
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pore structure engineering via hard-template synthesis: unlocking the high oxygen reduction reaction activity and stability of Fe–N@C electrocatalysts†
Giulia
Gianola‡
ab,
Mirtha A. O.
Lourenço‡
 *c,
Luca
Basile
ac,
Tiago
Morais
cd,
Luís
Mafra
c,
Candido
Pirri
ab,
Stefania
Specchia
a and
Juqin
Zeng
*ab
*c,
Luca
Basile
ac,
Tiago
Morais
cd,
Luís
Mafra
c,
Candido
Pirri
ab,
Stefania
Specchia
a and
Juqin
Zeng
*ab
aDepartment of Applied Science and Technology (DISAT), Politecnico di Torino, Turin, Italy. E-mail: juqin.zeng@polito.it
bIstituto Italiano di Tecnologia – IIT, Center for Sustainable Future Technologies (CSFT), Turin, Italy
cCICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, Aveiro, Portugal. E-mail: mirtha@ua.pt
dDepartment of Chemistry, University of Iceland, Science Institute, Reykjavik, Iceland
First published on 16th June 2025
Abstract
Developing efficient and durable iron–nitrogen–carbon (Fe–N@C) electrocatalysts with optimal pore architecture is crucial for advancing the oxygen reduction reaction (ORR) in fuel cells. In this study, we demonstrate how hard-templating with tailored silica scaffolds (SBA-15, KIT-6, and a dual SBA-15/KIT-6 template) can tune the pore structure of Fe–N@C materials. In these materials, the pore structure influences the formation and accessibility of active sites for the ORR. The mesoporous Fe–N@CMK-3 electrocatalyst, derived from SBA-15, exhibits the highest ORR activity (onset potential: 0.99 VRHE in alkaline media, and 0.82 VRHE in acid) due to its well-defined 2D hexagonal pores, which facilitate efficient oxygen diffusion. In contrast, the microporous Fe–N@CMK-8 (KIT-6-derived) exhibits lower ORR activity due to limited oxygen accessibility to the active sites. The dual-templated Fe–N@CMK-3/8 combines micro/mesoporosity to deliver balanced performance despite its lower surface area and pore volume resulting from the pore connectivity. All electrocatalysts initially follow a quasi-4e− ORR pathway, but their behavior changes during the long-term testing: Fe–N@CMK-8 shifts to the 2e− pathway despite its notably durable activity in acidic media; Fe–N@CMK-3 exhibits the best stability in terms of activity under alkaline conditions also with a slight shift to the 2e− pathway; Fe–N@CMK-3/8 excels in terms of selectivity sustaining a 4e− pathway along time with medium stability in the activity in both acid and alkaline media. These findings establish pore engineering as a powerful tool to tailor Fe–N@C electrocatalysts for specific operational environments, contributing to the development of high-performance non-precious metal catalysts for the ORR in proton exchange membrane and alkaline fuel cell applications.
New conceptsThis study presents a breakthrough in the design of iron–nitrogen–carbon (Fe–N@C) electrocatalysts by demonstrating how tailored pore architecture can be systematically controlled through hard-templating with silica scaffolds (SBA-15, KIT-6, and their combination). Unlike previous research that focused primarily on chemical composition, this work highlights the pivotal role of the pore structure in governing electrocatalytic performance, specifically activity, stability, and mass transport, in the oxygen reduction reaction (ORR). The innovation lies in linking distinct pore geometries to functional outcomes. Fe–N@CMK-3's 2D mesoporosity enhances activity, stability, and mass diffusion, particularly in alkaline media. Fe–N@CMK-8's microporous network shows much lower activity due to mass diffusion limit, and significant selectivity change despite superior retention in activity during stability test in an acidic environment. The dual-templated Fe–N@CMK-3/8 bridges these properties, balancing activity and stability in both acid and alkaline media through integrated micro/mesoporosity. This pore-engineering approach brings new insight to nanoscience by establishing structure–function relationships that guide the design of high-performance, non-precious metal catalysts. It sets a foundation for tuning catalytic behavior via physical architecture rather than solely chemical modification, advancing the rational design of nanomaterials for fuel cell technologies. |
1. Introduction
Nowadays, the main ways in which we produce energy are still powered by the combustion of fossil fuels such as coal, oil, and natural gas. Consequently, global greenhouse emissions keep increasing, resulting in irreversible climate changes.1 One of the most explored routes to address this crisis is the shift toward a more sustainable and green way to produce the energy we need. Among the possible alternatives, hydrogen-related technologies are emerging as a promising solution.2 Hydrogen fuel cells, in particular, offer a low-carbon option to conventional energy systems, generating electricity directly from the electrochemical reaction between hydrogen and oxygen.3 However, their commercialization is still hindered by the performance and cost of the electrocatalyst layer at the cathode side, where the ORR takes place. Because of its sluggish kinetics, a large amount of electrocatalyst is usually required to enhance the reaction's efficiency. Platinum-based electrocatalysts have long been the benchmark for the ORR.3,4 However, their high cost and limited availability have driven the scientific community to search for alternative materials.5 Among these, Fe–N@C electrocatalysts have gained attention due to their favorable activity, durability, and cost-effectiveness.6–8 A key factor influencing the performance of Fe–N@C electrocatalysts lies in their morphology and pore structure, which directly provides a high specific surface area for active site exposure, while ensuring efficient pathways for oxygen diffusion, minimizing mass transport limitations.9 In this regard, porous carbons have emerged as a valid material due to their tunable pore size and arrangement, excellent mechanical and chemical stability, graphitic framework, and high specific surface area.10–12 Micropores (<2 nm) play a key role in increasing the specific surface area, thus enabling a high density of exposed active sites. As studied by Jaouen et al.,13 micropores are essential to form active Fe–Nx sites, as they provide a confined environment that enhances nitrogen incorporation and site assembly during heat treatment, directly correlating microporous surface area with catalytic activity. However, excessive microporosity can restrict oxygen accessibility, limiting the performance of the electrocatalyst.14,15 This effect was further demonstrated by Zhang et al., where electrocatalysts with higher microporosity exhibited a significant drop in the limiting current density due to oxygen transport limitations caused by water flooding in micropores.16 To address these limitations, mesopores (2–50 nm) and macropores (>50 nm) are essential in providing diffusion pathways for efficient O2 transport and products removal. Pampel et al.17 demonstrated that mesopores significantly enhance ORR performance, increasing the half-wave potential by 60 mV compared to micropore-dominated systems. However, when going to the macropores range, the specific surface area decreases, resulting in a denser structure with fewer interconnected pores and finally limiting oxygen diffusion.17 To this aim, mesopores strike the ideal balance between surface area and transport efficiency, offering interconnected structures that facilitate reactant diffusion without compromising active site accessibility.18,19 Combining distinct pore sizes in a hierarchical structure enhances the catalytic performance benefiting from the high active site density of micropores, the efficient reactant diffusion through mesopores, and the rapid bulk transport enabled by macropores. This synergy results in superior electrocatalytic activity and durability as evidenced by Bae et al.20 in their rotating disk electrode (RDE) study. Their Fe–N@C electrocatalysts with hierarchical porosity exhibited enhanced ORR performance, including a higher onset potential of 0.994 V vs. the reversible hydrogen electrode (RHE), highlighting the role of hierarchical porosity in optimizing both active site utilization and reactant transport.Template-assisted synthesis has emerged as a widely employed strategy for achieving control over electrocatalyst morphology and porosity, enabling tailored nanoscale structural design.21–23 This approach typically involves three key steps: template preparation, template-driven material synthesis, and template removal. Template-assisted methods are broadly categorized into soft-templating and hard-templating (nanocasting) techniques.24,25 For carbon-based electrocatalysts, the nanocasting approach using hard templates consists of three fundamental stages: (1) template formation, (2) precursor infiltration and casting, and (3) template removal. A well-interconnected 3D pore network in the template is essential to ensure structural stability in the final replica. In stage 2 (e.g., via incipient wetness impregnation), complete pore-filling and efficient conversion into the desired composition are essential. Moreover, the template must be readily removed to obtain a high-fidelity replica with well-preserved structural integrity.26
Among hard templates for electrocatalyst synthesis, mesoporous silica stands out due to its chemical inertness and tunable pore sizes (2–50 nm).27,28 Its synthesis strategy relies on soft-templating, where silica precursors (e.g., tetraethyl orthosilicate – TEOS), and structure-directing agents (SDAs) form inorganic–organic composites via the Stöber process.29 In particular, using the non-ionic SDA Pluronic P123 [(PEO)20–(PPO)70–(PEO)20] under controlled conditions yields highly ordered templates like SBA-15 (2D hexagonal form, p6mm)30,31 and KIT-6 (cubic, Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d). Above 15 °C, P123 self-assembles into micelles driven by the hydrophobicity of its PPO blocks.28,29 The addition of cosurfactants (e.g., butanol)32 swells the PPO units, reducing micellar interfacial curvature and inducing a transition from hexagonal (SBA-15) to cubic (KIT-6) symmetry.32–34 These silica templates are well-suited for synthesizing mesoporous carbon catalysts due to their interconnected porosity, ensuring faithful architectural replication during precursor impregnation and template removal, thereby preventing pore collapse in the final material.26
d). Above 15 °C, P123 self-assembles into micelles driven by the hydrophobicity of its PPO blocks.28,29 The addition of cosurfactants (e.g., butanol)32 swells the PPO units, reducing micellar interfacial curvature and inducing a transition from hexagonal (SBA-15) to cubic (KIT-6) symmetry.32–34 These silica templates are well-suited for synthesizing mesoporous carbon catalysts due to their interconnected porosity, ensuring faithful architectural replication during precursor impregnation and template removal, thereby preventing pore collapse in the final material.26
This work employs SBA-15 and KIT-6 as hard templates for the synthesis of Fe–N@C electrocatalysts (Fe–N@CMK-3 and Fe–N@CMK-8, respectively) through nanocasting, using 1,10-phenantroline and iron(III) nitrate nonahydrate as N/C and Fe precursors. The goal is to investigate the impact of pore architecture on ORR activity. Additionally, a hybrid electrocatalyst (Fe–N@CMK-3/8) was developed by modifying the synthesis conditions of the silica template to combine the ordered hexagonal mesopores of SBA-15 with the 3D interconnectivity network of KIT-6.
