
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation and characterization of a highly porous, rigid cellulose-based hydrogel for biomedical and biotechnological applications
Jolanta
Liesiene
 a,
Sandra
Kiselioviene
b,
Audrius S.
Maruška
c and
Odeta
Baniukaitiene
*a
a,
Sandra
Kiselioviene
b,
Audrius S.
Maruška
c and
Odeta
Baniukaitiene
*a
aKaunas University of Technology, Department of Polymer Chemistry and Technology, Radvilenu pl. 19, LT-50254 Kaunas, Lithuania. E-mail: odeta.baniukaitiene@ktu.lt; Tel: +370 676 04891
bKaunas University of Technology, Food Institute, Radvilenu pl. 19, LT-50254 Kaunas, Lithuania
cVytautas Magnus University, Instrumental Analysis Open Access Centre, Universiteto g. 10, LT-53361 Kaunas, Lithuania
First published on 28th March 2025
Abstract
This study introduces a novel approach for preparing rigid, porous cellulose hydrogels using cellulose acetate as the starting material. The method relies on the slow hydrolysis of acetyl groups directly in an acetone/aqueous ammonia solution. The gradual pace of the process creates conditions favourable for reconstructing of inter- and intramolecular hydrogen bonding networks between the newly formed hydroxyl groups in the cellulose, resulting in a rigid three-dimensional structure. The hydrogels demonstrated excellent mechanical properties, with a compressive (Young's) modulus of up to 43 MPa and an elastic modulus of up to 0.23 MPa. X-ray analysis indicated that the cellulose hydrogels are semi-crystalline, with a crystallinity index of 43–45% and an average crystallite size of 4.3–4.5 nm. Wide-angle X-ray diffraction, along with FT-IR and Raman spectroscopy, confirmed that the gels belong to the cellulose II structural modification. The porous structure of the hydrogels was characterized using inverse size exclusion chromatography, revealing exclusion limits for linear polymers of up to 4 × 106 Da. Thanks to their enhanced mechanical properties and high porosity, crushed hydrogels show potential applications in column technologies for protein chromatography and heterogeneous biocatalysis processes with immobilized enzymes. In film form, the gels' elasticity makes them promising candidates for biomedical applications, such as wound dressings or artificial skin. Furthermore, the lyophilized gels create porous structures suitable for vascularization and bone tissue ingrowth, positioning them as ideal scaffolds for bone tissue engineering.
Introduction
Polymeric hydrogels are crucial for advancements in biotechnology, pharmacy, and biomedicine. Among these, cellulose-based hydrogels have garnered significant attention for their biocompatibility, non-toxicity, and biodegradability.1–5 Moreover, an abundance of hydroxyl groups provides cellulose with high chemical activity, facilitating the attachment of desired functional groups.Cellulose hydrogels can be fabricated in various sizes and morphologies, from nano- to macroparticles, and in diverse physical forms such as films, membranes, fibers, and rods, depending on the desired application. Thus, spherical cellulose nanogels, due to their versatile properties, are ideal for a broad range of applications, including drug delivery and biomedical purposes such as disease detection and diagnosis.3,6–8 Cellulose microgels can be used as emulsion stabilizers, with their preparation and application broadly described in reviews.9 Cellulose hydrogels in the form of films, membranes, or fibers have potential as wound dressings and artificial skin due to their specific benefits, such as biocompatibility and controlled drug delivery.10–12 Additionally, they offer potential applications in electronic devices.4,13,14
Cellulose hydrogels, particularly those fabricated as micro- to macroparticles, have gained significant attention in column technologies like liquid chromatography and immobilized heterogeneous biocatalysis. The preparation of cellulose hydrogels in spherical or irregularly shaped macroparticles received intense research interest in the eighties, driven by developments in biochemistry and contributing to progress in protein purification.15 Subsequently, cellulosic sorbents were improved and applied to protein chromatography in various types of processes.15–17 The structure of the sorbents and the amount of functional groups were optimized for the specific purified protein.18 Currently, cellulose-based chromatographic media are integral to nearly all technological schemes for the separation and purification of proteins and other biomaterials.17,19
Cellulose carriers, thanks to their porosity, biocompatibility, and simple functionalization, are highly suitable for enzyme immobilization and subsequent biocatalysis processes.20–23
Nowadays, the development of three-dimensional (3D) scaffolds for tissue regeneration is one of the current challenges in tissue engineering. Various materials are proposed for the fabrication of 3D scaffolds, with significant attention focused on natural polymers. Cellulose hydrogels, for instance, serve as a valuable starting material in developing bone tissue engineering aerogels.24–28 To enhance osteoconductive properties, the scaffolds are prepared by combining cellulose with mineral components such as hydroxyapatite, tricalcium phosphate, cuttlebone, or others.29–31 Aerogel preparation methods are based on drying cellulose hydrogels using lyophilization, supercritical CO2 treatment, or other techniques.32 A notable advantage of cellulose aerogels is that their preparation typically does not involve toxic substances, enhancing their suitability for use in pharmaceuticals, biomedicine, food, and cosmetics. Detailed reviews on the structure, properties, and applications of cellulose aerogels are extensively covered in the literature.33–35
There are different approaches to obtaining cellulose hydrogels, but the main principle is based on dissolving cellulose or its derivatives and subsequently crosslinking the polymer through various interactions. Different crosslinking techniques, such as chemical, physical, and polymerization methods, are used. The structural and mechanical characteristics of the gels depend on the concentration of cellulosic material, precipitation conditions, and the degree of crosslinking.36
One of the primary challenges in processing native cellulose is its limited solubility in conventional organic or inorganic solvents due to the extensive network of hydrogen bonds. Efforts to develop more effective methods for addressing this limitation are continually advancing. Consequently, in recent years, research has increasingly focused on using hydrolyzed low-molecular cellulose dissolved in NaOH solutions,35 various complex organic solvents, or ionic liquids. These methods are extensively discussed in detailed reviews4,37–39 and articles.40–42
Mechanical properties, such as mechanical stability and flexibility, are among the most important factors limiting the practical application of gels. Different application areas require specific mechanical characteristics of the gels. For example, in column technologies such as protein chromatography or enzymatic biocatalysis, the packing materials must withstand high pressure and changes in pH and ionic strength. Therefore, these materials must demonstrate substantial mechanical strength despite their high porosity. The gels in film or membrane form should be flexible and strong. Unfortunately, cellulose gels described in the literature are often characterized as soft gels.37,40,43 To improve their mechanical properties, cellulose hydrogels are crosslinked or reinforced by incorporating other polymers44–46 or mineral materials.47,48
The aim of these studies is to develop a novel method for producing rigid cellulose gels with a tunable porous structure and high mechanical properties. Cellulose diacetate, an industrial raw material used in the production of cellulose acetate fibers, served as the initial material. The method involves the gradual hydrolysis of acetyl groups directly within the solution. The kinetics of acetyl group hydrolysis and the correlation between gel contraction and the parameters of the hydrolysis process were investigated. The porous structure of the gels in the wet state was investigated by means of inverse size exclusion chromatography, while in the dry state by microcomputed tomography. The morphological structure of the cellulose gels was evaluated using wide-angle X-ray diffraction, Raman, and FT-IR spectral analysis. It was demonstrated that the porous structure of the gels, as well as their mechanical properties, can be regulated by varying the initial cellulose acetate concentration. Prerequisites for obtaining nanoporous structured gels with customizable porosity and high mechanical strength have been established.
Materials and methods
Materials
Cellulose diacetate with a degree of substitution (DS) of 2.4, containing 55% bound acetic acid, was kindly provided by DP Acetate Co, Lithuania. Aqueous ammonia with a concentration of 25%, chemically pure grade, was purchased from Stanlab, Poland. All other reagents were purchased from Sigma-Aldrich Co, Germany. All chemicals were used without any further purification.Gel formation
Cellulose gels were obtained by regenerating cellulose diacetate (CDA), which contains 55% bound acetic acid. CDA was dissolved in acetone, and the appropriate amount of aqueous ammonia was gradually added. After thorough mixing, the polymer solution was placed in a tightly sealed container at room temperature for 10 days (Fig. 1). During the hydrolysis reaction of acetyl groups, the polymer gradually lost its solubility forming a three-dimensional network. The resulting gel was washed with water to a neutral pH of 7. The sol–gel transition point was determined visually as the solution hardening time. To assess this, the container with the reaction mixture was shaken periodically to observe whether the solution had transformed into a gel. | ||
| Fig. 1 Scheme of the hydrogel preparation. | ||
Determination of bound acetic acid content
Approximately 0.7 g of completely dry CDA or cellulose gel was mixed with 20 mL of ethanol and maintained at a temperature of 60 °C for 30 minutes. Subsequently, 20 mL of 0.5 M NaOH solution was added, and the mixture was kept at 60 °C for an additional 15 minutes. After 24 hours, the solution was titrated with 0.5 M HCl using phenolphthalein as an indicator. Following the titration, an excess of 4 mL of 0.5 M HCl solution was added, allowed to react for 15 minutes, and then titrated with 0.1 M NaOH solution. The percentage of bound acetic acid A (%) was calculated using the formula:where: b – is the volume of 0.5 M HCl solution consumed for the titration of 20 mL of 0.5 M NaOH; a – is the volume of 0.5 M HCl solution consumed for the sample titration; d – is the volume of 0.1 M NaOH solution consumed for back titration; g – is the mass of the sample in grams, converted to dry matter.

Degree of substitution (DS) was calculated using the formula:
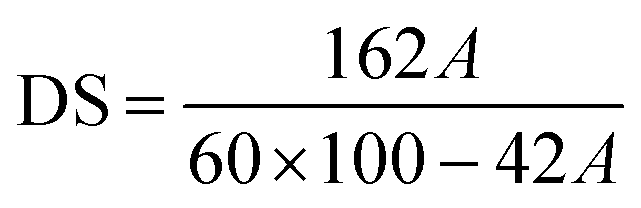
Wide angle X-ray diffraction analysis
Dried ground cellulose gel pressed into tablets was used for wide-angle X-ray diffraction structural analysis. X-ray structural analysis was performed with a DRON-G diffractometer. Cu-Kα radiation (λ = 0.154 nm) with a nickel filter was employed. The detector's angular step was 0.02°, the duration of intensity measurement per step was 0.5 s, the anode voltage UA was set to 30 kV, and the current strength was set to 20 mA. To eliminate noise, the recorded X-ray diffraction curves were processed using the Xfit program.FT-IR spectroscopy
FT-IR analysis was performed using the Bruker Tensor 37 apparatus. Samples for analysis and spectrum recording were prepared by pressing a mixture of 5 mg of dried cellulose derivative and 200 mg of dry KBr into transparent tablets. The crystallinity index of cellulose derivative denoted CI(FTIR) was calculated from the ratio of corresponding spectral peak intensities:| CI(FTIR) = 1376/2890 (%) |
Additionally, utilizing the intensity ratio of spectral peaks at 3440 and 2890 cm−1, the relative amount of hydrogen bonds (RAHB) in cellulose derivatives was calculated:
| RAHB = 3440/2890 (%) |
Raman spectroscopy
Raman spectroscopy was carried out using the Horiba Jobin Yvon Labram HR 800 apparatus with a 50-second exposure time and 7 accumulations. To mitigate fluorescence interference, an infrared laser (785 nm) was utilized. A Raman microscope objective with 50× magnification was employed.Evaluation of exclusion limits of the gels
Exclusion limits for linear polymers were evaluated by inverse size exclusion chromatography using a Waters HPLC system with a refractive index detector. In this approach, 0.1% standard dextran solutions (Pharmacia, Sweden) with known molecular masses were passed through a column (270 × 4.6 mm I.D.) packed with crushed gel particles of 63–100 μm under gel chromatography conditions, and their elution times Ve were recorded.49 The void volume Vo of the column was determined using a starch derivative with a molecular mass of 6 × 106 Da, while the total column volume Vt was determined using a glucose solution. The flow rate was 0.2 mL min−1. Exclusion limits were determined from the calibration graph log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MM = f(Kav), where Kav is the distribution coefficient and MM represents the molecular mass of dextran. The dextran distribution coefficient Kav was calculated as follows:
MM = f(Kav), where Kav is the distribution coefficient and MM represents the molecular mass of dextran. The dextran distribution coefficient Kav was calculated as follows:
Micro-computed tomography
The morphological parameters of the lyophilized gel were assessed via micro-computed tomography (μCT; n = 4), utilizing a μCT40 system from Scanco Medical AG in Bruttisellen, Switzerland. A cylindrical sample of lyophilized gel, with a diameter of 10 mm and a height of 8 mm, was utilized for the analysis. The scans were conducted with the following parameters: energy, 45 kVp; integration time, 600 ms; frame averaging, 2×; and nominal resolution, 10 μm. The data were filtered using a constrained 3D Gaussian filter to partially suppress the noise in the images (σ = 0.8, support = 1). Both 2D and 3D images were generated using image reconstruction software provided by the manufacturer. Quantitative evaluation of the structural parameters of the samples was performed using Scanco 6.0 evaluation software.Hepler hardness
Hardness, according to Hepler (cone hardness), was assessed by measuring the resistance of the sample to the penetration of a specialized cone using a Hepler consistometer. The conical needle was tapered at an angle of 53°08′, ensuring that the depth of penetration was always equal to the diameter of the circle formed by the embedded part of the cone contacting the surface of the sample. The apex of the cone was trimmed by 0.2 mm, so the depth of penetration measured by the indicator needs to be increased by 0.2 mm.The gel hardness was determined using tablet-shaped samples. The sample was placed on a measuring table, and the cone tip was gently pressed against the sample. The initial readings h0 of the micrometer indicator were recorded. Subsequently, a weight of predetermined size was hung on the lever, creating a force F of 7 N acting on undried samples. After 30 seconds, the lever with the weight was lifted, and the indicator reading h1 was recorded. Using the formulas below, Δh (mm), the contact surface area S (mm2) between the cone and the sample, and the hardness HH (N mm−2) of the sample were calculated:
| Δh = h1 − ho + 0.2 |


The hardness dimension is force per unit area, N mm−2, and the index next to H indicates the force value and duration. The final result was calculated as the arithmetic average of 10 measurements.
Compresion test
The compression test for cellulose gels was conducted using a Zwick/Roell dynamometer, which allows the sample to be compressed at various speeds and forces. Cylindrical specimens were prepared for the compression test. The specimens were positioned between the plates of compression machine, centered according to the direction of the acting force (10 N), and compressed at a speed of 50 mm min−1. The relative deformation ε was calculated using the formula:where: x – is the deformation, contraction of the sample, in m; l – is the length of the sample, in m.

The isotropic compression bulk modulus K (elasticity parameter) was calculated using the formula:

Young's modulus E (compression modulus) is a physical quantity that characterizes a material's resistance to crushing or stretching. It is defined as the ratio of stress to the relative elongation (deformation) using the formula:

The product of the modulus compression and the cross-sectional area is called the tensile-compressive strip stiffness ξ. It quantitatively assesses the strip's ability to resist the effects of deformable load.
| ξ = S·E (m2 Pa) |
The final result was calculated as the arithmetic average of 5 measurements.
Bending test
The bending test for cellulose gels was conducted using a Zwick/Roell dynamometer. Rectangular specimens were formed for the bending test. The resistance to bending was determined using a three-point bending setup. The rectangular specimen was positioned on two supports, and an external of 10 N force was applied at the centre of the specimen at a speed of 50 mm min−1. The elastic modulus E for the body placed on two supports was calculated using the formula:where: F – is the applied force, in N; l – is the distance between supports, in m; I – is the moment of inertia of the cross-section, in m4; yc – is the deflection of the specimen, in m. I was calculated for a cross-section with height h (m) and width b (m):
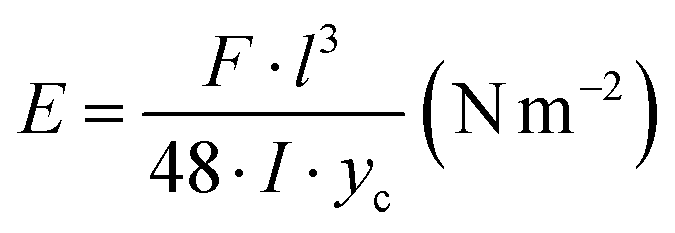

The final result was the arithmetic average of 5 measurements.
Statistical data analysis
The mean values of experiments and standard deviation values were calculated by using MS Excel software package.![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) arithmetic average was calculated as follows:
arithmetic average was calculated as follows:where: n – is the number of measurements; xi – is the measuring value obtained by i experiment.

The standard deviation (SD) was calculated using the following formula:
 – is the arithmetic average of the measurement values.
– is the arithmetic average of the measurement values.
Results and discussion
Preparation of cellulose-based gel
Cellulose gel was prepared from cellulose diacetate (CDA) by continuous gentle hydrolysis of the acetyl groups, following the scheme outlined in Fig. 2. To obtain a rigid cellulose gel, the hydrolysis process was carried out by gradually adding aqueous ammonia to the CDA solution in acetone. Careful control of the process rate prevented rapid precipitation of the hydrolyzed cellulose, creating conditions for the formation of new hydrogen bond network. This control is essential for achieving a well-defined gel structure with desired properties. | ||
| Fig. 2 Scheme of the reaction. | ||
During the hydrolysis of the acetyl groups, the solubility of the cellulose derivative gradually decreased in the reaction mixture. This led to the slow precipitation of cellulose macromolecules and the formation of new bonds between the newly formed hydroxyl groups. A sol–gel transition occurred through the reconstruction of inter- and intramolecular hydrogen bonding networks. The gel obtained through this process maintained the volume of the initial solution with minimal shrinkage. Inter- and intramolecular hydrogen bonding between the hydroxyl groups of cellulose molecules contributes to the formation of a rigid gel structure.
The cellulose diacetate used, with a degree of substitution (DS) of 2.4, is soluble in acetone. Cellulose acetates with a lower DS are soluble in solvents of higher polarity, with cellulose monoacetate being soluble even in water. However, as the DS decreases further, cellulose acetates lose their solubility both in water and common organic solvents. The gel formation method we used exploited this phenomenon of solubility of acetylated cellulose derivatives. The CDA was dissolved in acetone, followed by the gradual addition of an aqueous ammonia solution, avoiding the precipitation of the cellulose from the solution. After thorough mixing, the polymer solution was left in a sealed container at room temperature until the hydrolysis reaction was completed. The released ammonium acetate was washed from the gel with water.
The gel produced in this study constitutes regenerated cellulose, which is of higher chemical activity than native cellulose. The hydroxyl groups of regenerated cellulose are more accessible to reactants. This property is particularly significant in applications requiring the attachment of functional groups, such as the preparation of chromatographic sorbents or carriers for enzyme immobilization. During the formation of the porous structure of the regenerated cellulose gel, it was found that the properties of the obtained product depend on many factors, especially on the composition of the reaction mixture. One of the main factors is the concentration of the polymer in the solution used for gel formation. Lower concentrations of CDA in the solution lead to larger pores in the gel structure, as the distance between macromolecules increases. Additionally, the concentration of ammonia in the solution directly influences the gel structure, impacting the speed of the hydrolysis reaction and the resulting gel contraction.
To assess the impact of ammonia concentration in the solution on the reaction rate, the kinetics of hydrolysis of acetyl groups were investigated (Fig. 3).
 | ||
| Fig. 3 Kinetic of hydrolysis of CDA acetyl groups. Composition of reaction mixture: 1 – NH4OH/acetone = 0.23 v/v, NH4OH/CDA = 3 v/w; (y = 60.14 e−0.0105x, R2 = 0.9672); 2 – NH4OH/acetone = 0.33 v/v, NH4OH/CDA = 4 v/w; (y = 44.44 e−0.0111x, R2 = 0.9732); 3 – NH4OH/acetone = 0.59 v/v, NH4OH/CDA = 6 v/w; (y = 43.58 e−0.0254x, R2 = 0.9542). | ||
The results indicate that the initial hydrolysis of acetyl groups occurs rapidly. The higher the concentration of ammonia, the more intense the hydrolysis is. For instance, at an NH4OH/acetone ratio of 0.59 v/v, a sol–gel transition occurs within just 5 hours, while at a ratio of 0.23 v/v, gel formation takes place only after 7 days. However, to obtain a gel with the largest possible pores, hydrolysis must be carried out slowly, as rapid hydrolysis is accompanied by gel contraction.
