF-8 framework, confirming the complete decomposition of the organic linker. Instead, new peaks emerge in the lower wavenumber region (∼500–600 cm−1), which can be attributed to the Zn–O stretching vibrations of well-crystallized wurtzite-phase ZnO.6 Additionally, Fe-related vibrations may also contribute to this region, suggesting the formation of Fe3O4 in the calcined Fe-ZIF-8 samples.22 The progressive disappearance of ZIF-8 specific peaks and the emergence of metal oxides-specific vibrations confirm the structural transformation of Fe-ZIF-8 into Fe3O4/ZnO heterostructures with increasing calcination temperatures.
N stretching and C–N bending vibrations, respectively, which are indicative of the imidazole ring. In the fingerprint region (900–1350 cm−1), multiple sharp peaks appear due to the in-plane and out-of-plane bending of the imidazole ring. The bands in the range of ∼600–800 cm−1 are associated with the Zn–N bond, indicative of the metal–ligand coordination in the ZIF-8 structure. Upon calcination at 300 °C, the intensity of the C–H stretching bands decreases, indicating the partial decomposition of the organic linkers. Similarly, the CN stretching and C–N bending peaks weaken significantly. This reduction in intensity suggests the onset of thermal degradation of the ZIF-8 framework, although some organic components remain intact. At 500 °C, the spectra exhibit a complete disappearance of peaks associated with the ZIF-8 framework, confirming the complete decomposition of the organic linker. Instead, new peaks emerge in the lower wavenumber region (∼500–600 cm−1), which can be attributed to the Zn–O stretching vibrations of well-crystallized wurtzite-phase ZnO.6 Additionally, Fe-related vibrations may also contribute to this region, suggesting the formation of Fe3O4 in the calcined Fe-ZIF-8 samples.22 The progressive disappearance of ZIF-8 specific peaks and the emergence of metal oxides-specific vibrations confirm the structural transformation of Fe-ZIF-8 into Fe3O4/ZnO heterostructures with increasing calcination temperatures.
The XPS analysis provides critical insights into the surface composition and element states of the ZnO and Fe3O4/ZnO heterostructures derived from ZIF-8 and Fe-ZIF-8, respectively. The survey spectrum (Fig. 5(a)) reveals the presence of key elements, including Zn, O, and a minor amount of residual C in both Fe-ZIF-8 500 and ZIF-8 500, with additional Fe 2p peaks observed only in Fe-ZIF-8 500. This confirms the successful doping of Fe into the ZIF-framework, followed by the formation of Fe3O4/ZnO heterostructures. The absence of nitrogen in both spectra further validates the decomposition of the organic imidazolate framework, which aligns with the thermal decomposition of the ZIF-8 structure. The high-resolution Zn 2p spectra (Fig. 5(b)) of ZIF-8 500 display two distinct peaks at binding energies of 1022.7 eV (Zn 2p3/2) and 1045.8 eV (Zn 2p1/2), corresponding to the Zn2+ oxidation state in ZnO.32,52 The narrow peak shape and binding energy values confirm the presence of crystalline ZnO in the material, indicative of its transformation during calcination. In Fe-ZIF-8 500, these peaks shift slightly to 1021.9 eV and 1044.9 eV, suggesting some changes in the chemical environment of Zn2+ due to Fe doping. The Fe 2p spectrum (Fig. 5(c)) shows two prominent peaks at 710.6 eV (Fe 2p3/2) and 723.9 eV (Fe 2p1/2), with further splitting observed at 711.1 and 714.8 eV for Fe 2p3/2 and at 724.2 and 726.3 eV for Fe 2p1/2. These peaks correspond well to the mixed-valence states of Fe2+ and Fe3+.32 Additionally, a satellite peak at 718.4 eV supports the formation of Fe3O4. The O 1s spectra (Fig. 5(d)) is deconvoluted into three components, representing lattice oxygen (Olatt), defect oxygen (Odef), and adsorbed oxygen (Oads). In ZIF-8 500, the Olatt peak is located at 530.9 eV, representing Zn–O bonds, confirming the formation of ZnO. On the other hand, Odef and Oads overlap at 532.5 eV. In Fe-ZIF-8 500, the peak positions for Olatt, Odef, and Oads shift slightly to lower binding energies, indicating the influence of Fe doping on the local chemical environment of oxygen atoms. The Olatt peak shifts to 529.8 eV, confirming the presence of Fe–O bonds of Fe3O4, while the Odef peak is observed at 531.6 eV, and the Oads peak appears at 532.4 eV. The relative percentages of these oxygen types in Fe-ZIF-8 500 (based on area ratios) are 40.3% for Olatt, 44.5% for Odef, and 15.2% for Oads. The increased proportions of Odef and Oads in Fe-ZIF-8 500 suggest that Fe incorporation and the calcination process create additional defect sites and enhance surface reactivity. Huong et al. further suggested that such interactions introduce impurity defects (e.g., Fe2+/Fe3+ levels) below the conduction band edge of ZnO, facilitating electron transfer processes between ZnO and Fe3O4.32 This charge transfer mechanism reduces electron–hole recombination and improves the formation of reactive oxygen species, thereby enhancing the photocatalytic efficiency of the Fe3O4/ZnO heterostructure.
 |
| | Fig. 5 XPS spectra of ZIF-8 500 and Fe-ZIF-8 500: (a) survey spectrum, (b) Zn 2p, (c) Fe 2p, and (d) O 1s. | |
3.2. Photocatalytic performance
3.2.1. Photocatalytic degradation of TC.
The photocatalytic performance of Fe-ZIF-8 calcined at different temperatures (300, 500, 700, and 900 °C) for TC removal is presented in Fig. 6(a). The photolysis (conducted without a photocatalyst) shows negligible TC degradation, indicating the need of a catalyst for effective removal. In the initial dark adsorption phase, both uncalcined Fe-ZIF-8 and Fe-ZIF-8 300 demonstrate superior adsorption capacities, significantly reducing TC concentrations. This performance is attributed to the retention of the porous ZIF-8 framework, characterized by its high surface area and functional groups that facilitate efficient adsorption. As reported by Yang et al., the high adsorption performance of Fe-doped ZIF-8 is due to the synergistic effects of surface complexation, electrostatic attraction, π–π interactions, and hydrogen bonding.53 Calcination at higher temperatures reduces the adsorption capacity due to the collapse of the MOF framework, resulting in structural changes and phase transitions. The decrease is more significant for Fe-ZIF-8 700 and Fe-ZIF-8 900, where particle agglomeration decreases surface area and pore accessibility. Under light irradiation, Fe-ZIF-8 500 demonstrates the highest photocatalytic degradation efficiency among all samples. This superior performance is attributed to the formation of porous and crystalline Fe3O4/ZnO heterostructures, which promote efficient charge separation and prolonged electron–hole lifetimes. The Langmuir–Hinshelwood (L–H) rate expression has been used to evaluate the relationship between the degradation rate of TC by Fe-ZIF-8 samples. The linear plot of ln (C0/Ct) versus irradiation time (Figure not shown) signified that the photodegradation of TC over the Fe-ZIF-8 samples follows the pseudo-first-order kinetic model. Table 2 shows the values for the degradation rate, k, and relative coefficients (R2) from the model. The uncalcined Fe-ZIF-8 sample exhibits a degradation rate of 6.76 × 10−3 min−1. This low degradation is because of the high bandgap energy of Fe-ZIF-8 and TC adsorption. The degradation rate increases with increase in calcination temperature. The Fe-ZIF-8 sample calcined at 500 °C has the highest degradation rate of 8.21 × 10−3 min−1, confirming its superior photocatalytic activity in degrading TC molecules among all samples. As the calcination temperature was further increased beyond 500 °C, the degradation rates declined. This reduction is attributed to particle agglomeration and sintering, which significantly decrease the surface area and reduce the availability of active sites.
 |
| | Fig. 6 (a) Photocatalytic performance of Fe-ZIF-8 at different calcination temperature for TC removal, and (b) radical scavenging experiment. | |
Table 2 Degradation rates of uncalcined and calcined Fe-ZIF-8 at different temperatures for TC degradation
| Samples |
Degradation rate, k (×10−3 min−1) |
R
2
|
| Fe-ZIF-8 |
6.76 |
0.978 |
| Fe-ZIF-8 300 |
7.48 |
0.969 |
| Fe-ZIF-8 500 |
8.21 |
0.994 |
| Fe-ZIF-8 700 |
7.60 |
0.978 |
| Fe-ZIF-8 900 |
7.10 |
0.992 |
The degradation of TC primarily occurs through photogenerated charge carriers (electrons, e−, and holes, h+) and reactive oxygen species such as superoxide (˙O2−), hydroxyl (˙OH), and hydroperoxyl (˙HO2) radicals. To investigate the role of these species in the photodegradation process, a radical scavenging experiment was conducted on Fe-ZIF-8 500, as shown in Fig. 6(b). The results indicate a significant reduction in degradation efficiency when BQ was added, highlighting the vital role of ˙O2− radical in the photocatalytic degradation of TC. Photogenerated electrons interact with O2 molecules adsorbed on the catalyst surface, generating ˙O2− that actively oxidizes TC molecules. In contrast, the addition of EDTA and IPA did not notably affect the photocatalytic degradation, suggesting that h+ and ˙OH radicals have a limited contribution to the overall photocatalytic reaction.
A proposed photocatalytic mechanism for TC photodegradation using Fe-ZIF-8 500 is illustrated in Fig. 7(a). TC molecules are initially adsorbed onto the surface of Fe-ZIF-8 500. Upon visible light irradiation, Fe-ZIF-8 500 absorbs photon energy equal to or greater than its bandgap (∼3.27 eV). This leads to the excitation of electrons from the valence band (VB) to the conduction band (CB) of both ZnO and Fe3O4, leaving behind h+ in the VB (eqn (4)). At the Fe3O4/ZnO interface, photogenerated electrons in the CB of Fe3O4 transfer to the CB of ZnO due to favorable energy band alignment.32 This process enhances charge separation and reduces electron–hole recombination (eqn (5)). Additionally, impurity defects (Fe2+/Fe3+ states) at the interface act as intermediate electronic states, facilitating efficient electron transfer between Fe3O4 and ZnO. The accumulated electrons in the CB of ZnO and Fe3O4 reduce adsorbed O2 molecules on the catalyst surface to form ˙O2− radicals (eqn (6)). Simultaneously, h+ in the VB of ZnO and Fe3O4 directly oxidize TC molecules or react with surface OH− or H2O to generate ˙OH radicals (eqn (7)–(9)). The ˙O2− radicals are highly reactive and attack the TC molecules, leading to their degradation (eqn (10)). The ˙OH radicals formed in small amounts also contribute to the degradation of TC, though their role is minimal compared to ˙O2− radicals (eqn (11)). The intermediate products are further oxidized by ˙O2−, ˙OH, and h+, resulting in complete mineralization into CO2, H2O, and inorganic ions.
| | | ZnO/Fe3O4 + hv (photons) → eCB− + hVB+ | (4) |
| | | eCB(Fe3O4)− → eCB(ZnO)− | (5) |
| | | hVB+ + TC → CO2 + H2O + Degraded products | (7) |
| | | ˙O2− + TC → CO2 + H2O + Degraded products | (10) |
| | | ˙OH + TC → CO2 + H2O + Degraded products | (11) |
 |
| | Fig. 7 Illustration of the photocatalytic mechanisms in Fe3O4/ZnO heterostructures for (a) TC degradation and (b) H2O2 production, under light irradiation. | |
3.2.2. Photocatalytic H2O2 production.
The photocatalytic performance of Fe-ZIF-8 500 for H2O2 production under visible light irradiation is illustrated in Fig. 8. A significant enhancement in H2O2 production efficiency is observed due to the formation of Fe3O4/ZnO heterostructures. Over the course of 3 h, Fe- ZIF-8 500 demonstrates a markedly higher H2O2 concentration compared to ZIF-8 500, with a consistent increase in production rate across all time intervals. After 1 h of irradiation, Fe-ZIF-8 500 achieves an H2O2 concentration of 684.5 μM, nearly double that of ZIF-8 500, which only reaches around 391.1 μM. This trend continues after 3 h, where Fe-ZIF-8 500 reaches 919.2 μM, while ZIF-8 500 shows a notable decline, likely due to limited charge carrier generation or decomposition of H2O2.4 The superior performance of Fe-ZIF-8 500 can be attributed to the formation of Fe3O4/ZnO heterostructures (Fig. 7(b)). These heterostructures promote efficient charge separation, reducing recombination rates of photogenerated electron–hole pairs. The photogenerated electrons effectively reduce dissolved O2 molecules adsorbed on the catalyst surface, producing ˙O2− radicals (eqn (4)–(6)), which subsequently form H2O2via electron transfer pathways (eqn (12) and (13)). Simultaneously, photogenerated h+ in the VB of ZnO oxidize BA to benzaldehyde (BAL) (eqn (14)). BA acts as a sacrificial agent, effectively quenching h+ and donating more electrons to facilitate the oxygen reduction reaction.4| | | HO2˙ + e− + H+ → H2O2 | (13) |
| | | C6H5CH2OH + 2h+ → C6H5CHO + 2H+ | (14) |
 |
| | Fig. 8 Photocatalytic performance of ZIF-8 500 and Fe-ZIF-8 500 for H2O2 production. | |
These synergistic processes highlight the enhanced photocatalytic efficiency of Fe-ZIF-8 500, making it a promising material for H2O2 production under visible light irradiation.
4. Conclusions
This study successfully developed Fe3O4/ZnO heterostructures derived from Fe-doped ZIF-8 using a precipitation and thermal decomposition approach. Fe3+ doping into the ZIF-8 framework facilitated the formation of Fe3O4 during calcination under air, resulting in heterostructures with enhanced photocatalytic properties. Characterization by UV-vis DRS revealed improved visible-light absorption and a narrowed bandgap, while XRD confirmed the crystalline Fe3O4/ZnO heterostructures and phase transitions during calcination. TGA and SEM-EDX analyses identified 500 °C as the optimal calcination temperature, as it preserves porous structures with homogeneously distributed Fe, Zn, and O. FTIR confirmed the emergence of Zn–O and Fe–O bonds, while XPS reveals mixed-valence states of Fe2+ and Fe3+ and interfacial interactions promoting charge separation. Fe-ZIF-8 calcined at 500 °C demonstrated the highest photocatalytic performance, achieving 74.8% TC degradation with a degradation rate of 8.21 × 10−3 min−1 and a significant H2O2 yield of 919.2 μM under visible light irradiation. Mechanistic studies highlighted the role of defect states and favorable band alignment at the Fe3O4/ZnO interface in enhancing charge separation and generating reactive oxygen species, particularly ˙O2− radicals. These findings highlight the potential of Fe3O4/ZnO heterostructures as promising photocatalysts for sustainable wastewater treatment and green chemical synthesis, with future work aimed at improving their performance, stability, and recyclability.
Author contributions
Sumiyyah Sabar: writing – original draft, visualization, investigation, funding acquisition, conceptualization, methodology. Afiqah M. Jamil: investigation, writing – review & editing. Yifan Zhao: investigation, writing – review & editing. Enis Nadia Md Yusof: conceptualization, writing – review & editing. Raphaël Schneider: conceptualization, writing – review & editing. Abdul Rahman Mohamed: writing – review & editing. Yasutaka Kuwahara: writing – review & editing. Kohsuke Mori: writing – review & editing. Hiromi Yamashita: supervision, conceptualization, methodology, resources.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its ESI.†
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors would like to thank Universiti Sains Malaysia (USM) and Osaka University (OU) for the research facilities. In addition, we also would like to thank USM for the financial support under the USM Short Term Grant (R501-LR-RND002-0000000213-0000).
References
- Y. Amangelsin, Y. Semenova, M. Dadar, M. Aljofan and G. Bjørklund, Antibiotics, 2023, 12, 440 CrossRef CAS PubMed.
- L. Xu, H. Zhang, P. Xiong, Q. Zhu, C. Liao and G. Jiang, Sci. Total Environ., 2021, 753, 141975 CrossRef CAS PubMed.
- X. Chen, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, J. Mater. Chem. A, 2020, 8, 1904–1910 RSC.
- Y. Zhao, Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Appl. Catal., B, 2024, 351, 123945 CrossRef CAS.
- X. Han, T. Zhang, Y. Cui, Z. Wang, R. Dong, Y. Wu, C. Du, R. Chen, C. Yu, J. Feng, J. Sun and S. Dong, Molecules, 2023, 28, 2422 CrossRef CAS PubMed.
- V. P. Viswanathan, V. T. Balakrishnan, N. N. Adarsh, B. Varghese and S. Mathew, Crystals, 2023, 13, 47 CrossRef CAS.
- N. S. Abdul Mubarak, K. Y. Foo, R. Schneider, R. M. Abdelhameed and S. Sabar, Mater. Today Commun., 2023, 37, 107228 CrossRef CAS.
- X. Lei, Y. Cao, Q. Chen, X. Ao, Y. Fang and B. Liu, Colloids Surf., A, 2019, 568, 1–10 CrossRef CAS.
- K. Liu, N. Wang, J. Li, F. Du, B. Hou and R. Zhang, J. Ind. Eng. Chem., 2023, 128, 586–596 CrossRef CAS.
- M. A. Nazir, S. Ullah, M. U. Shahid, I. Hossain, T. Najam, M. A. Ismail, A. Ur Rehman, M. R. Karim and S. S. A. Shah, Sep. Purif. Technol., 2025, 356, 129828 CrossRef CAS.
- Y. Liu, H. Cheng, M. Cheng, Z. Liu, D. Huang, G. Zhang, B. Shao, Q. Liang, S. Luo, T. Wu and S. Xiao, Chem. Eng. J., 2021, 417, 127914 CrossRef CAS.
- A. Elaouni, M. El Ouardi, M. Zbair, A. BaQais, M. Saadi and H. Ait Ahsaine, RSC Adv., 2022, 12, 31801–31817 RSC.
- H. Dai, X. Yuan, L. Jiang, H. Wang, J. Zhang, J. Zhang and T. Xiong, Coord. Chem. Rev., 2021, 441, 213985 CrossRef CAS.
- Y. Du, R. Z. Chen, J. F. Yao and H. T. Wang, J. Alloys Compd., 2013, 551, 125–130 CrossRef CAS.
- G. Mani, A. V. Kumar and S. Mathew, RSC Sustainability, 2023, 1, 2081–2091 RSC.
- S. Wu, J. Liu, H. Wang and H. Yan, Int. J. Energy Res., 2019, 43, 697–716 CrossRef CAS.
- A. Shahzad, F. Ahmad, S. Atiq, M. Saleem, O. Munir, M. A. Khan, S. M. Bin Arif, Q. U. Ain, S. Sarwar, M. Asim and U. Habib, J. Energy Storage, 2024, 87, 111447 CrossRef.
- M. Zahid Hussain, Z. Yang, Z. Huang, Q. Jia, Y. Zhu, Y. Xia, M. Z. Hussain, Z. Yang, Z. Huang, Y. Zhu, Y. Xia and Q. Jia, Adv. Sci., 2021, 8, 2100625 CrossRef PubMed.
- Y. Su, S. Li, D. He, D. Yu, F. Liu, N. Shao and Z. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 11989–11998 CrossRef CAS.
- J. Abdi, Colloids Surf., A, 2020, 604, 125330 CrossRef CAS.
- S. Varangane, C. S. Vennapoosa, A. Tiwari, S. K. Nataraj, T. P. Yendrapati and U. Pal, Appl. Organomet. Chem., 2022, 36, e6815 CrossRef CAS.
- A. Mehrehjedy, P. Kumar, Z. Ahmad, P. Jankoski, A. S. Kshirsagar, J. D. Azoulay, X. He, M. K. Gangishetty, T. D. Clemons, X. Gu, W. Miao and S. Guo, ACS Omega, 2024, 9, 49239 CrossRef CAS PubMed.
- M. T. Thanh, T. V. Thien, P. D. Du, N. P. Hung and D. Q. Khieu, J. Porous Mater., 2018, 25, 857–869 CrossRef CAS.
- Y. Yin, J. Liu, Z. Wu, T. Zhang and Z. Li, New J. Chem., 2021, 45, 3095–3101 RSC.
- Q. Ding, D. Xu, J. Ding, W. Fan, X. Zhang, Y. Li and W. Shi, J. Colloid Interface Sci., 2021, 603, 120–130 CrossRef CAS PubMed.
- A. Mariappan, P. Mannu, T. Thiruppathiraja, T. T. T. Nga, S. Lakshmipathi, C. L. Dong, R. K. Dharman and T. H. Oh, Chem. Eng. J., 2023, 476, 146720 CrossRef CAS.
- C. Cheng, J. Wang, Z. Zhao, C. Chen, S. Cui, Y. Wang, L. Pan, Y. Ni and C. Lu, J. Alloys Compd., 2022, 896, 163064 CrossRef CAS.
- R. M. Mohamed and A. Shawky, Opt. Mater., 2022, 124, 112012 CrossRef CAS.
- P. Dhiman, G. Rana, A. Kumar, G. Sharma, D. V. N. Vo and M. Naushad, Environ. Chem. Lett., 2022, 20, 1047 CrossRef CAS.
- I. Ahmad, R. Bousbih, A. Mahal, W. Q. Khan, M. Aljohani, M. A. Amin, N. N. A. Jafar, M. S. Jabir, H. Majdi, A. S. Alshomrany, M. Shaban, I. Ali and H. Bayahia, Mater. Sci. Semicond. Process., 2024, 180, 108578 CrossRef CAS.
- N. D. Dien, T. T. H. Pham, X. H. Vu, V. T. Xuan, T. T. T. Nguyen, T. T. Trang, N. Van Hao, P. T. Nga, T. T. Kim Chi, T. T. H. Giang and N. D. Toan, RSC Adv., 2024, 14, 28244–28259 RSC.
- P. Thi Lan Huong, N. Van Quang, N. Thi Huyen, H. Thu Huong, D. Anh Tuan, M. Trung Tran, Q. Vinh Tran, T. Ngoc Bach, N. Tu and V. D. Dao, Sol. Energy, 2023, 249, 712–724 CrossRef CAS.
- P. D. Du, N. T. Hieu and T. V. Thien, J. Nanomater., 2021, 9988998, 12 Search PubMed.
- X. Chen, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, ACS Appl. Energy Mater., 2021, 4, 4823–4830 CrossRef CAS.
- C. Matsubara, N. Kawamoto and K. Takamura, Analyst, 1992, 117, 1781 RSC.
- Y. Zhao, Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Catal. Today, 2024, 431, 114558 CrossRef CAS.
- O. İloğlu and H. A. Yurtsever, J. Mater. Sci.: Mater. Electron., 2024, 35, 1–16 CrossRef.
- X. Yu, D. Xu, X. Jiang, J. Zhang, Z. Ni and M. Wang, J. Mol. Struct., 2023, 1289, 135861 CrossRef CAS.
- T. Wang, Y. Wang, M. Sun, A. Hanif, H. Wu, Q. Gu, Y. S. Ok, D. C. W. Tsang, J. Li, J. Yu and J. Shang, Chem. Sci., 2020, 11, 6670–6681 RSC.
- B. Yu, F. Wang, W. Dong, J. Hou, P. Lu and J. Gong, Mater. Lett., 2015, 156, 50–53 CrossRef CAS.
- Y. Sun, H. Zhang, Y. Lv, S. An and R. Wang, RSC Adv., 2024, 14, 17498–17506 RSC.
- Z. Esania, H. Younesi, M. Nowrouzi and H. Karimi-Maleh, J. Water Process Eng., 2022, 47, 102750 CrossRef.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- S. Sridharan, S. V. Perumalsamy, J. Kulandaivel, G. Nammalwar, H. Parangusan and D. Ponnamma, Ionics, 2024, 30, 1137–1150 CrossRef CAS.
- M. Athar, P. Rzepka, D. Thoeny, M. Ranocchiari and J. Anton Van Bokhoven, RSC Adv., 2021, 11, 38849–38855 RSC.
- Z. Abbasi, E. Shamsaei, S. K. Leong, B. Ladewig, X. Zhang and H. Wang, Microporous Mesoporous Mater., 2016, 236, 28–37 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- J. Liu, J. He, L. Wang, R. Li, P. Chen, X. Rao, L. Deng, L. Rong and J. Lei, Sci. Rep., 2016, 6(1), 1–11 CrossRef PubMed.
- C. Huang, C. Hao, Z. Ye, S. Zhou, X. Wang, L. Zhu and J. Wu, Nanoscale, 2019, 11, 10114–10128 RSC.
- R. M. Mohamed and A. A. Ismail, Sep. Purif. Technol., 2021, 266, 118360 CrossRef CAS.
- E. Shamsaei, F. Basquiroto de Souza, K. Sagoe-Crentsil and W. Duan, Microporous Mesoporous Mater., 2022, 332, 111702 CrossRef CAS.
- Y. Song, F. Gu, Z. Wang and D. Han, ACS Appl. Nano Mater., 2022, 5, 15298–15309 CrossRef CAS.
- H. Yang, S. Hu, H. Zhao, X. Luo, Y. Liu, C. Deng, Y. Yu, T. Hu, S. Shan, Y. Zhi, H. Su and L. Jiang, J. Hazard. Mater., 2021, 416, 126046 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Afiqah
M. Jamil
a,
Yifan
Zhao
b,
Enis Nadia
Md Yusof
a,
Raphaël
Schneider
*a,
Afiqah
M. Jamil
a,
Yifan
Zhao
b,
Enis Nadia
Md Yusof
a,
Raphaël
Schneider