The textural properties of these three electrocatalysts, identical in composition but different in pore structure, were characterized by powder X-ray diffraction (PXRD), nitrogen adsorption–desorption analysis, thermogravimetric analysis (TGA), scanning transmission electron microscopy (STEM), Raman spectroscopy, elemental analysis, and total reflection X-ray fluorescence (TXRF). These studies correlate structural features with ORR performance, evaluated by a rotating ring disk electrode (RRDE) in alkaline and acidic media.
2. Experimental section
2.1. Chemicals
Pluronic P123 poly(ethylene glycol)-block-poly(propylene glycol)-block-(ethylene glycol) ((PEO)20–(PPO)70–(PEO)20, average Mn ≈ 5800), tetraethoxysilane (TEOS, purity ≥ 98%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, purity ≥ 98.0%), 1-butanol (purity ≥ 99.4%), and 1,10-phenanthroline (purity ≥ 99.0%) were purchased from Sigma-Aldrich. Ethanol (purity ≥ 99.8%), hydrochloric acid (37 wt%), sodium hydroxide pellets (purity 98% up to 100.5%), and isopropanol (IPA, purity ≥ 99.9%) were purchased from Honeywell. All reagents were used as received without further purification. Perchloric acid (HClO4, ACS reagent, 70%) and potassium hydroxide solution (c(KOH) = 1 mol l−1 (1 N), reag. USP, ready-to-use volumetric solution for titration, Titripur®) were purchased from Sigma-Aldrich. Electrolytic solutions were prepared using KOH and HClO4 diluted with ultrapure deionized water from a Milli-pore Milli-Q system with resistivity >18 MΩ cm. A 5 wt% suspension of Nafion® (Ion Power, Inc.) was used as the ionomer. Nitrogen and oxygen gases were supplied in cylinders by Nippon Gases with 5.0 purity.2.2. Hard-silica template synthesis
Three types of mesoporous silica were synthesized, each exhibiting a distinct pore organization: (i) a 2D-hexagonal structure (designated SBA-15), (ii) a combination of 2D-hexagonal and 3D-cubic structures (designated SBA-15/KIT-6), and (iii) a 3D-cubic structure (designated KIT-6).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P123:HCl:H2O = 1:0.022:0.7:130. Specifically, 8.0 g of Pluronic P123 was dissolved in 146.25 g of distilled water and 4.43 g of 37% HCl under vigorous stirring at 35 °C. Once fully dissolved, 13.0 g of TEOS was added to the clear, homogeneous solution. The resulting mixture was stirred at ∼1000 rpm at 35 °C for 24 h. Subsequently, the mixture was transferred to a Teflon-lined reactor and heated at 80 °C under static conditions for 24 h. After filtration, the product was dried overnight at 140 °C. Template removal was achieved through solvent extraction using an ethanol/HCl mixture at 80 °C, followed by calcination at 550 °C for 5 h.
:P123:HCl:H2O:BuOH = 1:0.017:1.83:195:1.31. Specifically, 4.836 g of Pluronic P123 was dissolved in 175 g of distilled water and 9.516 g of concentrated HCl (37%) under vigorous stirring. Subsequently, 4.84 g of 1-butanol was added, and the mixture was stirred at 35 °C for 1 h. Afterward, 10.408 g of TEOS was added dropwise, and the mixture was stirred for another 24 h at the same temperature. The reaction mixture was then transferred to a Teflon-lined reactor and subjected to thermal treatment at 100 °C under static conditions for 24 h. The resulting white powder was hot-filtered without washing and dried overnight at the same temperature. The Pluronic template was removed using the same ethanol/HCl extraction and calcination procedure described previously.
d phase was synthesized following the protocol established by Tae-Wan Kim et al.36 using the molar composition TEOS:P123:HCl:H2O:BuOH = 1.5:0.017:1.83:195:1.7. The procedure involved dissolving 6.0 g of Pluronic P123 in 144 g of distilled water and 7.47 g of concentrated HCl (37%) under vigorous stirring. Then, 5.17 g of 1-butanol was added, and the mixture was stirred at 35 °C for 1 h. Subsequently, 12.8 g of TEOS was added dropwise, and the solution was stirred for 24 h at the same temperature. The mixture was transferred to a Teflon-lined reactor and thermally treated at 100 °C under static conditions for another 24 h. The Pluronic template was removed using the same method as previously described.
:P123:HCl:H2O = 1:0.022:0.7:130. Specifically, 8.0 g of Pluronic P123 was dissolved in 146.25 g of distilled water and 4.43 g of 37% HCl under vigorous stirring at 35 °C. Once fully dissolved, 13.0 g of TEOS was added to the clear, homogeneous solution. The resulting mixture was stirred at ∼1000 rpm at 35 °C for 24 h. Subsequently, the mixture was transferred to a Teflon-lined reactor and heated at 80 °C under static conditions for 24 h. After filtration, the product was dried overnight at 140 °C. Template removal was achieved through solvent extraction using an ethanol/HCl mixture at 80 °C, followed by calcination at 550 °C for 5 h.
:P123:HCl:H2O:BuOH = 1:0.017:1.83:195:1.31. Specifically, 4.836 g of Pluronic P123 was dissolved in 175 g of distilled water and 9.516 g of concentrated HCl (37%) under vigorous stirring. Subsequently, 4.84 g of 1-butanol was added, and the mixture was stirred at 35 °C for 1 h. Afterward, 10.408 g of TEOS was added dropwise, and the mixture was stirred for another 24 h at the same temperature. The reaction mixture was then transferred to a Teflon-lined reactor and subjected to thermal treatment at 100 °C under static conditions for 24 h. The resulting white powder was hot-filtered without washing and dried overnight at the same temperature. The Pluronic template was removed using the same ethanol/HCl extraction and calcination procedure described previously.
d phase was synthesized following the protocol established by Tae-Wan Kim et al.36 using the molar composition TEOS:P123:HCl:H2O:BuOH = 1.5:0.017:1.83:195:1.7. The procedure involved dissolving 6.0 g of Pluronic P123 in 144 g of distilled water and 7.47 g of concentrated HCl (37%) under vigorous stirring. Then, 5.17 g of 1-butanol was added, and the mixture was stirred at 35 °C for 1 h. Subsequently, 12.8 g of TEOS was added dropwise, and the solution was stirred for 24 h at the same temperature. The mixture was transferred to a Teflon-lined reactor and thermally treated at 100 °C under static conditions for another 24 h. The Pluronic template was removed using the same method as previously described.
2.3. Electrocatalyst synthesis
Three mesoporous electrocatalysts were synthesized to investigate the influence of different mesoporous silica hard templates on the structure and electrocatalytic activity of the resulting carbon-based materials. The synthesis followed a consistent protocol, varying only the silica source. The electrocatalysts were designated as Fe–N@X, where X represents the carbon derived from the silica template used (CMK-3 for the SBA-15, CMK-3/8 for the SBA-15/KIT-6, and CMK-8 for the KIT-6). In a typical procedure, 404 mg of Fe(NO3)3·9H2O was dissolved in 10 mL of a water/ethanol mixture (1:1 by volume) using sonication. This clear solution was added to 20 mL of water/ethanol (1:1) containing 500 mg of the synthesized mesoporous silica (SBA-15, SBA-15/KIT-6, or KIT-6) and 540.6 mg of 1,10-phenanthroline. The resulting mixture was stirred vigorously at 80 °C for 24 h to ensure thorough dispersion and complete solvent evaporation. The orange powder obtained was finely ground using a mortar and pestle and transferred to a vertical quartz tube furnace. The furnace was sealed, purged with a constant flow of nitrogen for 20 min, and then heated to 900 °C at a rate of 5 °C min−1 under continuous nitrogen flow. The target temperature was maintained for 3 hours to complete the pyrolysis. The resulting material was etched by stirring in 15 mL of 6 M NaOH at 60 °C for 24 h to remove the silica hard template. Afterward, the mixture was filtered, washed until neutral pH, and dried overnight. To eliminate any residual Fe and/or oxide nanoparticles, the material underwent acid etching in 50 mL of concentrated HCl (37 wt%) for 3 h at room temperature. The final product was filtered, washed, and dried.
2.4. Materials characterization
PXRD data were collected using a Panalytical Empyrean diffractometer equipped with Cu-Kα radiation in transmission mode, employing focusing mirror geometry. The instrument was operated at 45.0 kV and 40.0 mA, with data acquired in 2θ step-scans of 0.026°. Raman spectra were acquired in a Raman-FT Bruker RFS/100S instrument equipped with a Nd:YAG (1064 nm) laser at 25 mW and 4 cm−1 for 3000 scans. Thermogravimetric analysis (TGA) was performed on a Hitachi NEXTA STA 300 instrument, with a heating rate of 5 °C min−1 in an air atmosphere, covering a temperature range of 25–800 °C. Scanning transmission electron microscopy (STEM) images were obtained using a Hitachi® HD2700 Type B microscope operating at an acceleration potential of 200 kV. Samples were dispersed in ethanol using sonication, and a droplet of the suspension was deposited onto a lacey carbon-coated copper grid (Agar Scientific®). The grid was then dried at room temperature followed by vacuum drying. Nitrogen adsorption–desorption isotherms were measured at 77 K (−196 °C) using a Gemini V 2.00 instrument (Model 2380). Specific surface areas were determined using the Brunauer–Emmett–Teller (BET) method, while pore size distributions were calculated using the Barrett–Joyner–Halenda (BJH) method. Elemental analysis was used to obtain the quantities of carbon, hydrogen and nitrogen elements. The analysis was performed using a TruSpec20 Micro 630-200-200 instrument. The combustion furnace was operated at 1075 °C, while the afterburner was maintained at 850 °C. Detection was achieved via infrared absorption for carbon, hydrogen, and sulfur, and thermal conductivity for nitrogen. The Fe concentration was determined after the sample digestion. For this, 2.5 mg of the sample was treated with freshly prepared Aqua Regia (1:3 HNO3:HCl) under reflux conditions for 4 h. After digestion, the sample was diluted in 40 mL of ultrapure water and filtered using a nylon syringe filter (0.45 µm). The Fe concentration was measured by TXRF analysis with a Bruker S2 Picofox, employing Y as an internal standard. The method has a detection limit of 0.011 ppm.
2.5. Electrochemical tests
Electrochemical characterization studies were conducted at room temperature using a Biologic VSP-300 potentiostat and a Pine Research MSR Rotator for the RRDE setup. The electrocatalyst-based inks were prepared by ultrasonic dispersion in isopropanol (IPA) mixed with a 5 wt% Nafion® suspension (Ion Power, Inc.), ensuring an ionomer-to-carbon (I/C) ratio of 0.2. The resulting electrocatalyst inks were deposited onto a glassy carbon disk (active area: 0.247 cm2) with a loading of 0.4 mg cm−2. A carbon rod was employed as the counter electrode, while a hydroflex reversible hydrogen electrode (Gaskatel) was used as the reference electrode. Measurements were performed in both 0.1 M KOH (alkaline) and 0.1 M HClO4 (acidic) electrolytes, saturated with either nitrogen or oxygen as per the testing protocol. All reported potentials were referenced to the reversible hydrogen electrode (RHE). Cyclic voltammetry (CV) was carried out within a potential range of 0–1.1 VRHE at a scan rate of 100 mV s−1 in oxygen- and nitrogen-saturated electrolytes. Linear sweep voltammetry (LSV) was conducted in a three-electrode configuration (disk/reference/counter) in O2-saturated electrolyte, scanning 0–1.1 VRHE at a 10 mV s−1 rate with the disk rotating at 400, 900, 1600, and 2500 rpm. In a four-electrode setup (disk/ring/reference/counter), LSV measurements were performed in O2-saturated electrolyte at 1600 rpm, scanning the disk electrode from 0 to 1.1 VRHE at 10 mV s−1, with the ring potential fixed at 1 VRHE. iR-drop corrections were applied to all measurements through the series resistance (Rs) values obtained from electrochemical impedance spectroscopy (EIS) measurements. EIS data were collected at 1 VRHE with an AC amplitude of 10 mV across a frequency range of 100 Hz to 1 MHz. LSV curves were corrected for background contributions by subtracting the corresponding LSV recorded in nitrogen-saturated electrolyte under identical conditions, eliminating capacitive effects from the reduction current. Electrocatalyst durability was assessed in oxygen-saturated electrolyte by cycling the potential between 0.6 V and 1 V, holding each value for 3 seconds per cycle, and repeating the process 10000 times.37 LSV measurements were performed after the aging protocol to evaluate ORR activity degradation.
3. Results and discussion
3.1. Textural properties of the silica hard templates
The textural properties of the sacrificial hard templates used to produce the Fe–N@C samples were analyzed by PXRD, N2 adsorption–desorption isotherms at −196 °C, STEM and TGA. Fig. 1 presents the XRD patterns of the SBA-15, SBA-15/KIT-6, and KIT-6 templates. The SBA-15 sample exhibits a highly ordered 2D hexagonal pore arrangement with p6mm symmetry, showing reflections at 2θ corresponding to the [100], [110], and [200] planes.35 The KIT-6 sample reveals the well-resolved (211) and (220) reflections, consistent with the cubic Iad mesoporous silica structure.34,36,38 Additionally, the intermediate diffractogram of the SBA-15/KIT-6 sample displays features of both 3D and 2D-structured materials, indicating its dual structural nature. This duality, further confirmed by STEM images (Fig. S2, ESI†), arises from employing more acidic conditions during KIT-6 synthesis, facilitating the formation of 2D-hexagonal pores alongside the 3D cubic structure.36 The XRD unit cell size and the interplanar spacing d100 were determined and the results are shown in Table 1.
 | ||
| Fig. 1 PXRD of the synthesized sacrificial silica templates. | ||
| Material | d 100 (nm) | a 0 (nm) | S BET (m2 g−1) | V P (cm3 g−1) | d P (nm) | b (nm) |
|---|---|---|---|---|---|---|
| a Unit cell parameter calculated from the equation: a0 = 2d100√3. b Pore diameter obtained from the BJH method. c Pore wall thickness calculated as (b = a0 − dP). | ||||||
| SBA-15 | 11.85 | 13.69 | 734 | 0.76 | 8.1 | 5.6 |
| SBA-15/KIT-6 | 11.39 | 13.16 | 555 | 0.40 | 7.4 | 5.8 |
| KIT-6 | 11.25 | 12.99 | 757 | 1.06 | 5.6 | 7.4 |
Fig. S1 (ESI†) and Table 1 present the results of −196 °C N2 adsorption–desorption isotherms for the silica templates. As shown in Fig. S1a (ESI†), the synthesized silicas exhibit a type IV isotherm with an H1 hysteresis loop, consistent with the IUPAC classification typically associated with mesoporous silicas.39 By applying the BET and BJH theories to the −196 °C N2 adsorption/desorption isotherms it is possible to determine the specific surface area (SBET), pore volume (Vp) (Table 1), and pore size distribution (Fig. S1b, ESI†). Fig. S1b (ESI†) reveals that the synthesized silicas exhibit a narrow pore size distribution. The pore diameters fall within the mesoporous range, between 5.6 and 8.1 nm (Fig. S1b, ESI† and Table 1). The SBET of SBA-15, SBA-15/KIT-6, and KIT-6 are approximately 734, 555, and 757 m2 g−1, respectively. The sample with a dual pore structure exhibits the lowest pore volume (0.40 cm3 g−1), while KIT-6 shows the highest one (1.06 cm3 g−1). By integrating these findings with data from PXRD, the pore wall thickness of the materials can also be calculated. The summarized results are presented in Table 1. The results are in agreement with those observed in the literature.34–36
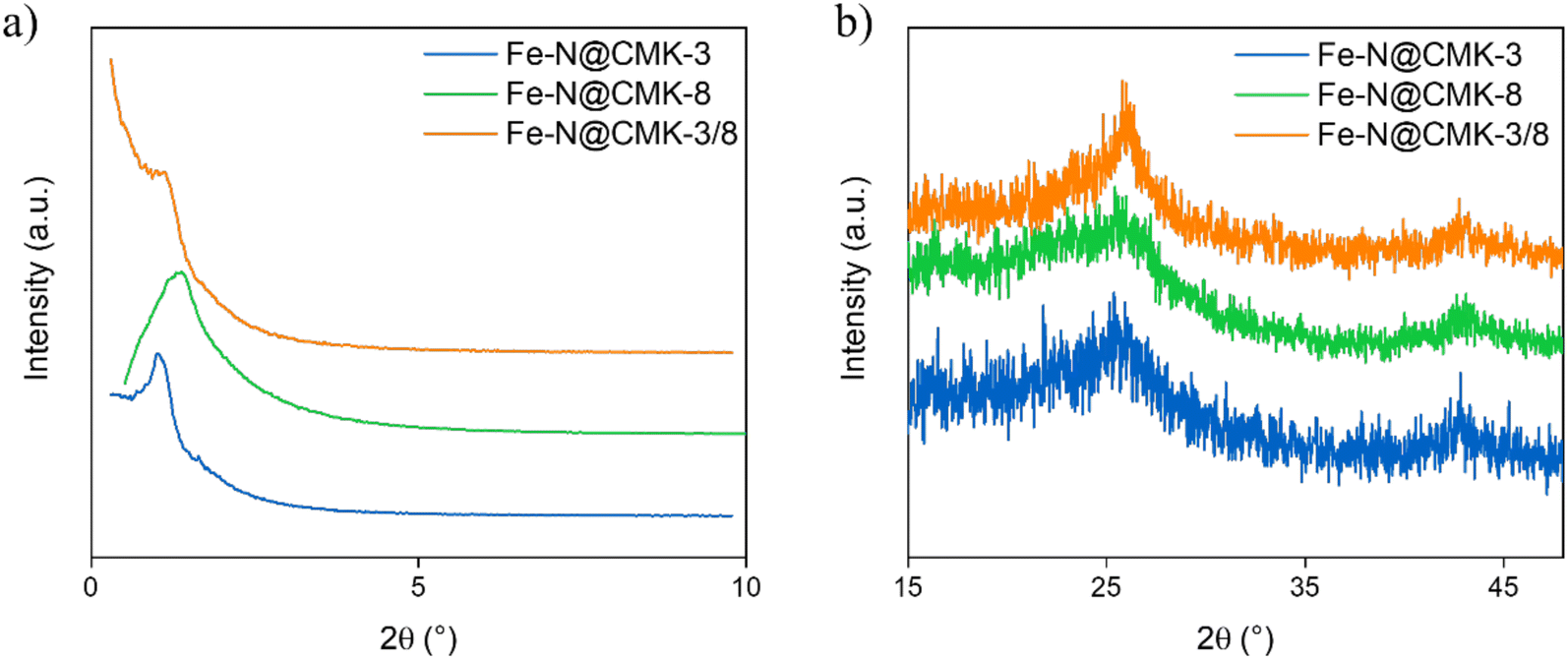 | ||
| Fig. 2 (a) Low-angle PXRD of the Fe–N@C electrocatalysts acquired under transmission mode; (b) high-angle PXRD of the Fe–N@C electrocatalysts obtained under reflection mode. | ||
TEM and SEM images of the silica hard-template materials were acquired to further support the PXRD and N2 sorption isotherms results. Fig. S2 (ESI†) presents TEM images of the materials, including the top view (1st row) and lateral view (2nd row) of the SBA-15, KIT-6, and SBA-15/KIT-6 samples. These images demonstrate the formation of a hexagonal pore network (p6mm) for SBA-15,35 a cubic pore arrangement (Ia3d symmetry) for KIT-634 and a combination of both p6mm and Ia3d structures in the SBA-15/KIT-6 samples. This information is further supported by SEM images (Fig. S2, 3rd row, ESI†).
3.2. Textural properties of the Fe–N@C electrocatalysts
PXRD diffractograms of the electrocatalysts Fe–N@CMK-3, Fe–N@CMK-3/8, and Fe–N@CMK-8, synthesized using SBA-15, SBA-15/KIT-6, and KIT-6 silica hard templates, respectively, were analyzed to investigate the mesostructure of the electrocatalyst pores (Fig. 2a), as well as the presence of graphitized carbon and Fe nanoparticles (Fig. 2b).All Fe–N@C replicas exhibit a similar pore arrangement, showing a single reflection between 2θ = 1.0° and 2θ = 1.4°, corresponding to the (100) or (211) planes, depending on the silica hard template used (Fig. 2a), less evidenced in the Fe–N@CMK-3/8 sample. These replicas demonstrate a reduced unit cell (a0) size compared to their respective silica templates (Tables 1 and 2). The Fe–N@CMK-3 sample exhibits the highest pore organization, as demonstrated by the more defined 100 reflection (Fig. 2a). The absence of additional diffraction patterns indicates that complete formation of cubic or hexagonal pore arrangements during the nanocasting process was not achieved. At higher diffraction angles (Fig. 2b), two sharp peaks at approximately 2θ = 25° and 43° are observed, attributed to graphitic carbon and corresponding to the (002) and (101) diffraction planes, respectively. The presence of graphitic carbon in the samples is further confirmed by Raman spectroscopy (Fig. S3, ESI†). The removal of Fe nanoparticles appears to be effective, as indicated by the absence of their diffraction peaks.
| Material | d 100 (nm) | a 0 (nm) | S BET (m2 g−1) | S micro (m2 g−1) | S meso (m2 g−1) | V P (cm3 g−1) | d P (nm) | b (nm) |
|---|---|---|---|---|---|---|---|---|
| a Unit cell parameter calculated from the equation: a0 = 2d100√3. b t-Plot micropore area. c BJH adsorption cumulative surface area of pores between 1.7 nm and 300 nm width. d Single point adsorption total pore volume of pores. e Average pore diameter obtained from the BJH method (adsorption brand). f Pore wall thickness calculated as (b = a0 − dP). | ||||||||
| Fe–N@CMK-3 | 11.14 | 13.09 | 1230 | 279 | 694 | 0.88 | 3.8 | 9.29 |
| Fe–N@CMK-3/8 | 9.39 | 10.85 | 720 | 194 | 383 | 0.51 | 4.5 | 6.35 |
| Fe–N@CMK-8 | 8.31 | 9.60 | 1313 | 322 | 650 | 0.81 | 3.0 | 6.60 |
The −196 °C N2 adsorption/desorption isotherms of the carbon replicas in Fig. 3a exhibit distinct adsorption branches compared to those of the silica hard templates. These isotherms exhibit characteristics that are a blend of type I(b), indicating a predominance of micropores and ink-bottle-shaped pores, as demonstrated by the rapid adsorption increase at low p/p0, and type IV(a), with a final saturation plateau reduced to a subtle inflection point, or type II, commonly associated with nonporous or macroporous adsorbents.39 All Fe–N@C electrocatalysts exhibit H4 hysteresis loops, with the more pronounced uptake at low p/p0 attributed to the filling of micropores. H4 loops are typically associated with certain micro–mesoporous carbon materials. Although the silica templates exhibit differences in pore size distribution (Fig. S1b, ESI†), Fig. 3b highlights the presence of mesopores within the range of 1.7 to 5 nm, with the highest population of the pores near 3.4 nm. The Fe–N@CMK-3 sample exhibits the most narrowly distributed pore sizes. The BET specific surface area (SBET) and the t-plot micropore surface area (Smicro) increase in the order of Fe–N@CMK-3/8, Fe–N@CMK-3, and Fe–N@CMK-8. Additionally, although the sample prepared using silica with dual porosity exhibits a similar curve type to Fe–N@CMK-3, its SBET is nearly half that of the electrocatalysts derived from silicas with hexagonal or cubic pore arrangements. Fe–N@CMK-3 and Fe–N@CMK-8 also show the highest total pore volume value (Table 2).
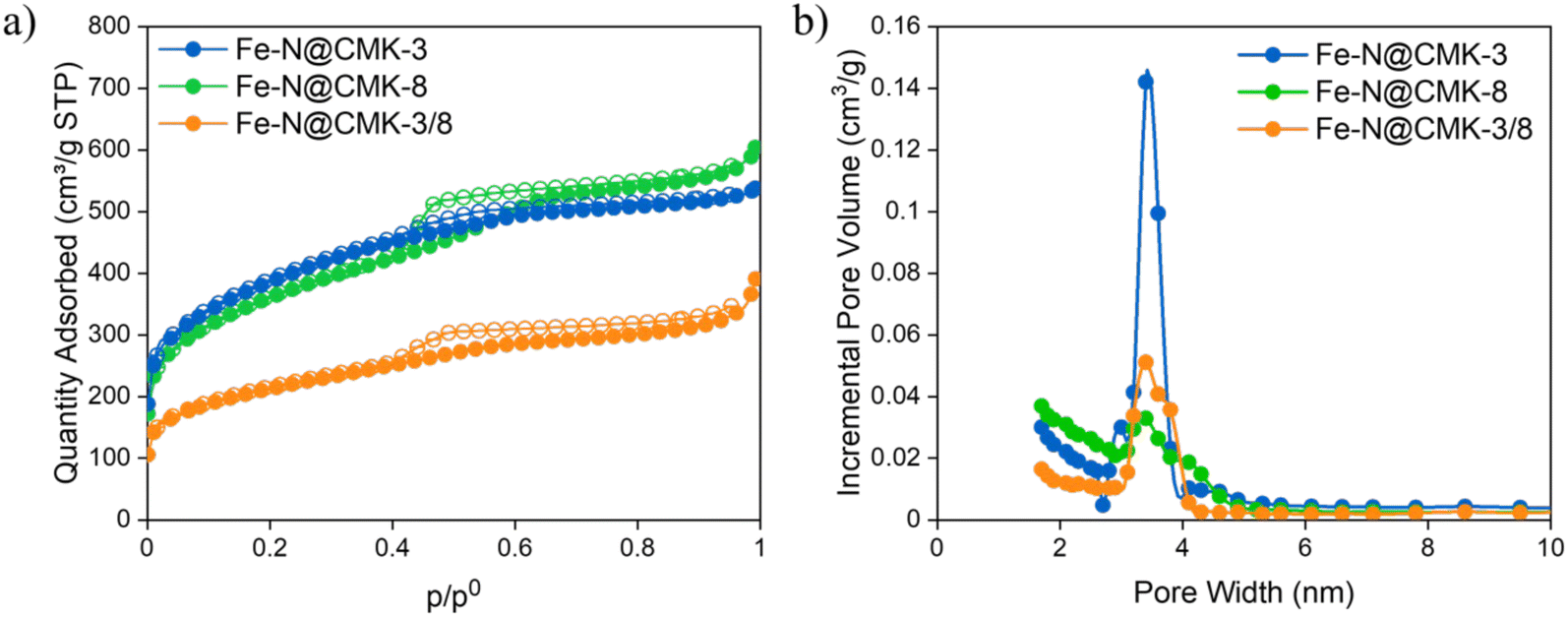 | ||
| Fig. 3 (a) −196 °C N2 adsorption–desorption isotherms of the Fe–N@C catalysts. Full symbols are related to the adsorption branches; empty symbols correspond to desorption ones. (b) Pore size distribution curves. | ||
TEM and SEM images (Fig. 4) further validate the XRD results. The Fe–N@CMK-3 and Fe–N@CMK-8 samples exhibit well-organized porosity, whereas the Fe–N@CMK-3/8 sample, derived from dual-type mesoporous silica, primarily consists of amorphous carbon. This may be due to the SBA-15/KIT-6 silica having blocked or non-existent interconnections between micro- and mesopores, preventing the diffusion of carbon precursors into the pores. Consequently, upon silica removal, the structure collapses, failing to retain the pore organization of the original silica matrix. Moreover, all samples contain carbon nanotubes, which are more prominent in the latter sample. EDS analysis (Fig. S4, ESI†) shows the presence of Fe and Si in all samples, with the latter attributed to residual silica.
 | ||
| Fig. 4 TEM images of (a) Fe–N@CMK-3, (b) Fe–N@CMK-8 and (c) Fe–N@CMK-3/8 electrocatalysts; and SEM images of the (d) Fe–N@CMK-3, (e) Fe–N@CMK-8 and (f) Fe–N@CMK-3/8 electrocatalysts. | ||
The thermal stability of the electrocatalysts was assessed using TGA, with the results shown in Fig. S5 (ESI†). All materials exhibit a similar degradation pattern, remaining thermally stable up to 400 °C and undergoing two main stages of thermal degradation. The first weight loss, ranging from ∼5 wt% to ∼15 wt%, occurs below 100 °C and is attributed to the evaporation of moisture and adsorbed gases. The second weight loss, observed within the temperature range of 400–650 °C, corresponds to the degradation of carbon and nitrogen species. The residual mass, accounting for 13–16 wt%, is likely associated with the remaining silica and iron oxide formed during degradation in the air atmosphere.
Table S1 in the ESI† presents the C, N, and H contents of the pyrolyzed Fe–N@C electrocatalysts, as determined by elemental analysis. The nitrogen atoms from the 1,10-phenanthroline are retained within the structure of the Fe–N@C samples after pyrolysis at 900 °C. Among the materials, Fe–N@CMK-8 exhibits the highest nitrogen content (3.95 wt%), followed by Fe–N@CMK-3 (3.74 wt%) and Fe–N@CMK-3/8 (2.88 wt%). Despite Fe–N@CMK-8 having the highest nitrogen content, it has the lowest carbon content (45.21 wt%) among the three synthesized samples, while Fe–N@CMK-3/8 and Fe–N@CMK-3 have similar carbon contents (∼60 wt%). The remaining composition, attributed to Si, Fe, and O atoms, is higher in the Fe–N@CMK-8 catalyst. Additionally, TXRF analysis was used to quantify the iron (Fe) content (Table S1, ESI†), revealing comparable Fe concentrations across all samples, ranging from 1.06 ± 0.014 to 1.19 ± 0.011 wt%. The N/Fe atomic ratios were determined by combining elemental analysis (for nitrogen content) with TXRF measurements (for iron content), determining values of 14.9 for Fe–N@CMK-8, 12.6 for Fe–N@CMK-3, and 9.8 for Fe–N@CMK-3/8. It is important to note that not all nitrogen in the samples is necessarily coordinated to iron. Some nitrogen is coordinated to Fe, while the majority of nitrogen is likely bonded to carbon within the matrix. No diffraction peaks corresponding to crystalline iron species were detected in the PXRD patterns, suggesting the presence of Fe–Nx species that are commonly stable under highly acidic leaching solution.
3.3. Influence of mesostructure on the ORR performance of Fe–N@C electrocatalysts
Fig. S6 (ESI†) shows cyclic voltammetry results in N2-saturated and O2-saturated HClO4 and KOH electrolytes. Reduction peaks are present in the oxygen atmosphere only, indicating the absence of active redox species responsible for unwanted reactions under inert conditions. Reduction peaks are in the 0.4–0.7 VRHE range for the acidic media and at 0.7–0.9 VRHE in the alkaline case, aligning with previous literature results for Fe–N@C electrocatalysts.40–42Fig. 5c and d display LSV curves achieved at 1600 RPM in both alkaline and acidic electrolytic solutions. The Fe–N@CMK-3 and Fe–N@CMK-3/8 electrocatalysts demonstrate outstanding ORR performance in both media, characterized by higher current densities and more positive onset potentials (0.99 VRHE and 0.97 VRHE in alkaline media, 0.82 VRHE and 0.80 VRHE in acidic electrolyte, respectively). This performance similarly underscores the crucial role of the SBA-15 template in providing a linear and unidirectional mesoporous structure, which facilitates the reactants’ and products’ mass transport. Fe–N@CMK-3/8's superior performance reflects an optimal balance between the 2D hexagonal and 3D interconnected pore structures. While its high ORR activity may seem unexpected, given that N2 adsorption–desorption analysis indicates the dual-templated catalyst has the lowest specific surface area (Table 2), its structural properties reveal a different story. As shown by XRD and N2-adsorption–desorption isotherms at −196 °C measurements (Table 2), the electrocatalyst features larger average pore diameters, as determined by the BJH method. This suggests the structure achieves a trade-off between lower porosity and larger pores, influencing electrocatalytic activity. In contrast, the electrocatalyst derived from the KIT-6 template exhibits the lowest performance, reflected in its reduced current densities and less positive onset potentials (0.96 VRHE in 0.1 M KOH and 0.77 VRHE in 0.1 M HClO4). Although the interconnected 3D framework provides a high pore volume and improves accessibility (refer to Fig. 3 and Table 2), it typically contains a greater fraction of micropores compared to SBA-15, as highlighted in the pore size distribution curve. These micropores can restrict mass transport because diffusion is limited in smaller pores, reducing ORR activity.15,16,43–45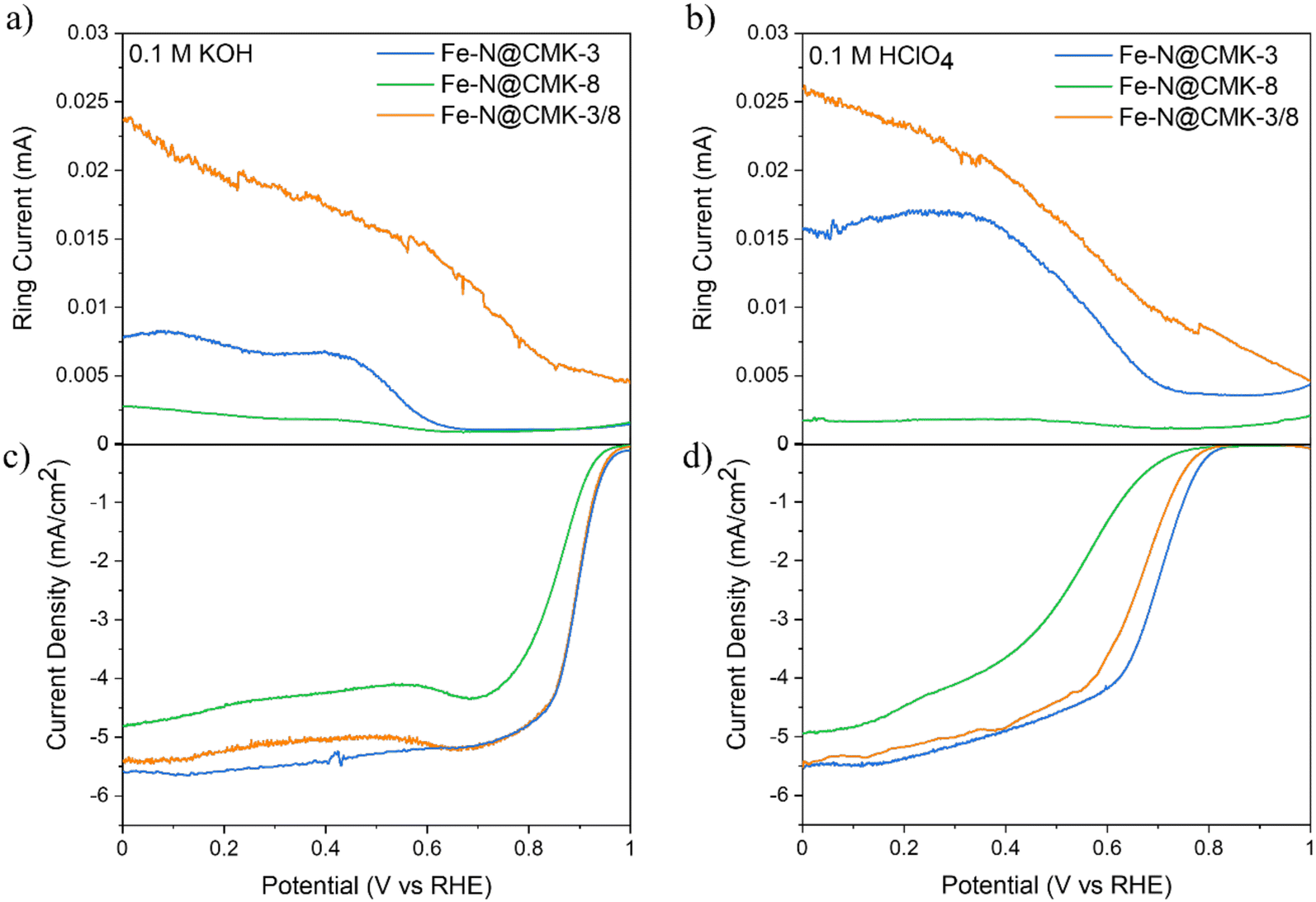 | ||
| Fig. 5 ORR electrocatalytic activity recorded at 1600 rpm: (a) ring current in 0.1 M KOH. (b) Ring current in 0.1 M HClO4. (c) Disk current density in 0.1 M KOH. (d) Disk current density in 0.1 M HClO4. | ||
To further analyze the electrocatalytic behavior of the materials under study, Fig. S7 (ESI†) presents LSV curves recorded at different rotation speeds in both alkaline and acidic media. At high potentials, the current density is unaffected by rotation speed, indicating that electron-transfer kinetics is the rate-determining step. As the potential decreases, the impact of rotation becomes evident. The increase in the limiting current density with higher rotation rates is consistent with enhanced mass transport due to forced convection, in accordance with the Levich equation (eqn (1)).
| ilim = 0.62nFAD2/3ν−1/6ω1/2C | (1) |
485 C mol−1), A is the area of electrode (cm2), D is the oxygen diffusion coefficient (cm2 s−1), ν is the kinematic viscosity of the solution (cm2 s−1), ω is the angular velocity of the rotating electrode (rad s−1), and C is the bulk concentration of the reactant (mol cm−3). While the Levich equation describes a purely diffusion-controlled regime, the Koutecký–Levich (KL) equation (eqn (2)) extends the model by incorporating both kinetic and mass transport contributions. | (2) |
 | (3) |
 | (4) |
I d is the current measured at the disk electrode, Ir is the current recorded at the ring one, and N is the collection efficiency (N = 0.4). The ring currents, associated with the reduction of intermediate species in the indirect ORR pathway, are significantly lower than those observed at the disk electrode. This suggests that the ORR primarily follows the direct pathway, indicating a high selectivity toward the 4-electron reduction reaction. The selectivity data, represented by the electron transfer number and peroxide yield, are calculated using eqn (3) and (4) and presented in Fig. 6.
 | ||
| Fig. 6 ORR selectivity by RRDE measurement in (a) alkaline and (b) acidic environments. | ||
From the detailed analysis, the Fe–N@CMK-8 sample appears to be slightly better in terms of selectivity with respect to the other two counterparts. Nevertheless, each electrocatalyst can still be regarded as a strong candidate for the ORR, as their selectivity remains consistent with results reported in other studies on Fe–N@CMK-based electrocatalysts.22,46,47 Moreover, oxygen reduction follows a quasi-direct reduction pathway across all potential ranges, in agreement with the KL results reported in Fig. S8 (ESI†). This suggests that the reaction mechanism is stable and does not shift between pathways over the tested potential range. Consequently, the active sites on the electrocatalyst surface are consistently promoting the same reduction pathway, indicating uniformity in their properties and behavior under the given conditions.
The durability of the electrocatalysts was evaluated through potential cycling between 0.6 VRHE and 1.0 VRHE for 10000 cycles. Fig. 7 presents LSV curves recorded at 1600 rpm before and after 10000 potential cycles, illustrating the catalysts’ electrochemical stability, while Table 3 quantifies the degradation by reporting the shifts in onset potential (ΔEon) and half-wave potential (ΔE1/2) in both alkaline and acidic environments.
 | ||
| Fig. 7 LSV recorded before (0 cycles) and after (10k cycles) the durability test in both (a) alkaline and (b) acid electrolytes. | ||
| Material | 0.1 M KOH | 0.1 M HClO4 | ||
|---|---|---|---|---|
| ΔEon (V) | ΔE1/2 (V) | ΔEon (V) | ΔE1/2 (V) | |
| Fe–N@CMK-3 | 0.03 | 0.02 | 0.07 | 0.09 |
| Fe–N@CMK-3/8 | 0.05 | 0.05 | 0.05 | 0.12 |
| Fe–N@CMK-8 | 0.07 | 0.08 | 0.01 | 0.03 |
In alkaline environments, all electrocatalysts exhibit moderate activity degradation, ascribed to the reduced aggressive nature of hydroxide ions on active sites. In acidic media, among the tested materials, Fe–N@CMK-3 and Fe–N@CMK-3/8 exhibit steep potential shifts, suggesting the dissolution of unstable sites in acidic solutions under potential cycling. In contrast, Fe–N@CMK-8 demonstrates the lowest degradation in acidic media, with only a 0.01 V drop in onset potential and a 0.03 V shift in half-wave potential. This improved stability may be linked to its higher micropore specific surface area (Table 2) and smaller average pore diameter, which allows the confinement of Fe–Nx active sites within a more protective carbon matrix. Micropores are known to provide a stabilization effect by limiting direct exposure of active sites to acidic electrolytes.48 This explains why Fe–N@CMK-3 and Fe–N@CMK-3/8 experience slightly greater losses than Fe–N@CMK-8, which benefits from the protective nature of micropores. However, this scenario changes when the long-term selectivity is taken into consideration. The stability in ORR selectivity was evaluated by monitoring the variation in electron transfer number (Δn) and peroxide yield (ΔH2O2/ΔHO2−) before and after 10k cycles (Fig. S9, ESI†). The degradation of ORR selectivity was particularly evident for Fe–N@CMK-8, which exhibited the highest Δn in both alkaline and acidic media, with a consequent increase in the HO2−/H2O2 production. This behavior suggests that micropores can help preserve Fe–Nx sites against acidic corrosion, while the nature of these sites could change during potential cycling, shifting the selectivity to the 2e− pathway. Fe–N@CMK-3, with predominantly mesoporous channels, exhibited intermediate behavior: its selectivity slightly decreased, especially in acidic media. In contrast, Fe–N@CMK-3/8, featuring a hierarchical pore structure, preserved its selectivity excellently over time. Combining the moderate loss in activity and excellent preservation in selectivity during potential cycling, Fe–N@CMK-3/8 outperforms the other two counterparts in terms of stability, highlighting the important role of the balance between microporosity and mesoporosity in the catalyst longevity.
Concisely, the comprehensive study highlights the crucial role of pore structure optimization in designing Fe–N@C electrocatalysts with both high activity and stability, reinforcing the importance of material engineering strategies for their application in fuel cells.
3.4. Comparison with porous Fe–N@C electrocatalysts
To better emphasize the interconnected role of electrochemical and porosity properties for the ORR, Table 4 provides a comparative analysis of Fe–N@C electrocatalysts from the literature against Fe–N@CMK-3. This comparison highlights the critical role of structural parameters, including BET specific surface area, micropore area, pore volume, and electron transfer number, in defining electrocatalytic performance.| Material | Electrochemical properties | Porosity properties | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Electrolyte | E on (VRHE) | E 1/2 (VRHE) | n | S BET (m2 g−1) | S micro (m2 g−1) | V p (cm3 g−1) | d p (nm) | Ref. | |
| Fe–N-HMC | 0.1 M KOH | 0.942 | 0.856 | 3.99@0.8 VRHE | 733 | — | 0.73 | 2.0 | 49 |
| MgO@Phen-Fe-800-3/1 | 0.1 M HClO4 | 0.940 | 0.80 | 4 | 760 | 470 | 1.60 | 0.6 | 52 |
| Meso-Fe–N–C | 0.1 M KOH | — | 0.846 | 3.9@0.45 VRHE | 494.7 | — | — | 6.4 | 53 |
| h-FeNC | 0.1 M HClO4 | — | 0.777 | 3.93 | 2454 | — | 1.90 | 3.4 | 54 |
| FeSNC-3 | 0.1 M KOH | 1.014 | 0.866 | 3.96@0.5 VRHE | 990 | 687 | 0.56 | 2.2 | 50 |
| FeNC-F3 | 0.1 M KOH | 1.0 | 0.905 | 3.95@0.5 VRHE | 1318 | 201 | 1.35 | 4.1 | 55 |
| DL-FeNC | 0.1 M KOH | 0.998 | 0.903 | 3.96@0.5 VRHE | 565 | 263 | 0.58 | 10.1 | 51 |
| Fe–N@CMK-3 | 0.1 M KOH | 0.99 | 0.89 | 3.9@0.5 VRHE | 1230 | 279 | 0.88 | 3.8 | This work |
| 0.1 M HClO4 | 0.80 | 0.69 | 3.8@0.5 VRHE | ||||||
Among several electrocatalysts available in the literature, Fe–N-HMC,49 with a SBET of 733 m2 g−1, achieves an onset potential of 0.942 VRHE and a half-wave potential of 0.856 VRHE, which are slightly lower than those observed for Fe–N@CMK-3 (0.99 VRHE and 0.89 VRHE, respectively). This suggests that Fe–N@CMK-3 benefits from a more developed mesoporous network, improving mass transport and electrocatalytic activity. FeSNC-350 exhibits the highest specific micropore area (687 m2 g−1), indicating a high density of active sites. However, its high Smicro, together with the small pore diameter (2.2 nm), could limit oxygen transport, as highlighted by its lower half-wave potential (0.866 VRHE). On the other hand, DL-FeNC,51 with a smaller SBET of 565 m2 g−1, exhibits a high onset potential (0.998 V), suggesting favorable active site accessibility. Nevertheless, its larger pore diameter (10.1 nm) may reduce the density of active sites, which could impact long-term catalytic stability. Overall, Fe–N@CMK-3 demonstrates a superior balance between specific surface area, microporosity, and mesoporosity, ensuring both high electrocatalytic activity and effective mass transport. This comparison underscores the importance of rational template selection in tuning Fe–N@C electrocatalysts for optimized ORR performance, positioning Fe–N@CMK-3 as a highly competitive candidate among non-precious metal electrocatalysts for fuel cells.
4. Conclusions
This study highlights the critical role of pore structure engineering in developing Fe–N@C electrocatalysts for the oxygen reduction reaction. By employing a nanocasting approach using SBA-15, KIT-6, and a hybrid SBA-15/KIT-6 as a hard-template, we demonstrate that pore architecture governs electrocatalytic activity, selectivity, and durability. Fe–N@CMK-3 exhibited the highest ORR activity due to its well-defined mesoporous structure, balancing mass transport and active site accessibility, while Fe–N@CMK-8, which shows unexpected microporosity, enhanced selectivity and active site density but limited mass transport. The dual-templated derived Fe–N@CMK-3/8 catalyst combined 2D and 3D porosity for intermediate performance. Durability tests revealed media- and porosity-dependent stability: Fe–N@CMK-8 excelled in terms of ORR activity under acidic conditions due to the confinement conferred by its microporous framework, which the selectivity of active sites significantly changed. Conversely, Fe–N@CMK-3 maintained high ORR activity in alkaline media along with a slight shift of reaction pathway. In comparison, the dual porous Fe–N@CMK-3/8 exhibits the best stability particularly in terms of selectivity in both acid and alkaline media.These findings establish clear design principles for optimizing the micropore/mesopore ratio in Fe–N@C electrocatalysts. Future studies should refine synthesis methods to further enhance active site utilization, and long-term durability under fuel cell operating conditions, and explore conductive supports to improve commercial viability. This research contributes to the ongoing efforts to develop cost-effective, non-precious metal catalysts for next-generation fuel cells.
Author contributions
Giulia Gianola: investigation, formal analysis, and writing – original draft. Mirtha A. O. Lourenço: conceptualization, formal analysis, writing – original draft, writing – review & editing, supervision, and funding acquisition. Luca Basile: investigation. Tiago Morais: investigation. Luís Mafra: writing – review & editing and funding acquisition. Candido Pirri: supervision and funding acquisition. Stefania Specchia: conceptualization, writing – review & editing and funding acquisition. Juqin Zeng: conceptualization, writing – original draft, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (DOI https://doi.org/10.54499/UIDB/50011/2020), UIDP/50011/2020 (DOI https://doi.org/10.54499/UIDP/50011/2020) & LA/P/0006/2020 (DOI https://doi.org/10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). This work has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement 865974). L. B. acknowledges Politecnico di Torino for supporting his stay@University of Aveiro. T. M. acknowledges FCT for the Doctoral grant (2024.03066.BD). M. A. O. L. acknowledges FCT for the Researcher position CEECIND/01158/2021 (DOI: https://doi.org/10.54499/2021.01158.CEECIND/CP1659/CT0022) and the funding from the European Union's Horizon Europe research and innovation program under the ERA-PF grant agreement no 101090287. J. Z. received a fund under the National Recovery and Resilience Plan (NRRP), Project code: IR0000027, Concession Decree No. 128 of 21/06/2022 adopted by the Italian Ministry of Research, CUP: B33C22000710006, Project title: iENTRANCE.References
- S. Kabir, K. Artyushkova, A. Serov, B. Kiefer and P. Atanassov, Binding Energy Shifts for Nitrogen-Containing Graphene-Based Electrocatalysts - Experiments and DFT Calculations, Surf. Interface Anal., 2016, 48(5), 293–300, DOI:10.1002/sia.5935.
- A. C. Lewis, Optimising Air Quality Co-Benefits in a Hydrogen Economy: A Case for Hydrogen-Specific Standards for NOx Emissions, Environ. Sci.: Atmos., 2021, 1(5), 201–207, 10.1039/d1ea00037c.
- L. Huang, S. Zaman, X. Tian, Z. Wang, W. Fang and B. Y. Xia, Advanced Platinum-Based Oxygen Reduction Electrocatalysts for Fuel Cells, Acc. Chem. Res., 2021, 54(2), 311–322, DOI:10.1021/acs.accounts.0c00488.
- G. Gavello, J. Zeng, C. Francia, U. A. Icardi, A. Graizzaro and S. Specchia, Experimental Studies on Nafion® 112 Single PEM-FCs Exposed to Freezing Conditions, Int. J. Hydrogen Energy, 2011, 36(13), 8070–8081, DOI:10.1016/j.ijhydene.2011.01.182.
- D. J. Berger, Fuel Cells and Precious-Metal Catalysts, Science, 1999, 286, 49 CrossRef CAS.
- P. Trogadas, T. F. Fuller and P. Strasser, Carbon as Catalyst and Support for Electrochemical Energy Conversion, Carbon, 2014, 5–42, DOI:10.1016/j.carbon.2014.04.005.
- R. Othman, A. L. Dicks and Z. Zhu, Non Precious Metal Catalysts for the PEM Fuel Cell Cathode, Int. J. Hydrogen Energy, 2012, 357–372, DOI:10.1016/j.ijhydene.2011.08.095.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, A Review on Non-Precious Metal Electrocatalysts for PEM Fuel Cells, Energy Environ. Sci., 2011, 3167–3192, 10.1039/c0ee00558d.
- G. Gianola, A. Cosenza, C. Roiron, C. F. Pirri, S. Specchia, P. Atanassov and J. Zeng, Effect of Silica Leaching Treatment during Template-Assisted Synthesis on the Performance of FeNC Catalysts for Oxygen Reduction Reaction, Electrochim. Acta, 2025, 525, 146085, DOI:10.1016/j.electacta.2025.146085.
- X. Cui, J. Shi, L. Zhang, M. Ruan and J. Gao, PtCo Supported on Ordered Mesoporous Carbon as an Electrode Catalyst for Methanol Oxidation, Carbon, 2009, 47(1), 186–194, DOI:10.1016/j.carbon.2008.09.054.
- J. Lee, J. Kim and T. Hyeon, Recent Progress in the Synthesis of Porous Carbon Materials, Adv. Mater., 2006, 2073–2094, DOI:10.1002/adma.200501576.
- G. Gianola, N. Garino, M. Bartoli, A. Sacco, C. F. Pirri and J. Zeng, Microwave-Assisted Synthesis of N/S-Doped CNC/SnO2 Nanocomposite as a Promising Catalyst for Oxygen Reduction in Alkaline Media, Mater. Chem. Phys., 2023, 308, 128205, DOI:10.1016/j.matchemphys.2023.128205.
- F. Jaouen, M. Lefèvre, J. P. Dodelet and M. Cai, Heat-Treated Fe/N/C Catalysts for O2 Electroreduction: Are Active Sites Hosted in Micropores?, J. Phys. Chem. B, 2006, 110(11), 5553–5558, DOI:10.1021/jp057135h.
- H. Wang and H. Dai, Strongly Coupled Inorganic–Nano-Carbon Hybrid Materials for Energy Storage, Chem. Soc. Rev., 2013, 42(7), 3088–3113, 10.1039/c2cs35307e.
- R. L. Zornitta, F. J. García-Mateos, J. J. Lado, J. Rodríguez-Mirasol, T. Cordero, P. Hammer and L. A. M. Ruotolo, High-Performance Activated Carbon from Polyaniline for Capacitive Deionization, Carbon, 2017, 123, 318–333, DOI:10.1016/j.carbon.2017.07.071.
- G. Zhang, R. Chenitz, M. Lefèvre, S. Sun and J. P. Dodelet, Is Iron Involved in the Lack of Stability of Fe/N/C Electrocatalysts Used to Reduce Oxygen at the Cathode of PEM Fuel Cells?, Nano Energy, 2016, 29, 111–125, DOI:10.1016/j.nanoen.2016.02.038.
- J. Pampel and T. P. Fellinger, Opening of Bottleneck Pores for the Improvement of Nitrogen Doped Carbon Electrocatalysts, Adv. Energy Mater., 2016, 6(8), 1–8, DOI:10.1002/aenm.201502389.
- H. W. Liang, X. Zhuang, S. Brüller, X. Feng and K. Müllen, Hierarchically Porous Carbons with Optimized Nitrogen Doping as Highly Active Electrocatalysts for Oxygen Reduction, Nat. Commun., 2014, 5, 4973, DOI:10.1038/ncomms5973.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, High-Performance Electrocatalysts for Oxygen Reduction Derived from Polyaniline, Iron, and Cobalt, Science, 2011, 332(6028), 443–447 CrossRef CAS PubMed.
- S. Bae, J. Park, S. Bong, J. S. Park, B. Jeong and J. Lee, Pore Surface Engineering of FeNC for Outstanding Power Density of Alkaline Hydrazine Fuel Cells, Chem. Eng. J., 2024, 479, 147522, DOI:10.1016/j.cej.2023.147522.
- L. Osmieri, A. H. A. Monteverde Videla, M. Armandi and S. Specchia, Influence of Different Transition Metals on the Properties of Me–N–C (Me = Fe, Co, Cu, Zn) Catalysts Synthesized Using SBA-15 as Tubular Nano-Silica Reactor for Oxygen Reduction Reaction, Int. J. Hydrogen Energy, 2016, 41(47), 22570–22588, DOI:10.1016/j.ijhydene.2016.05.223.
- A. Serov, K. Artyushkova and P. Atanassov, Fe–N–C Oxygen Reduction Fuel Cell Catalyst Derived from Carbendazim: Synthesis, Structure, and Reactivity, Adv. Energy Mater., 2014, 4, 1301735, DOI:10.1002/aenm.201301735.
- A. H. A. Monteverde Videla, L. Osmieri, M. Armandi and S. Specchia, Varying the Morphology of Fe–N–C Electrocatalysts by Templating Iron Phthalocyanine Precursor with Different Porous SiO2 to Promote the Oxygen Reduction Reaction, Electrochim. Acta, 2015, 177, 43–50, DOI:10.1016/j.electacta.2015.01.165.
- V. Malgras, Q. Ji, Y. Kamachi, T. Mori, F. K. Shieh, K. C. W. Wu, K. Ariga and Y. Yamauchi, Templated Synthesis for Nanoarchitectured Porous Materials, Bull. Chem. Soc. Jpn., 2015, 1171–1200, DOI:10.1246/bcsj.20150143.
- Y. Xie, D. Kocaefe, C. Chen and Y. Kocaefe, Review of Research on Template Methods in Preparation of Nanomaterials, J. Nanomater., 2016, 2016, 302595, DOI:10.1155/2016/2302595.
- A. Lu and F. Schüth, Nanocasting Pathways to Create Ordered Mesoporous Solids, C. R. Chim., 2005, 8(3–4), 609–620, DOI:10.1016/j.crci.2004.10.020.
- A. Wang, Y. Guo, F. Muhammad and Z. Deng, Controlled Synthesis of Lead-Free Cesium Tin Halide Perovskite Cubic Nanocages with High Stability, Chem. Mater., 2017, 29(15), 6493–6501, DOI:10.1021/acs.chemmater.7b02089.
- D. Gu and F. Schüth, Synthesis of Non-Siliceous Mesoporous Oxides, Chem. Soc. Rev., 2014, 7, 313–344, 10.1039/c3cs60155b.
- W. Stober; A. Fink and D. Ernst BohnControlled Growth of Monodisperse Silica Spheres in the Micron Size Range 1, 1968, vol. 26 Search PubMed.
- U. Mandal, A. Adhikari, S. Dey, S. Ghosh, S. K. Mondal and K. Bhattacharyya, Excitation Wavelength Dependence of Solvation Dynamics in a Supramolecular Assembly: PEO-PPO-PEO Triblock Copolymer and SDS, J. Phys. Chem. B, 2007, 111(21), 5896–5902, DOI:10.1021/jp0689722.
- P. Alexandridis, J. F. Holzwarth and A. T. Hatton, Micellization of Poly(Ethylene Oxide)-Poly(Propylene Oxide)-Poly(Ethylene Oxide) Triblock Copolymers in Aqueous Solutions: Thermodynamics of Copolymer Association, Macromolecules, 1994, 27(9), 2414–2425 CrossRef CAS.
- Y. Wan and D. Zhao, On the Controllable Soft-Templating Approach to Mesoporous Silicates, Chem. Rev., 2007, 2821–2860, DOI:10.1021/cr068020s.
- S. G. Wainwright, C. M. A. Parlett, R. A. Blackley, W. Zhou, A. F. Lee, K. Wilson and D. W. Bruce, True Liquid Crystal Templating of SBA-15 with Reduced Microporosity, Microporous Mesoporous Mater., 2013, 172, 112–117, DOI:10.1016/j.micromeso.2013.01.016.
- F. Kleitz, S. H. Choi and R. Ryoo, Cubic Ia3d Large Mesoporous Silica: Synthesis and Replication to Platinum Nanowires, Carbon Nanorods and Carbon Nanotubes, Chem. Commun., 2003,(17), 2136–2137, 10.1039/b306504a.
- R. Guillet-Nicolas, F. Bérubé, M. Thommes, M. T. Janicke and F. Kleitz, Selectively Tuned Pore Condensation and Hysteresis Behavior in Mesoporous SBA-15 Silica: Correlating Material Synthesis to Advanced Gas Adsorption Analysis, J. Phys. Chem. C, 2017, 121(39), 24505–24526, DOI:10.1021/acs.jpcc.7b06745.
- T. W. Kim, F. Kleitz, B. Paul and R. Ryoo, MCM-48-like Large Mesoporous Silicas with Tailored Pore Structure: Facile Synthesis Domain in a Ternary Triblock Copolymer-Butanol-Water System, J. Am. Chem. Soc., 2005, 127(20), 7601–7610, DOI:10.1021/ja042601m.
- H. Zhang, L. Osmieri, J. H. Park, H. T. Chung, D. A. Cullen, K. C. Neyerlin, D. J. Myers and P. Zelenay, Standardized Protocols for Evaluating Platinum Group Metal-Free Oxygen Reduction Reaction Electrocatalysts in Polymer Electrolyte Fuel Cells, Nat. Catal., 2022, 5(5), 455–462, DOI:10.1038/s41929-022-00778-3.
- E. Maruccia, M. A. O. Lourenço, T. Priamushko, M. Bartoli, S. Bocchini, F. C. Pirri, G. Saracco, F. Kleitz and C. Gerbaldi, Nanocast Nitrogen-Containing Ordered Mesoporous Carbons from Glucosamine for Selective CO2 Capture, Mater. Today Sustainability, 2022, 17, 100089, DOI:10.1016/j.mtsust.2021.100089.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87(9–10), 1051–1069, DOI:10.1515/pac-2014-1117.
- X. Yan, Y. Yao and Y. Chen, Highly Active and Stable Fe–N–C Oxygen Reduction Electrocatalysts Derived from Electrospinning and In Situ Pyrolysis, Nanoscale Res. Lett., 2018, 13, 0–6, DOI:10.1186/s11671-018-2635-x.
- S. Jerez, A. Pedersen, M. Ventura, L. Mazzoli, M. I. Pariente, M. Titirici, J. A. Melero and J. Barrio, Fe[Sbnd]N Doped Carbon Materials from Oily Sludge as Electrocatalysts for Alkaline Oxygen Reduction Reaction, Electrochim. Acta, 2024, 483, 144045, DOI:10.1016/j.electacta.2024.144045.
- C. Chen, Y. Wu, X. Li, Y. Ye, Z. Li, Y. Zhou, J. Chen, M. Yang, F. Xie, Y. Jin, C. Jones, N. Wang, H. Meng and S. Chen, Modulating Fe Spin State in FeNC Catalysts by Adjacent Fe Atomic Clusters to Facilitate Oxygen Reduction Reaction in Proton Exchange Membrane Fuel Cell, Appl. Catal., B, 2024, 342, 123407, DOI:10.1016/j.apcatb.2023.123407.
- H. Singh, S. Zhuang, B. Ingis, B. B. Nunna and E. S. Lee, Carbon-Based Catalysts for Oxygen Reduction Reaction: A Review on Degradation Mechanisms, Carbon, 2019, 1, 160–174, DOI:10.1016/j.carbon.2019.05.075.
- X. Chen, N. Wang, K. Shen, Y. Xie, Y. Tan and Y. Li, MOF-Derived Isolated Fe Atoms Implanted in N-Doped 3D Hierarchical Carbon as an Efficient ORR Electrocatalyst in Both Alkaline and Acidic Media, ACS Appl. Mater. Interfaces, 2019, 11(29), 25976–25985, DOI:10.1021/acsami.9b07436.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Isolated Single Iron Atoms Anchored on N-Doped Porous Carbon as an Efficient Electrocatalyst for the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2017, 56(24), 6937–6941, DOI:10.1002/anie.201702473.
- J. H. Zagal, S. Specchia and P. Atanassov, Mapping Transition Metal-MN4 Macrocyclic Complex Catalysts Performance for the Critical Reactivity Descriptors, Curr. Opin. Electrochem., 2021, 27, 100683, DOI:10.1016/j.coelec.2020.100683.
- M. M. Hossen, K. Artyushkova, P. Atanassov and A. Serov, Synthesis and Characterization of High Performing Fe–N–C Catalyst for Oxygen Reduction Reaction (ORR) in Alkaline Exchange Membrane Fuel Cells, J. Power Sources, 2018, 375, 214–221, DOI:10.1016/j.jpowsour.2017.08.036.
- X. Tao, R. Lu, L. Ni, V. Gridin, S. H. Al-Hilfi, Z. Qiu, Y. Zhao, U. I. Kramm, Y. Zhou and K. Müllen, Facilitating the Acidic Oxygen Reduction of Fe–N–C Catalysts by Fluorine-Doping, Mater. Horiz., 2022, 9(1), 417–424, 10.1039/d1mh01307f.
- T. Zhu, W. Lyu, Y. Wang, X. Mi and Y. Liao, Hierarchical Meso-Micro Porous Fe–N–C Derived from Tripolycyanamide-Based Microporous Polymer as Efficient Electrocatalyst for Oxygen Reduction Reaction, J. Colloid Interface Sci., 2023, 633, 265–274, DOI:10.1016/j.jcis.2022.11.095.
- B. Li, Q. Li and X. Wang, Iron/Iron Carbide Coupled with S, N Co-Doped Porous Carbon as Effective Oxygen Reduction Reaction Catalyst for Microbial Fuel Cells, Environ. Res., 2023, 228, 115808, DOI:10.1016/j.envres.2023.115808.
- Y. Wang, L. Wu, K. Qu, X. Wang, L. Wang and B. Li, Highly Dense Atomic Fe–N4 Derived from Double-Layer MOF as Efficient Electrocatalysts for Enhanced Oxygen Reduction in Zn–Air Battery and Microbial Fuel Cell, Fuel, 2024, 373, 132326, DOI:10.1016/j.fuel.2024.132326.
- Y. Zhan, H. Zeng, F. Xie, H. Zhang, W. Zhang, Y. Jin, Y. Zhang, J. Chen and H. Meng, Templated Growth of Fe/N/C Catalyst on Hierarchically Porous Carbon for Oxygen Reduction Reaction in Proton Exchange Membrane Fuel Cells, J. Power Sources, 2019, 431, 31–39, DOI:10.1016/j.jpowsour.2019.05.051.
- Y. Zhou, Y. Yu, D. Ma, A. C. Foucher, L. Xiong, J. Zhang, E. A. Stach, Q. Yue and Y. Kang, Atomic Fe Dispersed Hierarchical Mesoporous Fe–N–C Nanostructures for an Efficient Oxygen Reduction Reaction, ACS Catal., 2021, 11(1), 74–81, DOI:10.1021/acscatal.0c03496.
- R. Dun, M. Hao, Y. Su and W. Li, Fe–N-Doped Hierarchical Mesoporous Carbon Nanomaterials as Efficient Catalysts for Oxygen Reduction in Both Acidic and Alkaline Media, J. Mater. Chem. A, 2019, 7(20), 12518–12525, 10.1039/c9ta01807g.
- C. Shao, L. Wu, Y. Wang, K. Qu, H. Chu, L. Sun, J. Ye, B. Li and X. Wang, An Open Superstructure of Hydrangea-like Carbon with Highly Accessible Fe–N4 Active Sites for Enhanced Oxygen Reduction Reaction, Chem. Eng. J., 2022, 429, 132307, DOI:10.1016/j.cej.2021.132307.
Footnotes |
| † Electronic supplementary information (ESI) available: The −196 °C N2 adsorption–desorption isotherms and STEM images of the mesoporous silicas; the Raman spectra, EDS analysis, TGA analysis, and elemental analysis of the Fe–N@C electrocatalysts; and CVs, LSV at different rotation speeds, and KL plots of the Fe–N@C electrocatalysts. See DOI: https://doi.org/10.1039/d5nh00300h |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |