
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular engineering of gold nanocluster properties via peptide ligand charge state and topological modulation†
Pakawat
Toomjeen
 ,
Unnop
Srikulwong
,
Adulvit
Chuaephon
,
Witthawat
Phanchai
,
Cherdpong
Choodet
and
Theerapong
Puangmali
*
,
Unnop
Srikulwong
,
Adulvit
Chuaephon
,
Witthawat
Phanchai
,
Cherdpong
Choodet
and
Theerapong
Puangmali
*
Department of Physics, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: theerapong@kku.ac.th
First published on 26th May 2025
Abstract
The functionalization of gold nanoclusters (AuNCs) with peptides offers a promising strategy for tuning their electronic and optical properties, making them suitable for applications in bioimaging, sensing, and photodynamic therapy. However, the influence of peptide structure, charge state, and length on ligand-to-metal charge transfer (LMCT) and electronic transitions is not yet fully comprehended. In this study, we employ density functional theory (DFT) calculations to systematically investigate the role of linear and cyclic peptides in modulating the optical and electronic properties of AuNCs. In addition, interfragment charge transfer (IFCT) analysis is performed to quantify the charge redistribution between the peptide ligands and the AuNC core. Our findings reveal that zwitterionic peptides exhibit the most significant LMCT, leading to red-shifted absorption peaks and enhanced charge delocalization, while canonical and cyclic peptides display more localized electronic states with reduced charge transfer. Moreover, longer peptide chains, particularly in zwitterionic forms, facilitate increased electronic coupling with the AuNC core, amplifying their optical response. Despite variations in the peptide structure, the AuNC core remains structurally stable, ensuring consistent ligand–core electronic interactions. The IFCT results further confirm that peptide length and structural forms strongly influence charge transfer dynamics, with tetrapeptides exhibiting greater charge redistribution compared to tripeptides. These insights provide a fundamental foundation for the rational design of peptide-functionalized AuNCs with tailored optical and electronic properties. The ability to fine-tune the peptide structure to optimize charge transfer makes these nanoclusters highly promising for biomedical applications, including fluorescence imaging, targeted drug delivery, and molecular sensing. This study advances our understanding of the interactions between peptides and AuNCs and provides the basis for future experimental validation and application-driven modifications.
1 Introduction
Gold nanoclusters (AuNCs) have emerged as a unique class of nanomaterials due to their exceptional quantum size effects, which impart distinct optical and electronic properties, making them highly promising for bioimaging, sensing, and therapeutic applications.1 Unlike larger gold nanoparticles,2–6 AuNCs, typically smaller than 2 nm, lack surface plasmon resonance but exhibit tunable fluorescence, high biocompatibility, and excellent stability in physiological environments, making them particularly advantageous for biomedical applications.7 In addition, their physicochemical properties, such as efficient renal clearance and extended blood circulation, further enhance their suitability for drug delivery and therapeutic interventions. The fluorescence of AuNCs can be fine-tuned through surface ligand modifications, a key factor in optimizing their performance in biomedical applications.The physicochemical properties and reactivity of AuNCs are highly sensitive to their ligand environment. Various ligand-exchange strategies have been explored to alter the structural and optical properties of AuNCs.8,9 Among thiolate-protected AuNCs, such as Aun(SR)m, ligand-dependent structural transformations have been observed, directly influencing their electronic properties.10 The nature of thiolate ligands plays a crucial role in determining the stability and optical characteristics of AuNCs.11–13 A well-known example is Au10(SP)8, which consists of a tetrahedral Au4 core stabilized by two trimeric Au3 motifs, forming a core-shell-like structure.14,15 This structural organization dictates that the optical and electronic transitions of AuNCs arise from interactions between the AuNC core and the ligand shell. Peptide-functionalized AuNCs, particularly those stabilized with cysteine (Cys), have attracted interest due to their biocompatibility, tunability, and ability to enhance fluorescence intensity when functionalized with amino acids such as methionine (M), tyrosine (Y) and leucine (L).16,17
The functionalization of AuNCs with peptides provides an opportunity to modulate their electronic and optical properties through charge transfer interactions between peptide ligands and the AuNC core.18 Peptide-based ligands introduce electron-rich functional groups (e.g., thiol, carboxyl, and amine groups), which influence the ligand-to-metal charge transfer (LMCT) and alter the fluorescence behavior of AuNCs. Despite advances in peptide-functionalized AuNCs, challenges remain, including low quantum yield and susceptibility to enzymatic degradation, which limit their practical application in bioimaging and sensing.19–23 While linear peptides offer greater flexibility, they often suffer from low stability and bioavailability. In contrast, cyclic peptides exhibit greater resistance to enzymatic degradation, enhanced affinity for molecular targets, and greater structural rigidity, making them attractive for nanoparticle functionalization.24–26 Despite these advantages, a direct comparative investigation of linear vs. cyclic peptides in AuNC functionalization remains limited, necessitating further exploration of their impact on AuNC properties.
In this study, we employ time-dependent density functional theory (TD-DFT) calculations to investigate how peptide charge states, sequence variations, and structural forms (linear vs. cyclic) influence the electronic and optical properties of peptide-functionalized AuNCs. A small thiolate-coordinated Au10(SR)8 cluster (Fig. 1) is used as a representative system to reduce computational complexity. The study systematically examines (i) the effect of different amino acid sequences (CMM, CYY, CLL, CFF, CWW, CAA, and CKK), (ii) the influence of peptide charge states (zwitterionic vs. canonical) and (iii) comparisons between linear and cyclic peptides with identical sequences. The optical absorption spectra are calculated using TD-DFT, while interfragment charge transfer (IFCT) analysis is employed to evaluate the charge transfer dynamics between peptide ligands and the AuNC core. The results provide new insights into the structure–function relationships of peptide-functionalized AuNCs, guiding the rational design of NCs with customized optical and electronic properties.
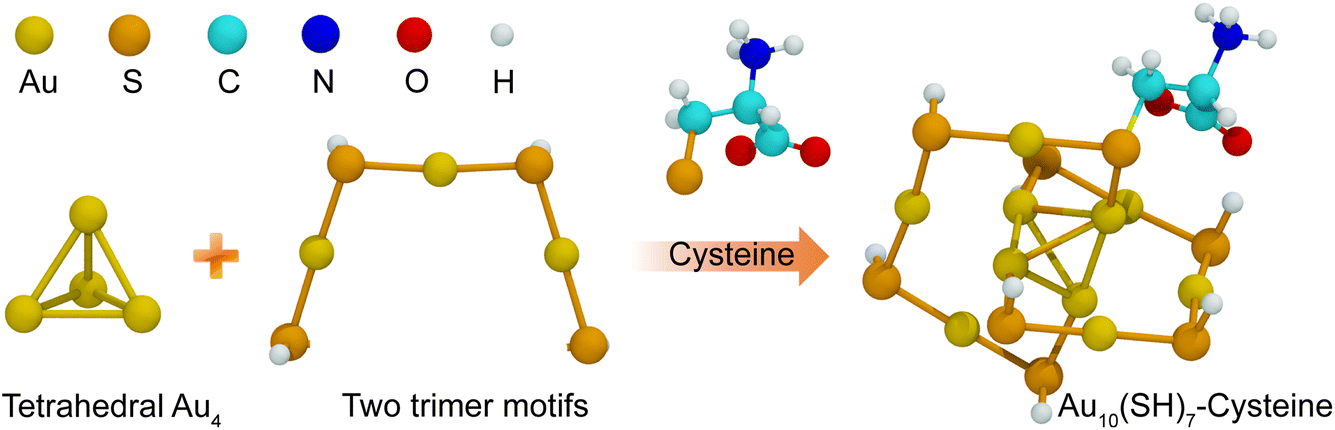 | ||
| Fig. 1 Schematic representation of the structural composition of the Au10(SH)8 nanocluster. The tetrahedral Au4 core is stabilized by two trimeric Au3 motifs, forming a compact and stable gold framework. A cysteine ligand introduces a thiol (–SH) functional group, which coordinates to the gold surface, leading to the formation of a cysteine-functionalized AuNC. Atomic species are color-coded for clarity: gold (Au, yellow), sulfur (S, orange), carbon (C, cyan), nitrogen (N, blue), oxygen (O, red), and hydrogen (H, white). | ||
2 Computational methodology
2.1 Structure and optimization
The AuNC model investigated in this study consists of ten gold atoms and adopts a structural morphology consistent with the highly stable Au10(SR)8 cluster, as reported by Liu et al.15 This model comprises a tetrahedral Au4 core, stabilized by two trimeric Au3 staple motifs –[SR–Au–SR–Au–SR–Au–SR]–. To construct peptide-modified AuNCs, cysteine residues were used as anchoring sites via their thiol (–SH) functional groups, enabling covalent attachment to the AuNC surface. A total of seven Au10(SR)8 (R = peptide) systems were modeled with the following peptide sequences: CAA, CLL, CFF, CYY, CKK, CMM, and CWW, with CYY serving as a reference system. Their structures are as illustrated in Fig. 2. Additionally, cyclic peptide analogs c(CAA), c(CLL), c(CFF), c(CYY), c(CKK), c(CMM), and c(CWW) were included to assess the impact of peptide topology on the electronic and optical properties. These specific peptide sequences were chosen due to their availability in experimental studies, allowing a direct comparison between computational predictions and measured optical properties.17 Furthermore, the study of a single ligand modification on the AuNC surface serves as a representative model to understand the broader impact of peptide functionalization on the electronic and optical properties of AuNC clusters. Previous studies27 have demonstrated that ligand-induced modifications primarily influence the surface electronic structure and charge distribution of AuNCs, which in turn dictate their optical absorption characteristics. Therefore, the findings of this work can be extrapolated to more complex systems with multiple ligand modifications. To further examine the influence of the length of the peptide, the extended peptide sequences CAAA, CLLL, CFFF, CYYY, CKKK, CMMM, and CWWW, along with their cyclic counterparts c(CAAA), c(CLLL), c(CFFF), c(CYYY), c(CKKK), c(CMMM), and c(CWWW), were incorporated into the study. | ||
| Fig. 2 Chemical structures and amino acid sequences of the zwitterionic forms of seven different linear and cyclic peptides: CAA, CLL, CFF, CYY, CKK, CMM, and CWW for linear peptides, and c(CAA), c(CLL), c(CFF), c(CYY), c(CKK), c(CMM), and c(CWW) for their cyclic counterparts. | ||
The Au10(SR)8 model represents the smallest thiolate-protected AuNC allowing systematic modification with diverse ligands while maintaining computational efficiency. Structural optimizations were performed using DFT with the Coulomb attenuated hybrid Becke three-parameter Lee–Yang–Parr (CAM-B3LYP) functional,28 which is suited for describing long-range interactions critical for accurately modeling charge transfer and electronic transitions in ligand-protected metal clusters. CAM-B3LYP was chosen for its ability to accurately describe charge transfer excitations and maintain consistency in ground- and excited-state calculations. Its suitability has been demonstrated in peptide–gold systems.29 Given the small size and rigidity of Au10(SR)8 nanoclusters, especially when using scalar relativistic ECPs such as LANL2DZ, the influence of the functional on core geometry is minimal. Although CAM-B3LYP lacks explicit dispersion correction, its use is justified for compact AuNC systems where the core structure is stabilized by strong Au–S and Au–Au interactions. Previous studies have shown that dispersion effects minimally impact geometry or charge transfer in similar gold–ligand nanoclusters with short covalently anchored ligands.30,31 Thus, the trends observed in this work are expected to remain reliable without DFT-D3 correction. A mixed basis set approach was adopted to balance computational cost and accuracy, applying the 6-31G(d) basis set32 for C, H, O, S and N atoms to adequately describe valence electron interactions while ensuring reliable geometry optimization. For Au atoms, the set of LANL2DZ effective core potential basis set33 was employed to account for relativistic effects, which are essential to accurately capture the electronic structure of heavy elements. Geometry optimizations were performed without symmetry constraints to ensure unbiased determination of equilibrium structures, allowing a more realistic representation of ligand-induced distortions.
2.2 Time-dependent density functional theory calculations
The excited-state properties of peptide-functionalized AuNCs were investigated using TD-DFT within the vertical excitation approximation, as implemented by Gaussian16.34 To maintain consistency with the ground state calculations, the functional CAM-B3LYP28 was used, which provides reliable predictions of charge transfer excitations. The 6-31G(d) basis set was used for non-metal atoms, while the LANL2DZ basis set was applied to Au atoms. Spin–orbit coupling (SOC) was not included due to the use of scalar-relativistic ECPs; however, prior studies have shown that SOC induces only minor spectral shifts and does not significantly affect the overall optical trends.35 To accurately capture electronic transitions associated with peptide moieties in the 200–300 nm region,36 a total of 100 excited states (nstates = 100) were included in TD-DFT calculations. To isolate the intrinsic effects of the peptide structure and charge state, all calculations were performed in vacuum. Although solvent environments can shift absolute excitation energies, previous studies have shown that key trends in LMCT and charge transfer remain consistent, supporting the validity of vacuum phase modeling for comparative analysis37–39 The predicted UV-vis absorption spectra were generated by convoluting transition energies and oscillator strengths with a Gaussian broadening factor of 0.15 eV to simulate experimental spectral line shapes. The molar absorption coefficient (ε) was plotted as a function of the excitation wavelength (λ) in nanometers, following a standard approach using Gaussian distributions.40 To further analyze the electronic structure, molecular orbitals were visualized using Multiwfn (version 3.8)41 and Visual Molecular Dynamics (VMD) (version 1.9.3).422.3 Interfragment charge transfer analysis
To quantify electronic charge redistribution between the Au core and the peptide ligands, IFCT analysis was performed using Multiwfn (version 3.8).41,43 This method allows the electronic system to be decomposed into distinct fragments, providing information on LMCT and metal-to-ligand charge transfer (MLCT) contributions. For IFCT calculations, the nanocluster was divided into two fragments: (i) the tetrahedral Au4 core, serving as the primary electron acceptor, and (ii) the thiolate-protected peptide ligands, acting as electron donors. The transition densities obtained from the TD-DFT calculations were used as input to determine fragment-based Mulliken and Löwdin population charges for each electronic excitation. The extent of charge transfer was quantified by integrating the transition density matrix between the acceptor (Au core) and donor (peptide-functionalized ligand shell) fragments.To gain deeper insights into charge delocalization effects, we generated difference density plots of key excited states using Multiwfn and visualized them in VMD (version 1.9.3).42 This analysis allowed us to assess the impact of the charge state, sequence, and topology of the peptide on electronic interactions within the AuNC system. The results provide a detailed understanding of how peptide modifications influence charge transfer mechanisms, shedding light on the structure–property relationships of peptide-functionalized AuNCs.
3 Results and discussion
3.1 Structural validation of gold nanocluster
Validating the structural stability of small thiolate protected AuNCs, such as Au10(SR)8, is crucial to ensuring the reliability of computational models and their agreement with experimental data. DFT has been widely applied to predict the structures of various AuNCs, often yielding results that align closely with experimental findings from X-ray diffraction and UV-vis spectroscopy.44–46 However, despite extensive theoretical investigations, Au10(SH)8 remains less explored due to the limited availability of experimental data. To address this, we performed DFT calculations to validate the structure of Au10(SH)8, using hydrogen (–H) as a model ligand replacement. The optimized geometry consists of a tetrahedral Au4 core stabilized by two staple motifs (2 × [SR–Au–SR–Au–SR–Au–SR]), consistent with previous structural analyzes of thiolate-protected AuNCs.14 To further evaluate the computational model, we analyzed the electronic structure and simulated UV-vis spectra, as illustrated in Fig. 3. | ||
| Fig. 3 (a) UV-vis absorption spectrum of the Au10(SH)8 nanocluster, with oscillator strengths (f) indicated by green vertical lines. (b) Kohn–Sham energy diagram of Au10(SH)8, highlighting two dominant transitions (vertical arrows). The density of states (DOS) is color-coded to represent contributions from different atomic orbitals: total DOS (grey), Au(6s + 6p) (red), Au(5d) (orange), S(3p) (green), and H (cyan). | ||
The computed UV-vis spectrum of Au10(SH)8 (Fig. 3(a)) exhibits two dominant absorption peaks at 359.3 and 516.0 nm, with oscillator strengths of 0.19 and 0.12, respectively. Since direct experimental data for Au10(SH)8 is not available, we compared our results with experimental spectra of structurally similar Au10 nanoclusters protected by different ligands, such as Au10(TBBT)10 (ref. 47) and [Au10(MesCH2Bimy)6Br3]Br.48 Experimental data indicate that Au10(TBBT)10 exhibits an absorption peak at 355 nm, while [Au10(MesCH2Bimy)6Br3]Br shows peaks at 366 nm and 474 nm, with a shoulder at 505 nm. This study isolates the influence of peptide structure, length, and charge state using a vacuum-phase framework, enabling consistent comparison of LMCT and optical transitions. While minor spectral deviations may arise from omitting solvent effects,37 the preserved relative trends indicate that the peptide-induced electronic modulation is accurately captured. The discrepancy between our simulated and experimental UV-vis spectra highlights the strong influence of ligands on the optical transitions of AuNCs. While Au10(SH)8 shows a peak at 359 nm, experimental clusters exhibit redshifted features due to ligand-induced orbital hybridization, charge redistribution, and enhanced LMCT. Ligands such as TBBT and NHCs stabilize low-energy transitions, leading to spectral shifts consistent with previous studies. These results demonstrate that ligand–core interactions play a critical role in modulating the optical properties of gold nanoclusters. Despite slight shifts as a result of ligand differences, our computed absorption peaks are in close agreement with these experimental trends. This consistency suggests that the computational model accurately captures the core structure and optical behavior of Au10 nanoclusters, reinforcing its validity for studying ligand-modified systems.48
To further investigate the electronic structure of Au10(SH)8, we analyzed Kohn–Sham (KS) energy level diagrams and density of states (DOS) (Fig. 3(b)). The two primary absorption peaks correspond to distinct electronic transitions. The peak at 359 nm originates from transitions involving HOMO−3 and HOMO−1 to LUMO, with a transition energy of 5.8 eV, while HOMO−2 has a relatively low contribution, as shown in the electronic transitions in Table S1 in ESI.† This transition mainly involves electron transfer from the Au(5d) orbitals (orange region in the DOS plot) to Au(6s + 6p) orbitals (red region). The second absorption peak at 516 nm corresponds to the transition from HOMO to LUMO, defining the HOMO–LUMO gap (Egap) of 4.7 eV. The KS orbitals indicate that HOMO, HOMO−1, and HOMO−3 are predominantly localized around the gold core and staple motifs, while LUMO is more delocalized across the entire Au core. These findings validate the computational model for predicting the optical properties of thiolate-protected AuNCs, providing a fundamental reference for ligand-induced spectral tuning. Given the strong influence of ligand modifications on charge distribution and absorption properties, we expanded our investigation to include peptide-functionalized Au10 nanoclusters to examine how variations in peptide sequence and topology influence their electronic and optical properties.
3.2 Optical properties of peptides-functionalized AuNC
The optical properties of AuNCs are highly sensitive to surface functionalization, particularly when modified by peptides. Understanding the impact of linear and cyclic peptides on these properties is essential to tailor AuNCs for bioimaging applications. Peptide-functionalized AuNCs exhibit distinct optical behaviors due to differences in peptide structure, sequence, and bonding interactions with the Au core. In this section, we investigate the effects of linear and cyclic tripeptide ligands on the optical properties of AuNCs, focusing on structural stability and implications for optical absorption.Ligand-induced structural distortions play a crucial role in modulating the optical properties of AuNC.27 To assess the impact of peptide functionalization, we examine the key bond lengths between the gold core (Aucore), gold surface atoms (Aushell), and the sulfur (S) ligands. Fig. 4(a) provides a schematic representation of the Au10(SH)8 nanocluster, highlighting these critical bonds. The computed structure closely aligns with the experimental data, yielding a cluster diameter of approximately 1.25 nm, consistent with previous reports.17 Thiol binding sites, labeled configuration 1 (Conf. 1) and configuration 2 (Conf. 2), as depicted in Fig. S1,† represent possible anchor positions for peptide ligands. Unless otherwise specified, all subsequent results correspond to configuration 1. Fig. 4(b) presents a quantitative comparison of key bond lengths, including Aucore–Aucore, Aucore–S, and Aushell–S interactions for Au10(SH)8 functionalized with various linear and cyclic tripeptides. These values, summarized in Table 1, provide insight into the relationship between structural modifications and electronic behavior.
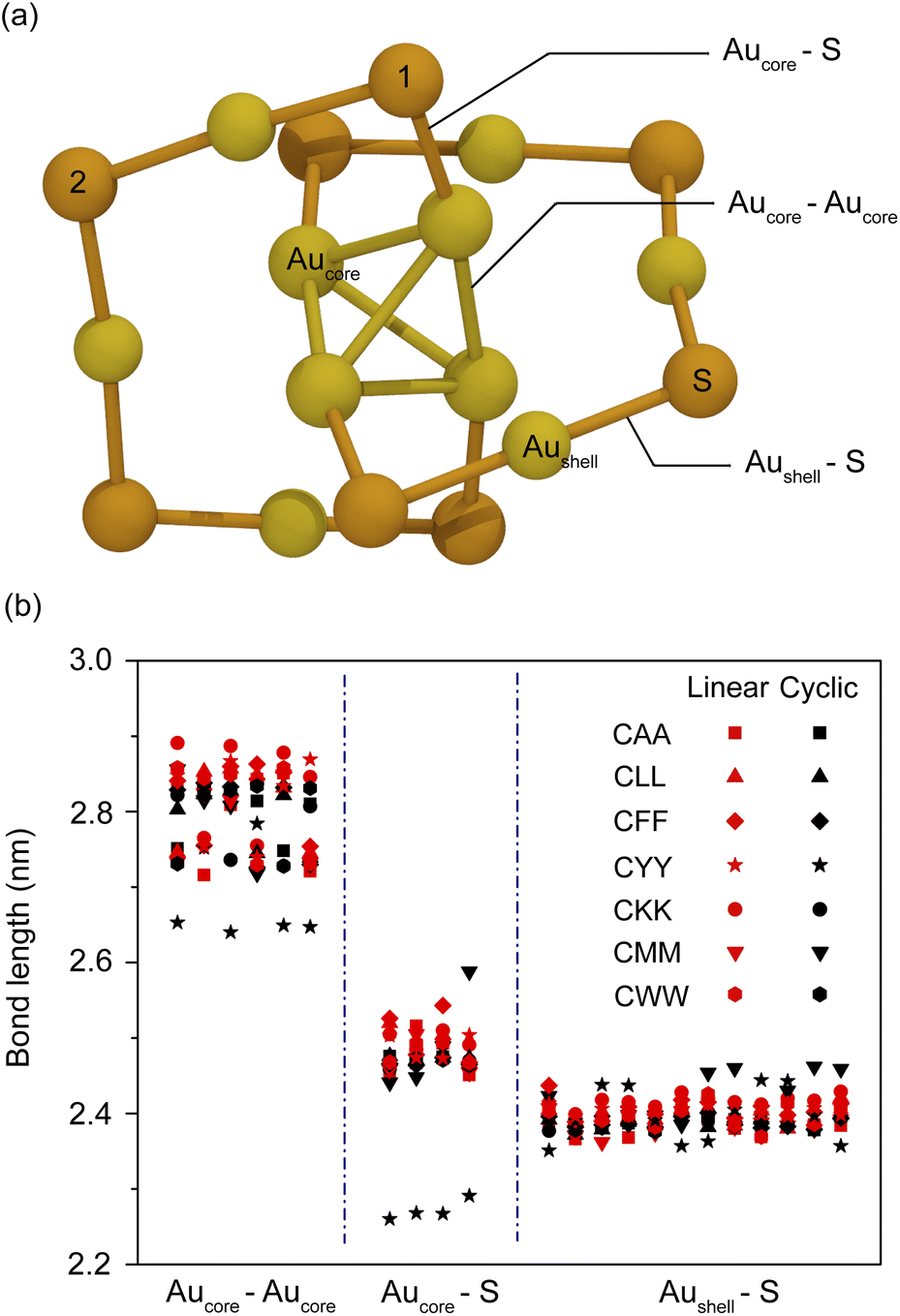 | ||
| Fig. 4 (a) Schematic representation of the Au10(SH)8 nanocluster, highlighting key bond interactions classified into three types: Aucore–Aucore, Aucore–S, and Aushell–S. Hydrogen atoms are omitted for clarity. Sulfur atoms labeled 1 and 2 represent anchoring sites for thiol bonds, corresponding to configurations 1 and 2. (b) Bond length comparison of linear (red symbols) and cyclic (black symbols) peptide-functionalized Au10(SH)7 nanoclusters for 14 different peptides: CAA, CLL, CFF, CYY, CKK, CMM, CWW, and their cyclic counterparts c(CAA), c(CLL), c(CFF), c(CYY), c(CKK), c(CMM), and c(CWW). | ||
| Peptides | Average bond length (nm) | ||
|---|---|---|---|
| Aucore–Aucore | Aucore–S | Aushell–S | |
| CAA | 2.80 | 2.48 | 2.38 |
| CLL | 2.81 | 2.49 | 2.40 |
| CFF | 2.82 | 2.50 | 2.41 |
| CYY | 2.82 | 2.49 | 2.40 |
| CKK | 2.84 | 2.50 | 2.42 |
| CMM | 2.80 | 2.48 | 2.39 |
| CWW | 2.81 | 2.48 | 2.40 |
| c(CAA) | 2.79 | 2.47 | 2.39 |
| c(CLL) | 2.79 | 2.47 | 2.39 |
| c(CFF) | 2.80 | 2.47 | 2.39 |
| c(CYY) | 2.69 | 2.27 | 2.40 |
| c(CKK) | 2.79 | 2.47 | 2.39 |
| c(CMM) | 2.83 | 2.49 | 2.42 |
| c(CWW) | 2.80 | 2.47 | 2.39 |
Among functionalized systems, Au10(SH)7c(CYY) exhibits significantly shorter Aucore–Aucore and Aucore–S bond lengths compared to its linear counterpart. This suggests enhanced structural compactness and stronger metallic bonding within the core, which is expected to influence electronic transitions and absorption properties. In contrast, configuration 2 (Fig. S2†) shows a less pronounced difference between linear and cyclic peptides, indicating that peptide-induced modifications are configuration dependent. The optimized structures shown in Fig. S3† further confirm that Au10(SH)7c(CYY) maintains a tetrahedral Td symmetry for Aucore, reinforcing the tightly packed nature of the cyclic peptides. These results indicate that due to their rigid and compact structures, cyclic peptides may significantly influence the electronic and optical characteristics of ligand-stabilized nanoclusters.
The UV-vis absorption spectra of peptide-functionalized AuNCs are presented in Fig. 5, with additional simulated spectra for other systems provided in Fig. S4.† This comparison highlights the influence of the conformation of the peptide (linear vs. cyclic) and protonation states (zwitterionic vs. canonical), which are known to vary depending on environmental conditions.49 These spectral variations provide key insights into how structural modifications impact electronic transitions and optical behavior. As illustrated in Fig. 5(a), the absorption spectra reveal distinct differences between AuNCs functionalized with linear and cyclic CYY peptides. Linear peptides, both in the zwitterionic and canonical forms, exhibit sharper absorption peaks at 350 and 550 nm. In contrast, cyclic peptides induce a broader and slightly red-shifted absorption band around 400 nm, with a shoulder peak appearing near 650 nm. This redshift correlates with the shorter bond lengths observed in cyclic peptides, which enhance electronic coupling between the gold core and the ligands, lowering the energy of the electronic transitions and shifting absorption to longer wavelengths. The increased delocalization of electronic states in cyclic peptides, resulting from their constrained structure, probably contributes to the observed broadening of spectral features. The spectral shift observed in Fig. 5(a) for tyrosine-functionalized AuNCs may stem from the aromatic group's π-conjugation, which can enhance charge delocalization and potentially affect LMCT efficiency. Prior studies have shown that aromatic ligands promote electronic interactions, often resulting in redshifted absorption.50–52 To assess this, we analyzed the correlation between Aromatic Fluctuation Index (FLU), IFCT, and absorption peaks for aromatic (Phe, Tyr, Trp) and non-aromatic peptides (Fig. S5†). Results show weak correlations overall. For aromatics, FLU–absorption and FLU–IFCT correlations were −0.12 and −0.09, respectively. For non-aromatics, FLU–absorption (0.22) and FLU–IFCT (−0.27) indicate slight trends. These results suggest that aromaticity alone does not strongly govern LMCT or optical behavior, and other structural and electronic factors likely dominate.
 | ||
| Fig. 5 Simulated UV-vis absorption spectra of Au10(SH)7-peptide nanoclusters functionalized with different peptides. (a) Comparison of linear CYY, cyclic CYY, and linear canonical CYY peptide-functionalized Au10(SH)7 nanoclusters. (b) Spectra of Au10(SH)7 functionalized with linear tripeptides: CAA, CLL, CFF, CYY, CKK, CMM, and CWW. (c) Spectra of Au10(SH)7 functionalized with cyclic tripeptides: c(CAA), c(CLL), c(CFF), c(CYY), c(CKK), c(CMM), and c(CWW). (d) Comparison of Au10(SH)7 functionalized with linear canonical tripeptides: canonical CAA, CLL, CFF, CKK, CMM, and CWW. | ||
Fig. 5(b) further explores sequence-dependent effects on absorption spectra, comparing AuNC functionalized with different tripeptide ligands (CAA, CLL, CFF, CYY, CKK, CMM and CWW). Although all peptide sequences exhibit a primary absorption band near 350 nm, subtle shifts are observed, particularly in the second peak near 550 nm, which undergoes a noticeable red-shift compared to the Au10(SH)8 reference system. This shift is most evident in cyclic peptides (except c(CYY)) and linear canonical peptides, as illustrated in Fig. 5(c) and (d), respectively. These findings suggest that while the sequence and structure of the peptides (linear vs. cyclic) contribute to minor variations in the optical response, they do not drastically alter the fundamental electronic transitions of Au10(SH)8. Instead, the primary spectral modifications arise from differences in peptide conformation, electronic coupling, and ligand-induced structural rearrangements.
3.3 Influence of peptide charge states
In biological environments, the charge state of peptides is primarily governed by the surrounding pH as protonation and deprotonation processes occur in both terminal and side-chain functional groups. These variations lead to different peptide forms, including zwitterionic, canonical, and neutral states, each of which can significantly impact the structural and electronic properties of peptide-functionalized AuNCs. Given the increasing use of AuNCs in biological applications, including imaging, sensing, and therapeutic delivery, a detailed understanding of how peptide charge states influence the electronic structure and optical properties is essential for optimizing their performance. In particular, key electronic interactions between the peptide ligands and the AuNC core are expected to vary depending on the charge state of the peptide. To investigate these effects, we examine the frontier molecular orbitals (FMOs), the HOMO–LUMO gaps, and the charge transfer dynamics of peptide-functionalized AuNCs across different charge states.Fig. 6 demonstrates the effect of various charge states, including zwitterionic, canonical, or neutral, of a CYY peptide on the electronic structure of AuNC, influenced by these different charge forms of the peptide. Other peptide systems are depicted in Fig. S6–S12.† FMO analysis reveals that zwitterionic peptides exhibit a narrower HOMO–LUMO gap, facilitating enhanced charge delocalization and LMCT due to their intrinsic dipole moments.53 In contrast, canonical and neutral peptides display wider HOMO–LUMO gaps, indicating a more localized electronic structure with reduced charge transfer. This effect is further supported by UV-vis spectroscopy, which shows red-shifted absorption peaks for zwitterionic peptides, suggesting stronger metal–ligand interactions, whereas canonical and neutral peptides maintain higher energy transitions due to weaker electronic coupling.54 Additional studies using X-ray photoelectron spectroscopy (XPS) and cyclic voltammetry (CV) further support these observations. XPS measurements indicate shifts in binding energy, consistent with enhanced electron density redistribution in zwitterionic peptides, while CV analysis reveals increased electrochemical activity, reinforcing the observed charge transfer trends.55 The observed trends align with charge transfer and electrostatic stabilization mechanisms, where zwitterionic peptides generate strong dipole moments that enhance electron density distribution across the ligand and AuNC core, leading to lower excitation energies and red-shifted optical transitions.56 These findings are consistent with ligand-induced electronic structure modification theory, which predicts that ligands with intrinsic charge separation exhibit stronger electronic hybridization with metal surfaces, thereby modulating the optical and electronic properties of AuNCs.57
 | ||
| Fig. 6 FMOs and HOMO–LUMO energy gaps of Au10(SH)7CYY in its zwitterionic, canonical, and cyclic (c(CYY)) peptide forms. The FMOs of the HOMO and LUMO are shown for configurations 1 and 2, along with the corresponding HOMO–LUMO gaps. Blue and red isosurfaces represent the positive and negative regions of the orbitals, respectively (isosurface density threshold: 0.03 a.u.). The calculated energy gaps are indicated for each configuration. Atom color coding: sulfur (orange), gold (yellow), carbon (cyan), nitrogen (blue), oxygen (red), and hydrogen (white). | ||
Table 2 presents a more detailed comparison of the electronic transition characteristics of peptide-functionalized AuNCs, focusing on the impact of zwitterionic, canonical, and neutral charge states on HOMO–LUMO gaps, oscillator strengths, and dominant electronic transitions. Additional details on electronic transition characteristics for other peptides are provided in Tables S2–S8.† The data confirm that zwitterionic peptides exhibit lower HOMO–LUMO gaps, leading to stronger LMCT and greater charge delocalization. This is particularly evident in the transition assignments, where zwitterionic peptides display highly mixed electronic states with greater transition densities, facilitating efficient excited-state charge redistribution. In contrast, canonical and neutral peptides, with their more localized electronic structures, exhibit higher-energy transitions and lower oscillator strengths, suggesting weaker electronic coupling between the ligand and Au core. Notably, transition analysis also reveals differences in the nature of electronic excitations, discussed in the following section. These findings suggest that zwitterionic ligands contribute to a broader absorption spectrum with enhanced optical activity, whereas canonical and neutral ligands result in sharper, blue-shifted transitions due to their more constrained electronic environments.
| Peptide | λ (nm) | f | Transitions (CI%) |
|---|---|---|---|
| Zwitterionic | 352.9 | 0.21 | H−12 → L (57.3%) |
| CYY | H−11 → L (17.0%) | ||
| (Conf. 1) | H−10 → L (11.5%) | ||
| 547.4 | 0.10 | H−6 → L (94.9%) | |
| Zwitterionic | 354.2 | 0.25 | H−12 → L (36.6%) |
| CYY | H−10 → L (35.1%) | ||
| (Conf. 2) | 548.3 | 0.12 | H−6 → L (94.9%) |
| Canonical | 344.5 | 0.01 | H−7 → L (53.1%) |
| CYY | H−2 → L (20.0%) | ||
| (Conf. 1) | 545.9 | 0.10 | H → L (96.7%) |
| Canonical | 350.5 | 0.14 | H−7 → L (41.2%) |
| CYY | |||
| (Conf. 2) | 536.6 | 0.15 | H → L (96.5%) |
| c(CYY) | 399.6 | 0.08 | H−1 → L+2 (43.6%) |
| (Conf. 1) | H−6 → L (14.5%) | ||
| H−8 → L (12.0%) | |||
| 687.0 | 0.04 | H → L+6 (74.8%) | |
| H → L+7 (16.4%) | |||
| c(CYY) | 354.3 | 0.22 | H−6 → L+2 (45.9%) |
| (Conf. 2) | H−4 → L (21.9%) | ||
| 553.6 | 0.12 | H → L (96.7%) |
Fig. 7 provides deeper insight into the electronic transitions of peptide-functionalized AuNCs, represented by Kohn–Sham (KS) orbital energy diagrams and FMOs. The transitions between key orbitals, denoted by vertical arrows, illustrate the nature of electronic excitations across different peptide charge states. In the case of zwitterionic CYY peptides (Fig. 7(a)), the dominant transitions occur from HOMO−12, HOMO−11, and HOMO−10 to LUMO, corresponding to d → sp interband transitions, which contribute to the absorption peak at 353 nm. Furthermore, a significant HOMO−6 → LUMO transition (sp → sp intraband transition) gives rise to the absorption peak at 547 nm, aligning with the data in Table 2. These transitions indicate stronger electronic coupling between the peptide and the AuNC core in the zwitterionic state, leading to greater charge delocalization and lower excitation energy requirements.
 | ||
| Fig. 7 Electronic structure and FMOs of Au10(SH)7-CYY (configuration 1) nanoclusters with different peptide conformations: (a) linear (zwitterionic form) CYY, (b) canonical CYY, and (c) cyclic c(CYY). The energy levels of KS orbitals are shown with corresponding energy values (eV), and vertical arrows indicate electronic transitions responsible for the observed absorption peaks. The relative contributions of different atomic orbitals are represented by color-coded lines: red (Au 6s + 6p), blue (Au 5d), green (S 3p), and black (other contributions). Molecular orbitals (MOs) are illustrated with atom-specific color coding: sulfur (purple), gold (yellow), carbon (cyan), nitrogen (blue), oxygen (red), and hydrogen (white). Blue and red isosurfaces represent the positive and negative regions of the orbitals, with an isosurface density threshold of 0.03 a.u. | ||
A comparison with canonical CYY peptides (Fig. 7(b)) reveals subtle shifts in absorption characteristics. Here, electronic transitions at 345 and 545 nm arise primarily from HOMO−7 and HOMO−2 to LUMO, as well as HOMO to LUMO transitions, suggesting that, while charge transfer remains significant, it is less pronounced than in the zwitterionic state. The effect of peptide cyclization is even more pronounced in cyclic CYY peptides, where transitions shift toward longer wavelengths (Fig. 7(c)). Primary excitations involve HOMO−6 and HOMO−8 (d band) to LUMO, as well as HOMO−1 to LUMO+2 (sp → sp intraband transition), with additional transitions from HOMO to LUMO+6 and LUMO+7, resulting in absorption peaks at 400 nm and 647 nm, respectively. The observed red-shift in absorption for cyclic peptides suggests that structural rigidity stabilizes molecular orbitals, lowering excitation energies, and extending the absorption range. This highlights the critical role of the peptide conformation in modulating the electronic and optical properties of AuNCs.
The orbital diagrams of KS in Fig. 7 further illustrate the relative contributions of the atomic orbitals to the FMOs, highlighting that the gold atoms (orbitals 6s + 6p and 5d dominate the FMOs in all peptide charge states, consistent with Fig. S13–S39).† However, the peptide moiety significantly contributes to both HOMO and LUMO, reinforcing the critical role of peptide functionalization in modifying the electronic structure of the nanocluster. This orbital hybridization between the peptide and the AuNC core introduces new electronic states, which influence the optical absorption and charge-transfer mechanisms. In summary, zwitterionic peptides enhance charge transfer and electronic delocalization, whereas cyclic peptides provide structural stability and maintain consistent molecular orbital configurations. Canonical peptides exhibit intermediate behavior that balances flexibility and stability.
3.4 Peptide length-dependent optical properties
The length and conformation of the peptide ligands play a crucial role in modulating the electronic and optical properties of AuNC functionalized with peptides by influencing the charge distribution, electronic transitions, and ligand–core interactions.58 Structural variations in the peptide ligands can introduce ligand-induced distortions, modify the electronic environment of the gold core, and alter fundamental optical properties such as HOMO–LUMO energy gaps and absorption spectra. These modifications are particularly relevant in peptide-functionalized AuNCs, where changes in peptide length impact the metal–ligand hybridization strength, ultimately determining excited-state behavior and charge-transfer dynamics. Fig. 8 provides a detailed analysis of these effects, illustrating how the structure of the peptide affects the length of the bonds, the HOMO–LUMO gaps, and the shifts of the absorption peak. The data highlight how the flexibility of the ligand, charge localization, and electronic excitation energy influence the peptide-driven tuning of AuNC properties. | ||
| Fig. 8 (a) Correlation between the HOMO–LUMO gap and absorption peak position for Au10(SH)7-peptide nanoclusters functionalized with various peptides, including linear, canonical, and cyclic configurations. The HOMO–LUMO gap (black circles, left y-axis) and absorption peak positions (blue squares, right y-axis) are plotted for each peptide, with different peptide sequences and topologies represented by distinct symbols along the x-axis. (band c) Comparison of average bond lengths in AuNCs for configuration 1 and configuration 2, respectively, showing Aucore–Aucore, Aucore–S, and Aushell–S bond distances for different linear (zwitterionic), canonical, and cyclic peptides. | ||
Fig. 8(a) presents the variations in the HOMO–LUMO gap and the positions of absorption peaks of AuNCs functionalized with different peptides, focusing on the 500–650 nm spectral region, which is particularly sensitive to subtle structural changes in the cluster geometry.59–62 Among the different peptide structures, cyclic peptides c(CYY) and c(CYYY) exhibit the most pronounced red shifts in their absorption peaks, with maxima at 687 nm and 573 nm, respectively. This substantial shift suggests that enhanced electronic delocalization and increased orbital overlap within the ligand play a significant role in modifying the optical properties. Despite this red shift, the HOMO–LUMO gap remains relatively unchanged, indicating that the absorption peak shift is primarily driven by stronger electronic interactions and charge delocalization rather than a direct reduction in the energy difference between the HOMO and LUMO. In contrast, linear peptide configurations exhibit lower HOMO–LUMO gaps, with values around 2.6 eV, while the lowest observed gap (∼1.8 eV) corresponds to longer peptide chains. The influence of peptide length on charge transfer was further examined through linear regression analysis (Tables S9–S10†). A strong inverse correlation was observed between peptide length and HOMO–LUMO gap, indicating that longer peptides promote charge delocalization and enhance electronic coupling with the Au core. Notably, peptides containing alanine, leucine, methionine, and tyrosine showed the highest correlations (R2 ≈ 0.98–0.99), while tryptophan shows a weaker correlation (R2 = 0.64), implying additional structural influences. Furthermore, solvent-accessible surface area (SASA) showed no significant correlation with the HOMO–LUMO gap (R2 = 0.08, p = 0.15), highlighting that electronic coupling is primarily governed by direct ligand–metal interactions. This trend suggests that elongated peptide chains facilitate charge redistribution, thereby promoting greater electronic coupling between the ligand and the gold core. These findings align with previous studies demonstrating that ligand-induced distortions can significantly alter electronic density and optical properties by modulating the metal–ligand hybridization strength.63 Furthermore, the correlation analysis, as shown in the heatmap in Fig. S40,† reveals that configuration 1 exhibits stronger charge transfer-absorption coupling, while configuration 2 depends more on electronic structure variations, emphasizing the impact of peptide conformation on charge delocalization and optical behavior in AuNCs.
Fig. 8(b) and (c) further investigate the structural impact of peptide ligands on the Au10(SH)7-peptide nanocluster by analyzing key bond lengths, including Aucore–Aucore, Aucore–S, and Aushell–S interactions. The results indicate that the average bond lengths remain relatively stable in the configurations of linear, canonical, and cyclic peptides, suggesting that variations in the conformation of the peptide do not induce significant structural distortions in the Au10(SR)8 core. This high structural stability implies that, despite differences in the peptide structure, the overall geometric integrity of the nanocluster is preserved. The minor bond length variations observed reinforce the rigidity of the Au–S bonding framework, which helps maintain a stable core–shell configuration regardless of the ligand flexibility. The consistent stability of the core aligns with the absorption spectra trends in Fig. 8(a), where no drastic changes in optical properties are observed in different peptide conformations.20 This suggests that the interactions between the core and the ligand play a more dominant role in the definition of the optical response than minor geometric variations in the structure of the peptide. Although electronic interactions between peptides and the nanocluster affect charge delocalization and electronic transitions, the fundamental electronic structure of the Au core remains unaltered.64
3.5 Mechanistic insights into charge transfer
The charge transfer dynamics between the AuNC core and peptide ligands play a crucial role in determining their electronic and optical properties. Increased LMCT generally correlates with a decrease in the HOMO–LUMO gap, a trend commonly observed in metal–ligand charge transfer systems.65 However, a key finding from our study reveals that a reduced HOMO–LUMO gap does not always lead to red-shifted optical absorption, indicating that additional structural and orbital factors play a crucial role in governing spectral shifts. As shown in Fig. 9, IFCT analysis provides a systematic approach to segment the nanocluster into distinct fragments, including the Au core, shell motifs and individual amino acid residues. This method allows for a detailed evaluation of the charge movement between these regions during electronic transitions, offering insight into both the directionality and magnitude of electron transfer. In zwitterionic forms of tripeptides and tetrapeptides, charge transfer occurs predominantly between the Au core and the ligands, with variations in magnitude depending on the peptide structure. Notably, tetrapeptides exhibit slightly enhanced charge transfer compared to tripeptides, reflecting their increased ligand contributions to electronic transitions and greater electronic delocalization.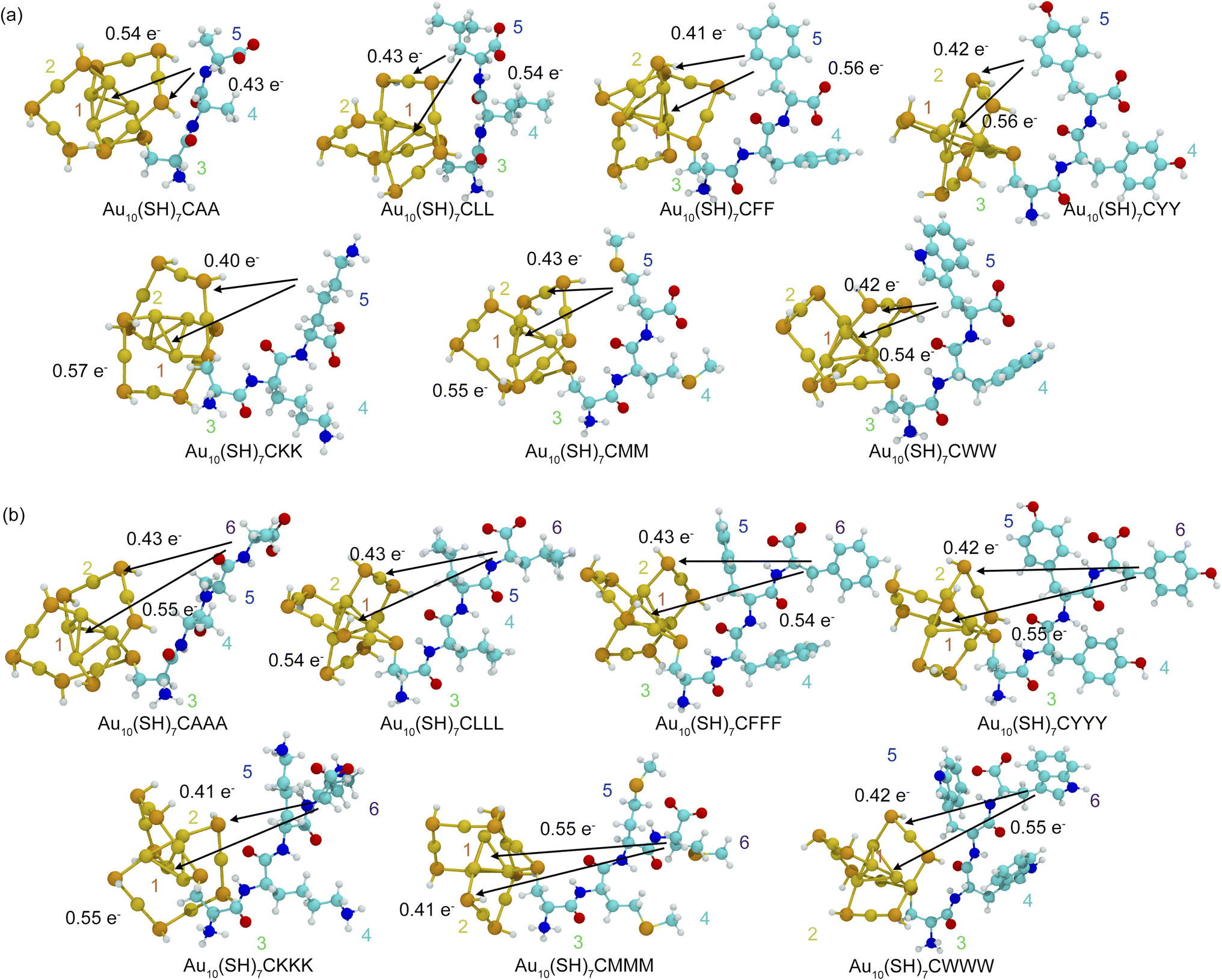 | ||
| Fig. 9 Charge transfer analysis during electron excitation in the zwitterionic forms of (a) Au10(SH)7-tripeptide and (b) Au10(SH)7-tetrapeptide, investigated using interfragment charge transfer. Arrows indicate the direction of charge transfer between molecular fragments. Fragment colors are coded as follows: (1) Aucore (red), (2) Aushell motifs (orange), (3) cysteine (green), and (4)–(6) other amino acid residues (cyan, blue, and purple, respectively). | ||
A more detailed examination of charge localization reveals that peptide length and structure strongly influence charge transfer patterns in peptide-functionalized AuNCs. In tripeptides, charge transfer is highly localized, predominantly originating from the terminal residue to the Au core. This is evident in zwitterionic CYY, where a significant electron transfer of 0.55689e− occurs from Fragment 5 to Fragment 1. In contrast, tetrapeptides display a more distributed charge transfer, which involves contributions from additional residues. For example, in zwitterionic CYYY, a charge transfer of 0.5475e− from Fragment 6 to the Au core highlights the enhanced electronic delocalization facilitated by the extended peptide chain. The directional nature of charge transfer, which occurs predominantly toward the Au core and shell motifs, further underscores the role of gold atoms as efficient electron acceptors, stabilizing LMCT. These findings emphasize the critical influence of the peptide structure and length in modulating charge redistribution, providing a fundamental framework for designing peptide-functionalized AuNCs with tunable electronic and optical properties.
Table 3 presents the net transferred electrons in AuNCs functionalized with peptides, highlighting variations in LMCT between different peptide conformations. Among CYY systems, the zwitterionic form exhibits the highest LMCT, with a significant charge transfer of 0.55689e− from fragment 5 to fragment 1, indicating a strong electronic coupling between the peptide ligand and the Au core. This suggests that the terminal residue plays a dominant role in mediating charge redistribution, probably due to its high electron density and polar functional groups, such as thiols, which enhance charge donation to the Au core. In contrast, canonical and cyclic CYY configurations exhibit considerably lower LMCT values, with charge transfer as low as −0.00028e−, indicating weaker electronic interactions between the ligands and the AuNC core. This trend aligns with the greater electronic rigidity of canonical and cyclic peptides, where localized electronic states limit charge delocalization. The observed differences in charge-transfer behavior suggest that zwitterionic peptides exhibit superior electronic communication with the Au core, making them more effective in modulating optical and electronic properties.
| LMCT | CYY (zwitterionic) | CYY (canonical) | c(CYY) | |||
|---|---|---|---|---|---|---|
| Conf. 1 | Conf. 2 | Conf. 1 | Conf. 2 | Conf. 1 | Conf. 2 | |
| Fragment2 → Fragment1 | 0.00000 | 0.00003 | 0.07783 | 0.12513 | 0.00001 | 0.11805 |
| Fragment3 → Fragment1 | 0.00012 | 0.00010 | 0.05205 | −0.00463 | −0.00024 | −0.00077 |
| Fragment4 → Fragment1 | 0.00908 | 0.00867 | 0.00000 | −0.00049 | −0.00051 | 0.00022 |
| Fragment5 → Fragment1 | 0.55689 | 0.54267 | −0.00028 | −0.00007 | 0.01599 | 0.00017 |
| Fragment3 → Fragment2 | 0.00009 | 0.00008 | 0.03262 | −0.01039 | −0.00069 | −0.00791 |
| Fragment4 → Fragment2 | 0.00680 | 0.00657 | −0.00007 | −0.00071 | −0.00061 | 0.00009 |
| Fragment5 → Fragment2 | 0.41747 | 0.41136 | −0.00031 | −0.00008 | 0.01607 | 0.00011 |
| Fragment4 → Fragment3 | 0.00015 | 0.00046 | −0.00005 | −0.00001 | −0.01469 | 0.00001 |
| Fragment5 → Fragment3 | 0.00891 | 0.02881 | −0.00009 | 0.00000 | 0.51663 | 0.00001 |
| Fragment5 → Fragment4 | 0.00037 | 0.00105 | 0.00000 | 0.00000 | 0.11260 | 0.00000 |
Based on the trends observed in Table 3, Table 4 extends the charge transfer analysis to tetrapeptides, revealing a strong correlation between peptide length and LMCT efficiency. The data demonstrate that longer peptide chains facilitate greater charge transfer, particularly in zwitterionic forms, due to increased orbital overlap and electronic delocalization. For example, CYYY exhibits a high LMCT value of 0.5475e− (Fragment 6 → Fragment 1), indicating that the additional amino acid residue enhances the transfer of charge to the Au core. The presence of multiple electron-donating functional groups in tetrapeptides contributes to their enhanced ability to stabilize electronic excitations, reinforcing the role of peptide structure in modulating charge dynamics. Furthermore, the structural stability of the AuNC core, as indicated by the bond length data from previous sections, ensures consistent electronic interactions regardless of peptide length or conformation. This stability supports the observed LMCT trends, as minor structural distortions do not significantly impact charge delocalization mechanisms. These findings further confirm that zwitterionic tetrapeptides facilitate enhanced charge transfer compared to shorter peptides.
| Peptide | LMCT | S0 → S1 | |
|---|---|---|---|
| Conf. 1 | Conf. 2 | ||
| CAA | Fragment5 → Fragment1 | 0.53757 | 0.53553 |
| Fragment5 → Fragment2 | 0.42999 | 0.38751 | |
| CLL | Fragment5 → Fragment1 | 0.54346 | 0.56909 |
| Fragment5 → Fragment2 | 0.42777 | 0.38197 | |
| CFF | Fragment5 → Fragment1 | 0.55952 | 0.54425 |
| Fragment5 → Fragment2 | 0.41256 | 0.40372 | |
| CYY | Fragment5 → Fragment1 | 0.55689 | 0.54267 |
| Fragment5 → Fragment2 | 0.41747 | 0.41136 | |
| CKK | Fragment5 → Fragment1 | 0.56618 | 0.53734 |
| Fragment5 → Fragment2 | 0.40041 | 0.39957 | |
| CMM | Fragment5 → Fragment1 | 0.54695 | 0.54941 |
| Fragment5 → Fragment2 | 0.42618 | 0.39847 | |
| CWW | Fragment5 → Fragment1 | 0.54464 | 0.01116 |
| Fragment5 → Fragment2 | 0.42311 | 0.00699 | |
| CAAA | Fragment6 → Fragment1 | 0.54869 | 0.53491 |
| Fragment6 → Fragment2 | 0.43308 | 0.42303 | |
| CLLL | Fragment6 → Fragment1 | 0.54062 | 0.54397 |
| Fragment6 → Fragment2 | 0.42721 | 0.40784 | |
| CFFF | Fragment6 → Fragment1 | 0.54125 | 0.55116 |
| Fragment6 → Fragment2 | 0.43210 | 0.41603 | |
| CYYY | Fragment6 → Fragment1 | 0.54750 | 0.54208 |
| Fragment6 → Fragment2 | 0.42227 | 0.4065 | |
| CKKK | Fragment6 → Fragment1 | 0.55428 | 0.53021 |
| Fragment6 → Fragment2 | 0.40537 | 0.40942 | |
| CMMM | Fragment6 → Fragment1 | 0.54598 | 0.53686 |
| Fragment6 → Fragment2 | 0.41413 | 0.40378 | |
| CWWW | Fragment6 → Fragment1 | 0.55116 | 0.45885 |
| Fragment6 → Fragment2 | 0.41603 | 0.36242 | |
Conclusions
This study provides a comprehensive analysis of how the structure, charge state, and length of the peptide influence the electronic and optical properties of AuNCs. Our findings underscore the crucial role of LMCT in modulating the optical response of peptide-functionalized AuNCs. Specifically, zwitterionic peptides exhibit enhanced LMCT, resulting in red-shifted absorption peaks and greater electronic delocalization, whereas canonical and cyclic peptides display more localized electronic structures with reduced charge transfer efficiency. The analysis of frontier molecular orbitals (FMOs) and HOMO–LUMO gaps further confirms that peptide charge states significantly impact charge transfer dynamics, ultimately affecting the optoelectronic behavior of AuNCs. Furthermore, this study reveals that the length and conformation of the peptide play a critical role in charge-transfer efficiency. Longer peptides, particularly in their zwitterionic forms, exhibit greater LMCT due to increased orbital overlap and electronic delocalization, thereby enhancing their optical response and electronic coupling with the Au core. These characteristics make them promising candidates for applications in bioimaging, photodynamic therapy, and molecular sensing. Despite these charge-transfer variations, the structural stability of the AuNC core remains largely unaffected across different peptide configurations, ensuring consistent electronic interactions between the ligands and the nanocluster core. The insights from this study contribute to the rational design of peptide-functionalized AuNCs with tunable electronic and optical properties. By systematically exploring how peptide charge states, lengths, and conformations modulate charge transfer and electronic interactions, these findings provide a fundamental framework for engineering peptide-stabilized nanoclusters. These insights guide the rational design of peptide-functionalized AuNCs and underscore the need for future experimental studies to validate ligand effects for precise optical and electronic tuning.Data availability
Because of the extensive amount of data associated with the calculation results in this paper and the restrictions imposed by our institution's data-sharing policies, it is not feasible to upload all the data to a public network. However, the relevant calculation data supporting the findings of this study are available within the article and its ESI.† Raw data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
P. T.: conceptualization, investigation, formal analysis, methodology, data curation, visualization, writing – original draft, writing – review & editing; U. S.: formal analysis, writing – original draft, writing – review & editing; A. C.: formal analysis, writing – original draft, writing – review & editing; W. P.: formal analysis, writing – original draft, writing – review & editing; C. C.: formal analysis, writing – original draft, writing – review & editing; T. P.: conceptualization, investigation, formal analysis, funding acquisition, methodology, project administration, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research of Khon Kaen University. PT would also like to express our gratitude to the Science Achievement Scholarship of Thailand for financial support.Notes and references
- W. Phanchai, J. Thonghlueng and T. Puangmali, ACS Appl. Nano Mater., 2024, 7, 5554–5563 CrossRef CAS.
- S. Koowattanasuchat, S. Ngernpimai, P. Matulakul, J. Thonghlueng, W. Phanchai, A. Chompoosor, U. Panitanarak, Y. Wanna, T. Intharah, K. Chootawiriyasakul, P. Anata, P. Chaimnee, R. Thanan, C. Sakonsinsiri and T. Puangmali, RSC Adv., 2023, 13, 1301–1311 RSC.
- W. Phanchai, U. Srikulwong, A. Chuaephon, S. Koowattanasuchat, J. Assawakhajornsak, R. Thanan, C. Sakonsinsiri and T. Puangmali, ACS Appl. Nano Mater., 2022, 5, 9042–9052 CrossRef CAS.
- P. Matulakul, D. Vongpramate, S. Kulchat, A. Chompoosor, R. Thanan, P. Sithithaworn, C. Sakonsinsiri and T. Puangmali, ACS Omega, 2020, 5, 17423–17430 CrossRef CAS PubMed.
- C. Choodet, P. Toomjeen, W. Phanchai, P. Matulakul, R. Thanan, C. Sakonsinsiri and T. Puangmali, RSC Adv., 2019, 9, 17592–17600 RSC.
- W. Phanchai, U. Srikulwong, A. Chompoosor, C. Sakonsinsiri and T. Puangmali, Langmuir, 2018, 34, 6161–6169 CrossRef CAS PubMed.
- L. V. Nair, R. V. Nair and R. S. Jayasree, Dalton Trans., 2016, 45, 11286–11291 RSC.
- B. Zhang, J. Chen, Y. Cao, O. J. H. Chai and J. Xie, Small, 2021, 17, 2004381 CrossRef CAS PubMed.
- T. M. Carducci, R. E. Blackwell and R. W. Murray, J. Phys. Chem. Lett., 2015, 6, 1299–1302 CrossRef CAS PubMed.
- S. Hossain, D. Hirayama, A. Ikeda, M. Ishimi, S. Funaki, A. Samanta, T. Kawawaki and Y. Negishi, Aggregate, 2023, 4, e255 CrossRef CAS.
- A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2015, 119, 20179–20187 CrossRef CAS.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- D. M. Chevrier, L. Raich, C. Rovira, A. Das, Z. Luo, Q. Yao, A. Chatt, J. Xie, R. Jin and J. Akola, et al. , J. Am. Chem. Soc., 2018, 140, 15430–15436 CrossRef CAS PubMed.
- D.-E. Jiang, W. Chen, R. L. Whetten and Z. Chen, J. Phys. Chem. C, 2009, 113, 16983–16987 CrossRef CAS.
- C. Liu, Y. Pei, H. Sun and J. Ma, J. Am. Chem. Soc., 2015, 137, 15809–15816 CrossRef CAS PubMed.
- W. Yang, W. Guo, J. Chang and B. Zhang, J. Mater. Chem. B, 2017, 5, 401–417 RSC.
- S. Xu, W. Li, X. Zhao, T. Wu, Y. Cui, X. Fan, W. Wang and X. Luo, Anal. Chem., 2019, 91, 13947–13952 CrossRef CAS PubMed.
- S. M. van de Looij, E. R. Hebels, M. Viola, M. Hembury, S. Oliveira and T. Vermonden, Bioconjug. Chem., 2021, 33, 4–23 CrossRef PubMed.
- H. Santhakumar, R. V. Nair, D. M. Govindachar, G. Periyasamy and R. S. Jayasree, ACS Sustain. Chem. Eng., 2023, 11, 2102–2114 CrossRef CAS.
- Y. Lin, P. Charchar, A. J. Christofferson, M. R. Thomas, N. Todorova, M. M. Mazo, Q. Chen, J. Doutch, R. Richardson and I. Yarovsky, et al. , J. Am. Chem. Soc., 2018, 140, 18217–18226 CrossRef CAS PubMed.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- G.-B. Qi, Y.-J. Gao, L. Wang and H. Wang, Adv. Mater., 2018, 30, 1703444 CrossRef PubMed.
- Y. Huan, Q. Kong, H. Mou and H. Yi, Front. Microbiol., 2020, 11, 2559 Search PubMed.
- K. Han, W.-Y. Zhang, J. Zhang, Q. Lei, S.-B. Wang, J.-W. Liu, X.-Z. Zhang and H.-Y. Han, Adv. Funct. Mater., 2016, 26, 4351–4361 CrossRef CAS.
- S. Chen, Q. Lei, S.-Y. Li, S.-Y. Qin, H.-Z. Jia, Y.-J. Cheng and X.-Z. Zhang, Biomaterials, 2016, 92, 25–35 CrossRef CAS PubMed.
- J. Damjanovic, J. Miao, H. Huang and Y.-S. Lin, Chem. Rev., 2021, 121, 2292–2324 CrossRef CAS PubMed.
- P. Toomjeen, U. Srikulwong, A. Chuaephon, W. Phanchai, C. Choodet and T. Puangmali, ACS Appl. Nano Mater., 2024, 7, 28358–28370 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- D. M. Govindachar and G. Periyasamy, Chem. Phys. Lett., 2020, 753, 137612 CrossRef CAS.
- S. Havenridge and C. M. Aikens, J. Phys. Chem. A, 2023, 127, 9932–9943 CrossRef CAS PubMed.
- K. Z. Milowska and J. K. Stolarczyk, Phys. Chem. Chem. Phys., 2016, 18, 12716–12724 RSC.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- S. Chiodo, N. Russo and E. Sicilia, Chem. Phys., 2006, 125, 104107 CAS.
- M. e. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson, H. Nakatsuji, et al., Gaussian 16, 2016 Search PubMed.
- A. Shayeghi, L. Pašteka, D. Götz, P. Schwerdtfeger and R. Schäfer, Phys. Chem. Chem. Phys., 2018, 20, 9108–9114 RSC.
- K. Hu, W. Xiong, C. Sun, C. Wang, J. Li, F. Yin, Y. Jiang, M.-R. Zhang, Z. Li and X. Wang, et al. , CCS Chem., 2020, 2, 42–51 CrossRef CAS.
- C. M. Aikens, J. Phys. Chem. A, 2009, 113, 10811–10817 CrossRef CAS PubMed.
- K. A. Barakat, T. R. Cundari, H. Rabaâ and M. A. Omary, J. Phys. Chem. B, 2006, 110, 14645–14651 CrossRef CAS PubMed.
- P. Yuan, R. Zhang, E. Selenius, P. Ruan, Y. Yao, Y. Zhou, S. Malola, H. Häkkinen, B. K. Teo and Y. Cao, et al. , Nat. Commun., 2020, 11, 2229 CrossRef CAS PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView Version 6, Semichem Inc., Shawnee Mission KS, 2019 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Lu, J. Chem. Phys., 2024, 161, 082503 CrossRef CAS PubMed.
- J. Zhang, Y. Zhou, K. Zheng, H. Abroshan, D. R. Kauffman, J. Sun and G. Li, Nano Res., 2018, 11, 5787–5798 CrossRef CAS.
- P. Liu, W. Han, M. Zheng, W. Li, J. Ren, A. Tlahuice-Flores and W. W. Xu, J. Phys. Chem. A, 2021, 125, 5933–5938 CrossRef CAS PubMed.
- J. Wei, J.-F. Halet, S. Kahlal, J.-Y. Saillard and A. Muñoz-Castro, Inorg. Chem., 2020, 59, 15240–15249 CrossRef CAS PubMed.
- L. D. Plath, H. Abroshan, C. Zeng, H. J. Kim, R. Jin and M. E. Bier, J. Am. Soc. Mass Spectrom., 2022, 33, 521–529 CrossRef CAS PubMed.
- P. A. Lummis, K. M. Osten, T. I. Levchenko, M. Sabooni Asre Hazer, S. Malola, B. Owens-Baird, A. J. Veinot, E. L. Albright, G. Schatte and S. Takano, et al. , JACS Au, 2022, 2, 875–885 CrossRef CAS PubMed.
- M. Petukh, S. Stefl and E. Alexov, Curr. Pharm. Des., 2013, 19, 4182–4190 CrossRef CAS PubMed.
- J. L. Balan, J. A. Morato-Márquez, J. G. Torres-Torres, J. L. Cabellos and F. Ortiz-Chi, R. Soc. Open Sci., 2023, 10, 230908 CrossRef CAS PubMed.
- Y. Wang, Z. Liu, A. Mazumder, C. G. Gianopoulos, K. Kirschbaum, L. A. Peteanu and R. Jin, J. Am. Chem. Soc., 2023, 145, 26328–26338 CrossRef CAS PubMed.
- M. Sugiuchi, Y. Shichibu, T. Nakanishi, Y. Hasegawa and K. Konishi, Chem. Commun., 2015, 51, 13519–13522 RSC.
- A. Shimizu, Y. Ishizaki, S. Horiuchi, T. Hirose, K. Matsuda, H. Sato and J.-i. Yoshida, J. Org. Chem., 2020, 86, 770–781 CrossRef PubMed.
- A. M. Rijs, G. Ohanessian, J. Oomens, G. Meijer, G. von Helden and I. Compagnon, Angew. Chem., Int. Ed., 2010, 49, 2332–2335 CrossRef CAS PubMed.
- J. L. Cabellos, D. J. Mowbray, E. Goiri, A. El-Sayed, L. Floreano, D. De Oteyza, C. Rogero, J. E. Ortega and A. Rubio, J. Phys. Chem. C, 2012, 116, 17991–18001 CrossRef CAS.
- S. Huo, Y. Jiang, Z. Jiang, R. F. Landis, X.-J. Liang and V. M. Rotello, Nanoscale, 2018, 10, 7382–7386 RSC.
- T. Higaki, Q. Li, M. Zhou, S. Zhao, Y. Li, S. Li and R. Jin, Acc. Chem. Res., 2018, 51, 2764–2773 CrossRef CAS PubMed.
- S. Kundu, B. Ghosh, S. Nandi, M. Ghosh, A. Pyne, J. Chatterjee and N. Sarkar, ACS Appl. Bio Mater., 2020, 3, 4282–4293 CrossRef CAS PubMed.
- Y. Li, M. Zhou and R. Jin, Adv. Mater., 2021, 33, 2006591 CrossRef CAS PubMed.
- Z.-M. Zhu, Y. Zhao, H. Zhao, C. Liu, Y. Zhang, W. Fei, H. Bi and M.-B. Li, Nano Lett., 2023, 23, 7508–7515 CrossRef CAS PubMed.
- Q. Yao, Y. Cao, T. Chen and J. Xie, Atomically Precise Nanochemistry, 2023, pp. 57–85 Search PubMed.
- S. M. Farkhani, P. Dehghankelishadi, A. Refaat, D. V. Gopal, A. Cifuentes-Rius and N. H. Voelcker, Prog. Mater. Sci., 2024, 142, 101229 CrossRef.
- S. Zhu, X. Wang, Y. Cong and L. Li, ACS Omega, 2020, 5, 22702–22707 CrossRef CAS PubMed.
- K. Konishi, M. Iwasaki and Y. Shichibu, Acc. Chem. Res., 2018, 51, 3125–3133 CrossRef CAS PubMed.
- M. Pillegowda and G. Periyasamy, RSC Adv., 2016, 6, 86051–86060 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5na00324e |
| This journal is © The Royal Society of Chemistry 2025 |