The sol–gel transition point is observed before the complete hydrolysis of the ester bonds. Depending on the amount of ammonia in the mixture, gel formation occurs within a range of 15–39% bound acetic acid. This is due to differences in the solubility of the cellulose derivative in reaction mixtures of different polarity. After the gel is formed, the hydrolysis of the acetyl groups continues, but at a slower rate. As the hydrolysis of acetyl groups progresses, the rigidity of the gel gradually increases.
After evaluating the time required for gel formation, the hydrolysis rate of acetyl groups, and the impact of ammonia concentration on gel characteristics such as rigidity and contraction, three samples of cellulose gel were prepared: Granocel-500, Granocel-2000, and Granocel-4000 with a water content that ranged from 92% to 97% (Table 1).
| Hydrogel | CDA, w/v,% | Acetone, v,% | NH4OH, v,% |
|---|---|---|---|
| Granocel-500 | 9.4 | 70 | 30 |
| Granocel-2000 | 8.4 | 64.5 | 35.5 |
| Granocel-4000 | 6.5 | 62.5 | 37.5 |
Supermolecular structure of the gels
According to wide-angle X-ray scattering data the produced gels are classified as cellulose II. The diffraction peaks 2θ ≈ 12.4°, 19.8° and 21.5° are assigned for the crystal planes (101), (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (002), respectively, and are characteristic of cellulose II.50 Cellulose I (cotton cellulose) crystal planes (101), (10) and (002) are assigned for the diffraction peaks 2θ ≈ 14.9°; 16.5° and 22.6°, respectively, (Fig. 4 and 5). From Fig. 5, we can see that CDA is an amorphous material, while the obtained gels are semi-crystalline materials.
) and (002), respectively, and are characteristic of cellulose II.50 Cellulose I (cotton cellulose) crystal planes (101), (10) and (002) are assigned for the diffraction peaks 2θ ≈ 14.9°; 16.5° and 22.6°, respectively, (Fig. 4 and 5). From Fig. 5, we can see that CDA is an amorphous material, while the obtained gels are semi-crystalline materials.
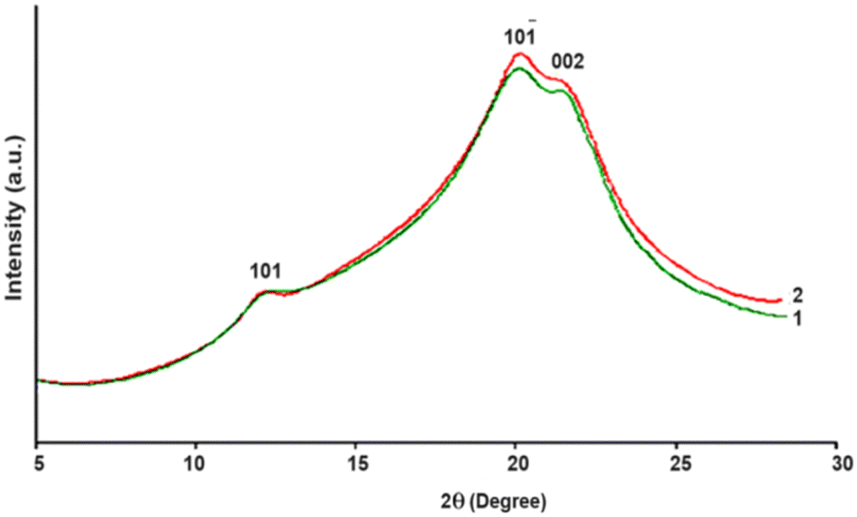 | ||
| Fig. 4 X-ray diffraction patterns of regenerated cellulose gels: 1 – Granocel-4000, 2 – Granocel-2000. | ||
 | ||
| Fig. 5 X-ray diffraction patterns of different cellulosic materials: 1 – Granocel-4000, 2 – CDA, 3 – native cotton cellulose. | ||
The crystallinity index CI(XRD) of the investigated materials and the size of the crystallites were calculated from the X-ray diffraction (XRD) graphs according to the most popular peak height method proposed by Jayme and Knolle:51,52

The results are presented in Table 2. The calculated CI from XRD of the prepared Granocel gels is approximately 43–45%. However, it is important to note that the peak height method used produces values that are significantly higher than those obtained by other methods.52
| Cellulose | CI(XRD) (%) | Crystallite size (nm) |
|---|---|---|
| Granocel-500 | 45 | 4.3 |
| Granocel-2000 | 43 | 4.5 |
| Granocel-4000 | 43 | 4.4 |
| Native cotton cellulose | 72 | 5.4 |
The size of the crystallites in the 002 plane characterizes the crystallites in a horizontal position, i.e. along the axis of the cellulose macromolecule. The crystallite size of cellulose derivatives for crystallographic plane 002 was calculated according to the Scherrer formula:

From the obtained results, it is evident that the Granocel gels of regenerated cellulose have significantly lower crystallinity compared to cotton cellulose. Moreover, the crystallite sizes of the regenerated cellulose are smaller compared to those of cellulose modification I.
The results confirm that the gelation process was accompanied by precipitation and crystallization, characterized by the gradual arrangement of cellulose macromolecules into regular crystallite structures, as observed by Pereira et al.54
FT-IR spectral analysis. FT-IR spectral analysis allows to identify the structure of the compound as well as to determine the predominant bonds.
At 3440 cm−1 a broad spectral peak of high intensity indicates the stretching vibrations of –OH groups (Fig. 6). These groups are connected by hydrogen bonds, which is why the absorption band is wide. At 2890 cm−1, vibrations of –CH group valence bonds are observed. The relative amount of hydrogen bonds was calculated from the ratio of absorption bonds 1376 to 2890 cm−1. While the amount of –OH groups is variable, the amount of –CH groups in cellulose gels remains constant.
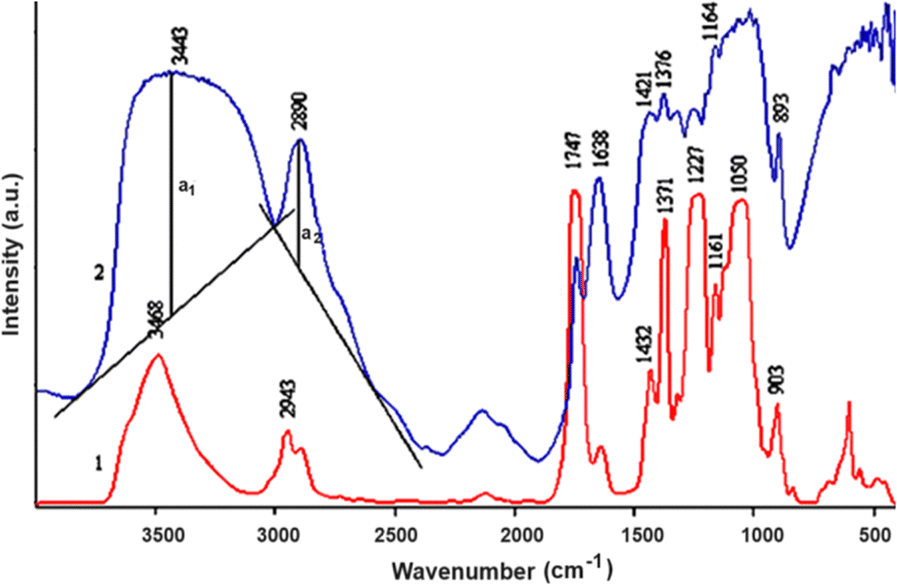 | ||
| Fig. 6 FT-IR spectra: 1 – CDA, 2 – Granocel-500. | ||
The peak at 2943 cm−1 in the FT-IR spectrum of CDA indicates asymmetric valence vibrations of the –CH3 (–CH2–) group. Meanwhile, a prominent peak at 1747 cm−1 in the FT-IR spectrum of CDA corresponds to the asymmetric valence vibrations of the carbonyl group. In Fig. 6, this carbonyl peak is also present in the spectra of regenerated cellulose but is less intense than in CDA. This may be due to the oxidation of hydroxyl groups or incomplete hydrolysis of acetyl groups. The peak at 1227 cm−1 in the spectrum of CDA represents asymmetric valence vibrations of the C–O–C bond, while the peak at 1050 cm−1 corresponds to symmetric valence vibrations of the C–O–C bond.
The crystallinity index CI(FTIR) of cellulose derivatives was also calculated from the FT-IR spectrum (Table 3).
| Sample | CI(FTIR) (%) | Relative amount of hydrogen bonds |
|---|---|---|
| Granocel-500 | 38 | 2.1 |
| Granocel-2000 | 33 | 2.0 |
| Granocel-4000 | 32 | 1.8 |
| Cotton cellulose | 71 | 2.3 |
| CDA | — | 1.4 |
It is known that the crystallinity index can vary significantly depending on the measurement method used. Determining the crystallinity index with FT-IR spectroscopy provides only relative values because the FT-IR spectrum always includes contributions from both crystalline and amorphous regions.52 Therefore, crystallinity estimated from FT-IR spectral analysis differs from that estimated from X-ray diffraction analysis. The largest discrepancy in the calculated crystallinity index is observed in regenerated cellulose gels, whereas the difference is less pronounced in native cotton cellulose. In both cases, crystallinity index calculations from both FT-IR spectra and X-ray diffraction patterns show that the crystallinity of regenerated cellulose gels is significantly lower than that of cotton cellulose.
The calculated relative amount of hydrogen bonds (Table 3) indicates that after hydrolysis of acetyl groups, a dense network of hydrogen bonds is formed between the newly generated OH groups.
FT-IR spectra are useful not only for identifying which bonds or groups are prevalent in materials but also for distinguishing between different types of cellulose formation. In this case, based on the shifts of the corresponding absorption bands, we can determine that the obtained regenerated cellulose is of cellulose type II. For cellulose I, the spectral peak at 1430 cm−1 identifies the deformational vibrations of the –CH2 group, whereas for cellulose II, this peak shifts to 1420 cm−1. Additionally, the peak identifying the vibrations of the C-1 group is observed at 896 cm−1 for cellulose I and at 893 cm−1 for cellulose II (Fig. 7).
 | ||
| Fig. 7 FT-IR spectra: 1 – cotton cellulose, 2 – Granocel-4000. | ||
Raman spectroscopy. Raman spectroscopy is of particular importance with compared to IR and NMR spectroscopy in determining the conformation of molecules and the formation of hydrogen bonds in cellulose. Fig. 8 and 9 show the Raman spectra of regenerated cellulose gels Granocel-500, -2000, -4000 and cotton cellulose.
 | ||
| Fig. 8 Raman spectra of cellulose gels and cotton cellulose: 1 – Granocel-500, 2 – Granocel-2000, 3 – Granocel-4000, 4 – cotton cellulose. | ||
 | ||
| Fig. 9 Raman spectra at high (a) and low (b) frequency region: 1 – cotton cellulose, 2 – Granocel-4000. | ||
The peaks at 896 and 1096 cm−1 in the Raman spectra reflect mixed vibrations of C–C–C, C–H–O bonds, and C–O bonds in the glucose ring, respectively (Fig. 8 and 9a). In cotton cellulose, the C–C–C and C–H–O bond oscillations at 896 cm−1 are less intense compared to those in regenerated cellulose gels containing cellulose II. In cotton cellulose, the peak at 2896 cm−1 is attributed to the vibrations of the methylene group. In cellulose II, this peak shifts to 2886 cm−1, indicating a shift toward shorter wavelengths. In the fingerprint region, for cellulose II, the peak at 1462 cm−1 is attributed to the C–O–H group, while peaks at 1376 and 1262 cm−1 correspond to the in-plane and out-of-plane vibrations of the methylene group, respectively. Peaks at 1117 and 1096 cm−1 are assigned to symmetric and asymmetric C–O–C vibrations of glycosidic groups. Vibrations of C–C, C–O, and C–O–C bonds in anhydrous glucopyranose (which forms the cellulose skeleton), as well as vibrations of H–C–C, H–C–O, C–O–H, and –CH2 groups, are observed in the region beyond 600 cm−1. The assignments of these groups are not unambiguous because the vibrations of most groups are not purely distinct.55,56 In cellulose II, the system of hydrogen bonds is more complex compared to cellulose I.50 Differences in hydrogen bond formation between cellulose I and cellulose II result in corresponding shifts or changes in signal intensity in Raman spectroscopy, which help distinguish one modification from another. The signal attributed to the rotation of methylene groups is useful for distinguishing cellulose I from cellulose II: the peak at 1294 cm−1 in cellulose I shifts to 1265 cm−1 in regenerated cellulose (Fig. 9a). In the Raman spectrum of cellulose I, the peaks at 1412 and 1480 cm−1 indicate two stereochemically nonequivalent CH2OH groups rotating around the C(5) and C(6) atoms of the cellulose chain. In cellulose II, which contains only one type of CH2OH group, these two scissor vibrations of methylene groups merge into a single signal at 1462 cm−1.
Changes in modifications of the glucopyranose ring are particularly visible in the low-frequency region (Fig. 9b). In the cellulose II spectrum, the intensity of the signal at 378 cm−1 decreases, while the intensity of the signal at 350 cm−1 increases compared to the cellulose I spectrum. This change is attributed to the conformational transition of the glucopyranose ring from cellulose I to cellulose II.55
Porous structure of wet gels
The porous structure of cellulose gels in their wet state was investigated using inverse size exclusion chromatography. The gels were mechanically cut in water using a blender-type device and then sieved to collect the 63–100 μm fraction. Molecules of known molecular mass were passed through a column packed with the gels, and their elution time was recorded. The exclusion limits for linear polymers, expressed as molecular mass (Da), were found to be 3 × 103–5 × 105, 5 × 104–2 × 106, and 3 × 104–4 × 106 for Granocel-500, -2000, and -4000, respectively. Notably, for globular polymers like proteins, the exclusion limits are typically higher than for linear macromolecules. The results showed that Granocel-500 is suitable for the chromatography of polymers with a molecular mass up to 5 × 105, while Granocel-4000 can accommodate macromolecules with a molecular mass up to 4 million. High pore size is especially important for enzyme immobilization and affinity chromatography when the ligands are large molecules.Porous structure of lyophilized gels
Polymeric gels are often used as starting materials for obtaining 3D aerogel scaffolds for bone tissue engineering. However, such scaffolds must exhibit specific morphological characteristics that are critical to supporting cell attachment, proliferation, differentiation, and the formation of new tissue. Porosity, generally between 50% and 90%, is essential to provide enough space for cell infiltration, vascularization, and the diffusion of gases, nutrients, and waste products. The scaffold's framework thickness is crucial for mechanical stability and should typically range from 100 to 300 μm. The pores should be interconnected and typically range from 200 to 1200 μm in size to support bone ingrowth and vascularisation.57,58Fig. 10 illustrates representative 2D and 3D images of Granocel, obtained by micro-computed tomography, emphasizing its porous structure. The structural parameters were identified via 3D analysis, revealing an interconnected porous structure in lyophilized Granocel. The analysis indicated a mean pore diameter of 750 μm, a framework volume percentage of 25%, a porosity of 75%, a specific surface area of 15 mm−1, and an average framework thickness of 210 μm.
 | ||
| Fig. 10 2D and 3D micro-CT images of lyophilized cellulose gel Granocel-2000. | ||
The obtained results suggest that Granocel gels have the potential to be used in bone tissue engineering due to their morphological characteristics, which are suitable for successful vascularization and bone tissue formation. Additionally, the technology adheres to the principles of Green Biomaterials59 by producing 3D scaffolds that are non-toxic, biocompatible, and biodegradable.
Mechanical properties of cellulose gels
| Sample | Relative deformation (%) | Bulk modulus 10−6 (N m−2) | Young's modulus (MPa) | Stiffness (m2 Pa) |
|---|---|---|---|---|
| Granocel-500 | 25.7 ± 0.3 | 2.8 ± 0.3 | 43 | 30000 |
| Granocel-2000 | 34.5 ± 0.5 | 1.8 ± 0.2 | 41 | 25641 |
| Granocel-4000 | 43.6 ± 0.6 | 1.3 ± 0.3 | 41 | 23437 |
The compression test and the calculated parameters indicate that the Granocel-500 gel exhibits the highest compression resistance, with a relative deformation of approximately 26%. The parameters characterizing material strength and resistance to compression, such as the bulk modulus, Young's modulus and material stiffness, are highest for this sample. Mechanical properties of the hydrogels depend on the concentration of cellulosic material. Lower concentration results in softer gel. The softest one is Granocel-4000, obtained using CDA concentration equal to 6.5 w/v%. This corresponds to approx. 5.4% of cellulose in the gel after CDA hydrolysis.
The cellulose hydrogels, particularly Granocel-500 and Granocel-2000, exhibit exceptional flexibility. Unlike typical hydrogels, which have an elastic modulus within the 100–102 kPa range and can easily break or slump under their own weight,60 these cellulose hydrogels show significantly improved characteristics. They possess an elastic modulus approximately twice that of conventional gels, indicating enhanced structural integrity and flexibility. This flexibility is also evident when the hydrogels are in thin film form, as demonstrated in Fig. 11, which showcases their remarkable ability to maintain elasticity when bent between the fingers.
 | ||
| Fig. 11 Demonstrative flexibility of cellulose hydrogel. | ||
Discussion on mechanical properties of cellulose based hydrogels
Cellulose gels are generally classified as soft gels.37,40,43 To enhance their mechanical properties, cellulose hydrogels are crosslinked. Various types of networks can be created within cellulose hydrogels. The strategies for cellulose-based hydrogel networking are discussed in a broad review.37 When crosslinking is achieved through ionic or intermolecular interactions, such as hydrogen bonding, dipole–dipole interactions, dipole-induced interactions, and hydrophobic interactions, physical hydrogels are formed. In contrast, chemically crosslinked hydrogels feature a three-dimensional structure stabilized by covalent bonds between polymer chains, typically using epichlorohydrin or other bifunctional compounds. Chemical hydrogels exhibit greater mechanical stability than physical ones.The mechanical properties of crosslinked cellulose hydrogels are influenced by network density.37,61 Single-crosslinked cellulose hydrogels generally have limited mechanical strength. To reinforce these gels, additional crosslinking is performed. Double-crosslinked hydrogels consist of cellulose connected by multiple crosslinking mechanisms, such as a combination of covalent and physical interactions, two distinct chemical crosslinking methods, or two different types of physical interactions.
Zhao et al. developed double-crosslinked cellulose hydrogels using sequential chemical and physical crosslinking and compared their mechanical properties with those of single-crosslinked hydrogels.61 Their findings showed that double-crosslinked cellulose hydrogels were mechanically superior. The compression modulus increased from 0.01 MPa for single covalently crosslinked gels to 0.61 MPa for double-crosslinked gels, while the tensile modulus increased from 0.03 MPa to 4.9 MPa.
Isobe et al. prepared cellulose hydrogels from an aqueous lithium bromide solution with tunable mechanical properties.43 Compression tests demonstrated that these cellulose hydrogels covered a wide range of mechanical properties, with the compressive Young's modulus adjustable from 30 kPa to 1.3 MPa by varying the initial cellulose concentration.
Cellulosic hydrogels can also be reinforced by combining them with other polymers through grafting or physical interpenetration. The mechanical properties of grafted or interpenetrating hydrogels strongly depend on the polymer composition. Yang et al. evaluated the mechanical properties of single and multiple-network hydrogels.62 They found that cellulose hydrogels crosslinked with epichlorohydrin had a compressive modulus of up to 107 kPa, whereas cellulose-polyacrylamide interpenetrating hydrogels reached 460 kPa. The authors also reported a significant improvement in the elastic modulus, which was determined to be 54 kPa for cellulose hydrogels and over 121 kPa for the interpenetrating material.
A comparison of cellulose-based hydrogels found in the literature (Table 5) reveals that Granocel hydrogels exhibit superior mechanical characteristics. The compression modulus of Granocel gels reaches up to 43 MPa - approximately 30 to 70 times greater than those prepared by other researchers, even using more complex methods. The elastic modulus of Granocel gels reaches up to 0.23 MPa in bending tests, whereas the elastic modulus of typical hydrogels is only up to 0.1 MPa.60 Zhao et al.'s double-crosslinked hydrogels exhibit very high elasticity, but their compression modulus remains low (Table 5).61
| Hydrogel | Compression modulus, MPa | Elastic modulus, MPa |
|---|---|---|
| a Cellulose/polyacrylamide interpenetrating gel.62 | ||
| Single crosslinked61 | 0.01 | 0.03 |
| Double crosslinked61 | 0.61 | 4.9 |
| Crosslinked with EClH62 | 0.107 | |
| From LiBr solution43 | 1.3 | |
| Cellulose/PAAa | 0.46 | 0.12 |
| Granocel-500 | 43 | 0.23 |
| Granocel-2000 | 41 | 0.22 |
| Granocel-4000 | 41 | 0.10 |
The superior mechanical properties of Granocel gels result from a fundamentally different cellulose hydrogel preparation strategy, which is based on the reconstruction of cellulose's own inter- and intramolecular network. As is well known, native cellulose possesses a strong network of inter- and intramolecular bonds, which prevents its dissolution in water despite its hydrophilic nature (due to the abundance of hydroxyl groups). In our method, the gradual hydrolysis of acetyl groups of cellulose acetate in solution promotes the reconstruction of inter- and intramolecular hydrogen bonding networks between the newly formed hydroxyl groups in the cellulose, leading to the formation of a dense network and a rigid three-dimensional gel structure.
Conclusions
Highly porous, rigid cellulose gels were developed through a novel process involving the slow hydrolysis of acetyl groups in cellulose acetate directly in an acetone/aqueous ammonia solution. The slow rate of the process creates conditions favourable for reconstructing of inter- and intramolecular hydrogen bonding networks between the newly formed hydroxyl groups in the cellulose, leading to the formation of a rigid three-dimensional structure. The sol–gel transition point was shown to depend on the composition of the reaction mixture. Both the porous structure and the mechanical properties of the gels can be tuned by adjusting the concentrations of the cellulosic material and ammonia.Structural analyses of the obtained gels confirmed that gelation is accompanied by precipitation and crystallization, characterized by the gradual arrangement of cellulose macromolecules into regular structural crystallites. Wide-angle X-ray diffraction, FT-IR, and Raman spectroscopy identified the gels as semi-crystalline materials with a cellulose II structure, and crystallite sizes of 4.3 to 4.5 nm. The gels exhibited exclusion limits for linear polymers of up to 4 × 106 Da.
The mechanical properties of the gels, such as hardness and resistance to bending and compression, classify them as rigid porous materials derived from biopolymers. They demonstrated a compressive (Young's) modulus of up to 43 MPa and an elastic modulus of up to 0.23 MPa in bending tests.
Thanks to their enhanced mechanical properties and high porosity, crushed gels show promise for applications in column technologies for protein chromatography and in heterogeneous biocatalysis processes with immobilized enzymes. Additionally in film form, the elasticity of the gels makes them suitable for biomedical applications such as wound dressings or artificial skin. Moreover, the cellulose gels are excellent foundational materials for bone tissue engineering, as they can create porous structures after lyophilization that support vascularization and bone tissue ingrowth.
Author contributions
J. Liesiene: conceptualization, supervision, writing – review & editing. S. Kiselioviene: investigation, formal analysis, writing – original draft. A. S. Maruska: conceptualization, investigation. O. Baniukaitiene: supervision, investigation, formal analysis, writing – review & editing.Data availability
The data supporting the findings of this study are stored in a secure repository. Data that are not included in this article are not publicly available but can be provided by the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Notes and references
- X. Li, C. Wan, T. Tao, H. Chai, Q. Huang, Y. Chai and Y. Wu, Cellulose, 2024, 31, 61 CrossRef CAS.
- D. K. Patel, E. Jung, S. Priya, S. Y. Won and S. S. Han, Carbohydr. Polym., 2024, 323, 121408 CrossRef CAS PubMed.
- A. Sannino, C. Demitri and M. Madaghiele, Materials, 2009, 2, 353 CrossRef CAS.
- S. Taokaew, Gels, 2023, 9, 546 CAS.
- P. Zou, J. Yao, Y. N. Cui, T. Zhao, J. Che, M. Yang, Z. Li and C. Gao, Gels, 2022, 8, 364 CAS.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 54, 631 Search PubMed.
- X. Li, G. Jiang, G. Wang, J. Zhou, Y. Zhang and D. Zhao, Cellulose, 2024, 31, 61 CAS.
- W. Tian, X. Gao, J. Zhang, J. Yu and J. Zhang, Carbohydr. Polym., 2022, 277, 118863 CAS.
- K. S. Lefroy, B. S. Murray and M. E. Ries, Cellulose, 2021, 28, 647 CAS.
- I. De Luca, P. Pedram, A. Moeini, P. Cerruti, G. Peluso, A. Di Salle and N. Germann, Appl. Sci., 2021, 11, 1713 CrossRef CAS.
- S. Kiselioviene, O. Baniukaitiene, V. Harkavenko, N. A. Babenko and J. Liesiene, Cellul. Chem. Technol., 2016, 50, 915 CAS.
- K. Zhang, Y. Wang, Q. Wei, X. Li, Y. Guo and S. Zhang, Gels, 2021, 7, 115 CAS.
- D. Kasprzak and M. Galiński, J. Solid State Electrochem., 2021, 25, 2549 CAS.
- H. Liao, J. Na, W. Zhou, S. Hur, P. M. Chien, C. Wang, L. Wang, Y. Yamauchi and Z. Yuan, Nano Energy, 2023, 116, 108769 CAS.
- P. Gemeiner, M. J. Benes and J. Stamberg, Chem. Pap., 1990, 43, 805 Search PubMed.
- J. Aniulyte, J. Liesiene and B. Niemeyer, J. Chromatogr. B:Anal. Technol. Biomed. Life Sci., 2006, 831, 24 CrossRef CAS PubMed.
- A. Jungbauer, J. Chromatogr. A, 2005, 1065, 3 CrossRef CAS.
- J. Liesiene, K. Racaityte, M. Morkeviciene, P. Valancius and V. Bumelis, J. Chromatogr. A, 1997, 764, 27 CrossRef CAS.
- T. Yao, J. Song, Y. Gan, L. Qiao and K. Du, J. Chromatogr. A, 2022, 1677, 463297 CrossRef CAS PubMed.
- J. Bryjak, J. Aniulyte and J. Liesiene, Carbohydr. Res., 2007, 342, 1105 CAS.
- M. M. Jaworska, J. Bryjak and J. Liesiene, Cellulose, 2009, 16, 261 CAS.
- K. Labus, A. Turek, J. Liesiene and J. Bryjak, Biochem. Eng. J., 2011, 56, 232 CAS.
- A. Rekuć, B. Jastrzembska, J. Liesiene and J. Bryjak, J. Mol. Catal. B: Enzym., 2009, 57, 216 CrossRef.
- T. Budtova, Cellulose, 2019, 26, 81 CrossRef CAS.
- A. Iglesias-Mejuto, B. Magariños, T. Ferreira-Gonçalves, R. Starbird-Pérez, C. Álvarez-Lorenzo, C. P. Reis, I. Ardao and C. A. García-González, Carbohydr. Polym., 2024, 324, 121536 CrossRef CAS PubMed.
- P. Parajuli, S. Acharya, J. L. Shamshina and N. Abidi, Cellulose, 2021, 28, 7559 CrossRef CAS.
- B. Schroeter, V. P. Yonkova, N. A. M. Niemeyer, I. Jung, I. Preibisch, P. Gurikov and I. Smirnova, Cellulose, 2021, 28, 223 CrossRef CAS.
- Z. Wei, C. Wu, R. Li, D. Yu and Q. Ding, Cellulose, 2021, 28, 7497 CAS.
- A. Palaveniene, K. Songailiene, O. Baniukaitiene, S. Tamburaci, C. Kimna, F. Tihminlioğlu and J. Liesiene, Int. J. Biol. Macromol., 2020, 152, 1194 Search PubMed.
- P. Daugela, M. Pranskunas, G. Juodzbalys, J. Liesiene, O. Baniukaitiene, A. Afonso and P. Sousa Gomes, J. Tissue Eng. Regener. Med., 2018, 12, 1195 CAS.
- M. Janmohammadi, Z. Nazemi, A. O. M. Salehi, A. Seyfoori, J. V. John, M. S. Nourbakhsh and M. Akbari, Bioactive Mater., 2022, 26, 137 Search PubMed.
- K. Ganesan, T. Budtova, L. Ratke, P. Gurikov, V. Baudron, I. Preibisch, P. Niemeyer, I. Smirnova and B. Milow, Materials, 2018, 11, 2144 CrossRef PubMed.
- N. Buchtová and T. Budtova, Cellulose, 2016, 23, 2585 CrossRef.
- H. Al Abdallah, J. H. Tannous and B. Abu-Jdayil, Cellulose, 2024, 31, 2001 CAS.
- T. Budtova and P. Navard, Cellulose, 2016, 23, 5 CAS.
- C. Chang and L. Zhang, Carbohydr. Polym., 2011, 84, 40 CrossRef CAS.
- B. R. Estevam, I. D. Perez, Â. M. Moraes and L. V. Fregolente, Mater. Today Chem., 2023, 34, 101803 CrossRef CAS.
- M. Gericke, J. Trygg and P. Fardim, Chem. Rev., 2013, 113, 4812 CAS.
- S. H. Zainal, N. H. Mohd, N. Suhaili, F. H. Anuar, A. M. Lazim and R. Othaman, J. Mater. Res. Technol., 2021, 10, 935 CAS.
- Y. Xia, X. Li, Y. Yuan, J. Zhuang and W. Wang, Cellulose, 2022, 29, 1473 CAS.
- F. Xu and B. U. Cho, Cellulose, 2022, 29, 1527 CAS.
- J. Zhou, C. Chang, R. Zhang and L. Zhang, Macromol. Biosci., 2007, 7, 804 CAS.
- N. Isobe, T. Komamiya, S. Kimura, U. J. Kim and M. Wada, Int. J. Biol. Macromol., 2018, 117, 625 CrossRef CAS PubMed.
- C. Chang, K. Han and L. Zhang, Polym. Adv. Technol., 2011, 22, 1329 CrossRef CAS.
- X. Shen, J. L. Shamshina, P. Berton, G. Gurau and R. D. Rogers, Green Chem., 2016, 18, 53 RSC.
- B. Yang, W. Q. Hua, L. Li, Z. H. Zhou, L. Xu, F. G. Bian, X. Ji, G. J. Zhong and Z. M. Li, J. Appl. Polym. Sci., 2019, 136, 47811 CrossRef.
- Y. Chen, X. Hou, R. Kang, Y. Liang, L. Guo, W. Dai, K. Nishimura, C. Te Lin, N. Jiang and J. Yu, J. Mater. Chem. C, 2018, 6, 12739 CAS.
- Q. Li, L. Ibrahim, W. Zhou, M. Zhan and Z. Yuan, Cellulose, 2021, 28, 5257 CAS.
- A. Maruška, A. Šėrys, J. Liesienė, J. Urbonavičienė and A. Žygas, J. Chromatogr. A, 1992, 596, 157 CrossRef.
- D. L. Kaplan, Biopolymers from Renewable Resources, 1998 Search PubMed.
- S. Y. Oh, I. Y. Dong, Y. Shin, C. K. Hwan, Y. K. Hak, S. C. Yong, H. P. Won and H. Y. Ji, Carbohydr. Res., 2005, 340, 2376 CrossRef CAS.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 Search PubMed.
- A. M. Hindeleh and D. J. Johnson, Polymer, 1972, 13, 423 CrossRef CAS.
- A. Pereira, H. Duarte, P. Nosrati, M. Gubitosi, L. Gentile, A. Romano, B. Medronho and U. Olsson, Cellulose, 2018, 25, 3205 CAS.
- K. Schenzel, H. Almlöf and U. Germgård, Cellulose, 2009, 16, 407 CAS.
- K. Schenzel and S. Fischer, Lenzinger Ber., 2004, 83, 64 CAS.
- K. M. Ferlin, M. E. Prendergast, M. L. Miller, D. S. Kaplan and J. P. Fisher, Acta Biomater., 2016, 32, 161 CAS.
- B. Cinici, S. Yaba, K. Kurt, H. C. Yalcin, L. Duta and O. Gunduz, Biomimetics, 2024, 9, 409 CrossRef CAS PubMed.
- N. Rabiee, M. R. Dokmeci, A. Zarrabi, P. Makvandi, M. R. Saeb, H. Karimi-Maleh and M. Radisic, Green Biomater., 2023, 1, 1 CrossRef.
- M. Czerner, L. S. Fellay, M. P. Suárez, P. M. Frontini and L. A. Fasce, Procedia Mater. Sci., 2015, 8, 287 CrossRef CAS.
- D. Zhao, J. Huang, Y. Zhong, K. Li, L. Zhang and J. Cai, Adv. Funct. Mater., 2016, 26, 6279 CrossRef CAS.
- B. Yang, W. Hua, L. Li, Z. Zhou, L. Xu, F. Bian, X. Ji, G. Zhong and Z. Li, J. Appl. Polym. Sci., 2019, 136, 47811 CrossRef.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |